

Key Points
Validation of the WHO 2017 CMML categories supports distinguishing MP-CMML from MDS–CMML.
The prognostic value of the new 3-tiered blast-based CMML stratification scheme is limited.
Abstract
The 2017 revision of the World Health Organization (WHO) classification includes substantial changes to the subclassification of chronic myelomonocytic leukemia (CMML): (1) a 3-tiered blast-based scheme including a novel “CMML-0” category replacing a 2-tiered system in place since 2001 and (2) 2 CMML subtypes, myelodysplastic (MDS-CMML) and myeloproliferative (MP-CMML), based on a white blood cell count cutoff of 13 × 109/L. The clinical utility of this subclassification scheme, particularly the expansion of blast-based subgroups, has not been validated. In this study, a large single-institution CMML patient cohort (n = 629) was used to assess the prognostic impact of the newly proposed categories. Patients were risk stratified according to the CMML-specific Prognostic Scoring System (CPSS) and the MD Anderson Prognostic Scoring System. MP-CMML patients had significantly shorter overall survival (OS; P < .0001; hazard ratio: 0.53, 95% confidence interval: 0.42-0.65) and median duration to acute myeloid leukemia (AML) transformation (P < .0001; 15.2 vs 22.0 months) compared with MDS-CMML patients. The CMML-0 group included 36.4% patients with higher risk CPSS categories and 11.2% of patients with high-risk cytogenetics. Among treatment-naïve patients (n = 499), there was a marginal difference in OS between the CMML-0 and CMML-12017 subgroups (P = .0552). The WHO 2017 blast-based categories were not associated with AML-free survival. Incorporation of the WHO 2017 blast-based subgroups in a modified CPSS scheme had a neutral effect and did not improve its prognostic strength. Our data support the inclusion of MP-CMML and MDS-CMML subtypes in the WHO 2017 revision. Although of some utility in MP-CMML, the 3-tiered blast-based system is not well supported in this study.
Visual Abstract
Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal myeloid neoplasm defined by relative (≥10%) and absolute monocytosis (≥1 × 109/L) in the peripheral blood (PB) with features of both a myeloproliferative neoplasm and a myelodysplastic syndrome (MDS). Because of the heterogeneous clinical and morphologic features of CMML, its subclassification has undergone multiple iterations throughout the years. The original French-American-British (FAB) group classified CMML as an MDS and divided it into 2 subtypes based on the white blood cell (WBC) count: MDS-CMML (WBC count <13 × 109/L) and myeloproliferative CMML (MP-CMML; WBC count ≥13 × 109/L).1
The prognosis of patients with CMML remains generally poor, with a median overall survival (OS) of 2 to 3 years and a 20% to 30% risk of transformation to acute myeloid leukemia (AML), even though some patients can have a protracted disease course with slow or limited progression.2-5 Several risk-stratification systems have been proposed for CMML patients, including the MD Anderson Prognostic System (MDAPS) and the CMML-specific Prognostic Scoring System (CPSS).2,3,5,6 These systems have been shown to have similar predictive value.7 The CPSS has become widely used and includes 4 variables: World Health Organization (WHO) subgroups (based on 2008 criteria), FAB subtype, erythrocyte transfusion dependency or hemoglobin level, and karyotype. Clonal cytogenetic abnormalities are seen in 20% to 30% of CMML patients. Three CPSS cytogenetic risk groups were proposed by Such et al.8 Tang et al subsequently proposed a modification to the CPSS cytogenetic risk groups.9 Recent studies have attempted to integrate clinicopathologic features and cytogenetic and molecular findings into CPSSs.6,7,10
In 200111 and 2008,12 the WHO classification scheme recognized CMML as a distinct entity under the MDS/myeloproliferative neoplasm category and further subclassified it into 2 groups based on PB and/or bone marrow (BM) blast/promonocyte percentages: CMML-1 (PB <5%; BM <10%) and CMML-2 (PB, 6% to 19%; BM, 10% to 19%) with no significant emphasis on the previously recognized FAB subtypes. The 2017 revision of the WHO classification13,14 diverges from the 2001 and 2008 iterations in 2 important ways. First, it reincorporates FAB-defined MP-CMML and MDS-CMML subtypes. Second, it introduces a novel 3-tiered blast-based subgrouping scheme that includes a “CMML-0” category (PB <2% and/or BM <5%). Given the modest concordance rate for morphologic quantification of blasts/promonocytes in CMML,15 adding a blast-based tier requires that the potential benefits clearly outweigh the potential compromises to reproducibility. To our knowledge, neither the WBC-based subtypes nor the blast-based subgroups introduced in the WHO 2017 scheme have been systematically validated in large-scale studies.
In this study, we assessed a large single-institution CMML patient cohort to determine whether the 3-tiered CMML subgroups as proposed in the WHO 2017 system provide superior predictive power in terms of OS and AML-free survival compared with the 2-tiered system in place since 2001. We also sought to determine whether incorporating the FAB-based CMML subtypes provides added clinical value.
