

Among 85 genes that maintain mitotic competence in quiescent eukaryotic cells, Nem1 protects nuclei from autophagy by regulating lipin.
Abstract
Quiescent (G0 phase) cells must maintain mitotic competence (MC) to restart the cell cycle. This is essential for reproduction in unicellular organisms and also for development and cell replacement in higher organisms. Recently, suppression of MC has gained attention as a possible therapeutic strategy for cancer. Using a Schizosaccharomyces pombe deletion-mutant library, we identified 85 genes required to maintain MC during the G0 phase induced by nitrogen deprivation. G0 cells must recycle proteins and RNA, governed by anabolism, catabolism, transport, and availability of small molecules such as antioxidants. Protein phosphatases are also essential to maintain MC. In particular, Nem1-Spo7 protects the nucleus from autophagy by regulating Ned1, a lipin. These genes, designated GZE (G-Zero Essential) genes, reveal the landscape of genetic regulation of MC.
INTRODUCTION
Switching from active mitosis to quiescence (G0) is an important longevity strategy for cell survival during times of limited nutrients, but only if the capacity to return to growth and division [vegetative (VE)] phase is assured. Therefore, it is apparent that mechanisms must exist to protect and maintain mitotic competence (MC) in G0 phase cells. Understanding these mechanisms is of great importance, since disabling MC could offer a new therapeutic approach for cancer (1, 2).
The fission yeast, Schizosaccharomyces pombe (S. pombe), is an excellent model to study these mechanisms, since switching between the two phases can easily be regulated by nitrogen availability in the media (3–5). When the nitrogen source (NH3Cl, hereafter N) is eliminated from the synthetic minimal medium Edinburgh minimal medium 2 (EMM2), cells divide twice without growth to form small and round cells. If cells can switch mating type or meet the opposite mating type, they can mate to sporulate; otherwise, they enter the G0 phase. In nature, heritable mating type or even sterile fission yeast cells commonly exist (6), so their main survival strategies under nutrient deficiency are not only to mate so as to sporulate but also to form “bachelor” G0 cells with long life (7). These G0 cells, in the absence of mate switching, could be a better model for the G0 phase of mammalian cells. G0 cells return to the VE phase when N is replenished.
RESULTS
Active recycling in nitrogen-deprived G0 cells
Nitrogen-deprived G0 cells can maintain high MC for weeks without medium change (Fig. 1A). The medium contains glucose, salt, vitamins, and minerals, so even though these cells cannot grow and divide because of the lack of N, metabolic activity remains high, as reported previously (8). Active movement of cytoplasmic particles was observed in cells under nitrogen deprivation (−N), whereas under glucose deprivation (−glc), no movement was seen, even though high MC was maintained in both conditions (Fig. 1B). One metabolic activity under −N might be to recycle the limited nitrogen available inside cells, since amino acid degradation seemed to be activated under −N. We observed cellular nitric oxide using a fluorescent dye 4-amino-5-methylamino-20, 70-difluorofluorescein diacetate (DAF-FM DA) (Fig. 1C) (9). This dye reacts with nitric oxide produced by arginine catabolism. In wild-type (WT) cells, nitric oxide fluorescence was observed 6.8 times more strongly in vacuoles of G0 cells than in VE cells, suggesting that amino acid degradation is activated under −N. Such fluorescence under −N was eliminated in the deletion-mutant strain of Aut12, a vacuolar fusion protein required for fusing endosomes or autophagosomes to vacuoles (10, 11), implying that active vesicle transport is necessary for this degradation. Consistently, amounts of most free amino acids decreased rapidly right after −N (Fig. 1D). However, the amount of glutamate began to recover after 12 hours of −N. Since it has been reported that environmental nitrogen (ammonium ion) is fixed inside cells to produce glutamate, and because this reaction is regarded as “the starting point of nitrogen biosynthesis” (12), the recovery of glutamate concentrations after continuous ammonium ion deprivation suggests the possibility of active recycling.
Fig. 1. Nitrogen-deprived cells maintained high MC with elevated cellular activity, apparently related to recycling nitrogen.
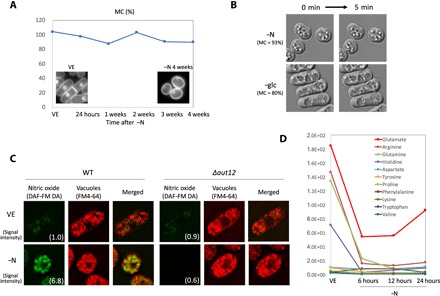
(A) Time course of MC in WT cells under nitrogen deprivation (−N). 4′,6-Diamidino-2-phenylindole (DAPI) images of VE and −N 4-week cells are shown as insets. (B) Differential interference contrast (DIC) images were taken at 5-min intervals of cells incubated 24 hours in EMM2−N (upper panels) and in EMM2–glc (lower panels). In both conditions, high MC was maintained (93 and 80%, respectively), but white particles observed inside cells showed position changes only in EMM2−N cells. (C) Fluorescence images of nitric oxide (green, DAF-FM DA), vacuoles (red, FM4-64), and merged images of WT cells and Δaut12, a deletion strain of the vacuolar fusion protein. The upper row shows VE cells, and the lower row shows cells 24 hours after −N. Signal intensity of DAF-FM DA was quantified and shown in the images, proportional to WT VE cells. (D) Time course changes of normalized peak areas of amino acids detected by quantitative metabolomics analysis, using liquid chromatography–mass spectrometry.
