

Key Points
Novel Tfrc-mutant mouse identified in ENU mutagenesis screen with stable receptor expression.
Flow imaging cytometry demonstrates microcytosis in mutants derives from dysfunctional receptor-mediated endocytosis of Tf-TfR complex.
Abstract
To identify novel regulators of erythropoiesis, we performed independent forward genetic screens using the chemical mutagen ENU in mice. Among progeny displaying microcytic red-cell phenotypes, 7 independent mouse strains harboring mutations within the transferrin receptor gene Tfrc were identified. Six of the mutants, including the previously described red blood cell 6 (RBC6) strain, displayed reduced erythroblast CD71 expression and midgestation lethality of homozygotes (E12.5-E14.5), and 1 novel strain, RBC21, displayed a variable phenotype with sustained CD71 expression and late homozygous lethality (E18.5). Standard iron studies were normal in the RBC21 mutant, but intracellular ferritin was significantly reduced. The microcytic phenotype seen in the RBC21 strain was the result of impaired binding of transferrin to the receptor. Neither RBC6 nor RBC21 responded to iron replacement therapy. These studies describe how point mutations of the transferrin receptor can cause a microcytic anemia that does not respond to iron therapy and would not be detected by routine iron studies, such as serum ferritin.
Visual Abstract
Introduction
The transferrin receptor (TfR, TfR1, CD71), encoded by the TFRC gene in humans, is an integral component of iron metabolism and erythrocyte production.1 The highest expression of TfR is seen in the bone marrow on developing erythroblasts, which require intake of circulating iron for hemoglobin synthesis.2,3 The 2 key ligands of TfR are transferrin (Tf) and the hemochromatosis protein (HFE), which both play prominent roles in iron metabolism and regulation, respectively.4 These ligands directly compete for overlapping binding sites within the ectopic helical domain of the receptor, composed of N-terminal amino acids 607 to 760 (mouse equivalent, 610-763) within exons 17 to 19.5 The binding affinities and conformational relationships of both ligands to TfR have been previously described.6,7 As a homodimer, TfR has 2 binding sites for the acceptance of 2 molecules of either Tf or HFE, or it can form a ternary structure with both simultaneously at a 1:1:1 ratio, but ultimately, the receptor has a much higher affinity for Tf, specifically iron-bound Tf (Fe-Tf), at biological potential of hydrogen (pH).5 This is due to the enclosed conformational change that Tf undergoes when iron is captured from the circulation. In contrast, TfR has a weak affinity for iron-free Tf, the conformation of which is more open and reduces the number of binding sites accessible by the receptor. After the binding of Tf to the membrane-bound receptor, the Tf-TfR complex undergoes receptor-mediated endocytosis within a clathrin-coated pit, facilitated by endocytic mediators, such as dynamin 2 (Dnm2).8 A reduction in endosomal pH (∼5.5) allows iron to disassociate from its carrier protein without disturbing Tf-TfR binding, and subsequently, iron enters the cell through the DMT-1 portal, where it can be used in the synthesis of heme or stored intracellularly in the form of ferritin.9 The iron-free Tf-TfR complex is then recycled to the cell surface, where Tf is released back into the circulation.
TfR-HFE interactions also play an important role in maintaining iron homeostasis.10 As the concentration of serum iron increases, so does Fe-Tf, which subsequently outcompetes HFE for TfR binding positions. Displaced HFE initiates a negative feedback loop by prompting the formation of an iron-regulatory complex around the liver-specific homolog TfR2.11 This complex involves the recruitment of proteins such as hemojuvelin (HJV) and BMP-6, which require HFE for stabilization. The regulatory complex initiates downstream signaling through TfR2 and other iron-sensing receptors and ultimately triggers production of the peptide hormone hepcidin.11 Hepcidin inhibits excessive iron from entering the circulation via blockage and degradation of ferroportin. At low iron concentrations, TfR is thought to sequester HFE to prevent it from inappropriately enacting on TfR2.10,12 In addition to regulation, TfR2 is also important for erythropoietic development through its cooperation with the erythropoietin receptor; however, the phenotypes of experimental animals lacking TfR2 are strikingly different to those with TfR defects.13
Inheritable mutations within genes encoding HFE, TfR2, or HJV are known to cause iron overload, or hemochromatosis.11 In contrast, there have been no definitive reported cases of red-cell diseases resulting from inherited mutations in TFRC in humans. Genome-wide association studies previously identified a homozygous mutation in the C-terminal cytoplasmic domain of TfR, resulting in primary immunodeficiency,14 but no erythroid-specific diseases resulting from mutations in TFRC have been confirmed to date. However, microcytic anemia with iron deficiency–like characteristics, often unresponsive to iron therapy, is a recognized clinical finding in humans, and many cases go unresolved.15 It is therefore possible that congenital mutations within other key proteins of the iron metabolism pathway, such as TfR, do exist in the population but go undetected. The generation of experimental animals harboring Tfrc mutations, particularly mice, typically results in iron-deficient erythropoiesis with various degrees of anemia and liver iron overload,1,8,16 but the pathological role of aberrant TfR endocytic cycling in iron therapy–resistant microcytic anemia and its clinical relevance remain poorly characterized. In a series of genome-wide ENU mutagenesis screens, we identified a collection of mutant mice with dominant microcytic red-cell phenotypes, all harboring Tfrc mutations within the Tf/HFE binding domain. Detailed phenotypical analyses of these mutant strains have offered greater insight into the key characteristics of TfR-mediated microcytic anemia and the molecular mechanisms underpinning red-cell pathologies.