Methods
Study group
We identified patients with CMML diagnosed and treated at the University of Texas MD Anderson Cancer Center (UTMDACC) between the years 2001 and 2017. All patients fulfilled the diagnostic criteria of CMML as defined in the WHO classification. All patients underwent BM evaluation that included microscopic examination, as well as karyotyping and molecular analysis to exclude the presence of Philadelphia chromosome/BCR-ABL1 or PDGFRA, PDGFRB, and FGFR1 rearrangements. A total of 634 CMML patients met the study inclusion criteria; 5 patients without adequate material for blast enumeration and/or without cytogenetic data were excluded from further analysis.
Using laboratory data at initial presentation, patients in the study group (n = 629) were subclassified into MDS-CMML and MP-CMML per aforementioned FAB criteria and subgrouped into blast-based categories as follows: CMML-0 (PB <2% and/or BM <5%); CMML-1 (PB 2% to 4% and/or BM 5% to 9%) (CMML-12017); and CMML-2 (PB 5% to 19% and/or BM 10% to 19% and/or Auer rods are present).14 Patients were also subgrouped per 2008 WHO classification definitions of CMML-1 (CMML-12008) and CMML-2. Blasts (including promonocytes) were enumerated in accord with published guidelines15 using a 500-cell differential count of BM aspirate smears and/or touch preparations. Four hundred ninety-nine patients were treatment-naïve upon initial presentation to UTMDACC.
Cytogenetic analysis was performed using conventional methods and reported in accordance with the 2017 International System for Human Cytogenetic Nomenclature as described previously.16 Cytogenetics risk groups were assigned per CPSS criteria8 and per proposed modification by Tang et al.9 Molecular studies included single gene PCR-based methods and next-generation sequencing analysis using clinically validated mutation screening panels.17 For ASXL1, only frameshift alterations and nonsense (not missense) single nucleotide variants were considered as mutations.
Most patients were observed actively and provided with supportive therapy as needed or were treated with hypomethylating agents. A subset of patients received allogeneic stem cell transplant or AML-type chemotherapy; these patients were censored on the date that such therapies were initiated for purposes of statistical analysis. Patients were risk-stratified according to the CPSS and MDAPS systems.5 CPSS risk groups were assigned using the hemoglobin level (alternate CPSS)2 instead of red blood cell transfusion dependency, in keeping with the intent of the alternate CPSS, as the former was a more readily available parameter in our database. This study was approved by the UTMDACC Institutional Review Board and conducted in accordance with the Declaration of Helsinki.
Statistical analysis
The associations between CMML categories and the factors of interest were assessed using the χ2, Wilcoxon rank-sum, and Kruskal-Wallis tests as appropriate. The times for OS and evolution to AML (AML-free survival) were computed from the date of diagnosis to the time of last follow-up or event occurrence (AML or death). Patients who were alive at the last follow-up date and whose CMML had not evolved to AML were censored (19 April 2017). Patients who underwent stem cell transplant were censored at the date of transplant. The Kaplan-Meier method was used to estimate OS times. Differences in survival between groups were assessed using the log-rank test. The cumulative incidence rate of evolution to AML was determined using the competing risk method, with the competing risk being death before developing AML. Differences in cumulative incidence rate between groups were assessed using Gray’s test.18
Associations between the prognostic factors of interest and OS or AML-free survival were assessed using Cox proportional hazards regression models, and associations between the prognostic factors of interest and the cumulative incidence outcomes were determined using the Fine and Gray (FG) proportional subdistribution hazards regression model.19 The multivariable hazards regression models included independent factors with P < .1 in the univariate analysis to determine the associations between the prognostic factors of interest and OS or AML transformation. Impact of variables was expressed in hazard ratio (HR) and associated 95% confidence intervals (CIs). Comparison of the prognostic strength of CPSS modifications to the original CPSS was estimated using the concordance probability estimates (CPEs) for the Cox proportional hazards model with ties.20 No adjustments for multiple testing were made. Recursive partitioning was used to determine the optimal BM blast percentage cutoff value to predict OS and AML-free survival.21 All statistical analyses were performed using SAS 9.4 for Windows (SAS Institute Inc., Cary, NC). Significance level was defined as 5% for all statistical tests.
Results
Comparative analysis of WBC-based CMML subtypes
Comparisons of patients with MDS-CMML and MP-CMML are summarized in Table 1. Patients in each subtype differed significantly in terms of blast-based subgroups, across both the WHO 2008 and 2017 schemes, with MP-CMML including a relatively higher proportion of patients with CMML-2. Similar differences were noted in terms of MDAPS and CPSS risk groups, with a higher proportion of patients in higher-risk categories having MP-CMML. We found a significant association between MP-CMML and RAS mutations. Comparisons of treatment-naïve patients showed similar results.
Table 1.
