

22q11.2 deletions, a genetic risk for schizophrenia, could be susceptible to Parkinson’s disease through elevated expression of α-synuclein.
Abstract
Individuals with chromosome 22q11.2 deletions are at increased risk of developing psychiatric conditions, most notably, schizophrenia (SZ). Recently, clinical studies have also implicated these recurrent 22q11.2 deletions with the risk of early-onset Parkinson’s disease (PD). Thus far, the multiple mouse models generated for 22q11.2 deletions have been studied primarily in the context of congenital cardiac, neurodevelopmental, and psychotic disorders. One of these is the Df1/+ model, in which SZ-associated and developmental abnormalities have been reported. We present the first evidence that the mouse model for the 22q11.2 deletion exhibits motor coordination deficits and molecular signatures (that is, elevated α-synuclein expression) relevant to PD. Reducing the α-synuclein gene dosage in Df1/+ mice ameliorated the motor deficits. Thus, this model of the 22q11.2 deletion shows signatures of both SZ and PD at the molecular and behavioral levels. In addition, both SZ-associated and PD-relevant deficits in the model were ameliorated by treatment with a rapamycin analog, CCI-779. We now posit the utility of 22q11.2 deletion mouse models in investigating the mechanisms of SZ- and PD-associated manifestations that could shed light on possible common pathways of these neuropsychiatric disorders.
INTRODUCTION
Microdeletions within the chromosome 22q11.2 region represent the most frequent pathogenic interstitial deletions found in humans, occurring in approximately 1 in 4000 live births (1). Previous studies have identified 22q11.2 deletions as a strong genetic risk factor for psychiatric conditions, most notably, schizophrenia (SZ) (2, 3). Thus, 22q11.2 deletions have been shown to confer susceptibility to SZ and related neurodevelopmental disorders in humans, and mouse models of 22q11.2 deletions have proven valuable in providing insights into the pathophysiological mechanisms underlying these conditions along developmental trajectories (4).
Recent studies have now reported an increasing number of individuals with 22q11.2 deletions who developed early-onset Parkinson’s disease (PD) (5–8), including those confirmed by postmortem brain studies (7). A genetic association study estimated that 22q11.2 deletions increase the risk of early-onset PD by 20-fold (9). To our knowledge, however, no study has yet addressed the pathological signatures possibly relevant to PD in a mouse model of 22q11.2 deletions.
We therefore investigated a mouse model of 22q11.2 deletion, Df1/+ (10), known to display anatomical and behavioral abnormalities, including defective interneuron migration (11), deficits in sensorimotor gating (12), and psychostimulant-induced hyperlocomotion (13). Using this model, we have reproduced key phenotypes relevant to psychotic symptoms and, for the first time, also demonstrated the presence of significant motor coordination deficits and elevated expression of α-synuclein and p62 proteins, major constituents of Lewy bodies that are well-accepted signatures relevant to PD (14, 15). Reducing the α-synuclein gene dosage in Df1/+ mice ameliorated the motor deficits. Furthermore, we report that both SZ-associated and PD-relevant deficits in this model were ameliorated by treatment with a rapamycin analog, CCI-779.
RESULTS
Df1/+ mice display both motor and psychiatric deficits
Df1/+ mice have been studied from SZ-associated dimensions in the context of developmental trajectories (11–13). On the basis of recent clinical findings that the 22q11.2 deletion is also associated with PD (5–9) and that motor deficits exist in adults with 22q11.2 deletions (16, 17), we examined whether Df1/+ mice might also display motor deficits. To assess motor functions of Df1/+ mice, we used the rotarod test, which has been used to evaluate motor coordination and motor learning in PD mouse models (18). Latency to fall from an accelerating rotarod was measured over nine trials at 3.5 months of age. Df1/+ mice stayed at a significantly shorter time on the rotating bar than wild-type (WT) control mice (F1,18 = 14.09, P = 0.0015) (Fig. 1A), demonstrating the existence of motor coordination deficits. Motor learning did not appear to be impaired, as the Df1/+ mice showed an increase in latency to fall over successive trials (Fig. 1A). The observed motor deficits in Df1/+ mice were not apparently due to impairment in basic locomotive function because voluntary locomotor activities in Df1/+ mice were equivalent to those in WT in the open-field box (0 to 60 min; Fig. 1B).
Fig. 1. Motor coordination deficits, psychostimulant hypersensitivity, and sensorimotor gating deficits in Df1/+ mice.
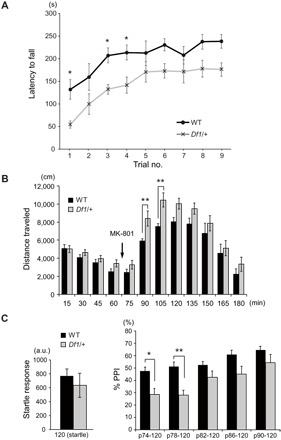
(A) Latency to fall (in seconds) from the rotarod was scored three times per day over three consecutive days. WT control (n = 10) and Df1/+ (n = 10) mice were tested at 3.5 months of age. Statistical significance [two-way analysis of variance (ANOVA) with repeated measures: F1,18 = 14.09, P = 0.0015]; *P < 0.05 (Bonferroni post hoc test). (B) Locomotor activities [distance traveled (in centimeters)] measured in the open-field box before and after MK-801 challenge were plotted in 15-min bins. WT (n = 10) and Df1/+ (n = 10) mice were tested at 2 months of age. Statistical significance (two-way ANOVA with repeated measures: F1,18 = 4.763, P = 0.0426); **P < 0.01 (Bonferroni post hoc test). (C) Acoustic startle response (left) and PPI (%) of startle response (right) were evaluated. WT (n = 10) and Df1/+ (n = 10) mice were tested at 2 months of age. Statistical significance (two-way ANOVA with repeated measures: F1,18 = 8.528, P = 0.0091); *P < 0.05 and **P < 0.01 (Bonferroni post hoc test). a.u., arbitrary units.