Materials and methods
Mice
Dominant ENU mutagenesis screens were performed in mice as described previously.17 G1 pedigrees displaying microcytosis were identified from 7 individual founder mice. Gene mapping followed by whole-exome sequencing was performed on each strain,8 which identified each mutation in the Tfrc gene, as listed in Table 1. Genotyping of the strains red blood cell 6 (RBC6; Tfrc+/L645R) or RBC21 (Tfrc+/R654H) was performed using bone marrow complementary DNA (cDNA) and the primers forward (5′-CAC ACC TGG CTT TCC TTC TT-3′) and reverse (5′-ATG AGG AAC CAG ACC GTT ATG-3′), followed by sequencing using BigDye Terminator reagents. All animal experiments were approved by the animal ethics committees of the Alfred Medical and Research Education Precinct and Monash University.
Table 1.
List of Tfrc-mutant mouse strains generated by ENU mutagenesis
| Mutant | Nucleotide mutation, cDNA | Amino acid change | Mean heterozygote MCV | Homozygote phenotype | CD71 expression, heterozygote |
|---|---|---|---|---|---|
| BL/6 | NA | NA | 46 | NA | Normal |
| RBC1 | T1849C | Y617H | 42 | Lethal (E10.5-E12.5) | Reduced |
| RBC4 | T2048A | Y686Stop | 42 | Lethal (E10.5-E12.5) | Reduced |
| RBC5 | T1925G | L642R | 41 | Lethal (E10.5-E12.5) | Reduced |
| RBC6* | T1934G | L645R | 43 | Lethal (E10.5-E12.5) | Reduced |
| RBC8, RBC17 | A2105G | H702R | 41 | Lethal (E10.5-E12.5) | Reduced |
| RBC21 | G1961A | R654H | 37 | Lethal (E16.5-E18.5) | Normal |
Mouse mutants with autosomal-dominant microcytic phenotypes, identified in independent ENU mutagenesis screens, were deemed RBC mutants, by order of discovery. Mutants were phenotyped by their mean corpuscular volume (MCV), calculated by an automated blood analyzer (Hemavet, Drew Scientific). Timed pregnancy studies determined embryonic lethality in each mutant strain. CD71 expression was determined by flow cytometry, performed on live Ter-119+ bone marrow erythroblasts, which compared CD71 intensity with that of WT littermates in each cohort.
NA, not applicable.
Blood and serum studies
Full blood examinations were performed on EDTA-treated whole blood collected from submandibular venepuncture and processed by the Hemavet automated blood analyzer (Drew Scientific, Miami Lakes, FL). Tf and ferritin assays were performed by enzyme-linked immunosorbent assay (ALPCO Diagnostics, Salem, MA). Serum iron and Tf saturation were quantified using a total iron binding capacity reagent set (Pointe Scientific, Inc., Canton, MI). Liver iron was assayed using an iron assay kit (Abcam, Cambridge, MA).
Tf uptake assay and flow imaging
Single-cell flow imaging was performed on whole bone marrow stained with conjugated antibodies Ter-119 and CD71 (BD Biosciences, San Jose, CA) on the Amins ImageStreamX Mark II flow cytometer (Merck, Darmstadt, Germany). A minimum of 2000 erythroblast (Ter-119+) events were captured for each run. Compensation and analysis were performed using Amnis IDEAS software (Merck). Early proerythroblasts and late erythroid cell subtypes were excluded from analysis based on Ter-119 expression. Focused cells were chosen based on a bright field intensity gradient of >40, followed by elimination of debris and doublets by the bright field aspect ratio vs area dot plot. Colocalization analysis generated a median bright intensity score for the florescent intensities of Ter-119 and CD71, where higher score indicated greater overlap of the 2 probes. Internalization analysis generated the mean internalization erode score using Ter-119 as the mask probe, where a score equal to 1.0 indicated the probe was membrane bound, and a score >1.0 correlated with increased internalization of the probe. Using these parameters, Tf uptake was assessed on bone marrow additionally stained with 5 mg/mL of Tf–Alexa Fluor 647 conjugate (Life Technologies, Scoresby, Australia) for 45 minutes at 4°C in the dark. Uptake was initiated by incubating cells at 37°C and halted at desired time points by placing cells in ice. Cells were then briefly acid washed (150 mmol/L of sodium chloride, 20 mmol/L of calcium dichloride, 20 mmol/L of sodium acetate; pH, 4.6) before cytometric analysis and imaging. Tf uptake was calculated at each time point by dividing the relative intensity of internalized Tf by the 0-minute noninternalized unwashed control.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from liver using TRIzol (Invitrogen) according to the manufacturer’s instructions, followed by cDNA amplification of 1 μg total RNA using a reverse transcription kit (Promega). Quantitative real-time polymerase chain reaction was performed on a LightCycler 480II (Roche Diagnostics) using the GoTaq qPCR Master Mix (Promega). Expression of genes was normalized to β-actin, and data are presented as relative expression compared with WT controls. Gene-specific primers were as follows: Hamp-1: forward (5′-AAG CAG GGC AGA CAT TGC GAT-3′) and reverse (5′-CAG GAT GTG GCT CTA GGC TAT GT-3′); β-Actin: forward (5′-CTG TCC CTG TAT GCC TCT G-3′) and reverse (5′-ATG TCA CGC ACG ATT TCC-3′).
