

Key Points
DCA, an inhibitor of PDKs, impedes glucose uptake and aerobic glycolysis in activated platelets.
DCA inhibits agonist-induced human and mouse platelet aggregation and arterial thrombosis without altering hemostasis in mice.
Abstract
Resting platelets rely on oxidative phosphorylation (OXPHOS) and aerobic glycolysis (conversion of glucose to lactate in the presence of oxygen) to generate adenosine triphosphate, whereas activated platelets exhibit a high level of aerobic glycolysis, suggesting the existence of metabolic flexibility in platelets. Mitochondrial pyruvate dehydrogenase kinases (PDK 1-4) play a pivotal role in metabolic flexibility by inhibiting pyruvate dehydrogenase complex. We determined whether metabolic reprogramming, diverting metabolism from aerobic glycolysis back to OXPHOS, would inhibit platelet function. PDKs activity in human and mouse platelets was inhibited with dichloroacetic acid (DCA), a potent inhibitor of all 4 forms of PDK. Human and mouse platelets pretreated with DCA exhibited decreased platelet aggregation to suboptimal doses of collagen, convulxin, thrombin, and adenosine diphosphate concomitant with decreased glucose uptake. Bioenergetics profile revealed that platelets pretreated with DCA exhibited decreased aerobic glycolysis in response to convulxin only. Furthermore, DCA inhibited ATP secretion, thromboxane A2 generation, and tyrosine phosphorylation of Syk and PLCγ2 in response to collagen or convulxin in human and mouse platelets (P < .05 vs vehicle treated). In the flow chamber assay, human and mouse blood pretreated with DCA formed smaller thrombi when perfused over collagen for 10 minutes at an arterial shear rate of 1500 s−1 (P < .05 vs control). Wild-type mice pretreated with DCA were less susceptible to thrombosis in the FeCl3-induced carotid and laser injury–induced mesenteric artery thrombosis models (P < .05 vs vehicle control), without altering hemostasis. Targeting metabolic plasticity with DCA may be explored as a novel strategy to inhibit platelet function.
Visual Abstract
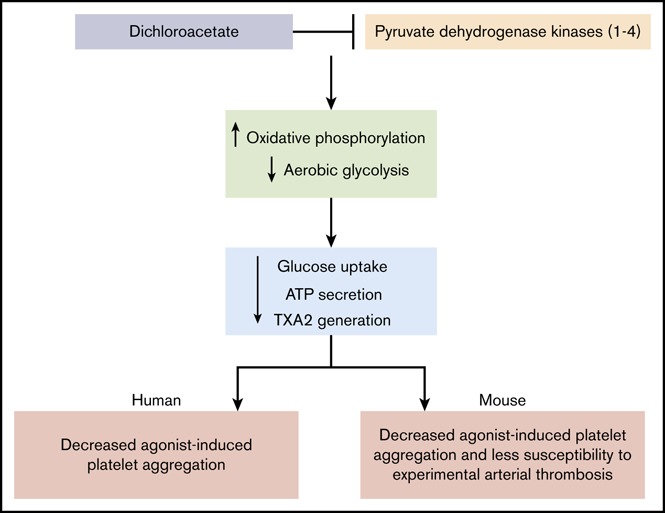
Introduction
Most of the processes associated with platelet activation in response to endothelial injury, including aggregation, integrin αIIbβ3 activation, and granule secretion, are energy consuming.1,2 Despite anucleate, platelets have functional mitochondria. For several years mitochondrial oxidative phosphorylation (OXPHOS) was considered the sole energy pathway for quiescent platelets.1 Bioenergetics profile studies in platelets have revealed that resting platelets rely on aerobic glycolysis (formation of lactate in the presence of oxygen, also known as Warburg effect) and mitochondrial OXPHOS for energy requirement.3 On the other hand, platelet activation is accompanied by an increase in the rate of aerobic glycolysis relative to OXPHOS, suggesting that metabolic plasticity exists in platelets.4,5
Mitochondrial pyruvate dehydrogenase (PDH) complex converts pyruvate (the end product of glycolysis in the cytosol) to acetyl coenzyme A, which is further oxidized to tricarboxylic acid cycle to produce energy (adenosine triphosphates [ATPs]) in the OXPHOS pathway.6 PDH complex activity is tightly regulated by pyruvate dehydrogenase kinases (PDKs 1-4).7 PDKs inhibit PDH complex activity by phosphorylating it on the E1α subunit8 and thereby divert the pyruvate flux from tricarboxylic acid cycle toward aerobic glycolysis (more lactate production). In other words, we can say that mitochondrial PDKs by inhibiting PDH activity regulate aerobic glycolysis. The role of PDKs in platelet function and thrombosis remains unclear.