Summary of patient characteristics across WHO 2017 WBC count–based subtypes, MDS- and MP-CMML
| Variable | MDS-CMML | MP-CMML | P* |
|---|---|---|---|
| Patients, N (%) | 306 (48.6) | 323 (51.4) | |
| Age, median (range), y | 69.9 (31.2-91.1) | 69.9 (24.8-87.6) | .9559 |
| Age group, n (%), y | |||
| <60 | 61 (19.9) | 55 (17.0) | .4987 |
| 60-69 | 152 (49.7) | 158 (48.9) | |
| ≥70 | 93 (30.4) | 110 (34.1) | |
| Sex, n (%) | |||
| Female | 93 (30.4) | 106 (32.8) | .5133 |
| Male | 213 (69.6) | 217 (67.2) | |
| WHO-2017, n (%) | |||
| CMML-0 | 122 (39.9) | 92 (28.5) | .0052 |
| CMML-1 | 101 (33.0) | 113 (35.0) | |
| CMML-2 | 83 (27.1) | 118 (36.5) | |
| WHO-2008, n (%) | |||
| CMML-1 | 223 (72.9) | 205 (63.5) | .0114 |
| CMML-2 | 83 (27.1) | 118 (36.5) | |
| PB parameters | |||
| WBC count, median (range), ×109/L | 7.3 (0.8-12.9) | 28.7 (13.0-223.1) | <.0001 |
| Hemoglobin concentration, median (range), g/dL | 10.6 (5.9-16.0) | 10.2 (5.1-16.4) | .0905† |
| Platelet count, median (range), ×109/L | 88.5 (2.0-1554) | 94.0 (5.0-912.0) | .2705 |
| Immature myeloid cells,‡ median (minimum-maximum) | 1.0 (0.0-34.0) | 8.0 (0.0-50.0) | <.0001 |
| LDH, median (range), IU/L | 537.0 (191-3400) | 749.0 (142-11060) | <.0001 |
| BM blast percentage, median (range) | 5.5 (0.0-19.0) | 6.0 (0.0-19.0) | .7885 |
| MDAPS group, n (%) | |||
| High | 47 (15.4) | 57 (17.6) | <.0001 |
| Intermediate-2 | 98 (32.0) | 186 (57.6) | |
| Intermediate-1 | 110 (35.9) | 66 (20.4) | |
| Low | 51 (16.7) | 14 (4.3) | |
| CPSS group, n (%) | |||
| HIGH | 16 (5.2) | 49 (15.2) | <.0001 |
| INT-2 | 85 (27.8) | 178 (55.1) | |
| INT-1 | 90 (29.4) | 96 (29.7) | |
| Low | 115 (37.6) | 0 (0) | |
| CPSS cytogenetic risk groups, n (%) | |||
| High | 58 (19.0) | 50 (15.5) | .0704 |
| Intermediate | 36 (11.8) | 58 (18.0) | |
| Low | 212 (69.3) | 215 (66.6) | |
| Tang cytogenetic risk groups, n (%) | |||
| High | 36 (11.8) | 36 (11.1) | .5856 |
| Intermediate | 58 (19.0) | 72 (22.3) | |
| Low | 212 (69.3) | 215 (66.6) | |
| Mutations, present, n (%) | |||
| TET2 | 40 (41.7) | 35 (38.9) | .6995 |
| ASXL1 | 26 (37.7) | 36 (50.7) | .1209 |
| RAS | 42 (17.4) | 88 (36.5) | <.0001 |
| TP53 | 5 (5.2) | 5 (5.6) | .9164 |
| RUNX1 | 15 (21.7) | 13 (18.3) | .6121 |
| Follow-up duration, median (range), mo | 23.6 (0.0-191.9) | 17.2 (0.0-147.0) | <.0001 |
| Death, n (%) | 183 (59.8) | 249 (77.1) | <.0001 |
| Survival time, median (95% CI), mo | 33.8 (28.8-40.0) | 21.1 (18.4-24.5) | <.0001 |
| Duration to AML transformation, median (range), mo | 22.0 (0.0-185.2) | 15.2 (0.0-138.9) | <.0001 |
| AML, n (%) | 49 (16.0) | 61 (18.9) | .3432 |
LDH, lactate dehydrogenase.
Kruskal-Wallis test and Wilcoxon rank-sum test for continuous factors and χ2 test for categorical factors as appropriate.
Full comparison conducted using treatment-naïve group (n = 499) showed comparable results with the following exception: P significant (.0251) in treatment-naïve group.
Sum of promyelocytes, myelocytes, and metamyelocytes in PB.
Comparative analysis of blast-based CMML subgroups
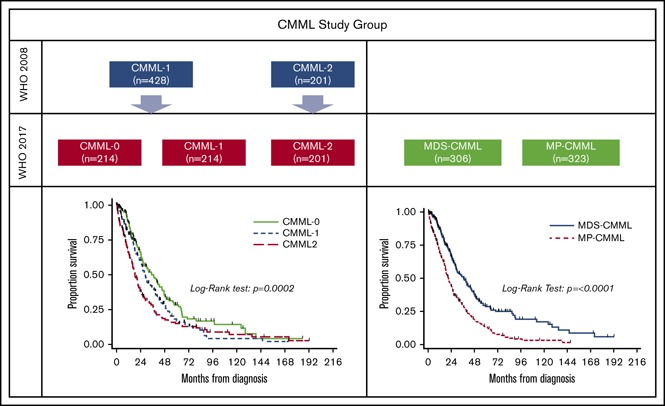
Among 428/629 (68%) CMML-12008 patients, 214 (50%) were subgrouped as CMML-0 (Figure 1). The CMML-0 and CMML-12017 categories were similar in terms of CPSS risk categories and across cytogenetic risk groups. Furthermore, the CMML-0 group included 36.4% patients categorized as CPSS high or intermediate-2, with 11.2% of patients with CPSS high-risk cytogenetics (6.5% per Tang risk groups). Significant differences between patients in the CMML-0 and CMML-12017 categories included FAB subtypes, platelet count, median number of circulating immature myeloid cells (IMCs), and serum LDH level. A significant difference was identified in the incidence of TET2 mutations between CMML-0 and CMML-12017, but the distribution of mutations in ASXL1, RAS (NRAS and KRAS), TP53, and RUNX1 across the 3 blast-based subgroups was otherwise similar. Comparisons of all patients in WHO 2017–defined blast-based subgroups are summarized in Table 2. Comparisons of treatment-naïve patients showed similar results.