Meanwhile, we successfully replicated behavioral deficits in Df1/+ mice associated with psychiatric manifestations previously reported for this and other 22q11.2 mouse models (4, 12, 13, 19, 20). More specifically, Df1/+ mice showed exacerbated responses to the psychostimulant MK-801, exhibiting higher locomotor activities than WT in the open-field box (F1,18 = 4.763, P = 0.0426) (60 to 180 min; Fig. 1B). Df1/+ mice also showed evidence of impaired sensorimotor gating, as demonstrated by reduced levels of prepulse inhibition (PPI) of acoustic startle responses compared with WT (F1,18 = 8.528, P = 0.0091) (Fig. 1C). These two changes are commonly regarded as mouse behavioral phenotypes relevant to psychiatric disorders, including SZ (4). A previous study reported a possibility that Df1/+ mice have altered hearing thresholds or hearing loss (21), which may confound interpretation of the PPI results. To address this issue, we quantified the responses to nonstartle sound stimuli (65 to 90 dB) delivered to each mouse during the PPI assays. The amplitude of responses to these stimuli in Df1/+ mice was equivalent to that in control mice (fig. S1), suggesting that both Df1/+ and control mice were equally capable of sensing rather small stimulus sound used in the assays and that attenuated PPI observed in Df1/+ mice was not likely due to hearing loss.
Expressions of α-synuclein and p62 are elevated in Df1/+ mice
As motor deficits possibly relevant to PD were observed, we next addressed the question of whether PD-related neuropathological and molecular changes may exist in Df1/+ mice. We conducted immunohistochemistry with markers for Lewy bodies (α-synuclein and p62), a pathological hallmark of PD. We analyzed the anterior cingulate cortex (ACC), caudate putamen (CPu), and substantia nigra pars compacta (SNc) because Lewy bodies are frequently observed in these regions of PD brains (14, 15). Functional deficits in the ACC and CPu are also linked to SZ (22, 23). Brain sections at the ACC, CPu, and SNc were costained with anti–calcium/calmodulin-dependent protein kinase II (CaMKII), anti–dopamine- and cAMP-regulated phosphoprotein 32 kDa (DARPP-32), and anti–tyrosine hydroxylase (TH) antibodies, respectively, to mark the most abundant neuronal cell type in each brain region. We analyzed brains at 3.5 and 8 months of age, which correspond to the earliest age of onset (that is, 20s) and to the mean age of onset (that is, 40s), respectively, of PD with 22q11.2 deletions (16, 17). We found that expression of α-synuclein was significantly elevated in these brain regions at 3.5 months of age (1.77-, 1.50-, and 1.61-fold increase in the ACC, CPu, and SNc, respectively, compared with WT) (Fig. 2A), and at 8 months of age (1.73-, 1.30-, and 1.50-fold increase in the ACC, CPu, and SNc, respectively, compared with WT) (fig. S2A; summarized in Table 1). In addition, expression of p62 was also increased, compared with WT (1.88-, 1.66-, and 1.17-fold increase in the ACC, CPu, and SNc, respectively, at 3.5 months of age; Fig. 2B; 2.42-fold increase in the ACC at 8 months of age; fig. S2B) (summarized in Table 1). However, we did not observe apparent signs of inclusion body formation. To further validate these findings, we quantified the fluorescence intensities of the cell type markers used for the assays. The relative fluorescence intensities per soma for each cell type marker were equivalent between WT and Df1/+ mice. The intensities of CaMKII in the ACC of WT versus Df1/+ mice were 1.00 ± 0.04 versus 0.98 ± 0.05 at 3.5 months of age (P > 0.5) and 1.00 ± 0.03 versus 0.98 ± 0.04 at 8 months of age (P > 0.5); the intensities of DARPP-32 in the CPu of WT versus Df1/+ mice were 1.00 ± 0.04 versus 1.01 ± 0.03 at 3.5 months of age (P > 0.99) and 1.00 ± 0.03 versus 0.99 ± 0.04 at 8 months of age (P > 0.99); and the intensities of TH in the SNc of WT versus Df1/+ mice were 1.00 ± 0.01 versus 1.01 ± 0.01 at 3.5 months of age (P > 0.99) and 1.00 ± 0.02 versus 0.97 ± 0.05 at 8 months of age (P > 0.5). Together, the data support the conclusion that the expression levels of α-synuclein and p62 levels are specifically elevated in Df1/+ mice when compared with control mice of the same age group.
Fig. 2. Elevated expression of α-synuclein and p62 proteins in Df1/+ mice.
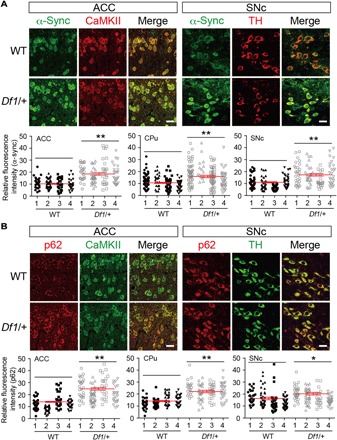
(A and B) The ACC, CPu, and SNc in Df1/+ mice (n = 4) and the WT littermates (n = 4) (3.5 months old) were immunostained with anti–α-synuclein (α-Sync) (A) or anti-p62 (B) antibodies, together with anti-CaMKII, anti–DARPP-32, and anti-TH antibodies, which label the most abundant neuronal cell type in the ACC, CPu, and SNc, respectively. Images were acquired by a confocal microscope using the same parameters across multiple specimens. Images in the ACC and SNc are shown. Scale bars, 20 μm. Relative fluorescence intensities (arbitrary units) of α-synuclein or p62 immunostaining were measured from each soma colabeled with the neuronal marker (30 to 50 soma per section) and plotted in the graph. Overall average fluorescence intensities (±SEM) calculated across three to four serial sections per animal from all animals used (n = 4 per group) are shown in red in the graph. *P < 0.05 and **P < 0.01 (two-tailed Mann-Whitney test).