85 G-zero essential genes required to maintain MC
We used a quiescent cell system that supplied normal energy sources and nutrients (glucose, salt, vitamins, and minerals), but which was devoid of nitrogen, except for that stored inside the cells. This system produces an extreme intracellular environment that necessitates nitrogen recycling to support critical cellular functions, an environment suitable to investigate genes essential to MC maintenance during the G0 phase.
To this end, we used 3280 haploid deletion mutants (13). These strains were first screened using a spot test with an automated laboratory system, whereby we identified 145 strains that were defective in colony formation upon N replenishment after previous −N incubation (Fig. 2A). Next, MC of these 145 strains was reconfirmed to eliminate false-positive strains. MC was expressed as a percentage of colonies formed on rich medium plates divided by the number of plated cells after −N incubation. We selected strains in which MC diminished less than 50% during 4 weeks of −N incubation, and for selected strains, polymerase chain reaction (PCR) was conducted to confirm gene deletions. As a result, 85 mitosis-incompetent strains were identified, and deleted genes were designated GZE (G-zero essential) genes, since they are nonessential for VE phase, but are requisite for MC in the G0 phase. These GZE genes are evolutionarily conserved in eukaryotes, as 89% are shared by budding yeast and 73% by humans.
Fig. 2. 85 genes proved essential to sustain MC during the G0 phase, and these were categorized into three classes.
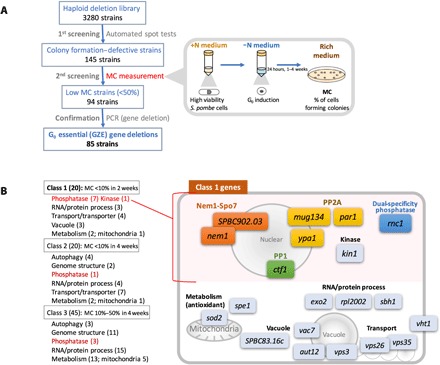
(A) Schematized identification process for GZE genes. (B) Genes belonging to each class and diagram of class 1 genes.
The 85 GZE genes were sorted into three classes based on the timing of MC loss (Fig. 2B): class 1 (20 genes; Table 1) was the most severe, with MC less than 10% after 2 weeks; class 2 (20 genes; table S1), MC less than 10% after 4 weeks; class 3 (45 genes; table S1), MC 10 to 50% after 4 weeks. All three classes included a significant number of genes related to protein phosphorylation signaling (Fig. 2B). Most were phosphatase-related genes rather than kinases (11:1), implying that dephosphorylation is a primary means of maintaining MC under −N. Class 2 and 3 gene deletions required long −N incubation (4 weeks) before MC loss, and in these classes, seven autophagy genes were identified. These strains failed to divide twice at G0 entry, resulting in elongated cell shapes, but MC was maintained more than 50% until 3 weeks (table S1). This result implies that autophagy is required for the small, round G0 cell shape but that morphological defects may not cause immediate MC loss. Similarly, a significant number of genes related to genome structure (13 genes) were identified, but only in classes 2 and 3, implying that genomic structure must also be modified to maintain MC during a protracted G0 phase.
Table 1. Class 1 genes with functional group, human and budding yeast orthologs, MC (%) at the indicated time points, average cell length 24 hours after −N, cell number increment 24 hours after −N, and major DNA peaks in VE and 24 hours after −N, measured by FACS.
| Functional group | Strain name | Ortholog | MC (%) |
Cell length |
Cell number |
Major DNA peak |
||||||||
| Human |
Budding yeast |
VE |
24 hours |
1 week |
2 weeks |
3 weeks |
4 weeks |
−N 24 hours (μm) |
Fold change |
VE |
-N 24 hours |
|||
| — | WT | — | — | 104.4 | 97.8 | 87.8 | 103.3 | 90.6 | 90.0 | 4.9 | 3.6 | 2C | 1C | |
| Protein phosphatase– related |
Nem1- Spo7 |
Δnem1 | CTDNEP1 | NEM1 | 96.7 | 80.0 | 5.0 | 3.3 | 2.0 | 2.8 | 5.4 | 3.0 | 2C | 1C |
| ΔSPBC902.03 | CNEP1R1 | SPO7 | 108.3 | 80.0 | 4.8 | 0.0 | 0.0 | 0.0 | 6.2 | 2.4 | 2C | 1C/2C | ||
| CWI | Δrnc1 | HNRNPK | PBP2 | 103.3 | 100.0 | 96.7 | 7.0 | 3.3 | 0.8 | 5.0 | 3.8 | 2C | 1C | |
| PP1 | Δctf1 | CSTF2T | RNA15 | 66.7 | 78.3 | 20.0 | 0.0 | 0.0 | 0.0 | 4.8 | 4.0 | 2C | 1C | |