Figure 1.

Distribution of CMML patients across categories in the 2008 and 2017 WHO classification schemes.
Table 2.
Summary of patient characteristics across WHO 2017 blast-based subgroups
| Variable | Entire group | CMML-0 | CMML-1 | CMML-2 | P* | ||
|---|---|---|---|---|---|---|---|
| Overall | CMML-0 vs 1 | CMML-0/1 vs 2 | |||||
| Patients, N (%) | 629 | 214 (34.0) | 214 (34.0) | 201 (32.0) | |||
| Age, median (range), y | 69.9 (24.8-91.1) | 69.9 (31.2-91.1) | 69.3 (24.8-87.6) | 70.3 (27.5-86.7) | .0686 | .0764 | .1446 |
| Age group, n (%), y | |||||||
| <60 | 116 (18.4) | 35 (16.4) | 53 (24.8) | 28 (13.9) | .0534 | .0850 | .1348 |
| 60-69 | 203 (32.3) | 73 (34.1) | 61 (28.5) | 69 (34.3) | |||
| ≥70 | 310 (49.3) | 106 (49.5) | 100 (46.7) | 104 (51.7) | |||
| Sex, n (%) | |||||||
| Female | 199 (31.6) | 70 (32.7) | 57 (26.6) | 72 (35.8) | .1215 | .1690 | .1221 |
| Male | 430 (68.4) | 144 (67.3) | 157 (73.4) | 129 (64.2) | |||
| FAB subtype, n (%) | |||||||
| MDS-CMML | 306 (48.6) | 122 (57.0) | 101 (47.2) | 83 (41.3) | .0052 | .0422 | .0114 |
| MP-CMML | 323 (51.4) | 92 (43.0) | 113 (52.8) | 118 (58.7) | |||
| PB parameters | |||||||
| WBC count, median (range), ×109/L | 13.2 (0.8-223.1) | 11.3 (0.8-149.8) | 13.3 (1.3-211.8) | 17.2 (1.1-223.1) | .0009 | .0287 | .0027 |
| Hemoglobin concentration, median (range), g/dL | 10.4 (5.1-16.4) | 10.7 (5.1-16.4) | 10.6 (5.9-15.7) | 9.9 (5.3-16.0) | .0119 | .4335 | .0041 |
| Platelet count, median (range), ×109/L | 92.0 (2.0-1554.0) | 110.0 (7.0-1554) | 88.0 (6.0-912.0) | 81.0 (2.0-606.0) | .0001 | .0091 | .0009 |
| Immature myeloid cells,† median (minimum-maximum) | 3.0 (0.0-50.0) | 1.0 (0.0-34.0) | 4.0 (0.0-50.0) | 6.0 (0.0-50.0) | <.0001 | <.0001 | <.0001 |
| LDH, median (range), IU/L | 610.0 (142.0-11 060.0) | 551.0 (142-11 060) | 662.5 (191-6372) | 662.5 (250-9997) | <.0001 | .0001 | .0005 |
| BM blast percentage, median (range) | 6.0 (0.0-19.0) | 2.0 (0.0-4.0) | 6.0 (0.0-9.0) | 12.0 (1.0-19.0) | <.0001 | <.0001 | <.0001 |
| MDAPS group, n (%) | |||||||
| High | 104 (16.5) | 0 (0) | 0 (0) | 104 (51.7) | <.0001 | .0057 | <.0001 |
| Intermediate-2 | 284 (45.2) | 88 (41.1) | 118 (55.1) | 78 (38.8) | |||
| Intermediate-1 | 176 (28.0) | 85 (39.7) | 73 (34.1) | 18 (9.0) | |||
| Low | 65 (10.3) | 41 (19.2) | 23 (10.7) | 1 (0.5) | |||
| CPSS group, n (%) | |||||||
| HIGH | 65 (10.3) | 5 (2.3) | 10 (4.7) | 50 (24.9) | <.0001 | .3673 | <.0001 |
| INT-2 | 263 (41.8) | 73 (34.1) | 71 (33.2) | 119 (59.2) | |||
| INT-1 | 186 (29.6) | 73 (34.1) | 81 (37.9) | 32 (15.9) | |||
| Low | 115 (18.3) | 63 (29.4) | 52 (24.3) | 0 (0) | |||
| CPSS cytogenetic risk groups, n (%) | |||||||
| High | 108 (17.2) | 24 (11.2) | 28 (13.1) | 56 (27.9) | .0001 | .8356 | <.0001 |
| Intermediate | 94 (14.9) | 33 (15.4) | 33 (15.4) | 28 (13.9) | |||
| Low | 427 (67.9) | 157 (73.4) | 153 (71.5) | 117 (58.2) | |||
| Tang cytogenetic risk groups, n (%) | |||||||
| High | 72 (11.4) | 14 (6.5) | 22 (10.3) | 36 (17.9) | .0013 | .3634 | .0003 |
| Intermediate | 130 (20.7) | 43 (20.1) | 39 (18.2) | 48 (23.9) | |||
| Low | 427 (67.9) | 157 (73.4) | 153 (71.5) | 117 (58.2) | |||
| Mutations, present, n (%) | |||||||
| TET2 | 75 (40.3) | 35 (47.9) | 18 (30.5) | 22 (40.7) | .1269 | .0422‡ | .9407 |
| ASXL1 | 62 (44.3) | 23 (40.4) | 18 (43.9) | 21 (50.0) | .6326 | .7251 | .3729 |
| RAS | 130 (27.0) | 37 (23.0) | 49 (29.3) | 44 (28.6) | .3720 | .1905§ | .5875 |
| TP53 | 10 (5.4) | 6 (8.2) | 2 (3.4) | 2 (3.7) | .3840 | .2476 | .5177 |
| RUNX1 | 28 (20.0) | 11 (19.3) | 10 (24.4) | 7 (16.7) | .6693 | .5445 | .5186 |
| Follow-up duration, median (range), mo | 19.6 (0.0-191.9) | 23.7 (0.0-185.2) | 21.6 (0.0-171.0) | 16.1 (0.0-191.9) | .0001 | .1532 | <.0001 |