Table 1. Expression of α-synuclein and p62 proteins in Df1/+ mice.
ND, not determined; ↑, up-regulated (>1.5×, compared with WT); ↗, up-regulated (<1.5×, compared with WT); →, unchanged (compared with WT).
| ACC | CPu | SNc | ||||
| α-Synuclein | p62 | α-Synuclein | p62 | α-Synuclein | p62 | |
| 2 months | ND | ↑ | ND | ↗ | ND | → |
| 3.5 months | ↑ | ↑ | ↑ | ↑ | ↑ | ↗ |
| 8 months | ↑ | ↑ | ↗ | → | ↑ | → |
Motor coordination deficits in Df1/+ mice are ameliorated by reducing α-synuclein gene dosage
Increased expression of α-synuclein has been linked to pathophysiology of PD; recent clinical studies have reported that prodromal stages of PD show increased levels of α-synuclein microaggregates, with no apparent sign of Lewy body formation (24, 25). Besides, overexpression of α-synuclein in mice serves as a model for PD (26–28). To investigate whether increased expression of α-synuclein in Df1/+ mice is pathophysiologically relevant to PD, we sought to test the impact of reducing the dosage of α-synuclein gene (Snca) on motor deficits in Df1/+ mice. We crossed Df1/+ mice with Snca–knockout (KO) mice to generate WT, Df1/+, Df1/+;Snca/+, and Snca/+ mice and tested their motor function in rotarod assays at 3.5 months of age. While Df1/+ mice showed motor coordination deficits (F3,28 = 9.552, P = 0.0250), the Df1/+;Snca/+ mice showed motor function equivalent to that of WT and Snca/+ mice (Fig. 3A).
Fig. 3. Genetic or pharmacological rescue of motor coordination deficits, psychostimulant hypersensitivity, and sensorimotor gating deficits in Df1/+ mice.
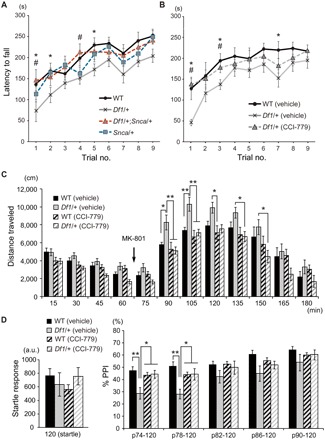
(A) Latency to fall (in seconds) from the rotarod was scored three times per day over three consecutive days. WT control (n = 8), Df1/+ (n = 8), Df1/+;Snca/+ (n = 8), and Snca/+ (n = 8) mice were tested at 3.5 months of age. Statistical significance (two-way ANOVA with repeated measures: F3,28 = 9.552, P = 0.0250); *P < 0.05 (Df1/+, as compared to WT) and #P < 0.05 (Df1/+, as compared to Df1/+;Snca/+) (Bonferroni post hoc test). (B) Latency to fall (seconds) from the rotarod was scored three times per day over three consecutive days. WT mice treated with vehicle (n = 10), Df1/+ mice treated with vehicle (n = 12), and Df1/+ mice treated with CCI-779 (n = 8) were tested at 3.5 months of age. Statistical significance (two-way ANOVA with repeated measures: F2,27 = 4.34, P = 0.0232); *P < 0.05 (Df1/+ mice treated with vehicle, as compared to WT) and #P < 0.05 (Df1/+ mice treated with CCI-779, as compared to Df1/+ mice treated with vehicle) (Bonferroni post hoc test). (C) Locomotor activities [distance traveled (in centimeters)] measured in the open-field box before and after MK-801 challenge were plotted in 15-min bins. WT mice treated with vehicle (n = 10), WT mice treated with CCI-779 (n = 10), Df1/+ mice treated with vehicle (n = 10), and Df1/+ mice treated with CCI-779 (n = 10) were tested at 2 months of age. Statistical significance (two-way ANOVA with repeated measures: F3,36 = 3.132, P = 0.0374); *P < 0.05 and **P < 0.01 (Bonferroni post hoc test). (D) Acoustic startle response (left) and PPI (%) of startle response (right) were evaluated. WT mice treated with vehicle (n = 10), WT mice treated with CCI-779 (n = 10), Df1/+ mice treated with vehicle (n = 10), and Df1/+ mice treated with CCI-779 (n = 10) were tested at 2 months of age. Statistical significance (two-way ANOVA with repeated measures: F3,36 = 4.347, P = 0.0103); *P < 0.05 and **P < 0.01 (Bonferroni post hoc test).
PD-relevant and SZ-associated changes found in Df1/+ mice are ameliorated by pharmacological treatment with a rapamycin analog
In the first half of the present study, we observed PD-relevant behavior (motor deficits) and molecular signatures (increase in the expression of α-synuclein and p62) in Df1/+ mice. To our knowledge, this is the first study that reports these PD-relevant changes in 22q11.2 mouse models. In parallel, we also observed SZ-associated behavioral changes (for example, psychostimulant hypersensitivity and deficits in sensorimotor gating), which have been previously reported by other groups as relevant to psychiatric disorders, including SZ (4, 12, 13, 19, 20).
We then questioned whether PD-relevant and SZ-associated changes found in Df1/+ mice might stem from common pathological pathways. Although this is an ambitious question for which long-term studies are needed in the future, elucidating pathological mechanisms in the 22q11.2 deletion model is critical in identifying potential targets for therapeutic interventions. To obtain initial clues to address this issue, we conducted a hypothesis-driven approach by selecting a specific pharmacological agent to test whether this could ameliorate either or both the PD-relevant and SZ-associated changes observed in the mouse model.