Herein, we determined whether diverting metabolism in platelets from aerobic glycolysis back to OXPHOS would inhibit platelet aggregation and arterial thrombosis. Global knockout or floxed PDK mice of all 4 isoforms (PDK1-4) do not exist. Therefore, we used dicholoroacetic acid (DCA), a small molecule that inhibits PDKs 1-4 activity and is known to divert metabolism from aerobic glycolysis back to mitochondrial OXPHOS.9-11 In the current study, we provide evidence that DCA inhibits platelet aggregation in response to suboptimal doses of various agonists and experimental arterial thrombosis that was associated with decreased glucose uptake and partial reduction in aerobic glycolysis.
Materials and methods
An expanded version of “Materials and methods” is available in the supplemental Data.
Animals and human samples
Mice (male and female, C57BL/6J background) used in the present study were 3 to 4 weeks (14-16 g) or 8 to 10 weeks (22-28 g) old. Platelets for infusion were isolated from 4- to 6-month-old donor wild-type (WT) mice. Human venous blood was obtained from healthy donors by the Declaration of Helsinki and approved by the Institutional Review Board, University of Iowa. All participants gave written informed consent. The University of Iowa Animal Care and Use Committee approved all experiments. In vivo studies were performed according to the current Animal Research: Reporting of In Vivo Experiment guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
In vitro platelet aggregation and secretion
Platelet-rich plasma (PRP) or washed platelets (0.2 × 109/mL) were stirred (1200 rpm) at 37°C for 2 minutes in a Chrono-log Whole Blood/Optical Lumi-Aggregometer (model 700-2) before the addition of agonists. Aggregation was induced with different agonists (convulxin, collagen, thrombin, and adenosine diphosphate [ADP]). Aggregation was measured as percent change in light transmission, where 100% refers to transmittance through the blank sample. Platelet-dense granule secretion was determined by measuring the release of ATP by adding luciferin-luciferase reagent. ATP release and aggregation were performed simultaneously in a Lumi-aggregometer at 37°C. To examine the effect of DCA on platelet aggregation and secretion, washed platelets or PRP was pretreated with vehicle or DCA at different doses (10 and 25 mM) for 10 minutes before treatment with various agonists.
Platelet bioenergetics
The 24-well format Seahorse extracellular flux analyzer, XF24 (Seahorse Bioscience, Chicopee, MA), a service provided by Metabolic Core at the University of Iowa, was used to measure bioenergetics (oxygen consumption rate [OCR] and extracellular acidification rate [ECAR]). Pooled platelets from 5 mice were diluted to a concentration of 1 × 108 in XF Dulbecco’s modified Eagle medium assay buffer (Dulbecco’s modified Eagle medium with 1 mM pyruvate, 5.5 mM d-glucose, 4 mM l-glutamine, pH 7.4) and were seeded onto Cell-Tak coated XF24 microplates, and mitochondrial stress test was performed as described.3,5 These assays were performed with a 10-minute pretreatment of platelets in the absence or presence of DCA (10 and 25 mM) with convulxin (25 ng/mL), thrombin (0.05 U/mL), and ADP (0.5 µM) as platelet agonists.
Bioflux flow chamber assay
Experiments were performed using a BioFlux 200 (Fluxion Biosciences) microfluidics flow chamber. The channels were coated with type I collagen fibrils (150 μg/mL) for 1 hour at room temperature and then blocked with 0.5% bovine serum albumin for 30 minutes. Platelets were isolated from citrated whole blood as described above, washed, labeled with the fluorescent dye (calcein AM; 2 µM), and reconstituted back with platelet-poor plasma and remaining blood containing leukocytes and red blood cells. An amount of 300 µL of whole blood pretreated with DCA or without DCA (vehicle) was perfused over the collagen-coated plate at shear stress 1500 s−1 for 10 minutes. Experiments were performed in triplicate using whole blood from human and mice. The fluorescently labeled platelets/thrombi on the collagen-coated surface were analyzed using ImageJ software from the National Institutes of Health (Bethesda, MD).
FeCl3 injury-induced carotid thrombosis
FeCl3 injury-induced carotid thrombosis was assessed by intravital microscopy as described.12-14 Briefly, 8- to 10-week-old mice were anesthetized using 100-mg/kg ketamine and 10-mg/kg xylazine. Platelets labeled with calcein green (2.5 × 109 platelets/kg body weight) were infused through the retro-orbital plexus. The common carotid artery was carefully exposed and kept moist by superfusion with warm (∼37°C) saline. Whatman paper (0.4 × 0.4 mm2) saturated with ferric chloride (7.5%) solution was applied topically for 3 minutes, and thrombus formation in the injured carotid vessel was monitored in real time using a Nikon upright microscope with a Plan Fluor 4×/0.2 objective. Thrombus formation in real time was recorded using a high-speed electron-multiplying camera for 30 minutes or until occlusion occurred. The time to form occlusive thrombus was considered as the time required for blood to stop flowing entirely for >1 minute. Videos were evaluated off-line using a Nikon computer-assisted image analysis program.