| Death, n (%) | 432 (68.7) | 131 (61.2) | 152 (71.0) | 149 (74.1) | .0119 | .0320 | .0435 |
| Survival time, median (95% CI), mo | 26.3 (24.3-29.2) | 35.0 (26.7-42.8) | 28.1 (24.5-32.3) | 19.4 (17.4-23.4) | <.0001 | .0110 | <.0001 |
| Duration to AML transformation, median (range), mo | 17.7 (0.0-185.2) | 22.6 (0.0-185.2) | 17.4 (0.0-171.0) | 13.4 (0.0-172.1) | <.0001 | .1045 | <.0001 |
| AML, n (%) | 110 (17.5) | 27 (12.6) | 42 (19.6) | 41 (20.4) | .0680 | .0486 | .1880 |
Full comparison conducted using treatment-naïve group (n = 499) showed comparable results with exceptions noted (see ‡ and §).
Kruskal-Wallis test and Wilcoxon rank-sum test for continuous factors and χ2 test for categorical factors as appropriate.
Sum of promyelocytes, myelocytes, and metamyelocytes in PB.
P not significant (.2026) in treatment-naïve group.
P is significant (.0376) in treatment-naïve group.
Outcome analysis and prognostic impact of CMML categories
To eliminate the potential confounding effect of treatment-induced bias, only treatment-naïve patients were considered in survival analyses. Univariate analysis results for variable association with OS are summarized in supplemental Table 1. Patients with MP-CMML had a significantly shorter OS compared with MDS-CMML patients (P < .0001; HR [95% CI], 1.89 [1.54-2.38]) (Figure 2). There was no difference in the cumulative incidence of AML between MP-CMML and MDS-CMML patients. When we further explored the potential effect of prognostic factors on OS within the MP-CMML group, the most notable (in terms of HR) included hemoglobin level and cytogenetics (data not shown). In this category, CMML-0 vs CMML-1 (P = .0278; HR [95% CI], 0.67 [0.47-0.96]) but not CMML-1 vs CMML-2 (P = .3738; HR [95% CI], 1.16 [0.84-1.61]) correlated with OS. These factors were independently associated with OS by multivariate Cox regression analysis (supplemental Table 2A). Notably, in the MDS-CMML group, the WHO 2008 but not the WHO 2017 subgroups correlated with OS by univariate analysis (P = .0408 and 0.1146, respectively); however, even WHO 2008 subgroups were not significant in multivariate analysis (supplemental Table 2B). In addition, none of the CMML prognostic variables independently correlated with AML-free survival with the exception of age (P = .0357).
Figure 2.

Outcome comparisons for MDS-CMML and MP-CMML. Kaplan-Meier plots for OS (A) and cumulative incidence of AML (B) in treatment-naïve patients stratified by WHO 2008 and 2017 WBC count (FAB-based MP and MDS subgroups)–based subgroups.
Whereas both the WHO 2008 and 2017 subgroups as a whole correlated with OS, the difference in OS between patients in the CMML-0 and CMML-12017 subgroups (P = .0552) was modest (Figure 3). Furthermore, neither the WHO 2008 nor the 2017 blast-based subgroups were associated with AML-free survival. We performed multivariate Cox regression analysis using the WHO 2008 and 2017 subgroups separately as independent variables alongside known CMML prognostic variables. Patients with CMML-0 and CMML-12017 had different OS independent of other factors, albeit the HR and 95% CI of CMML-0 vs CMML-12017 and CMML-22017 were comparable to those of CMML-1 vs CMML-2 in WHO 2008 (Tables 3 and 4) Furthermore, there was no significant difference between CMML-12017 and CMML-22017 by multivariate analysis suggesting that incorporation of CMML-0 mitigated the prognostic utility of CMML-12017.
Figure 3.

Outcome comparisons for 2008 and 2017 blast-based subgroups. Kaplan-Meier plots for OS (A,C) and cumulative incidence of AML (B,D) in treatment-naïve patients stratified by blast-based subgroups.
Table 3.