| PP2A | Δmug134 | ENSA | IGO1 | 110.0 | 106.7 | 50.0 | 8.3 | 5.0 | 5.0 | 10.5 | 2.4 | 2C | 2C | |
| Δpar1 | PPP2R5B | RTS1 | 93.3 | 88.3 | 48.3 | 0.0 | 0.0 | 0.0 | 5.0 | 2.2 | 2C | 2C | ||
| Δypa1 | PPP2R4 | RRD1 | 86.7 | 75.0 | 70.0 | 5.8 | 4.3 | 4.7 | 5.8 | 2.2 | 2C | 2C | ||
| Protein kinase | Δkin1 | MARK | KIN1 | 101.7 | 65.0 | 23.7 | 0.0 | 0.0 | 0.0 | 5.8 | 2.5 | 2C | 2C | |
| RNA/protein process |
Δexo2 | XRN1 | XRN1 | 41.7 | 61.7 | 8.8 | 0.0 | 0.0 | 0.0 | 10.1 | 1.7 | 2C | 2C | |
| Δrpl2002 | RPL18A | RPL20B | 105.0 | 106.7 | 46.7 | 1.2 | 5.8 | 0.0 | 4.4 | 3.5 | 2C | 1C | ||
| Δsbh1 | SEC61B | SBH1 | 61.7 | 63.3 | 5.0 | 1.7 | 0.0 | 1.2 | 4.6 | 4.1 | 2C | 1C | ||
| Vesicle transport |
Δvps3 | TGFBRAP1 | VPS3 | 115.0 | 45.0 | 1.0 | 0.0 | 0.0 | 0.0 | 5.1 | 2.4 | 2C | 1C | |
| Δvps26 | VPS26B | PEP8 | 113.3 | 63.3 | 25.0 | 1.7 | 3.3 | 1.7 | 6.6 | 1.6 | 2C | 2C | ||
| Δvps35 | VPS35 | VPS35 | 78.3 | 30.0 | 5.0 | 0.7 | 0.7 | 0.3 | 7.2 | 1.9 | 2C | 2C | ||
| Transporter | Δvht1 | SLC17 | VHT1 | 70.0 | 90.0 | 10.0 | 0.0 | 0.0 | 0.0 | 6.2 | 1.8 | 1C | 1C | |
| Vacuole | Δaut12 | MON1B | MON1 | 100.0 | 71.7 | 0.0 | 0.0 | 0.0 | 0.0 | 8.2 | 1.4 | 2C | 1C/2C | |
| Δvac7 | — | VAC7 | 108.3 | 71.7 | 0.0 | 0.0 | 0.0 | 0.0 | 8.5 | 2.1 | 2C | 2C | ||
| ΔSPBC83.16c | TTC39B | YKR018C | 103.3 | 90.0 | 7.5 | 0.0 | 0.0 | 0.0 | 4.9 | 3.6 | 2C | 1C | ||
| Metabolism (antioxidant) |
Δsod2 | SOD2 | SOD2 | 76.7 | 48.3 | 3.7 | 0.0 | 0.0 | 0.0 | 5.1 | 2.0 | 2C | 1C | |
| Δspe1 | ODCC1 | SPE1 | 90.0 | 83.3 | 75.0 | 4.2 | 1.0 | 1.8 | 5.0 | 3.1 | 1C/2C | 1C/2C | ||
For the 20 genes in class 1, Table 1 shows their human and budding yeast orthologs, MC, average cell length 24 hours after −N, cell number change (fold change) in 24 hours after −N, and major DNA peaks before and 24 hours after −N, measured by fluorescence-activated cell sorting (FACS). These 20 genes are also schematized in Fig. 2B by their reported functions (14). They include protein phosphatases and genes related to RNA/protein process (exo2, rpl2002, and sbh1), antioxidation [sod2 and spe1 (15)], biotin transporter (vht1), and intracellular vesicle transport (vps3, vps26, and vps35). The genes aut12, vac7, and SPBC83.16c may function in maintenance of vacuole structure during the G0 phase, since deletions showed abnormal vacuolar sizes and shapes in cells under −N (fig. S1). In these strains with abnormal vacuoles, DAF-FM DA fluorescence is reduced because of diminished arginine catabolism to nitric oxide, reflecting reduced amino acid degradation under −N (fig. S1). These genes may be required for proper nitrogen recycling.
Genes for the Nem1-Spo7 complex, the most essential signaling GZEs
As mentioned above, many genes in class 1 encode phosphatase-related proteins: the Nem1-Spo7 phosphatase complex (nem1, SPBC902.03), three protein phosphatase 2A (PP2A) regulators (par1; B’/B56 subunit, mug134; endosulfine, ypa1; protein tyrosine phosphatase activator), one PP1 regulator (ctf1), and one dual-specificity MAPK (mitogen-activated protein kinase) phosphatase regulator (rnc1). The only kinase was kin1, microtubule affinity-regulating kinase, deletion of which caused defective cytokinesis at G0 entry.
Among these signaling genes, Δnem1 and ΔSPBC902.03 promoted the most severe MC loss (Fig. 3A). Nem1 and SPBC902.03 ortholog, Spo7, form a phosphatase complex, with Nem1 as the catalytic subunit and Spo7 as the regulatory subunit. The complex regulates nuclear envelope morphology and phospholipid biosynthesis (16, 17). Nem1 highly resembles mammalian Dullard/CTDNEP1 (48% amino acid identity in the conserved C-terminal half), which is required for neural induction and nuclear membrane biogenesis (18, 19) and for modulating WNT and transforming growth factor–β (TGF-β) signalings (20, 21). In S. pombe, Nem1 protein localizes mainly in the nuclear membrane in both the VE and G0 phases (Fig. 3B), as does the nuclear membrane marker Cut11-mCherry (22).
Fig. 3. nem1 showed the most severe MC loss and manifested deformed nuclei that resembled those of the ned1-264 mutant.