Recently, elevated mammalian target of rapamycin (mTOR) activity has been shown to play a role in the pathophysiology of PD (29, 30) and SZ (31). As a proof of concept for this finding, pharmacologically inhibiting mTOR activity ameliorated PD-related phenotypes in PD models (29, 30). Notably, a recent proteomic analysis reported up-regulated expression of proteins in the mTOR signaling pathway in another 22q11.2 deletion mouse model called Df(16)A+/− (32). Thus, our working hypothesis for this study was that up-regulation of a pathway involving mTOR may commonly underlie the pathophysiologies of the expression of SZ- and PD-associated phenotypes in Df1/+ mice.
Treatment of Df1/+ mice with CCI-779, a rapamycin analog that inhibits mTOR activity, significantly rescued motor coordination deficits at 3.5 months of age (F2,27 = 4.34, P = 0.0232) (Fig. 3B). This treatment also normalized MK-801–induced hyperlocomotion (F3,36 = 3.132, P = 0.0374) (Fig. 3C) and PPI deficits (F3,36 = 4.347, P = 0.0103) (Fig. 3D) at 2 months of age. Thus, at least for these phenotypes, CCI-779 appears to ameliorate both PD-relevant and SZ-associated changes in Df1/+ mice.
To evaluate the pharmacological effect of CCI-779 in the brain, we quantified the levels of p62 protein in the ACC of Df1/+ mice after CCI-779 treatment. The treatment significantly attenuated the levels of p62 in Df1/+ mice to the levels equivalent to WT at 2 months of age (fig. S3), consistent with the effect of mTOR inhibition on the level of p62 expression (33). To further validate the effect of CCI-779 on mTOR inhibition, we also quantified the levels of phosphorylated S6 ribosomal protein in the ACC of Df1/+ mice (2 to 3 months of age) after CCI-779 treatment. The ratio of phospho-S6 to total S6 was significantly elevated by ~28% in Df1/+ mice compared with WT, and CCI-779 treatment suppressed it to the level equivalent to WT (fig. S4), indicating that CCI-779 can normalize elevated mTOR activity in Df1/+ mice.
DISCUSSION
Here, we showed for the first time that a 22q11.2 deletion mouse model (Df1/+ mice) displays significant motor coordination deficits, as well as elevated expression of α-synuclein and p62 proteins. Moreover, reducing the α-synuclein gene dosage in Df1/+ mice rescued deficits in motor coordination, demonstrating the causal role of elevated α-synuclein expression in the manifestation of this phenotype. The data provide a possible link between 22q11.2 deletions and PD-related pathobiology. In parallel, we also confirmed SZ-associated behavioral changes in Df1/+ mice, consistent with the previous findings by others (4, 12, 13, 19, 20). A rapamycin analog, CCI-779, ameliorated both motor coordination deficits and SZ-associated behavioral changes. These data suggest a possible common pathway that may underlie both SZ-associated and PD-relevant manifestations in the context of 22q11.2 deletions.
Motor dysfunction of 22q11.2 deletion mouse models has not been intensively studied before; most investigators have used these models to study behavioral changes thought to represent psychiatric conditions in conjunction with the developmental trajectory. A previous study that reported no apparent motor deficits in Df1/+ mice (12) was performed using mice on a mixed genetic background, which can result in variable animal behavioral phenotypes, including motor, mood, and cognitive functions (34). In contrast, the present study used a mouse model fully backcrossed to a congenic C57BL/6J background. A congenic background is recognized as important to delineate the effects of specific genomic alterations on animal behaviors.
We observed elevated levels of α-synuclein and p62 expression in brain regions, including the ACC, CPu, or SNc, but no apparent sign of Lewy body–like inclusions in the Df1/+ mice, as also reported in other currently available mouse models of PD (28). Recent clinical studies have reported that prodromal stages of PD show microaggregates of α-synuclein with no apparent sign of Lewy body formation, as well as increased p62 expression, in several brain regions, including the SNc, striatum, and frontal cortex (24). Increased expression of α-synuclein has also been reported for PD patients (35). Given these lines of evidence, the phenotypes observed in the Df1/+ mice might represent pathophysiological conditions relevant to prodromal PD. Alternatively, the motor coordination deficits observed in Df1/+ mice may provide us with an opportunity to understand mild motor dysfunctions frequently observed in patients with neurodevelopmental and psychiatric conditions, including SZ (36, 37), as well as those with 22q11.2 deletions (16, 17). Conversely, SZ-relevant phenotypes observed in Df1/+ mice may help us understand the mechanisms underlying the psychiatric symptoms frequently observed in PD. Note that nonmotor deficits, especially psychiatric symptoms, are clinically observed in individuals in prodromal stages of PD (38).
Although we observed motor coordination deficits in Df1/+ mice at 3.5 months of age with α-synuclein protein levels less than two times that of control mice, PD mouse models that overexpressed α-synuclein transgene generally had greater levels of the overexpression (1.5- to 3.4-fold at the protein level compared to the control) (26, 27) and exhibited later onset of motor deficits (after 7 months to 1 year of age) (27, 28). We postulate that the amount of α-synuclein overexpression necessary to cause motor deficits and the age of onset of PD-related motor phenotypes may differ, depending on the genetic mechanism of each model. For example, the Df1/+ mice tested in this study have a hemizygous deletion of ~30 genes, resulting in downstream elevated expression of not only α-synuclein but also p62 proteins and possibly other proteins as well. In addition, the 22q11.2 region notably contains genes (for example, PRODH and TXNRD2) that were suggested to affect motor functioning (39). In contrast, the genetic mutation of α-synuclein transgenic mice is restricted to the transgene integration sites on the mouse genome, presumably resulting in specific overexpression of the transgene. Besides the elevated α-synuclein levels, the genetic, biochemical, or circuit-wide changes in Df1/+ model versus α-synuclein transgenic mice may explain phenotypic differences among these models.