Statistical analysis
Where applicable, results are expressed as mean ± standard deviation. For statistical analysis, a 2-tailed Student t test was employed, unless stated otherwise, where P < .05 indicated significance (or as defined in the figure legends).
Results
Identification of microcytic mouse strains with dominant Tfrc mutations
A series of independent genome-wide ENU mutagenesis screens were performed in mice to identify novel genes or alleles regulating erythropoiesis, as previously described.17,18 Seven G1 progeny exhibiting a reduced MCV >3 standard deviations below the average population were isolated. Further breeding demonstrated the phenotype was fully penetrant and autosomal dominant in inheritance. Pedigrees were termed the RBC mutants, numbered in order of discovery (Table 1).
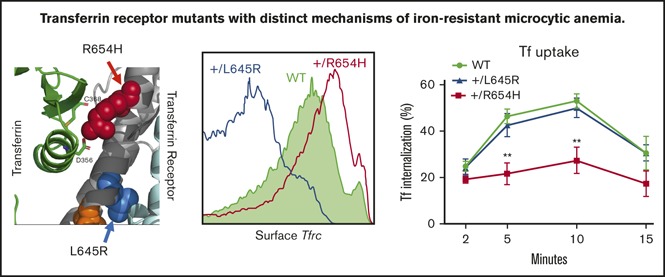
To identify the genetic mutation responsible for the microcytic phenotypes observed, gene mapping or whole-exome sequencing was used as previously described8,17 and confirmed by Sanger sequencing on bone marrow cDNA. Mutations within the Tfrc gene on chromosome 16 were identified in each pedigree. Two mutants were genetically identical (RBC8 and RBC17), resulting in 6 different Tfrc mouse strains (Table 1), including the previously reported RBC6 mouse (Tfrc+/L645R).8 Five of the 6 unique alleles were missense substitutions, and 1 was a premature stop codon (RBC4). All mutations were localized within exon 17, 18, or 19, resulting in substitutions of conserved amino acids (Figure 1A). Three-dimensional modeling of the murine TfR dimer, in the context of the Fe-Tf complex, showed several Tfrc mutations (RBC5, RBC6, RBC21) to be within the helical binding domain, either at or adjacent to known Tf/HFE binding sites (Figure 1B). Modeling predicted that many mutational sites played important structural and conformational roles within TfR, such as providing hydrophobic packing for the helical bundle (RBC5, RBC6), or otherwise produced an unstable truncated form (RBC4). One mutation, R654H (RBC21), was found to be directly involved in securing Tf to the receptor (Figure 1C) and was the only substitution predicted not to distort protein conformation, instead affecting the ligand binding capacity of Tf.
Figure 1.

Distribution of Tfrc mutations identified in ENU mutagenesis screens. (A) Amino acid sequence of the mouse (upper) and human (lower) ectopic helical domain of the Tfrc gene. Mutations identified in murine ENU mutagenesis screens are highlighted (red), with their corresponding RBC mutant strain. Known ligand binding positions include Tf binding (yellow), HFE binding (blue), and sites with overlapping Tf/HFE binding (gray). (B) Three-dimensional structure of the murine TfR-Tf complex (PD entry 3S9L) inspected by PyMOL (version 2.0.3). A cluster of Tfrc mutations (RBC5, orange; RBC6, blue; RBC21, red) is highlighted. (C) Closer inspection of the RBC21 mutation and its positioning within the TfR-Tf complex. The R654 amino acid (human equivalent, R651) is shown to have a direct Tf binding role via interactions with Tf amino acids C368 and D356. Other mutation sites (RBC5, RBC6) are suggested to play structural roles within the receptor.