Laser injury-induced mesenteric artery thrombosis
The Micropoint laser ablation system (Andor Technology) was used to make an injury in the mesenteric arterioles as described.12,15 Briefly, young mice (3 to 4 week [14-16 g] old males) were used to minimize fat surrounding the arterioles and facilitate focusing of the laser. Fluorescent platelets labeled with calcein green (1.5 × 109 platelets/kg body weight) were infused in anesthetized mice through the retro-orbital plexus. Infused platelets were isolated from adult (4-5 months) WT donor mice. Mesenteric arterioles having a diameter of ∼80 to 100 μm (with shear rates of ∼1300-1800 s−1) were used for the study. The specific illumination of the area of interest was carried out through the microscope eyepiece. The wavelength of light in the range of 365 to 400 nm with the maximum output of 50 to 500 μJ was used for illumination. The power and frequency of pulses were regulated by software and empirically defined. Thrombus growth in the injured vessel was monitored in real time by using a Nikon upright microscope with a Plan Fluor 10×/0.3 objective, and thrombus formation over time was recorded using a high-speed EM camera for 3 to 4 minutes. In our experimental setup, in the laser injury model, the thrombus grows to its maximum size in ∼1 minute and then gradually disintegrates over time. Videos were evaluated, and mean fluorescence intensity was calculated using a Nikon computer-assisted image analysis program.
Thromboxane (TXB2) Assay
The TXB2 assay was done as described.16 Briefly, PRP (0.2 × 109 platelets/mL) was treated with agonists for 3 minutes utilizing an aggregometer, and the reaction was stopped by quickly freezing the sample in a dry ice–methanol bath. Before performing the TXB2 measurement assay, samples were thawed and centrifuged at 15 000g for 3 minutes. The supernatants were diluted 1:50 with assay buffer. Levels of TXB2 were determined in triplicate using an immunoassay kit (Abcam Thromboxane B2 enzyme-linked immunosorbent assay kit) according to the manufacturer’s instructions.
Glucose uptake assay
A luminescence-based assay was used to measure glucose uptake in platelets as per the manufacturer’s instruction (glucose uptake-Glo assay kit from Promega). Briefly, washed platelets (2 × 105 in 50 μL of glucose-free tyrode buffer) in a 96-well clear bottom plate were preincubated in either the presence or absence of DCA (10 and 25 mM) and cytochalasin B (50 μM). To each well, 1 mM 2-deoxyglucose (2-DG) was added followed by addition of agonists (convulxin, 25 ng/mL; collagen, 0.46 μg/mL; thrombin, 0.05 U/mL; and ADP, 0.5 μM) to the specific wells. When 2-DG is added to cells, it is transported across the membrane and rapidly phosphorylated to 2-DG-6-phosphate (2DG6P) in the same manner as glucose and accumulates in the cell. After 15 minutes of stimulation, 25 µL of Stop buffer (acid detergent) was added to lyse the cells, terminate uptake, and destroy any reduced nicotinamide adenine dinucleotide phosphate within the cells. Then 25 µL of neutralization buffer was added to neutralize the acid, and the solution was shaken briefly. Luminescence produced by recombinant luciferase that is proportional to the concentration of 2DG6P was measured using a luminometer (Spectramax i3X) within 10 minutes after addition of detection reagent.
Statistical analysis
For statistical analysis, GraphPad Prism software, version 7.03, was used. Results are reported as the mean ± standard error of the mean (SEM). The statistical significance of the difference between means was assessed using the unpaired Student t test (for the comparison of 2 groups) or by repeated measure 2-way analysis of variance followed by Tukey's multiple comparisons test (for the comparison of >2 groups). P < .05 was considered significant.
Results
DCA inhibits agonist-induced platelet aggregation in vitro
To determine the role of aerobic glycolysis in platelet function, PDKs activity in human and mouse platelets was inhibited with DCA, a potent inhibitor of all 4 forms of PDK (1-4).7,17,18 Platelets were activated using different agonists (collagen, convulxin, thrombin, and ADP) that are known to initiate different pathways, which participate in integrin αIIβ3 signaling and thereby platelet aggregation. The dose of DCA was chosen from published studies.10,11,19,20 We found that human and mouse platelets pretreated with DCA showed a significantly impaired platelet aggregation response with suboptimal doses of various agonists, including collagen, convulxin, thrombin, and ADP, in a dose-dependent manner (P < .05, N = 4-5 per group; Figure 1). At higher doses, DCA is known to induce apoptosis in cells. To rule out the possibility that observed impaired platelet aggregation could be related to apoptosis, we determined phosphatidylserine (PS) by Annexin V assay. One of the earlier events of apoptosis is the translocation of PS from the inner side of the membrane to outside surface. Annexin V, which has a high affinity for PS, is routinely used for detection of apoptosis using flow cytometry. We found that DCA up to a dose of 100 mM did not induce apoptosis in human and mouse platelets (supplemental Figure 1).