Genetic evidence has shown that 22q11.2 deletions are a susceptibility factor for SZ and PD (2, 3, 9), and we tested deregulated mTOR as a candidate common pathway shared between these two under this genetic condition. However, this does not mean that SZ pathophysiology generally contributes to PD pathophysiology and vice versa. 22q11.2 deletions represent only a small portion of SZ susceptibility among the entire SZ populations, and many additional genetic alterations have been suggested to serve as SZ risk factors (2, 3). Likewise, 22q11.2 deletions represent only a small portion of PD susceptibility, and many others, including mutations in Parkin, PINK, PARK2, PARK7, or LRRK2 genes, serve as PD risk factors that are not considered as SZ susceptibility factors (9). Nonetheless, we predicted that 22q11.2 deletions may provide a unique opportunity to test whether there is some level of overlap, if any, in the pathophysiological mechanisms shared across these two distinct disease categories associated with 22q11.2 deletions. Elevated expression of α-synuclein and p62 proteins that we observed in Df1/+ brains could result from attenuated activity of the cellular recycling machinery due to elevated mTOR activity (33), and we detected a steady-state increase in mTOR activity in Df1/+ brains. Administration of the rapamycin analog in Df1/+ mice normalized mTOR activity and concomitantly rescued both the motor- and non–motor-related behavioral deficits. Together, these lines of evidence suggest that elevated mTOR activity causes, at least in part, both the motor and nonmotor deficits in Df1/+ mice.
To further delineate SZ and PD pathobiology associated with 22q11.2 deletions, future studies will need to investigate where, when, and how much of these pathological abnormalities (that is, elevated mTOR signaling or elevated expression of α-synuclein or p62 proteins) may occur within the human brains with 22q11.2 deletions. Depending on the cell types (for example, excitatory or inhibitory neuron), the brain regions (for example, ACC, CPu, or SNc), or the circuits (for example, corticolimbic, corticostriatal, or nigrostriatal pathway) that are principally affected, we predict seeing a range of phenotypic consequences, including SZ or PD.
There are several limitations to our study. First, motor dysfunctions observed in PD, including resting tremor and bradykinesia, are more complex than the motor coordination deficit we showed in the Df1/+ model. Df1/+ mice may not be suited to model these cardinal motor features of PD. Second, our current immunofluorescence analyses were designed to compare levels of α-synuclein or p62 proteins across genotypes within the same age group instead of following longitudinal changes in their expression levels for each genotype. Given that aggregate-prone proteins tend to accumulate during aging (14, 15), questions remain as to whether α-synuclein or p62 would show an accelerated increase in Df1/+ mice relative to WT mice and, if so, at which age this would begin. To address these questions, we will need to directly compare the expression levels over additional longitudinal age groups in one experimental cohort. Third, elevated α-synuclein expression levels alone may not fully explain pathophysiological mechanisms for early-onset PD associated with 22q11.2 deletions. Although human genetic studies and postmortem analyses have demonstrated the association of recurrent 22q11.2 deletions with early-onset PD (7, 9), the detailed mechanisms underlying this comorbidity remain to be elucidated. While a high proportion (that is, up to 30%) of patients with 22q11.2 deletions are shown to develop psychiatric conditions, a smaller proportion (~5%) of those with 22q11.2 deletions are estimated to develop PD (7). This suggests that additional pathogenic events, such as age-related stresses, environmental risk factors, or other genetic alterations, may interact with 22q11.2 deletions to contribute to the development of PD. For example, some chronic insults that would disrupt metabolic homeostasis of dopaminergic neurons (16), coupled with elevated α-synuclein in SNc due to 22q11.2 deletions, may culminate in dopaminergic neuron death, leading to full-blown PD pathology. In this regard, the present study provides initial evidence that 22q11.2 deletion mouse models may be useful to investigate how the biological disturbances elicited by 22q11.2 deletions could underlie both neurodevelopmental conditions, such as SZ, and neurological conditions, such as early-onset PD, at the mechanistic levels. The preliminary findings of the current study support the possibility that deregulated mTOR signaling may represent a joint mechanism, providing a working hypothesis for future research.
There is precedence that molecular mechanisms underlying both psychiatric and nonpsychiatric (for example, motor) symptoms are delineated in the same animal model. For example, the R6/2 model, a representative mouse model for Huntington’s disease with respect to its polyglutamine pathology, displays motor deficits and anhedonia-like manifestations: The mutant huntingtin protein with a polyglutamine stretch causes both motor and psychiatric symptoms through distinct molecular mechanisms (40). Likewise, we cautiously but optimistically propose that the animal models of 22q11.2 deletions will become important tools for studying mechanisms not only for neurodevelopmental conditions, such as SZ, but also for neurodegenerative pathologies, such as PD.
MATERIALS AND METHODS
Experimental design
On the basis of the clinical evidence linking human 22q11.2 deletions not only to SZ but also to early-onset PD (7, 9), we hypothesized that a mouse model of 22q11.2 deletions could be used to investigate pathobiological mechanisms underlying the early-onset PD associated with the 22q11.2 deletions. The Df1/+ mouse line (10, 12) was used as a representative model because this line has been extensively characterized to address the pathophysiologies of SZ. The Df1/+ mouse line was maintained on the C57BL/6J genetic background for >12 generations. Snca-KO mice were obtained from the Jackson Laboratory (stock #016123 maintained on C57BL/6NJ). Animal handling was performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Animal Research Committee at Kyoto University.
Behavioral assays
All mice were group-housed and maintained under standard conditions (12/12-hour light/dark cycle, lights on at 7:00; 22° ± 1°C; with food and water ad libitum). Mice to be used for behavioral assays were acclimated in the test room 1 day before the assay, and all the assays were performed between 15:00 and 19:00. Df1/+ and the littermate control mice (3.5 months old; male; n = 8 to 12 per group) were used for rotarod tests. Independent cohorts of mice (2 months old; male; n = 10 per group) were used for PPI tests, followed by locomotor tests 3 days later. Personnel performing the tests were blinded to the genotype of the mice.