TfR mutants with distinct phenotypes
Mice carrying the RBC21 allele (denoted TfrcR654H) displayed a unique phenotype among the RBC mutants and therefore became the major focus of our investigations. Similar to other Tfrc mutants described here and previously,17 Tfrc+/R654H mice displayed significantly reduced MCV, hemoglobin, and hematocrit, as well as increased red-cell count and red-cell distribution width, in comparison with WT littermates (Table 2). White-cell and platelet values were unchanged. Peripheral blood smears revealed microcytic red cells with unremarkable morphology in heterozygotes (Figure 2A). Surprisingly, serum ferritin and serum Tf, typically used to diagnose iron deficiency in humans, were normal in both Tfrc+/L645R and Tfrc+/R654H mice (Table 3). Total liver iron was also normal. However, red-cell ferritin concentrations were significantly reduced in both strains, indicating an intracellular iron deficit in both the Tfrc+/L645R and Tfrc+/R654H mutants (Table 3).
Table 2.
Full blood examination of the Tfrc+/R654H strain
| Mean ± SD | P | ||
|---|---|---|---|
| WT (n = 22) | Tfrc+/R654H (n = 18) | ||
| RBCs, × 1012/L | 9.57 ± 0.7 | 11.21 ± 1.2 | <.0001 |
| Hb, g/L | 14.2 ± 1.2 | 13.4 ± 1.4 | <.05 |
| HCT, % | 45.9 ± 2.5 | 41.3 ± 4.6 | <.0001 |
| MCV, fL | 47.6 ± 1.7 | 36.9 ± 2.5 | <.0001 |
| MCH, pg | 14.9 ± 0.4 | 12.0 ± 0.7 | <.0001 |
| MCHC, g/dL | 31.3 ± 1.3 | 32.6 ± 1.9 | n.s. |
| RDW, % | 18.7 ± 1.1 | 19.7 ± 1.0 | <.05 |
| WBCs, × 109/L | 8.69 ± 1.8 | 7.63 ± 2.5 | n.s. |
| Platelets, × 109/L | 972 ± 190 | 1057 ± 385 | n.s. |
| Reticulocytes, % | 5.2 ± 1.6 | 4.0 ± 2.0 | n.s. |
Blood parameters obtained from 7-week-old WT and heterozygous (Tfrc+/R654H) mice using automated blood analyzer (Hemavet). Two-tailed Student t test was used.
Hb, hemoglobin; HCT, hematocrit; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; n.s., not significant; RDW, red-cell distribution width; SD, standard deviation; WBC, white blood cell.
Figure 2.

Phenotype of the RBC21 mouse. (A) Peripheral blood smears of 7-week-old WT and RBC21 heterozygous (Tfrc+/R654H) mice (original magnification ×400; hematoxylin and eosin stain). Heterozygotes show microcytic red cells with mild pallor. (B) Flow cytometric analysis of CD71 expression levels, performed on live Ter-119+ bone marrow erythroblasts of WT, RBC6 (Tfrc+L645R), and RBC21 (Tfrc+/R654H) mice. (C) Timed pregnancies showing WT, RBC21 homozygous (TfrcR654H/R654H), and RBC6/RB21 double-heterozygous (TfrcL645R/R654H) embryos, dissected at embryonic days 14.5, 16.5, and 18.5. (D) Fetal liver live cell counts performed on WT and RBC21 homozygous (TfrcR654H/R654H) embryos at embryonic day 14.5 (n = 4). (E) Flow cytometric analysis of CD71 expression levels, performed on live Ter-119+ fetal liver cells of WT and RBC21 homozygous (TfrcR654H/R654H) embryos at embryonic day 14.5. (F) Survival curve of irradiated adult mice transplanted with WT or RBC21 homozygous (TfrcR654H/R654H) fetal liver cells, extracted from E14.5 embryos. Median survival of homozygous cohort, 13 days (n = 12). *P < .05. Tx, treatment.
Table 3.
Iron studies of Tfrc+/L645R and Tfrc+/R654H mice
| Mean ± SD | |||
|---|---|---|---|
| WT (n = 9) | Tfrc+/L645R (n = 6) | Tfrc+/R654H (n = 5) | |
| Serum Tf, g/L | 3.80 ± 1.2 | 4.24 ± 0.68 | 3.99 ± 0.87 |
| Serum ferritin, μg/L | 1021 ± 130.6 | 905 ± 228.8 | 908 ± 125.2 |
| RBC ferritin, μg/L | 60.43 ± 9.54 | 17.22 ± 1.21* | 28.70 ± 8.65* |
| Serum iron, μmol/L | 47.21 ± 2.23 | 38.09 ± 9.29* | 47.90 ± 27.6 |
| TIBC, μmol/L | 115.84 ± 11.7 | 106.68 ± 6.82 | 95.92 ± 15.7* |
| Tf saturation, % | 48.95 ± 2.59 | 35.85 ± 2.16* | 55.75 ± 2.33* |
| Liver iron, nmol/kg | 1.89 ± 0.25 | 1.64 ± 0.34 | 1.62 ± 0.46 |
Serum and tissue iron parameters obtained from 10-week-old WT, RBC6 (Tfrc+/L645R), and RBC21 (Tfrc+/R654H) mice. Two-tailed Student t test was used to compare values with WT.
TIBC, total iron binding capacity.
P < .05.