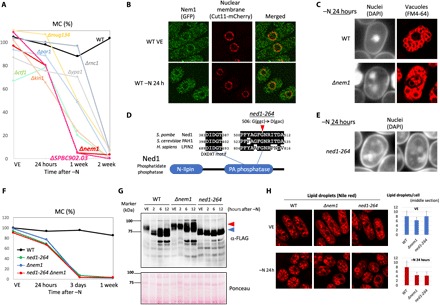
Western blot analysis also showed that Nem1 was required for Ned1 dephosphorylation after −N. (A) MC graph of class 1 genes related to phosphorylation signaling. (B) Fluorescence images of Nem1-GFP (green) and Cut11-mCherry (nuclear membrane, red) in WT cells in the VE phase and 24 hours after −N. (C) Fluorescence images of nuclei (DAPI) and vacuoles (FM4-64) in WT and Δnem1 cells. (D) Diagram of Ned1 protein. The mutation site of ned1-264 is indicated. (E) DAPI images of cell shape and nuclei in ned1-264 24 hours after −N. (F) MC graphs of the indicated strains. (G) Western blot analysis of Ned1-FLAG in WT, Δnem1, and ned1-264 in a 6% Phos-tag gel. Phos-tag traps phosphorylated proteins, reducing electrophoretic mobility. Samples were prepared from VE cells and 2, 6, and 12 hours after −N. Red and blue arrowheads indicate low and high electrophoretic mobility bands, respectively. (H) Fluorescence images of lipid droplets (Nile red) in WT, Δnem1, and ned1-264 cells in the VE phase and 24 hours after −N. Numbers of lipid droplets counted from midsection images of 20 cells for each strain were averaged and shown in right bar graphs with SD.
To better understand the severe MC loss in Δnem1, we observed cell morphology after −N. DAPI images showed deformed nuclei observed as several pieces, and FM4-64 (the fluorescent dye for vacuoles) images showed vacuoles located in the position of the nucleus (Fig. 3C). Since such a nuclear defect is hypothesized to damage MC maintenance, this defect was analyzed further.
It was reported that Nem1-Spo7 targets lipin, a phosphatidate lipid phosphatase catalyzing the hydrolytic dephosphorylation of a phosphatidate to a diacylglycerol. It controls cellular lipid homeostasis (17, 19, 23). Lipin in S. pombe is called Ned1, so it could also be required to maintain MC under −N as a Nem1 target. ned1 is an essential gene, so a deletion strain is not available. However, in a previous study, we identified 164 strains from a temperature-sensitive mutant library containing point mutations defective in MC maintenance under −N (24). We designated these mutated genes as “super housekeeping (SHK)” genes with regard to GZE, since they are required for both VE growth at high temperature and MC maintenance in the G0 phase. Among these mutants, we found a ned1 strain identified as SHK. This strain, designated ned1-264, had a point mutation changing Gly506 to aspartate in the conserved C-terminal phosphatidate phosphatase domain (Fig. 3D), located in the vicinity of the phosphatidate phosphatase catalytic motif DXDXT (19). As with Δnem1, ned1-264 cells showed deformed nuclei after −N (Fig. 3E). Also, they displayed a severe loss of MC after −N, to the same degree as Δnem1 (Fig. 3F). In addition, a double mutant of ned1-264 and Δnem1 showed an almost identical MC curve, implying that ned1 and nem1 are in the same MC regulation pathway.
To assess the genetic interaction between ned1 and nem1, Western blot analysis was conducted using WT, Δnem1, and ned1-264 strains, in which Ned1 was FLAG-tagged. Phos-tag gels (6%) were used to distinguish phosphorylated bands. In WT VE cells, the Ned1 band (~73 kDa) was detected as a low-mobility band (Fig. 3G, red arrowhead). This low-mobility band became two high-mobility bands (blue arrowhead) after λ protein phosphatase treatment, proving that it is phosphorylated (fig. S2). After −N, Ned1 becomes dephosphorylated within 2 hours, showing high electrophoretic mobility (Fig. 3G). The expression level of the Ned1 protein apparently increased after protracted −N (WT 6, 12 hours), but it remained dephosphorylated. On the other hand, dephosphorylation of Ned1 (shift from a low- to a high-mobility band) did not occur in Δnem1 2 hours after −N, providing evidence of the usual Nem1-Ned1 interaction. The increased expression of Ned1 protein at 6 and 12 hours was also detected in Δnem1, but the phosphorylated low-mobility band still remained. ned1-264 showed no low-mobility Ned1 band under either normal or −N condition, and no change of Ned1 expression level was detected, indicating that Gly506 in Ned1 is important for modification and subsequent control of expression level.
Since Ned1 is a phosphatidate lipid phosphatase, which directs phosphatidate metabolism to produce neutral storage lipids rather than structural phospholipids (25), its functional relevance in these mutants was examined. Cellular storage lipids were visualized as lipid droplets using a Nile red fluorescent dye (Fig. 3H). In VE cells, the number and shapes of lipid droplets were relatively similar among WT, Δnem1, and ned1-264 strains, with a slightly reduced number in Δnem1. Under −N, however, Δnem1 and ned1-264 cells showed about half the numbers of lipid droplets compared to WT. In these mutants, red fluorescence was observed in organelle membrane-like structures, indicating that lipid metabolism and usage under −N were disturbed in these mutants.
Defects in Δnem1 rescued by inhibition of the autophagy pathway
Nuclear defects observed in Δnem1 are suspected as the cause of MC loss. Therefore, electron micrographs were taken for further inspection. They showed vacuolated nuclei in Δnem1, very different in appearance from intact WT nuclei (Fig. 4A). Similar nuclear vacuolation was also observed in ΔSPBC902.03 cells (fig. S3).
Fig. 4. MC loss and nuclear defects of Δnem1 were rescued by deletions of autophagy genes.