Rotarod
Mice were tested for their ability to stay on a rotating bar (rotarod, Harvard Apparatus), which accelerated from 4 to 50 rpm over 5 min per trial. For each mouse, three trials were performed per day (30-min break between trials) for three consecutive days. Latency to fall (seconds) was scored in each trial.
PPI
To assess sensorimotor gating function, startle response and PPI were measured using a startle chamber (SR-LAB). Briefly, a prepulse-pulse trial started with a 50-ms null period, followed by a 20-ms prepulse white noise (74, 78, 82, 86, or 90 dB) and a 100-ms delay, and then, the startle stimulus (a 40-ms, 120-dB white noise) was presented. Six trial types (pulse-only trial and five types of prepulse-pulse trial) and six nonstartle stimuli (65, 74, 78, 82, 86, or 90 dB) were presented in a pseudorandomized order such that each trial type or nonstartle stimulus was presented once within a block of assay. %PPI = 100 − [(response on prepulse-pulse stimulus trials/startle response on pulse-only trials) × 100].
Locomotor
Each mouse was placed in the center of the open-field box (W × D × H = 40 cm × 40 cm × 27 cm), and the baseline activities were measured over 60 min. Subsequently, MK-801 was administered intraperitoneally [0.1 mg/kg body weight, intraperitoneally (ip)], and the locomotor activities were measured over 120 min to evaluate psychostimulant sensitivity. The distance traveled (in centimeters) was analyzed in 15-min bins using EthoVision (Noldus).
Immunohistochemistry
Mice (n = 4 per group; male; 2, 3.5, or 8 months of age) were perfused transcardially with 4% paraformaldehyde in phosphate-buffered saline while under anesthesia [ketamine (100 mg/kg body weight)/xylazine (10 mg/kg body weight) mixture, ip]. The brains were then serially cut into 50-μm-thick coronal sections using a vibratome (VT1200 S, Leica), and the sections from one cohort of mice (a littermate pair of WT and Df1/+ mice) were simultaneously immunostained for 16 hours at 4°C with primary antibodies anti-p62 (guinea pig, 1:400; Medical and Biological Laboratories), anti–α-synuclein (rabbit, 1:200; Immuno-Biological Laboratories), anti-CaMKII (mouse, 1:1000; StressMarq), anti–DARPP-32 (mouse, 1:1000; Calbiochem), and anti-TH (mouse, 1:3000; Abcam), followed by incubation with Alexa Fluor 488– or Alexa Fluor 546–conjugated secondary antibodies (1:500; Gibco). Images of one optical section (1 μm thick) were acquired via confocal microscopy (SP8, Leica; 40× objective lens; numerical aperture, 1.3) from sections containing either the ACC, dorsolateral part of CPu, or SNc. For each brain region, three to four serial sections were analyzed, and three to six nonoverlapping areas were randomly chosen for image acquisition in each section. The fluorescence intensities of the α-synuclein or p62 immunostaining were measured from each neuron marker–positive soma (30 to 50 somas per section) using ImageJ (NIH), with the background levels in adjacent regions being subtracted. The average immunofluorescence intensity per soma was calculated across all serial sections from every mouse used.
Western blot
Mice (male; 2 to 3 months of age; n = 6 per group) were acutely sacrificed by decapitation. The brain (the ACC) was then dissected quickly within 30 s, snap-frozen in liquid nitrogen, and subsequently lysed in radioimmunoprecipitation assay buffer containing 1% Triton X-100, 0.1% SDS, protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktail (Sigma-Aldrich). The extracted proteins were subjected to SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (0.45-μm pore, Millipore), which was incubated in a membrane-blocking agent (GE Healthcare) for 30 min and probed with anti-S6 ribosomal protein (rabbit, 1:1000; Cell Signaling Technology) or anti–phospho-S6 (Ser235/236) antibodies (rabbit, 1:1000; Cell Signaling Technology) and subsequently with horseradish peroxidase–conjugated anti-rabbit immunoglobulin G (1:10,000; GE Healthcare). Signals were detected with an ECL agent (GE Healthcare). Blotting images were acquired by ChemiDoc XRS imaging system (Bio-Rad), and the densitometric analysis was performed using ImageJ (NIH).
Pharmacological rescue
A water-soluble rapamycin analog, CCI-779 (Tocris), dissolved in saline (10 mg/kg body weight) or the vehicle alone was injected (intraperitoneally) once daily for eight consecutive days before behavioral testing. During the behavioral testing period, daily injection was performed 1 hour before behavioral assessment and was continued up to day 11 until all behavioral assays were completed.
Statistical analysis
Data were means ± SEM. Behavioral assay data were analyzed using a two-way ANOVA with repeated measures, followed by Bonferroni post hoc tests. Immunohistochemistry data without drug rescue were analyzed by Mann-Whitney test (two-tailed). Immunohistochemistry data with drug rescue and Western blot data were analyzed by Kruskal-Wallis test, followed by Dunn’s multiple comparisons tests. Statistical significance: *P < 0.05 and **P < 0.01.