Multivariate analysis for OS in treatment-naïve patients, WHO 2017 blast-based subgroups
| Variable | P | HR | 95% CI for HR | |
|---|---|---|---|---|
| WHO 2017 | ||||
| CMML-0 vs CMML-1 | .0212 | 0.732 | 0.561 | 0.954 |
| CMML-2 vs CMML-1 | .4136 | 1.118 | 0.856 | 1.459 |
| CMML-0 vs CMML-2 | .0019 | 0.655 | 0.501 | 0.855 |
| Hemoglobin concentration, g/dL | <.0001 | 0.862 | 0.819 | 0.908 |
| FAB subtype (MDS vs MP) | <.0001 | 0.513 | 0.410 | 0.640 |
| CPSS cytogenetic risk group | ||||
| High vs low | <.0001 | 2.470 | 1.826 | 3.342 |
| Intermediate vs low | .2721 | 1.202 | 0.866 | 1.669 |
| Age (<70 y vs ≥70 y) | .0093 | 0.745 | 0.596 | 0.930 |
Table 4.
Multivariate analysis for OS in treatment-naïve patients, WHO 2008 blast-based subgroups
| Variable | P | HR | 95% CI for HR | |
|---|---|---|---|---|
| WHO 2008 (CMML-1 vs CMML-2) | .0182 | 0.757 | 0.601 | 0.954 |
| Hemoglobin concentration | <.0001 | 0.864 | 0.820 | 0.910 |
| FAB subtype (MDS vs MP) | <.0001 | 0.517 | 0.414 | 0.645 |
| CPSS cytogenetic risk | ||||
| High vs low | <.0001 | 2.428 | 1.796 | 3.282 |
| Intermediate vs low | .3465 | 1.170 | 0.844 | 1.623 |
| Age (<70 y vs ≥70 y) | .0078 | 0.740 | 0.593 | 0.924 |
Using recursive partitioning, we sought further to determine the optimal BM blast percentage cutoff predictive of both OS and AML-free survival. The lowest node identified 8% as an optimal cutoff (P = .0048). This cutoff (≤8% vs >8%) correlated with OS (P = .0112; HR [95% CI], 1.30 [1.06-1.58]) and AML-free survival (P = .0032; FG-HR [95% CI], 1.76 [1.21-2.55]) when the entire study group was considered. However, when only the treatment-naïve group was considered, a cutoff of 10% (<10% vs ≥10%) (P = .0305; HR [95% CI], 1.29 [1.02-1.63]) correlated with OS; cutoffs of 8%, 7%, and 5% were not significant.
Impact of WHO 2017 subgroups on CPSS
The CPSS was devised using WHO 2008 subgroups, with variable scores of 0 or 1 assigned to CMML-1 and CMML-2, respectively. We postulated that if the CMML-0 subgroup is prognostically useful, its impact should be manifest through its incorporation into an established prognostic system such as CPSS. To test this hypothesis, we designed a modified CPSS system (CPSS_2017) in which a variable score of 0, 1, or 2 was assigned for each of CMML-0, CMML-1, and CMML-2, respectively. This modification resulted in a new possible overall score ranging from 0 to 6 (instead of 0-5). Pairwise comparisons were conducted using Cox proportional hazards model for OS and AML-free survival to determine optimal groupings of overall scores into risk groups. The best fit resulted in the following CPSS_2017 scheme: low (0-1), intermediate-1 (2-3), intermediate-2 (4-5), and high (6). The new scheme was highly prognostic (supplemental Tables 3A-B; supplemental Figure 1). However, using CPE as a measure of prognostic strength, CPSS_2017 and CPSS had comparable prognostic strength (0.6310 vs 0.2358). Similar results were obtained using numerous permutations to define risk groups based on overall scores from modified CPSS iterations. Together, these results suggest that the incorporation of the WHO 2017 3-tiered blast-based subgroups in CPSS has a neutral effect, neither abrogating nor improving the predictive strength of CPSS.
Discussion
In this study, we assessed the clinical validity of CMML categories proposed in the 2017 revision of the WHO classification on the basis of 2 integral parameters: FAB-based MDS/MP subtypes and blast-based subgroups. Our data confirm that distinguishing MDS-CMML from MP-CMML using a WBC count of 13 × 109/L is highly informative because patients with MP-CMML have significantly shorter duration of progression to AML and shorter OS compared with patients with MDS-CMML. As reported by others,2,10 MP-CMML patients in our study group tended to have higher-risk disease with shorter OS and shorter time to AML transformation compared with MDS-CMML patients. Furthermore, multivariate analysis showed that FAB subtype was an independent predictor of OS. Univariate analysis did not show correlation with the rate of AML transformation per se, which we postulate might be because of the fact that most patients with MP-CMML have a shorter OS duration resulting in death from complications of CMML before transformation to AML. Notably, we found a significant association between MP-CMML and RAS mutations (P < .0001) in our study group, as described by others.22 Clonal evolution studies have shown that RAS mutations are late events in CMML pathogenesis23; thus MP-CMML appears to represent a more advanced disease phenotype in which leukemic cells have acquired a constellation of features, most notably RAS mutations, that phenotypically manifest as proliferative disease with an increased risk of additional detrimental events leading to shorter AML-free and OS duration. In such instances, targeted inhibition of mutant RAS has shown promising results.24 In addition to its prognostic value, distinction between MDS-CMML and MP-CMML is likely to help tailor therapeutic options because MP-CMML patients might in principle benefit from targeted therapies to abrogate proliferation signals.