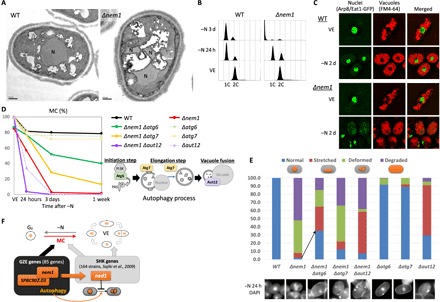
(A) Electron micrographs of WT and Δnem1 24 hours after −N. N, nuclei. (B) DNA contents of WT and Δnem1 in the VE phase, 24 hours and 3 days after −N, by FACS. (C) Fluorescence images of nuclear chromatin region–localized protein Arp8/Eat1-GFP (green), vacuoles (FM4-64, red), and merged images of WT and Δnem1 in the VE phase and 2 days after −N. (D) MC graphs of the indicated strains. A diagram of autophagy processes associated with the strains is shown. (E) Percentages of DAPI-stained nuclear shapes found in the indicated strain and DAPI images 24 hours after −N. Two hundred cells for each strain were counted. (F) Schematized diagram of the GZE (nem1-SPBC902.03) and SHK (ned1) genes.
This morphology was suggestive of nucleophagy, which degrades nuclei to potentially cause rapid MC loss. Actually, in Δnem1, FACS analysis indicated nuclear degradation within 3 days after −N (Fig. 4B), and green fluorescent protein (GFP)–tagged nuclear chromatin region protein [Arp8/Eat1-GFP (26)] accumulation was observed in vacuoles (Fig. 4C).
To test this idea, we created double-deletion mutants of Δnem1 with autophagy genes from different autophagy processes deleted. Intriguingly, a double deletion involving Δatg6 markedly improved the MC of Δnem1, from 3 to 52% on 3 days after −N (Fig. 4D). Double-deletion Δnem1 Δatg7 also rescued the MC of single Δnem1, but Δnem1 Δaut12 exacerbated it (Fig. 4D). Consistently, abnormal nuclear shapes caused by Δnem1 were ameliorated by double-deletion Δnem1 Δatg6 or Δnem1 Δatg7, but not by Δnem1 Δaut12 (Fig. 4E).
These results imply that deletions defective in processes upstream from nucleophagy can rescue MC, but those downstream cannot, since atg6 and atg7 are genes for the initial and elongation steps of autophagy, while aut12 participates in the last step (Fig. 4D, bottom right diagram). These results confirm that Nem1-Spo7 protects the nucleus from nucleophagy under −N. Similarly, defects of MC and nuclear shape in ned1-264 were also rescued in double mutant ned1-264 Δatg6 or ned1-264 Δatg7, but not in ned1-264 Δaut12 (fig. S4).
DISCUSSION
Genetic information generally exists in excess. Among ~5200 fission yeast genes, only ~1300 genes were identified as essential for growth and division (13, 14). Recently, however, it was reported that only 473 genes were sufficient to sustain and regenerate life of artificially produced bacteria (27). Here, we searched for genes exclusively required to sustain MC under −N and identified 85 of them. These genes are probably just the tip of the iceberg for maintaining MC under −N, but they appear to be the most essential. This minimal gene set indicates that recycling of proteins and RNA by anabolism, catabolism, and transport is necessary to ensure MC, in addition to metabolic processes involving small molecules and antioxidants in mitochondria and cytoplasm. Phosphatases play key roles in controlling MC in the early G0 phase. In contrast, nuclear chromatin functions that modify genome structure and autophagy are generally required well after long-term G0 quiescence. Autophagy has dual roles during pre-G0 division and MC maintenance under prolonged −N. Among the 85 GZE genes identified, the study of the Nem1-Spo7 phosphatase complex sheds particular light on the exquisite balance required to maintain MC. One such connection is between GZE (Nem1) and GZE (autophagy). Autophagy is induced under nutrient deficiency to degrade unnecessary cellular material for recycling, but our results concerning Nem1 deletion indicate that without proper protection, autophagy can be fatal. It is speculated that since disturbed lipid droplets in Δnem1 indicate improper lipid metabolism upon −N, nuclear membrane composition might also be disturbed in Δnem1, triggering nucleophagy. In addition, the Nem1 target, lipin/Ned1, is reported to down-regulate nuclear SREBPs (sterol regulatory element–binding proteins), which are regulators of the transcription program for lipogenesis and autophagy, under nutrient deficiency (28). Our results suggest a possible role for Nem1-Ned1 in controlling −N-induced autophagy by coupling and balancing transcriptional regulation and phospholipid metabolism.
As mentioned above, the other connection is between GZE (Nem1) and SHK (Ned1). We previously identified SHK genes required for MC maintenance in both the VE and G0 phases (24), and in this study, a genetic interaction between GZE (Nem1-Spo7) and SHK (Ned1) was revealed. This relationship suggests that the two gene groups may work as regulators and targets (Fig. 4F). GZE may function as a switch, adjusting to specific conditions, while SHK genes comprise an actual working unit regulated by that switch.
The GZE and SHK gene groups differ in relation to genes involved in protein phosphorylation signaling. Among the GZE genes, phosphatases outnumber kinases, but the opposite is true for SHK (29). It seems reasonable that under nutrient deficiency, crucial switches are regulated by ATP (adenosine triphosphate)–free phosphatases, rather than energy-requiring kinases. In addition to Nem1-Spo7, three PP2A-related strains (Δmug134, Δpar1, and Δypa1) were identified among class 1 GZE deletions, while a previous study identified cdc2-974 and cdc13-563 as SHK mutants (24). These strains show a similar phenotype, failing to conduct the second cell division at G0 entry. Since PP2A is a well-known phosphatase complex that regulates cell division via CDK (cyclin-dependent kinase) (30, 31), we speculate that the same sort of relationship, GZE regulating SHK, may also exist among these genes. A checkpoint to secure cell division machinery for MC may exist before the second mitotic division to complete G0 phase entry.