Supplementary Material
Acknowledgments
We thank E. Illingworth and K. Tanigaki for sharing Df1/+ mice and E. Illingworth and E. Sibille for the comments. Funding: This work was supported by the NIH (MH-094268 Silvio O. Conte Center, MH-092443, MH105660, and MH107730 to A. Sawa and MH101723 to A.S.B. and E.B.), the Department of Defense/Congressionally Directed Medical Research Program (W81XWH-11-1-0269 to T.T.), the Canadian Institutes of Health Research (MOP #97800 and MOP #111238 to A.S.B.), and Japan Society for the Promotion of Science (15H01285 and 16K01948 to T.S.). This work was also supported by grants from Stanley, S-R, RUSK, NARSAD, and Maryland Stem Cell Research Fund (to A. Sawa), Children’s Tumor Foundation Drug Discovery Initiative (to T.T.), University of Toronto McLaughlin Centre (MC-2014-01 to A.S.B.), the Canada Research Chairs in Schizophrenia Genetics and Genomic Disorders (to A.S.B.), and the Dalglish Chair in 22q11.2 Deletion Syndrome (to A.S.B.). E.B. has a Dalglish–22q11.2 Deletion Syndrome Fellowship. Author contributions: A. Sumitomo and T.T. designed the study. A. Sumitomo, K. Horike, and K. Hirai performed the data collection and processing. A. Sumitomo performed the statistical analysis. T.S., N.B., E.B., and F.C.N. provided expertise and analytical tools. N.B., E.B., A.S.B., A. Sawa, and T.T. wrote and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaar6637/DC1
Fig. S1. Amplitude of responses to nonstartle sound stimuli in Df1/+ mice during PPI assays.
Fig. S2. Elevated expression of α-synuclein and p62 in Df1/+ mice at 8 months of age.
Fig. S3. Elevated p62 expression in Df1/+ mice is normalized by CCI-779 administration.
Fig. S4. Increased mTOR activity in Df1/+ mice is normalized by CCI-779 administration.
REFERENCES AND NOTES
- 1.Scambler P. J., The 22q11 deletion syndromes. Hum. Mol. Genet. 9, 2421−2426 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Bassett A. S., Chow E. W. C., Weksberg R., Chromosomal abnormalities and schizophrenia. Am. J. Med. Genet. 97, 45−51 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schneider M., Debbané M., Bassett A. S., Chow E. W. C., Fung W. L. A., van den Bree M. B. M., Owen M., Murphy K. C., Niarchou M., Kates W. R., Antshel K. M., Fremont W., McDonald-McGinn D. M., Gur R. E., Zackai E. H., Vorstman J., Duijff S. N., Klaassen P. W. J., Swillen A., Gothelf D., Green T., Weizman A., Van Amelsvoort T., Evers L., Boot E., Shashi V., Hooper S. R., Bearden C. E., Jalbrzikowski M., Armando M., Vicari S., Murphy D. G., Ousley O., Campbell L. E., Simon T. J., Eliez S.; International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome , Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: Results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 171, 627−639 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meechan D. W., Maynard T. M., Tucker E. S., Fernandez A., Karpinski B. A., Rothblat L. A., LaMantia A.-S., Modeling a model: Mouse genetics, 22q11.2 deletion syndrome, and disorders of cortical circuit development. Prog. Neurobiol. 130, 1−28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaleski C., Bassett A. S., Tam K., Shugar A. L., Chow E. W. C., McPherson E., The co-occurrence of early onset Parkinson disease and 22q11.2 deletion syndrome. Am. J. Med. Genet. A 149A, 525−528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booij J., van Amelsvoort T., Boot E., Co-occurrence of early-onset Parkinson disease and 22q11.2 deletion syndrome: Potential role for dopamine transporter imaging. Am. J. Med. Genet. A 152A, 2937−2938 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Butcher N. J., Kiehl T.-R., Hazrati L.-N., Chow E. W. C., Rogaeva E., Lang A. E., Bassett A. S., Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: Identification of a novel genetic form of Parkinson disease and its clinical implications. JAMA Neurol. 70, 1359−1366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rehman A. F., Dhamija R., Williams E. S., Barrett M. J., 22q11.2 deletion syndrome presenting with early-onset Parkinson’s disease. Mov. Disord. 30, 1289−1290 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Mok K. Y., Sheerin U., Simón-Sánchez J., Salaka A., Chester L., Escott-Price V., Mantripragada K., Doherty K. M., Noyce A. J., Mencacci N. E., Lubbe S. J.; International Parkinson’s Disease Genomics Consortium (IPDGC), Williams-Gray C. H., Barker R. A., van Dijk K. D., Berendse H. W., Heutink P., Corvol J.-C., Cormier F., Lesage S., Brice A., Brockmann K., Schulte C., Gasser T., Foltynie T., Limousin P., Morrison K. E., Clarke C. E., Sawcer S., Warner T. T., Lees A. J., Morris H. R., Nalls M. A., Singleton A. B., Hardy J., Abramov A. Y., Plagnol V., Williams N. M., Wood N. W., Deletions at 22q11.2 in idiopathic Parkinson’s disease: A combined analysis of genome-wide association data. Lancet Neurol. 15, 585−596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindsay E. A., Botta A., Jurecic V., Carattini-Rivera S., Cheah Y.-C., Rosenblatt H. M., Bradley A., Baldini A., Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401, 379−383 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Toritsuka M., Kimoto S., Muraki K., Landek-Salgado M. A., Yoshida A., Yamamoto N., Horiuchi Y., Hiyama H., Tajinda K., Keni N., Illingworth E., Iwamoto T., Kishimoto T., Sawa A., Tanigaki K., Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc. Natl. Acad. Sci. U.S.A. 110, 17552−17557 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paylor R., McIlwain K. L., McAninch R., Nellis A., Yuva-Paylor L. A., Baldini A., Lindsay E. A., Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Hum. Mol. Genet. 