Figure 1.

DCA inhibits agonist-induced platelet aggregation. PRP or washed platelets from human (A) and mouse (B) were stimulated with suboptimal doses of convulxin, collagen, ADP, and thrombin. Results are expressed as the percentage change in light transmission concerning the blank (platelet-poor plasma/buffer without platelets), set at 100%. The upper panel in each bar graph denotes the representative aggregation curves (blue, control; black, 10 mM DCA; red, 25 mM DCA). For mouse studies, PRP or washed platelets were pooled from 3 to 4 mice (n = 5 individual experiments per group). For human studies, PRP or washed platelets are from the individual donors (n = 5 per group). Values are mean ± SEM. *P < .05 vs control (vehicle).
DCA inhibits glucose uptake in activated platelets
A recent study suggested that glucose uptake and metabolism contribute to platelet function.21 We determined whether the inhibitory effect of DCA on platelet aggregation is associated with glucose uptake. We found that DCA did not inhibit glucose uptake in quiescent washed platelets (Figure 2). Glucose uptake was increased by approximately twofold in response to suboptimal doses of agonists, including collagen, convulxin, thrombin, and ADP (Figure 2). Both human and mouse platelets pretreated with DCA showed a significant decrease in glucose uptake in response to suboptimal doses of various agonists, suggesting an association between decreased glucose uptake and reduced platelet aggregation.
Figure 2.

DCA inhibits glucose uptake in agonist-induced platelet activation. Measurement of 2-DG uptake in washed human (A) and mouse (B) platelets with a luminescence-based assay kit. Luminescence (produced by 2DG6P) was monitored in resting and platelets response to suboptimal dose of collagen, convulxin, thrombin, and ADP. For mouse studies, washed platelets were pooled from 3 to 4 mice (n = 5 individual experiments per group). For human studies, washed platelets are from the individual donors (n = 5 per group). Values are mean ± SEM. *P < .05 vs unstimulated platelets; #P < .05 vs control (vehicle).
DCA increases OXPHOS and inhibits aerobic glycolysis in convulxin-stimulated platelets
To determine whether the inhibitory effect of DCA on platelet aggregation and glucose uptake is associated with a metabolic reprogramming from aerobic glycolysis back to mitochondrial OXPHOS, metabolic profiling was performed using mouse platelets. Using Seahorse extracellular flux analyzer, the OCR and ECAR, which are indirect measures of mitochondrial OXPHOS and aerobic glycolysis, respectively, were calculated. The compounds (oligomycin, carbonylcyanide p-trifluoromethoxy phenylhydrazone [FCCP], and a mix of rotenone A/myxothiazol) were serially injected to measure ATP production, maximal respiration, and nonmitochondrial respiration, respectively. Convulxin (25 ng/mL) or vehicle was added to platelets at time 0. We found a significant increase in basal OCR and ECAR in convulxin-stimulated platelets when compared with control resting platelets (Figure 3). After 8 minutes, the ATP synthase (complex V) inhibitor, oligomycin (1 μg/mL), was injected to measure ATP production. Oligomycin targets the ATP synthase (complex V) and decreases the OCR, thus representing the portion of basal respiration rate that is being used to drive ATP production. As expected, we observed a decrease in OCR in all groups (Figure 3). To estimate the maximal OCR, the proton ionophore, FCCP (1 μM), was added, which resulted in enhancement of respiration in all groups. Convulxin-stimulated pretreated DCA platelets showed a dose-dependent increase in respiration compared with convulxin-stimulated platelets, suggesting an increase in OXPHOS. In other words, DCA reprogrammed the pyruvate flux from aerobic glycolysis back to OXPHOS most likely by inhibiting PDKs. To measure nonmitochondrial sources of oxygen consumption, the mitochondrial complex III inhibitors, rotenone A (10 μM)/myxothiazol (0.5 μM), were injected 16 minutes after FCCP, which caused a decrease in OCR in all the groups. Finally, we found that DCA inhibited ECAR in the convulxin-stimulated platelets in a dose-dependent manner (Figure 3B). Next, we determined whether DCA could inhibit aerobic glycolysis in response to suboptimal doses of thrombin (0.05 U/mL) and ADP (0.5 μg/mL). To our surprise, we found that thrombin or ADP-stimulated pretreated DCA platelets exhibited increased OCR, but comparable ECAR (supplemental Figure 2). Together, these results suggest that DCA inhibits aerobic glycolysis (decreased lactate) in platelet response to convulxin, but not to thrombin or ADP (at suboptimal doses), suggesting an association between glycoprotein VI (GPVI) signaling and aerobic glycolysis.