How cells maintain and execute MC is an interesting question from the standpoint of cancer stem cell therapy. The majority of cancer stem cells are in the G0 phase, thereby evading currently available therapies targeting dividing cells and maintaining malignant propagation. More than 40% of GZE genes identified here are reportedly linked to cancers (table S2). These two sets of genes, GZE and SHK, could reveal the landscape of genetic regulation of MC and may hold the key for more effective cancer therapy.
MATERIALS AND METHODS
Strains and yeast cell culture
Haploid deletion-mutant library sets were purchased from a commercial source (Bioneer) (13). The version 1 set (2814 strains including 2 duplicated strains) was initially purchased and upgraded with both version 2 (421 new strains and 127 upgraded strains) and version 3 (222 new strains and 325 upgraded strains) sets, resulting in 3455 unique strains. Upgraded strains replaced the older strains. The original strains contained three auxotrophic markers (ade6-M210/216, ura4-D18, and leu1-32), and these were removed by crossing with a WT strain (h-972) using a Biomek FX robot (Beckman Coulter). Mutants that failed to mate or form colonies were excluded (175 strains listed in table S3). The resulting 3280 prototrophic deletion-mutant strains included 64% of all protein-coding genes (3280 of 5124) and 84.9% of nonessential genes [based on Kim et al. (13), reporting 1260 genes as essential genes].
A C-terminally GFP-tagged nem1 strain was established by chromosomal integration under the native promoter, then crossed with the cut11-mCherry strain [a gift from T. Hayashi, Okinawa Institute of Science and Technology Graduate University (OIST), Japan]. A Δnem1 arp8/eat1-GFP strain was established by crossing Δnem1 and our laboratory stock, arp8/eat1-GFP strain (26).
Spot tests
For screening of the 3280 strains, spot tests were conducted on agar plates using Biomek FX (Beckman Coulter) and RoToR HDA (Singer Instruments) automation systems. Strains were spotted on EMM2−N agar plates and incubated under −N conditions. Since the agar contained trace quantities of nitrogen, nitrogen starvation required slightly longer incubation times. Three independent spot tests were conducted on EMM2−N using different incubation times (14 days and 1 and 2 months) (fig. S5A). After incubation, we respotted them in quadruplicate on EMM2 plates to replenish the nitrogen source and to check colony formation. These spot tests were for the first rough screening, which could have contained false positives, so additional MC assays were conducted afterward.
MC, changes in cell number, cell length measurements, and flow cytometry
Strains defective in colony formation in the first spot test were assayed for MC using liquid media. Nitrogen was depleted by changing the culture medium to EMM2−N using vacuum filtration with a nitrocellulose membrane (0.45-μm pore size) as described previously (24), and cells were incubated for designated periods under −N (fig. S5B). After incubation, cells were counted using a Multisizer 3 (Beckman Coulter), and MC was calculated as the percentage of colony formation on duplicate nutrient-rich YPD (yeast extract, peptone, and dextrose) plates streaked with 300 cells each. From this screening, strains that showed less than 50% MC after a 4-week incubation under −N were selected. For cell number changes, cell counts before and 24 hours after −N were determined using a Multisizer 3, and differences were calculated. For cell length, the long axes of 10 medium-sized cells were measured and averaged for each strain. To measure the DNA content of cells, flow cytometry was performed (3) using FACSCalibur (Becton Dickinson).
PCR deletion confirmation
Strains with MC <50% were checked by PCR using the primer sets listed in Kim et al. (13) to confirm proper gene deletions. PCR was performed with two primer sets for both 5′ and 3′ deletions. The 5′ deletions were checked with cp5, a gene-specific primer outside the deletion cassette and with either CPN10 or CPN1, which are primers that bind inside the cassette (fig. S6A). Similarly, the 3′ deletion was checked with cp3, as an outside primer, and either CPC3 or CPC1, as inside primers (fig. S6B). LA Taq DNA polymerase (Takara Bio Inc.) was used for PCR following the manufacturer’s instructions. Nine strains failed to show amplification bands, confirming deletion, and were removed (fig. S6). Genes associated with the remaining 85 deletions were considered GZE.
Light microscopy observation
Cell images using DIC, DAPI, FM4-64, DAF-FM DA, and Nile red staining were acquired using fluorescence microscopes [AxioPlan2 (Zeiss) and DeltaVision Elite (GE Healthcare)]. For nuclear staining, cells were fixed with 2% glutaraldehyde for 10 min on ice, washed three times with phosphate-buffered saline, and observed under a fluorescence microscope after mixing with DAPI (25 μg/ml) for staining. For vacuole staining, 1 μl of FM4-64 (1 μg/μl) (ThermoFisher) was added to 1 ml of cell culture, and cultures were incubated in the dark at room temperature for 45 min. After incubation, cells were washed in media without FM4-64, then incubated again for 45 min before observation. For nitric oxide staining, 5 μl of DAF-FM DA (1 μg/μl) (AdipoGen) was added to 1 ml of cell culture. Then, cultures were incubated in the dark at room temperature for 45 min. Cells were washed twice with media before observation. Signal intensity of DAF-FM DA was obtained using softWoRx software (GE Healthcare) and calculated by taking the average of 10 fluorescence points in each image. For lipid droplet staining, 1 μl of Nile red (1 μg/μl) (Wako) was added to 1 ml of cell culture, and cultures were incubated in the dark at room temperature for 5 min before observation.
Transmission electron microscopy
Cells were fixed with 2% glutaraldehyde in 100 mM phosphate buffer (pH 7.2) for 2 hours at 26°C, postfixed with 2% potassium permanganate overnight at 4°C, and embedded in Epon812 (TAAB). Ultrathin sections were stained in 2% uranyl acetate and Reynold’s lead citrate and viewed with a TEM JEM1230R (JEOL) operated at 100 kV.