10, 2645−2650 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Kimoto S., Muraki K., Toritsuka M., Mugikura S., Kajiwara K., Kishimoto T., Illingworth E., Tanigaki K., Selective overexpression of Comt in prefrontal cortex rescues schizophrenia-like phenotypes in a mouse model of 22q11 deletion syndrome. Transl. Psychiatry 2, e146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braak H., Del Tredici K., Invited article: Nervous system pathology in sporadic Parkinson disease. Neurology 70, 1916−1925 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Dickson D. W., Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2, a009258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butcher N. J., Marras C., Pondal M., Rusjan P., Boot E., Christopher L., Repetto G. M., Fritsch R., Chow E. W. C., Masellis M., Strafella A. P., Lang A. E., Bassett A. S., Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain 140, 1371−1383 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Boot E., Butcher N. J., Udow S. J., Marras C., Mok K. Y., Kaneko S., Barrett M. J., Prontera P., Berman B. D., Masellis M., Dufournet B., Nguyen K., Charles P., Mutez E., Danaila T., Jacquette A., Colin O., Drapier S., Borg M., Fiksinski A. M., Vergaelen E., Swillen A., Vogels A., Plate A., Perandones C., Gasser T., Clerinx K., Bourdain F., Mills K., Williams N. M., Wood N. W., Booij J., Lang A. E., Bassett A. S., Typical features of Parkinson disease and diagnostic challenges with microdeletion 22q11.2. Neurology 90, e2059–e2067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nuber S., Petrasch-Parwez E., Winner B., Winkler J., von Hörsten S., Schmidt T., Boy J., Kuhn M., Nguyen H. P., Teismann P., Schulz J. B., Neumann M., Pichler B. J., Reischl G., Holzmann C., Schmitt I., Bornemann A., Kuhn W., Zimmermann F., Servadio A., Riess O., Neurodegeneration and motor dysfunction in a conditional model of Parkinson’s disease. J. Neurosci. 28, 2471−2484 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long J. M., LaPorte P., Merscher S., Funke B., Saint-Jore B., Puech A., Kucherlapati R., Morrow B. E., Skoultchi A. I., Wynshaw-Boris A., Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics 7, 247−257 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Stark K. L., Xu B., Bagchi A., Lai W.-S., Liu H., Hsu R., Wan X., Pavlidis P., Mills A. A., Karayiorgou M., Gogos J. A., Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751−760 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Fuchs J. C., Zinnamon F. A., Taylor R. R., Ivins S., Scambler P. J., Forge A., Tucker A. S., Linden J. F., Hearing loss in a mouse model of 22q11.2 deletion syndrome. PLOS ONE 8, e80104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rushworth M. F. S., Buckley M. J., Behrens T. E. J., Walton M. E., Bannerman D. M., Functional organization of the medial frontal cortex. Curr. Opin. Neurobiol. 17, 220−227 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Simpson E. H., Kellendonk C., Kandel E., A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 65, 585−596 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrer I., Martinez A., Blanco R., Dalfó E., Carmona M., Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: Preclinical Parkinson disease. J. Neural Transm. 118, 821−839 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Giráldez-Pérez R. M., Antolín-Vallespín M., Muñoz M. D., Sánchez-Capelo A., Models of α-synuclein aggregation in Parkinson’s disease. Acta Neuropathol. Commun. 2, 176 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rockenstein E., Mallory M., Hashimoto M., Song D., Shults C. W., Lang I., Masliah E., Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 68, 568–578 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Chesselet M.-F., Richter F., Zhu C., Magen I., Watson M. B., Subramaniam S. R., A progressive mouse model of Parkinson’s disease: The Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9, 297–314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen C., Björklund T., Petit G. H., Lundblad M., Murmu R. P., Brundin P., Li J.-Y., A novel α-synuclein-GFP mouse model displays progressive motor impairment, olfactory dysfunction and accumulation of α-synuclein-GFP. Neurobiol. Dis. 56, 145–155 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Malagelada C., Jin Z. H., Jackson-Lewis V., Przedborski S., Greene L. A., Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166−1175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dehav B., Bové J., Rodríguez-Muela N., Perier C., Recasens A., Boya P., Vila M., Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535−12544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gururajan A., van den Buuse M., Is the mTOR-signalling cascade disrupted in schizophrenia? J. Neurochem. 129, 377−387 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Wesseling H., Xu B., Want E. J., Holmes E., Guest P. C., Karayiorgou M., Gogos J. A., Bahn S., System-based proteomic and metabonomic analysis of the Df(16)A+/− mouse identifies potential miR-185 targets and molecular pathway alterations. Mol. Psychiatry 22, 384−395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katsuragi Y., Ichimura Y., Komatsu M., p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672−4678 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Mao J.-H., Langley S. A., Huang Y., Hang M., Bouchard K. E., Celniker S. E., Brown J. B., Jansson J. K., Karpen G. H., Snijders A. M., Identification of genetic factors that modify motor performance and body weight using Collaborative Cross mice. Sci. Rep. 5, 16247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuchs J., Nilsson C., Kachergus J., Munz M., Larsson E.-M., Schüle B., Langston J. W., Middleton F. A., Ross O. A., Hulihan M., Gasser T., Farrer M. J., Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916−922 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Bernard J. A., Mittal V. A., Updating the research domain criteria: The utility of a motor dimension. Psychol. Med. 45, 2685−2689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walther S., Psychomotor symptoms of schizophrenia map on the cerebral motor circuit. Psychiatry Res. 233, 293−298 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Frandsen R., Kjellberg J., Ibsen R., Jennum P., Morbidity in early Parkinson’s disease and prior to diagnosis. Brain Behav. 4, 446−452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guna A., Butcher N. J., Bassett A. S., Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J. Neurodev. Disord. 7, 18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka M., Ishizuka K., Nekooki-Machida Y., Endo R., Takashima N., Sasaki H., Komi Y., Gathercole A., Huston E., Ishii K., Hui K. K.-W., Kurosawa M., Kim S.-H., Nukina N., Takimoto E., Houslay M. D., Sawa A., Aggregation of scaffolding protein DISC1 dysregulates phosphodiesterase 4 in Huntington’s disease. J. Clin. Invest. 127, 1438−1450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaar6637/DC1
Fig. S1. Amplitude of responses to nonstartle sound stimuli in Df1/+ mice during PPI assays.
Fig. S2. Elevated expression of α-synuclein and p62 in Df1/+ mice at 8 months of age.
Fig. S3. Elevated p62 expression in Df1/+ mice is normalized by CCI-779 administration.
Fig. S4. Increased mTOR activity in Df1/+ mice is normalized by CCI-779 administration.