Figure 3.

DCA inhibits aerobic glycolysis in convulxin-stimulated platelets. Pooled washed platelets from 5 mice were used to measure bioenergetics. OCR (A) and ECAR (B) were measured in platelets response to convulxin in the presence or absence of DCA (10 and 25 mM) with sequential injections of 1 μg/mL oligomycin, 1 μM proton ionophore FCCP, and 1 μM rotenone/0.5 μM myxothiazol. Convulxin was added at time 0 just before the microplates were put in the Seahorse extracellular flux analyzer. RP with or without DCA was used as a control. All the bioenergetic measurements were normalized to protein content per well. Values shown in bar graph are mean ± SEM, with n = 9 values per group. *P < .05 vs unstimulated platelets (no convulxin, blue bar); #P < .05 vs convulxin-stimulated platelets (violet bar). Convx, convulxin; Rot/Myx, rotenone/myxothiazol.
DCA inhibits TXB2 A2 generation, ATP secretion, and tyrosine phosphorylation of Syk and PLCγ2
Because DCA inhibited aerobic glycolysis in convulxin-stimulated platelets, we determined the effect of DCA on GPVI expression levels. We found that DCA by itself does not affect basal GPVI expression levels (supplemental Figure 3). We next determined whether DCA inhibits TXB2 A2 generation, ATP secretion, and kinases Syk and phospholipase C (PLCγ2), all of which are known to mediate collagen or convulxin-induced platelet activation. We found that human and mouse platelets pretreated with DCA and stimulated with suboptimal dose of collagen exhibited a significant decrease in TXB2 generation and ATP secretion (Figure 4). However, at the suboptimal dose (0.46 μg/mL), collagen did not affect α-granule release (assayed by P-selectin exposure; supplemental Figure 4). Furthermore, human and mouse platelets pretreated with DCA exhibited decreased phosphorylation of Syk and PLCγ2 in a dose-dependent manner in response to convulxin (P < .05, N = 5 per group; Figure 5).
Figure 4.

DCA inhibits TXB2 A2 production and ATP secretion in platelet response to collagen. The level of TXB2 A2 generation was measured in human (A, left) and mouse (B, left) RP and in platelet response to suboptimal dose of collagen (0.46 μg). PRP samples were preincubated in the presence or absence of DCA (10 and 25 mM), aspirin (100 μM), and indomethacin (20 μM). The aggregation was performed for 3 minutes, and the reaction was stopped by quickly freezing the sample in a dry ice–methanol bath. Samples were prepared, and the level of TXA2 was measured with an enzyme-linked immunosorbent assay kit. ATP secretion was measured by luciferase/luciferin-based reaction. Luminescence generated by secreted ATP from human (A, right) and mouse (B, right) platelets were monitored using a Lumi-aggregometer. Asp., aspirin; Ind., indomethacine; NS, nonsignificant.
Figure 5.

DCA inhibits phosphorylation of Syk and PLCγ2 in convulxin-stimulated platelets. Washed platelets (0.2 × 109/mL) from human (A) and mouse (B) were preincubated with DCA (10 and 25 mM) for 10 minutes at room temperature and stimulated with convulxin (25 ng/mL) for 10 minutes. The upper panels in A and B show representative images of immunoblots of phospho-Syk (Tyr 525/526), total Syk, phospho-PLCγ2 (Tyr1217), and total PLCγ2. Bottom panels indicate densitometry analysis of immunoblots using ImageJ software. The data are expressed as a ratio of phospho-Syk/total Syk and phospho-PLCγ2/total PLCγ2. Values are mean ± SEM, with n = 5 human or mice per group.