A key difference observed in the Tfrc+/R654H strain was increased expression of CD71 (TfR) on the surface of Ter-119+ bone marrow erythroblasts (Figure 2B). In all other Tfrc-mutant RBC strains, such as Tfrc+/L645R, CD71 expression was significantly reduced compared with WT littermates (Figure 2B). These data, combined with the 3-dimensional modeling, suggested that TfrcR654H erythroblasts express a structurally stable mutant TfR protein at the cell surface.
In contrast to other strains, TfrcR654H/R654H embryos displayed notably delayed homozygous lethality (Table 1). TfrcR654H/R654H embryos had an anemic phenotype detectable at E14.5 and died between E16.5 and E18.5 (Figure 2C). E14.5 TfrcR654H/R654H embryos showed severe pallor, edema, and underdeveloped craniofacial structures and had significantly fewer fetal liver cells compared with WT littermates (Figure 2D). Despite this, CD71 expression on Ter-119+ cells harvested from E14.5 TfrcR654H/R654H fetal livers was increased (Figure 2E). To determine which mutant allele was dominant, we intercrossed Tfrc+/L645R and Tfrc+/R654H heterozygous mice to generate TfrcL645R/R654H compound heterozygotes. Double-heterozygous embryos were found to be phenotypically identical to TfrcR654H/R654H embryos, with embryonic lethality observed between days E16.5 and E18.5 (Figure 2C). Thus, the TfrcR654H mutation retained partial function and was able to rescue the loss-of-function Tfrc+/L645R-mutant allele.
Given that the TfrcR654H mutation was germ line and affected all tissues requiring iron intake, fetal liver transplantations were performed to establish whether embryonic lethality in homozygotes was due to erythropoietic defects. WT or TfrcR654H/R654H fetal liver cells from E14.5 embryos were transplanted into lethally irradiated adult WT mice. Survival of the transplanted recipients was tracked over time (Figure 2F), and peripheral blood was analyzed after 7-week recovery (Table 4). As expected, WT fetal liver cells were able to rescue lethally irradiated mice, with complete donor reconstitution of all hematopoietic lineages. However, a majority of mice transplanted with TfrcR654H/R654H fetal liver cells died within 2 weeks as a result of severe anemia (Figure 2F). Furthermore, blood analysis of the single surviving TfrcR654H/R654H recipient demonstrated severe microcytic, hypochromic anemia (Table 4). Thus, the lethality of homozygous embryos could be attributed to an intrinsic defect in red-cell production.
Table 4.
Full blood examination of fetal liver–transplanted mice
| Mean ± SD | ||
|---|---|---|
| WT | TfrcR654H/R654H | |
| RBCs, × 1012/L | 11.72 ± 1.4 | 4.46 |
| Hb, g/dL | 15.4 ± 0.7 | 6.6 |
| HCT, % | 52.6 ± 3.2 | 17.5 |
| MCV, fL | 45.1 ± 2.9 | 35.9 |
| MCH, pg | 13.6 ± 1.3 | 14.8 |
| MCHC, g/dL | 30.1 ± 1.5 | 37.7 |
| RDW, % | 19.5 ± 1.6 | 20.0 |
| WBCs, × 109/L | 9.80 ± 0.9 | 5.66 |
| Lymphocytes, % | 78.9 ± 2.4 | 62.7 |
| Platelets, × 109/L | 970 ± 269 | >2972 |
Blood parameters obtained from mice 7 weeks posttransplantation after lethal irradiation and transplantation with either WT or homozygous (TfrcR654H/R654H) E14.5 fetal liver cells.
TfrcR654H mutation inhibits Tf binding and internalization
The reduced intracellular iron, despite elevated CD71 expression, suggested that the TfrcR654H mutation affected interactions with Tf. The binding capacity and internalization of Tf and TfR were analyzed in erythroblasts using the Amnis ImageStreamX flow imaging cytometer. We first validated CD71 expression levels on Ter-119+ erythroblasts harvested from each cohort using single-cell fluorescent imaging (Figure 3A). In comparison with WT cells, CD71 expression on Tfrc+/L645R erythroblasts was significantly reduced and displayed a lower CD71/Ter-119 overlap score (1.56 compared with 1.81 in WT; P = .01), indicating reduced CD71 expression. In Tfrc+/R654H erythroblasts, however, the median CD71/Ter-119 overlap was greater than the WT ratio (1.96; P = .01), consistent with the increased CD71 surface expression observed by flow cytometry (Figure 2B). Erythroblasts from WT, Tfrc+/L645R, and Tfrc+/R654H mice were subsequently stained with a Tf–Alexa Fluor conjugate for flow imaging to analyze Tf-TfR binding and internalization. After incubation with the ligand, both WT and Tfrc+/L645R erythroblasts showed an average of 20% Tf saturation across membrane-bound CD71 (Figure 3B). In contrast, Tfrc+/R654H erythroblasts demonstrated significantly reduced Tf conjugation to CD71 at the cell surface, despite increased CD71 expression (13.8%; P = .045), indicating that the TfrcR654H mutation reduced Tf binding to TfR.