Immunoblotting
Cell extracts from FLAG-tagged strains were prepared with a Multi-beads Shocker (Yasui Kikai) by the trichloroacetic acid (TCA) precipitation method. For lambda protein phosphatase (λPP) (New England BioLabs) treatment, NEBuffer for protein metallophosphatases (PMP) (New England BioLabs) with 1 mM PMSF (phenylmethylsulfonyl fluoride) (Wako) was used instead of TCA. Then, 1 μl of λPP was added per 20 μl of sample, containing 100 μg of protein, and incubated at 30°C for 30 min. After TCA precipitation, the same amounts of protein were loaded into each lane. SDS gels containing 6% Phos-tag (Wako) were used to perform electrophoresis, and samples were blotted onto nitrocellulose membranes. Anti-FLAG was used as the primary antibody, and horseradish peroxidase–conjugated secondary antibody and an enhanced chemiluminescence system (GE Healthcare) were used for signal expression. Signal detection used an LAS-3000 system (Fujifilm).
Metabolome analysis
WT cells were incubated at 26°C in EMM2 for VE and in EMM2−N for 6, 12, and 24 hours after shifting for −N, and metabolome samples were prepared as described previously (32, 33). At each time point, experiments were performed in triplicate. Samples were spiked with two internal standards, Pipes and Hepes, extracted, and separated by liquid chromatography on a ZIC-pHILIC column (Merck), and then measured using an LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific). MZmine 2 software was used for raw mass spectrum analysis (34). Detected amino acid peaks were identified by comparing mass/charge ratio values and retention times with standards.
Supplementary Material
Acknowledgments
We gratefully acknowledge T. Hayashi for the cut11-mCherry strain. We thank H. Zhang for experimental assistance and S. D. Aird for editing the manuscript. Funding: This work was supported by OIST. Author contributions: K.S. designed the experiments and wrote the manuscript. K.S. and Y.T. performed the experiments. L.U. supported the experiments. T.S. performed the electron microscopy visualization. T.P. performed the metabolome analyses. M.Y. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat5685/DC1
Fig. S1. Fluorescence images of nitric oxide (green, DAF-FM DA) and vacuoles (red, FM4-64) and merged images of indicated strains.
Fig. S2. A low-mobility band of Ned1-FLAG is phosphorylated.
Fig. S3. Electron micrograph of ΔSPBC902.03 at 24 hours after −N.
Fig. S4. ned1-264 showed MC loss and deformed nuclei, which were rescued by deletions of autophagy genes.
Fig. S5. Scheme of two screening procedures.
Fig. S6. PCR confirmation of gene deletion.
Table S1. Table of 85 GZE genes.
Table S2. List of cancer-related references for 85 GZE genes.
Table S3. Strains that failed to backcross or grow.
REFERENCES AND NOTES
- 1.Yoshida G. J., Saya H., Therapeutic strategies targeting cancer stem cells. Cancer Sci. 107, 5–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeishi S., Nakayama K. I., To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 107, 875–881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costello G., Rodgers L., Beach D., Fission Yeast Enters the Stationary Phase G0 State from Either Mitotic G1 or G2. Curr. Genet. 11, 119–125 (1986). [Google Scholar]
- 4.Su S. S., Tanaka Y., Samejima I., Tanaka K., Yanagida M., A nitrogen starvation-induced dormant G0 state in fission yeast: The establishment from uncommitted G1 state and its delay for return to proliferation. J. Cell Sci. 109 (Pt 6), 1347–1357 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Shimanuki M., Chung S.-Y., Chikashige Y., Kawasaki Y., Uehara L., Tsutsumi C., Hatanaka M., Hiraoka Y., Nagao K., Yanagida M., Two-step, extensive alterations in the transcriptome from G0 arrest to cell division in Schizosaccharomyces pombe. Genes Cells 12, 677–692 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Schlake T., Gutz H., Mating configurations in Schizosaccharomyces pombe strains of different geographical origins. Curr. Genet. 23, 108–114 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Yanagida M., Cellular quiescence: Are controlling genes conserved? Trends Cell Biol. 19, 705–715 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Mochida S., Yanagida M., Distinct modes of DNA damage response in S. pombe G0 and vegetative cells. Genes Cells 11, 13–27 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Kojima H., Urano Y., Kikuchi K., Higuchi T., Hirata Y., Nagano T., Fluorescent indicators for imaging nitric oxide production. Angew. Chem. Int. Ed. Engl. 38, 3209–3212 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Meiling-Wesse K., Barth H., Voss C., Barmark G., Murén E., Ronne H., Thumm M., Yeast Mon1p/Aut12p functions in vacuolar fusion of autophagosomes and cvt-vesicles. FEBS Lett. 530, 174–180 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Wang C.-W., Stromhaug P. E., Shima J., Klionsky D. J., The Ccz1-Mon1 protein complex is required for the late step of multiple vacuole delivery pathways. J. Biol. Chem. 277, 47917–47927 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofman-Bang J., Nitrogen catabolite repression in Saccharomyces cerevisiae. Mol. Biotechnol. 12, 35–73 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Kim D.-U., Hayles J., Kim D., Wood V., Park H.-O., Won M., Yoo H.-S., Duhig T., Nam M., Palmer G., Han S., Jeffery L., Baek S.-T., Lee H., Shim Y. S., Lee M., Kim L., Heo K.-S., Noh E. J., Lee A.-R., Jang Y.-J., Chung K.-S., Choi S.-J., Park J.-Y., Park Y., Kim H. M., Park S.-K., Park H.-J., Kang E.-J., Bai Kim H., Kang H.-S., Park H.-M., Kim K., Song K., Song K. B., Nurse P., Hoe K.