Others have identified different BM blast cutoff levels that associate with CMML outcomes. Recursive partitioning identified 8% as an optimal BM blast cutoff in the present study group. Coincidentally, our recursive partitioning result is very close to the 7% cutoff obtained by Elena et al10 using somewhat different statistical methodology. Their group adopted a threshold of 5% by reasoning that it was more easily applicable clinically. Notwithstanding, when we assessed these cutoffs in our treatment-naïve group, only 10% and none of the aforementioned cutoffs correlated with OS. When we evaluated blast-based subgroups separately within MP- and MDS-CMML patients, the prognostic impact of the 5% cutoff as adopted in the WHO 2017 scheme turned out to be most relevant within the MP-CMML subtype. As such, different cutoffs might have different implications in CMML subtypes, with CMML-0 being most informative in MP-CMML. However, because the latter is generally a worse CMML subtype compared with MDS-CMML, the incremental value of CMML-0 in this context does not outweigh the reproducibility limitations that such a narrow tier poses in a disease where blast enumeration typically requires a significant level of expertise. CMML encompasses a highly complex pathogenic landscape in which the neoplastic cell population typically spans multiple lineages (erythroid, granulocytic, megakaryocytic) and includes immature (blasts, promonocytes) and maturing cells. An increase in the proportion of immature cells is generally thought to indicate a skewed steady state in which the neoplastic cells have an increased proliferation rate and a diminished capacity to mature, together raising the risk for acquiring mutational events that tip the balance toward AML development. Conversely, the absence of an increase in blasts says nothing about the extent of the neoplastic burden, as measured indirectly by parameters such as WBC count or LDH, or underlying high-risk genomic lesions including complex cytogenetics. The proof that such disparate elements play an intertwined role in CMML is probably best illustrated by the number of CMML-specific prognostic systems that have shown comparable correlation with outcome despite using occasionally disparate parameters and cutoffs.7,25
In a departure from earlier versions, the 2017 WHO CMML subclassification scheme replaced the prior 2-tiered blast-based CMML categories with a 3-tiered scheme by splitting CMML-1 into CMML-0 and CMML-12017. The cited studies on which the change was based include 1 using registry-derived data,26 another that includes a limited set of 30 patients,27 and a third in which the authors actually reported that the optimal prognostic impact for BM blasts was achieved using a cutoff of 10%.5 Our data indicate that CMML-0 offers added prognostic value in the MP-CMML subset only, but its utility in the overall CMML patient population is limited. The proportion of blasts is of established importance in CMML prognosis as evidenced by the inclusion of this parameter, along a 2-tiered scheme, in essentially all CMML prognostic systems, even if the optimal cutoff for BM blast percentage remained elusive.2,3,5,6,10 Our data are in keeping with this general premise but show no added value from the introduction of CMML-0 as proposed in the WHO 2017 revision. Indeed, in our study group, 24 (11.2%) patients in the CMML-0 subgroup had high-risk CPSS cytogenetics, and 5 (2.3%) were in the high-risk CPSS group. Using a less conservative measure of discordance, among CMML-0 patients, 73 (34.1%) were in the intermediate-2 CPSS risk group, and 88 (41.1%) were in the intermediate-2 MDAPS risk group. Interestingly, none of the CMML-0 or CMML-12017 was in the high-risk MDAPS group, likely a reflection of the absence of cytogenetics in the MDAPS scheme. In addition, incorporation of the proposed WHO 2017 subgroups into CPSS showed no added prognostic value as measured by CPE.
The role of somatic mutations in CMML prognostic modeling has been controversial, and no molecular-based CMML categories exist as of yet in the WHO classification, although molecularly integrated prognostic schemes have shown added prognostic value particularly through inclusion of nonsense and frameshift ASXL1 mutations.6,10,28,29 In our study group, the most commonly detected mutations involved ASXL1 (44.3%), TET2 (40.3%), RAS (27%), and RUNX1 (20%). In contrast to other studies,6,29 TET2 mutation was associated with OS in our study, both in the treatment-naïve group (P = .0111; HR [95% CI], 2.36 [1.22-4.59]) and the entire study group (P = .0085). Only RUNX1 was associated with AML-free survival (P = .0470; HR [95% CI], 3.12 [1.01-9.09]), in keeping with the results reported by Kuo et al.30 We notably did not identify a prognostic role for ASXL1 in our study group. Our mutation data were only available on a subset of patients and notably lacked SETBP1 and SRSF2, an acknowledged limitation of our data set. As these limitations did not detract from the goals of this study, they also underscored the validity of CPSS and MDAPS, both of which are mutation-agnostic prognostication schemes. Indeed, the molecular data and their implication in our study group might after all be in keeping with the character of CMML wherein a multitude of broad factors (age, WBC count, blasts, cytogenetics) can effectively provide a measure of prognosis by reflecting the sum total of multiple underlying pathogenic factors.31
In summary, assessment of the 2017 WHO CMML categories supports a prominent role for distinguishing MDS-CMML from MP-CMML. The addition of CMML-0 to create a 3-tiered blast-based subgrouping scheme seems to be of limited value.
Supplementary Material
The full-text version of this article contains a data supplement.