DCA inhibits thrombus formation in vitro and in vivo
To examine whether DCA inhibits platelet aggregation in whole blood, in addition to washed platelets and PRP, we performed in vitro studies using microfluidics flow chamber assay. The surface area covered by fluorescently labeled platelets was determined to quantify the size of thrombi. Under arterial shear rate (1500 s−1), human and mouse blood pretreated with either 10 or 25 mM DCA (doses used for in vitro aggregation) did not inhibit thrombus formation (supplemental Figure 5). However, human and mouse blood pretreated with a dose of 50 mM DCA significantly formed smaller thrombi when perfused over collagen for 10 minutes at a shear rate of 1500 s−1 (P < .05, n = 3-4 per group; Figure 6). Furthermore, we found that WT mouse platelets pretreated with DCA adhere less to collagen in a dose-dependent manner (supplemental Figure 6). We next determined whether DCA inhibits thrombosis and hemostasis in vivo. To define the hemostatic phenotype, we measured the tail-transection bleeding time. Mice were divided into 2 groups, 1 pretreated with DCA (600 mg/kg) and another vehicle. The dose of DCA used in vivo was comparable to that used in microfluidics flow chamber assay. DCA at this dose did not induce apoptosis in mouse and human platelets in vitro (supplemental Figure 1). Tail bleeding time was comparable among groups, suggesting that DCA treatment does not alter hemostasis (Figure 7A). To define the effect of DCA in thrombosis, we determined platelet aggregation in vitro and in vivo by measuring time to complete occlusion in the FeCl3-induced carotid thrombosis model. PRP isolated after DCA infusion showed a significantly impaired aggregation response with collagen (P < .05, N = 5 per group; Figure 7B). In line with these observations, in another set of experiments, the time to vessel occlusion was significantly prolonged in the mice pretreated with DCA when compared with vehicle control group (19.2 ± 1.5 minutes vs 14.4 ± 0.6 minutes, P < .05, N = 8-10 mice per group; Figure 7C). To ensure the inhibitory effect of DCA on experimental thrombosis is generalizable to the broader context, we evaluated the role of DCA in modulating thrombosis in another assay, laser-induced mesenteric thrombosis model. We found that the mean fluorescence intensity was significantly decreased in the mice pretreated with DCA compared with vehicle control group (Figure 7D).
Figure 6.

DCA inhibits platelet aggregates/thrombus formation in vitro. The analysis was done using flow chamber system from Bioflux Microfluidics. Human (A) and mouse (B) whole blood pretreated with 50 mM DCA (lower channels) or control (upper channels) was perfused over a collagen-coated (150 μg/mL) surface for 10 minutes at a shear rate of 1500 s−1. Left panels show representative images taken using a 10× objective. Right panels show quantification of the surfaced area covered by fluorescent platelets after 10 minutes of perfusion. Four areas from different areas of flow chamber were analyzed from each blood sample. Data represent the mean percentage of surface area covered by fluorescence platelets ± SEM, with n = 5 per group for mice and 3 per group for the human.
Figure 7.

DCA inhibits experimental arterial thrombosis without altering tail bleeding time. (A) The tail-transection bleeding time was determined as the time taken for the initial cessation of bleeding after transection in vehicle or DCA-treated group. Each symbol represents a single mouse. Horizontal bar shows the mean of each group ± SEM, n = 15 per group. (B) Aggregation induced by collagen (0.5 µg/mL) in PRP from mice treated with DCA or vehicle. Upper panel denotes the representative aggregation curves (blue, vehicle; black, DCA). Values are represented as mean ± SEM, with n = 4 mice per group. (C) The left panel shows representative microphotographs (4× objective), and right panel shows time to occlusive thrombus in the FeCl3-induced carotid artery injury model. Each symbol represents a single mouse. Horizontal bars show the mean of each group ± SEM, with n = 8 to 10 per group. (D) The left panel shows representative microphotographs (10× objective), and right panel shows time mean fluorescence intensity in the laser-induced mesentery artery injury model. N = 10 to 12 vessels from 4 mice per group. *P < .05 compared with vehicle control. Platelets were labeled with calcein green. White line delineates vessel.
Discussion
Current potent antiplatelet therapies available, including abciximab, P2Y12 inhibitors, diminish the risk of thrombosis in patients with acute coronary syndrome but significantly increase the possibility of bleeding and are not suitable for long-term use. Therefore, new antiplatelet strategies are required to inhibit thrombosis with minimal bleeding risk. In this study, utilizing the mouse and human platelets, we provide evidence that targeting metabolic plasticity with small molecule DCA, an inhibitor of mitochondrial PDKs known to regulate aerobic glycolysis by controlling pyruvate flux, may be explored as a novel strategy to inhibit platelet function and arterial thrombosis.