-L., Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28, 617–623 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood V., Harris M. A., McDowall M. D., Rutherford K., Vaughan B. W., Staines D. M., Aslett M., Lock A., Bähler J., Kersey P. J., Oliver S. G., PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Res. 40, D695–D699 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenberg T., Knauer H., Schauer A., Büttner S., Ruckenstuhl C., Carmona-Gutierrez D., Ring J., Schroeder S., Magnes C., Antonacci L., Fussi H., Deszcz L., Hartl R., Schraml E., Criollo A., Megalou E., Weiskopf D., Laun P., Heeren G., Breitenbach M., Grubeck-Loebenstein B., Herker E., Fahrenkrog B., Fröhlich K.-U., Sinner F., Tavernarakis N., Minois N., Kroemer G., Madeo F., Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Siniossoglou S., Santos-Rosa H., Rappsilber J., Mann M., Hurt E., A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J. 17, 6449–6464 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santos-Rosa H., Leung J., Grimsey N., Peak-Chew S., Siniossoglou S., The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J. 24, 1931–1941 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satow R., Kurisaki A., Chan T.-c., Hamazaki T. S., Asashima M., Dullard promotes degradation and dephosphorylation of BMP receptors and is required for neural induction. Dev. Cell 11, 763–774 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Kim Y., Gentry M. S., Harris T. E., Wiley S. E., Lawrence J. C. Jr., Dixon J. E., A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 6596–6601 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka S. S., Nakane A., Yamaguchi Y. L., Terabayashi T., Abe T., Nakao K., Asashima M., Steiner K. A., Tam P. P. L., Nishinakamura R., Dullard/Ctdnep1 modulates WNT signalling activity for the formation of primordial germ cells in the mouse embryo. PLOS ONE 8, e57428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayata T., Ezura Y., Asashima M., Nishinakamura R., Noda M., Dullard/Ctdnep1 regulates endochondral ossification via suppression of TGF-β signaling. J. Bone Miner. Res. 30, 318–329 (2015). [DOI] [PubMed] [Google Scholar]
- 22.West R. R., Vaisberg E. V., Ding R., Nurse P., McIntosh J. R., cut11+: A gene required for cell cycle-dependent spindle pole body anchoring in the nuclear envelope and bipolar spindle formation in Schizosaccharomyces pombe. Mol. Biol. Cell. 9, 2839–2855 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith S. W., Weiss S. B., Kennedy E. P., The enzymatic dephosphorylation of phosphatidic acids. J. Biol. Chem. 228, 915–922 (1957). [PubMed] [Google Scholar]
- 24.Sajiki K., Hatanaka M., Nakamura T., Takeda K., Shimanuki M., Yoshida T., Hanyu Y., Hayashi T., Nakaseko Y., Yanagida M., Genetic control of cellular quiescence in S. pombe. J. Cell. Sci. 122, 1418–1429 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Makarova M., Gu Y., Chen J.-S., Beckley J. R., Louise Gould K., Oliferenko S., Temporal Regulation of Lipin Activity Diverged to Account for Differences in Mitotic Programs. Curr. Biol. 26, 237–243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tatebe H., Goshima G., Takeda K., Nakagawa T., Kinoshita K., Yanagida M., Fission yeast living mitosis visualized by GFP-tagged gene products. Micron 32, 67–74 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Hutchison C. A. III, Chuang R.-Y., Noskov V. N., Assad-Garcia N., Deerinck T. J., Ellisman M. H., Gill J., Kannan K., Karas B. J., Ma L., Pelletier J. F., Qi Z.-Q., Richter R. A., Strychalski E. A., Sun L., Suzuki Y., Tsvetanova B., Wise K. S., Smith H. O., Glass J. I., Merryman C., Gibson D. G., Venter J. C., Design and synthesis of a minimal bacterial genome. Science 351, aad6253 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Peterson T. R., Sengupta S. S., Harris T. E., Carmack A. E., Kang S. A., Balderas E., Guertin D. A., Madden K. L., Carpenter A. E., Finck B. N., Sabatini D. M., mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanagida M., Ikai N., Shimanuki M., Sajiki K., Nutrient limitations alter cell division control and chromosome segregation through growth-related kinases and phosphatases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3508–3520 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunter T., Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 80, 225–236 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Yanagida M., Kinoshita N., Stone E. M., Yamano H., Protein phosphatases and cell division cycle control. Ciba Found. Symp. 170, 130–140–140–136 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Pluskal T., Nakamura T., Villar-Briones A., Yanagida M., Metabolic profiling of the fission yeast S. pombe: Quantification of compounds under different temperatures and genetic perturbation. Mol. Biosyst. 6, 182–198 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Sajiki K., Pluskal T., Shimanuki M., Yanagida M., Metabolomic analysis of fission yeast at the onset of nitrogen starvation. Metabolites 3, 1118–1129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pluskal T., Castillo S., Villar-Briones A., Orešič M., MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat5685/DC1
Fig. S1. Fluorescence images of nitric oxide (green, DAF-FM DA) and vacuoles (red, FM4-64) and merged images of indicated strains.
Fig. S2. A low-mobility band of Ned1-FLAG is phosphorylated.
Fig. S3. Electron micrograph of ΔSPBC902.03 at 24 hours after −N.
Fig. S4. ned1-264 showed MC loss and deformed nuclei, which were rescued by deletions of autophagy genes.
Fig. S5. Scheme of two screening procedures.
Fig. S6. PCR confirmation of gene deletion.
Table S1. Table of 85 GZE genes.
Table S2. List of cancer-related references for 85 GZE genes.
Table S3. Strains that failed to backcross or grow.