Figure 3.

Analysis of TfR binding and internalization using Amnis flow imaging cytometry. (A) CD71/Ter-119 colocalization imaging of erythroblasts obtained from the bone marrow of WT, RBC6 (Tfrc+/L645R), and RBC21 (Tfrc+/R654H) mice. Intensity of CD71/Ter-119 overlap (yellow) generates the median bright detail similarity score. (B) Tf/CD71 colocalization imaging of erythroblasts from the 3 cohorts. Intensity of Tf/CD71 overlap (pale blue) generates the median bright detail similarity score, which is normalized to generate the mean percentage of Tf saturation. (C) Tf/Ter-119 internalization imaging of erythroblasts from each cohort at 0 minutes (before initiation of endocytosis). The mean internalization erode score of Tf uses Ter-119 as the mask. (D) Tf/Ter-119 internalization imaging of erythroblasts from each cohort at 5 minutes postinitiation of endocytosis. All scores represent 2000 Ter-119+ events captured in each cohort. (E) Total time course of Tf uptake, via endocytosis, calculated in Ter-119+ erythroblasts from each cohort. Mean ± (SD) shown in error bars. (F) Quantitative real-time polymerase chain reaction of liver Hamp-1 messenger RNA expression in each cohort. Mean ± (SD) shown (n = 6). *P < .05, **P < .01. MAD, median average deviation.
To investigate whether reduced Tf binding resulted in less intracellular Tf uptake, TfR uptake through receptor-mediated endocytosis was assessed by flow imaging over time using a Tf–Alexa Fluor conjugate. Internalization of Tf was quantified by the mean internalization erode score, which positively correlates with the percentage of endocytosed Tf. At 0 minutes (Figure 3C), Tf was detectable exclusively at the cell membrane in all 3 cohorts, resulting in a low internalization score (∼1.03). After 5 minutes (Figure 3D), a significant proportion of Tf was internalized by receptor-mediated endocytosis in WT erythroblasts (1.23), correlating with Tf uptake of 46% (Figure 3E). Tfrc+/L645R-mutant erythroblasts had a similar internalization erode score (1.19; P = .09) and Tf uptake of 42% (Figure 3E). In contrast, Tfrc+/R654H-mutant erythroblasts showed significantly reduced internalization (1.06; P = .02) and endocytosis of Tf (21%; Figure 3E) after 5 minutes.
Tf and HFE have a number of overlapping binding sites for TfR; however, based on the helical domain sequence (Figure 1A),6 the TfrcL645R and TfrcR654H mutations were not predicted to affect HFE binding. To confirm the mutations did not interfere with HFE binding to the receptor, we measured liver Hamp-1 messenger RNA expression as an indirect readout of the HFE-TfR interaction. Liver Hamp-1 expression was normal in both Tfrc+/L645R and Tfrc+/R654H mice compared with WT controls (Figure 3F). These results indicated the HFE binding regions on TfR were unaffected, and the red-cell phenotypes observed in these mice were not the result of increased hepcidin production.
Tfrc-mutant mice are resistant to iron therapy
To determine if Tfrc mutations respond to iron therapy, we treated Tfrc+/L645R and Tfrc+/R654H mice with iron dextran and reevaluated iron parameters 3 weeks later. Despite the significant increases that occurred to serum Tf, serum ferritin, and liver iron after administration, iron dextran treatment was unable to restore red-cell ferritin concentrations in either mutant strain (Table 5). Consequently, the reduced MCV and mean corpuscular hemoglobin persisted in both Tfrc+/L645R and Tfrc+/R654H mice (Table 5). Taken together, these results indicate that microcytic, hypochromic anemia caused by Tfrc mutations is nonresponsive to iron replacement therapy, regardless of CD71 expression (TfrcL645R) or endocytic function (TfrcR654H).
Table 5.
Iron studies of Tfrc+/L645R and Tfrc+/R654H mice after iron (dextran) treatment
| Mean ± SD | |||
|---|---|---|---|
| WT | Tfrc+/L645R | Tfrc+/R654H | |
| Serum Tf, g/L | 9.33 ± 2.44 | 8.22 ± 0.55 | 8.23 ± 3.90 |
| Serum ferritin, μg/L | 3427 ± 445 | 3220 ± 315 | 3939 ± 567 |
| RBC ferritin, μg/L | 148.2 ± 3.68 | 27.80 ± 7.9* | 38.64 ± 5.63* |
| Serum iron, μmol/L | 83.90 ± 9.91 | 77.03 ± 17.4 | 78.80 ± 2.59 |
| TIBC, μmol/L | 89.06 ± 12.1 | 79.04 ± 21.4 | 86.73 ± 15.6 |
| Tf saturation, % | 71.28 ± 15.1 | 64.2 ± 19.8 | 82.37 ± 19.8 |
| Liver iron, nmol/kg | 2.81 ± 0.73 | 3.72 ± 1.37 | 2.69 ± 0.54 |
| MCV, fL | 48.0 v 2.5 | 39.5 ± 1.5* | 35.7 ± 1.6* |
| MCH, pg | 14.7 ± 0.42 | 12.4 ± 0.51* | 11.2 ± 0.25* |
Blood and tissue iron parameters obtained from 10-week-old WT, RBC6 (+/L645R), and RBC21 (+/R654H) mice, 3 weeks after a single intraperitoneal injection of iron dextran. Two-tailed Student t test was used to compare values with the WT.