Resting platelets rely on both mitochondrial OXPHOS and aerobic glycolysis for energy demand, whereas activated platelets exhibit a high degree of aerobic glycolysis. It seems paradoxical that activated platelets prefer aerobic glycolysis instead of OXPHOS because aerobic glycolysis is less efficient and generates 2 ATP molecules per molecule of glucose, whereas OXPHOS generates up to 36 ATP molecules per molecule of glucose. Despite low efficiency, the rate of ATP generation is 100× faster in aerobic glycolysis compared with OXPHOS.22 Because glucose is sufficient, we speculated that an inefficient but accelerated rate of ATP generation might be well suited to fulfill the short-term requirements of high-energy demand of activated platelets to form the blood clot in the shortest time. Aerobic glycolysis regenerates NAD+, a cofactor necessary to maintain high glycolytic flux. It is also possible that upon platelet activation, increased aerobic glycolysis may facilitate faster glucose metabolism to meet the short but high-energy demand of activated platelets. We targeted PDKs, with a small molecule DCA, which is known to inhibit aerobic glycolysis.9,17,18 We found that human and mouse platelets pretreated with DCA inhibited platelet aggregation to suboptimal doses of various agonists, including collagen, convulxin, thrombin, and ADP in vitro. Also, DCA inhibited agonists-induced glucose uptake. Together these results suggest that DCA by regulating glucose uptake may inhibit platelet aggregation. Our results are consistent with a recent study by Fidler et al, which showed that glucose metabolism was essential for platelet function.21
To confirm whether an effect of DCA on agonist-induced platelet aggregation and decreased glucose uptake are associated with metabolic reprogramming (diverting metabolism from aerobic glycolysis back to OXPHOS), we measured ECAR and OCR. We found that DCA promoted OCR (OXPHOS) and partially inhibited ECAR (aerobic glycolysis) in platelets in response to suboptimal doses of convulxin. On the other hand, DCA promoted OCR in platelets in response to suboptimal doses of thrombin or ADP but surprisingly did not have any effect on ECAR. Together, these results suggest that the inhibitory effect of DCA on platelet aggregation induced by suboptimal doses of thrombin or ADP involve mechanisms that are independent of aerobic glycolysis. Our results also indicate that aerobic glycolysis could be the main source of ATP generation in platelets in response to collagen or convulxin and OXPHOS in the case of thrombin and ADP. Further studies are required to dissect the precise mechanisms involved in regulation of energy metabolism by different signaling pathways. We suggest that the inhibitory effect of DCA on collagen-induced platelet aggregation was in part due to decreased aerobic glycolysis rather than increased OXPHOS because of the following observations. First, the OXPHOS pathway is known to generate reactive oxygen species, which are known to promote platelet activation and aggregation.23 Although DCA treatment increased OXPHOS in platelets, it still inhibited platelet aggregation in response to collagen, convulxin, thrombin, and ADP, suggesting the inhibitory effect of DCA on platelet aggregation is not likely due to increased OXPHOS. Second, we found that DCA inhibited glucose uptake and partial aerobic glycolysis in activated platelets. These results are somewhat consistent with a previous study suggesting that DCA by decreasing lactate, and not other metabolites of aerobic glycolysis, significantly attenuates the effect of glucose on platelet inhibition.24
Our studies also suggest a potential link between aerobic glycolysis and GPVI signaling pathway. Collagen/GPVI signaling results in platelet shape change, increase in cytosolic calcium, granules release, and platelet activation, which is mediated by phosphorylation of Syk and PLCγ2, downstream of GPVI signaling pathway. We found that DCA partially inhibited aerobic glycolysis in platelets in response to convulxin or collagen that was associated with a significant decrease in TXB2 A2 generation, reduction in ATP release, and a decrease in phosphorylation of Syk and PLCγ2, all of which are known to regulate the collagen/GPVI signal transduction pathway. The possibility that DCA directly inhibits phosphorylation of Syk and PLCγ2 cannot be ruled out. Future studies using platelet-specific PDKs null mice may help in dissecting the precise mechanism by which DCA modulates GPVI signaling pathway. There could be additional mechanisms by which DCA may contribute to the collagen/GPVI signaling pathway. For example, increase in NADH/NAD+ ratio during glycolytic flux could also increase de novo synthesis of diacylglycerol, which in turn activates protein kinase C, known to participate in αIIbβ3 activation and platelet aggregation.
Finally, using human and mouse whole blood, we showed that DCA inhibited platelet aggregation/thrombus formation on the collagen matrix under arterial shear conditions in vitro. Furthermore, WT mice pretreated with DCA were less susceptible to arterial thrombosis in 2 in vivo models of thrombosis with unaltered hemostasis. The inhibitory effect of DCA on platelet function is similar to Revacept, a soluble dimeric glycoprotein GPVI-Fc fusion protein known to specifically inhibit collagen-induced platelet aggregation, with unaltered hemostasis.25 Although Revacept is currently in a phase 2 setting, its efficacy and side effects in large populations remain unclear. An advantage of DCA over GPVI inhibition with Revacept is that DCA can be used in a broader context. Several studies suggest that DCA can be used in combination with other therapies for the treatment of several types of tumors, metabolic disorders including diabetes, and hyperlipidemia that are risk factors for cardiovascular diseases.10,11,18,19
Our studies have a few limitations. First, the present study did not define the definitive role of PDKs in platelet function. The inhibitory effect of DCA on platelet aggregation and arterial thrombosis due to off-targeting in addition to PDKs cannot be ruled out. Future studies utilizing platelet-specific PDKs null mice are required to define the specific role of PDKs in platelet function. Second, we did not determine the inhibitory effect of DCA on other cell types that exhibit a high rate of aerobic glycolysis. For example, activated monocytes similar to activated platelets also exhibit a high rate of aerobic glycolysis compared with OXPHOS.3 Monocytes are known to contribute to arterial thrombosis by several mechanisms, including tissue factor expression, shedding microparticles from P-selectin–rich microdomains known to express tissue factor, PS.26 It is possible that some of the inhibitory effects of DCA on arterial thrombosis in vivo could be contributed by inhibiting aerobic glycolysis in monocytes. In summary, we provide evidence that targeting metabolic plasticity in platelets by DCA could be explored as a novel strategy to inhibit platelet aggregation and arterial thrombosis.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work is supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R35HL139926) (A.K.C.), the American Heart Association, Established Investigator Award 18EIA33900009 (A.K.C.), and the National Institutes of Health, National Institute on Aging (R01AG049784) (S.D.).