Authorship
Contribution: S.L. and J.D.K. were responsible for concept and study design; and S.L., D.S., P.W., S.P., G.G.-M., M.J.R., E.J.J., N.P., R.K.-S., H.D.G., S.H., Z.Z., L.J.M., H.M.K., and J.D.K. performed data collection, data analysis, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joseph D. Khoury, Department of Hematopathology, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, MS-072, Houston, TX 77030; e-mail: jkhoury@mdanderson.org.
References
- 1.Bennett JM, Catovsky D, Daniel MT, et al. . Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33(4):451-458. [DOI] [PubMed] [Google Scholar]
- 2.Such E, Germing U, Malcovati L, et al. . Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121(15):3005-3015. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg PL, Tuechler H, Schanz J, et al. . Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Germing U, Kündgen A, Gattermann N. Risk assessment in chronic myelomonocytic leukemia (CMML). Leuk Lymphoma. 2004;45(7):1311-1318. [DOI] [PubMed] [Google Scholar]
- 5.Onida F, Kantarjian HM, Smith TL, et al. . Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99(3):840-849. [DOI] [PubMed] [Google Scholar]
- 6.Patnaik MM, Padron E, LaBorde RR, et al. . Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes [published correction appears in Leukemia. 2013;27(10):2112]. Leukemia. 2013;27(7):1504-1510. [DOI] [PubMed] [Google Scholar]
- 7.Padron E, Garcia-Manero G, Patnaik MM, et al. . An international data set for CMML validates prognostic scoring systems and demonstrates a need for novel prognostication strategies. Blood Cancer J. 2015;5:e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Such E, Cervera J, Costa D, et al. . Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96(3):375-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang G, Zhang L, Fu B, et al. . Cytogenetic risk stratification of 417 patients with chronic myelomonocytic leukemia from a single institution. Am J Hematol. 2014;89(8):813-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elena C, Gallì A, Such E, et al. . Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128(10):1408-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vardiman JW, Pierre R, Bain B, et al. . Chronic Myelomonocytic Leukaemia. In: Jaffe ES, Harris LN, Stein H, Vardiman JW, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2001:49-52. [Google Scholar]
- 12.Orazi A, Bennett JM, Germing U, Brunning RD, Bain BJ, Thiele J. Chronic Myelomonocytic Leukemia. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008:76-79. [Google Scholar]
- 13.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 14.Orazi A, Bennett JM, Germing U, et al. . Chronic Myelomonocytic Leukemia. In: Swerdlow SHCE, Harris NL, Jaffe ES, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2017:82-86. [Google Scholar]
- 15.Goasguen JE, Bennett JM, Bain BJ, Vallespi T, Brunning R, Mufti GJ; International Working Group on Morphology of Myelodysplastic Syndrome. Morphological evaluation of monocytes and their precursors. Haematologica. 2009;94(7):994-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGowan-Jordan J, Simons A, Schmid M, eds. ISCN 2016: An International System for Human Cytogenomic Nomenclature. Basel, Switzerland: S. Karger; 2016. [Google Scholar]
- 17.Luthra R, Patel KP, Reddy NG, et al. . Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99(3):465-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16(3):1141-1154. [Google Scholar]
- 19.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94(446):496-509. [Google Scholar]
- 20.Gönen M, Heller G. Concordance probability and discriminatory power in proportional hazards regression. Biometrika. 2005;92(4):965-970. [Google Scholar]
- 21.Seibold H, Zeileis A, Hothorn T. Model-based recursive partitioning for subgroup analyses. Int J Biostat. 2016;12(1):45-63. [DOI] [PubMed] [Google Scholar]
- 22.Ricci C, Fermo E, Corti S, et al. . RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16(8):2246-2256. [DOI] [PubMed] [Google Scholar]
- 23.Itzykson R, Kosmider O, Renneville A, et al. . Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186-2198. [DOI] [PubMed] [Google Scholar]
- 24.Borthakur G, Popplewell L, Boyiadzis M, et al. . Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer. 2016;122(12):1871-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solary E, Itzykson R. How I treat chronic myelomonocytic leukemia. Blood. 2017;130(2):126-136. [DOI] [PubMed] [Google Scholar]
- 26.Schuler E, Schroeder M, Neukirchen J, et al. . Refined medullary blast and white blood cell count based classification of chronic myelomonocytic leukemias. Leuk Res. 2014;38(12):1413-1419. [DOI] [PubMed] [Google Scholar]
- 27.Storniolo AM, Moloney WC, Rosenthal DS, Cox C, Bennett JM. Chronic myelomonocytic leukemia. Leukemia. 1990;4(11):766-770. [PubMed] [Google Scholar]
- 28.Patnaik MM, Itzykson R, Lasho TL, et al. . ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28(11):2206-2212. [DOI] [PubMed] [Google Scholar]
- 29.Itzykson R, Kosmider O, Renneville A, et al. . Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428-2436. [DOI] [PubMed] [Google Scholar]
- 30.Kuo MC, Liang DC, Huang CF, et al. . RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23(8):1426-1431. [DOI] [PubMed] [Google Scholar]
- 31.Ball M, List AF, Padron E. When clinical heterogeneity exceeds genetic heterogeneity: thinking outside the genomic box in chronic myelomonocytic leukemia. Blood. 2016;128(20):2381-2387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.