P < .05.
Discussion
We conducted a genome-wide ENU mutagenesis screen to identify novel regulators of erythropoiesis in mice. Here, we describe a series of autosomal-dominant mutations of the Tfrc gene. The phenotype in each case was microcytic hypochromic anemia. Of the 6 unique Tfrc mutants identified, the RBC21 strain (Tfrc+/R654H) displayed a distinct phenotype, including sustained erythroblast CD71 expression and delayed homozygous lethality. This study offers a broader examination of hematological and developmental phenotypes caused by autosomal-dominant Tfrc mutations, as well proposed pathological mechanisms underlying those phenotypes.
Receptor-mediated endocytosis of the Tf-TfR complex is the key mechanism by which cells, particularly erythroblasts, obtain iron. Extensive studies in vitro have identified key motifs within the extracellular helical domain of the receptor (exons 17-19) that are indispensable for ligand binding and function.5,6 Studies of microcytic mice harboring point substitutions and deletions within Tfrc, including those identified in ENU mutagenesis screens,1,8,16 or mutations in upstream factors regulating TfR transcription19,20 typically reported a microcytic, hypochromic phenotype because of haploinsufficiency of the receptor. Through crystal structure modeling and CD71 flow cytometry, we found the Tfrc+/R654H mouse strain expressed a stable mutant protein at the cell surface. This provided a unique opportunity to study the consequences of defective TfR ligand binding and its effect on erythropoiesis and development in vivo.
The R654 amino acid (human equivalent, R651) is a key Tf binding hotspot within the TfR, the loss of which has been shown to severely diminish the Tf, but not HFE, binding affinity of the receptor.1,10 The use of novel cytometric tools, such as the Amnis ImageStreamX flow imager, provided visual and quantitative evidence of the importance of this binding site in iron transport and metabolism and ultimately demonstrated how a microcytic phenotype could persist in the Tfrc+/R654H mutant despite sustained CD71 expression levels. Interestingly, the pattern of ineffective Tf-TfR uptake observed in Tfrc+/R654H erythroblasts was comparable to a mouse model harboring a Dnm2 mutation, displaying defects in endocytosis.8 This suggested both Tf binding and receptor internalization were disrupted by the TfrcR654H mutation; however, there may be additional disruptions to intracellular mechanisms contributing to the phenotype of the animal. Indeed, other aspects of receptor-mediated endocytosis and endosomal iron transport could be further investigated using this method, because many intracellular erythroid metabolic programs remain poorly described.
Surprisingly, and in contrast to other mutants described here, TfrcR654H/R654H embryos survived until late embryogenesis, enabling the study of the requirements of Tfrc during development. Early embryonic lethality of homozygotes previously prevented these observations.8,16,20 Aside from the profound anemia and tissue hypoxia, gross malformations of the brain and craniofacial regions seen in E14.5 to E18.5 homozygotes and double TfrcL645R/R654H heterozygous embryos are consistent with previous reports that TfR cycling has a prominent role in neurological development.1 The opportunity to perform fetal liver transplantations using TfrcR654H/R654H cells allowed further assessment of the function of this Tfrc allele in adult hematopoiesis. Although homozygosity of the TfrcR654H mutation was ultimately incompatible with life, blood values of the long-surviving transplant recipient suggested reconstitution of nonerythroid cell lineage populations was still possible in the absence of TfR cycling. Lymphocyte development, for instance, may use alternative uptake pathways when TfR-mediated endocytosis is inefficient, given the observed role of iron in lymphoid maturation.21
Iron studies performed on Tfrc+/R654H and Tfrc+/L645R mice were normal except for Tf saturation, which was slightly elevated in the Tfrc+/R654H strain. This may be linked to CD71 expression status. Red-cell ferritin proved to be the most informative indicator of an iron metabolic defect in Tfrc mutants, which did not respond to iron dextran administration. Red-cell ferritin is frequently used to identify cellular iron deficits in microcytic mice when serum markers appear normal8,22 and has been shown to be more informative than ZnPP/heme ratios, for example.8 However, intraerythrocytic ferritin is not a typical clinical test of iron deficiency anemia.23 Despite the obvious defect in iron uptake by red cells, the predominant red-cell phenotype was microcytosis in the absence of hypochromia. This suggests that isolated microcytosis may be due to inherited defects in the TfR. Although mutations within the TFRC gene have not yet been associated with microcytosis in humans, the inclusion of intracellular iron measurements may prove to be highly valuable in the search for congenital mutations within iron internalization pathways, particularly in cases when iron therapy has failed. A greater understanding of the phenotypical characteristics associated with congenital iron metabolic defects in animal models, along with the development of novel tools and markers of iron deficiency, will progressively contribute to better clinical management strategies for patients with unresolved microcytic anemias.