Authorship
Contribution: M.K.N. designed the study, performed experiments, analyzed, interpreted results, and cowrote the manuscript; N.D. performed experiments; P.D. analyzed results; V.K.S. and S.D. helped with in vitro flow assays using the BioFlux 200 microfluidics flow chamber; O.R. helped with the human samples; and A.K.C. directed the project, designed the study, interpreted results, and cowrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anil K. Chauhan, Department of Internal Medicine, University of Iowa, 25 S. Grand Ave, 3120 Medical Labs, Iowa City, IA 52242; e-mail: anil-chauhan@uiowa.edu.
References
- 1.Doery JC, Hirsh J, Cooper I. Energy metabolism in human platelets: interrelationship between glycolysis and oxidative metabolism. Blood. 1970;36(2):159-168. [PubMed] [Google Scholar]
- 2.Karpatkin S. Studies on human platelet glycolysis. Effect of glucose, cyanide, insulin, citrate, and agglutination and contraction on platelet glycolysis. J Clin Invest. 1967;46(3):409-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chacko BK, Kramer PA, Ravi S, et al. . Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013;93(6):690-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kramer PA, Ravi S, Chacko B, Johnson MS, Darley-Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: implications for their use as bioenergetic biomarkers. Redox Biol. 2014;2:206-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravi S, Chacko B, Sawada H, et al. . Metabolic plasticity in resting and thrombin activated platelets. PLoS One. 2015;10(4):e0123597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289(24):16615-16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond). 2014;11(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem. 2009;389(2):157-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38(11):1124-1144. [DOI] [PubMed] [Google Scholar]
- 10.Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med. 1978;298(10):526-530. [DOI] [PubMed] [Google Scholar]
- 11.Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N Engl J Med. 1983;309(7):390-396. [DOI] [PubMed] [Google Scholar]
- 12.Prakash P, Kulkarni PP, Lentz SR, Chauhan AK. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood. 2015;125(20):3164-3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prakash P, Kulkarni PP, Chauhan AK. Thrombospondin 1 requires von Willebrand factor to modulate arterial thrombosis in mice. Blood. 2015;125(2):399-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhanesha N, Ahmad A, Prakash P, Doddapattar P, Lentz SR, Chauhan AK. Genetic ablation of extra domain A of fibronectin in hypercholesterolemic mice improves stroke outcome by reducing thrombo-inflammation. Circulation. 2015;132(23):2237-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prakash P, Nayak MK, Chauhan AK. P-selectin can promote thrombus propagation independently of both von Willebrand factor and thrombospondin-1 in mice. J Thromb Haemost. 2017;15(2):388-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagy B Jr, Jin J, Ashby B, Reilly MP, Kunapuli SP. Contribution of the P2Y12 receptor-mediated pathway to platelet hyperreactivity in hypercholesterolemia. J Thromb Haemost. 2011;9(4):810-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003;43(7):683-691. [PubMed] [Google Scholar]
- 18.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99(7):989-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 2010;102(12):1746-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vella S, Conti M, Tasso R, Cancedda R, Pagano A. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int J Cancer. 2012;130(7):1484-1493. [DOI] [PubMed] [Google Scholar]
- 21.Fidler TP, Campbell RA, Funari T, et al. . Deletion of GLUT1 and GLUT3 reveals multiple roles for glucose metabolism in platelet and megakaryocyte function. Cell Reports. 2017;20(4):881-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504-507. [DOI] [PubMed] [Google Scholar]
- 23.Jang JY, Min JH, Chae YH, et al. . Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid Redox Signal. 2014;20(16):2528-2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobzar G, Mardla V, Samel N. Lactate is a possible mediator of the glucose effect on platelet inhibition. Platelets. 2014;25(4):239-245. [DOI] [PubMed] [Google Scholar]
- 25.Ungerer M, Rosport K, Bültmann A, et al. . Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 2011;123(17):1891-1899. [DOI] [PubMed] [Google Scholar]
- 26.Swystun LL, Liaw PC. The role of leukocytes in thrombosis. Blood. 2016;128(6):753-762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.