Acknowledgments
The authors thank Rust Turakolov and Matthew Tinning at the Australian Genome Research Facility for their contributions to gene sequencing and bioinformatics, as well as support from the Australian Phenomics Network. The authors also thank Eva Orlowski and Alfred Medical Research and Education Precinct FlowCore for cytometry services and the Animal Research Laboratory (Monash University) and its technicians for animal husbandry.
This work was supported in part by Project Grant #382900 (S.M.J. and D.J.C.) and Program Grant #1016647 (B.T.K.) and Independent Research Institutes Infrastructure Support Scheme Grant #361646 (B.T.K.) from the Australian National Health and Medical Research Council and a Viertel Senior Medical Research Fellowship (D.J.C).
Authorship
Contribution: A.J.C., F.C.B., B.T.K., and D.J.C. designed and performed experiements; G.R. designed mutagenesis screens; C.J.M. prepared structural models; A.J.C, F.C.B., S.M.J., and D.J.C. prepared the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David J. Curtis, Australian Centre for Blood Diseases, Central Clinical School, Monash University, Level 1, Alfred Centre, 99 Commercial Rd, Melbourne, VIC 3004, Australia; e-mail: david.curtis@monash.edu.
References
- 1.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21(4):396-399. [DOI] [PubMed] [Google Scholar]
- 2.Marsee DK, Pinkus GS, Yu H. CD71 (transferrin receptor): an effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol. 2010;134(3):429-435. [DOI] [PubMed] [Google Scholar]
- 3.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci USA. 2009;106(41):17413-17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.West AP Jr, Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem. 2000;275(49):38135-38138. [DOI] [PubMed] [Google Scholar]
- 5.Giannetti AM, Björkman PJ. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J Biol Chem. 2004;279(24):25866-25875. [DOI] [PubMed] [Google Scholar]
- 6.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116(4):565-576. [DOI] [PubMed] [Google Scholar]
- 7.Aisen P. Transferrin receptor 1. Int J Biochem Cell Biol. 2004;36(11):2137-2143. [DOI] [PubMed] [Google Scholar]
- 8.Brown FC, Collett M, Tremblay CS, et al. . Loss of dynamin 2 GTPase function results in microcytic anaemia. Br J Haematol. 2017;178(4):616-628. [DOI] [PubMed] [Google Scholar]
- 9.Ponka P, Lok CN. The transferrin receptor: role in health and disease. Int J Biochem Cell Biol. 1999;31(10):1111-1137. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7(3):205-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9(3):217-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleming RE. Iron sensing as a partnership: HFE and transferrin receptor 2. Cell Metab. 2009;9(3):211-212. [DOI] [PubMed] [Google Scholar]
- 13.Forejtnikovà H, Vieillevoye M, Zermati Y, et al. . Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood. 2010;116(24):5357-5367. [DOI] [PubMed] [Google Scholar]
- 14.Jabara HH, Boyden SE, Chou J, et al. . A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat Genet. 2016;48(1):74-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hershko C, Camaschella C. How I treat unexplained refractory iron deficiency anemia. Blood. 2014;123(3):326-333. [DOI] [PubMed] [Google Scholar]
- 16.Lelliott PM, McMorran BJ, Foote SJ, Burgio G. Erythrocytic iron deficiency enhances susceptibility to plasmodium chabaudi infection in mice carrying a missense mutation in transferrin receptor 1. Infect Immun. 2015;83(11):4322-4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown FC, Scott N, Rank G, et al. . ENU mutagenesis identifies the first mouse mutants reproducing human β-thalassemia at the genomic level. Blood Cells Mol Dis. 2013;50(2):86-92. [DOI] [PubMed] [Google Scholar]
- 18.Rank G, Sutton R, Marshall V, et al. . Novel roles for erythroid ankyrin-1 revealed through an ENU-induced null mouse mutant. Blood. 2009;113(14):3352-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galy B, Ferring D, Minana B, et al. . Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood. 2005;106(7):2580-2589. [DOI] [PubMed] [Google Scholar]
- 20.Zhu BM, McLaughlin SK, Na R, et al. . Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood. 2008;112(5):2071-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ned RM, Swat W, Andrews NC. Transferrin receptor 1 is differentially required in lymphocyte development. Blood. 2003;102(10):3711-3718. [DOI] [PubMed] [Google Scholar]
- 22.Brown RG. Determining the cause of anemia. General approach, with emphasis on microcytic hypochromic anemias. Postgrad Med. 1991;89(6):161-164, 167-170. [DOI] [PubMed] [Google Scholar]
- 23.Lopez A, Cacoub P, Macdougall IC, Peyrin-Biroulet L. Iron deficiency anaemia. Lancet. 2016;387(10021):907-916. [DOI] [PubMed] [Google Scholar]