

Key Points
SC411 increases DHA in blood cell membranes and reduces home-managed SCD pain crisis and analgesic and opioid use at home to treat SCD pain.
The rate of sickle cell crisis was 53% lower for the pooled active groups vs placebo.
Abstract
Blood cell membranes in sickle cell disease (SCD) have low docosahexaenoic acid (DHA). DHA treatment reduces sickle cell crisis (SCC) rate and ameliorates the inflammation, oxidative stress, and hypercoagulable state of SCD. SC411 is a novel DHA ethyl ester formulation with a proprietary delivery platform (Advanced Lipid Technology) that enhances DHA bioavailability. The SCOT trial investigated the effect of 3 different doses of SC411 on clinical and biochemical endpoints in 67 children with SCD (5-17 years old). Seventy-six percent of subjects were also receiving hydroxyurea. After 4 weeks of treatment with SC411 at 20, 36, and 60 mg DHA/kg per day or placebo a statistically significant (P < .001) mean percentage increase of blood cell membrane DHA and eicosapentaenoic acid was seen vs baseline: 109.0% (confidence interval [CI], 46.7-171.3), 163.8% (CI, 108.3-219.2), 170.8% (CI, 90.2-251.4), and 28.6% (CI, 250.1 to 107.3), respectively. After 8 weeks of treatment, statistically significant changes vs placebo were also observed in D-dimer (P = .025) and soluble E-selectin (P = .0219) in subjects exposed to 36 mg/kg. A significant increase in hemoglobin was observed against placebo in subjects receiving 20 mg DHA/kg per day (P = .039). SC411 significantly reduced electronic diary recorded SCC, analgesic use at home, and days absent from school because of sickle cell pain. The lower rate of clinical SCC observed in the pooled active groups vs placebo did not reach statistical significance (rate ratio, 0.47; 95% CI, 0.20-1.11; P = .07). All tested doses were safe and well tolerated. This trial was registered at www.clinicaltrials.gov as #NCT02973360.
Visual Abstract
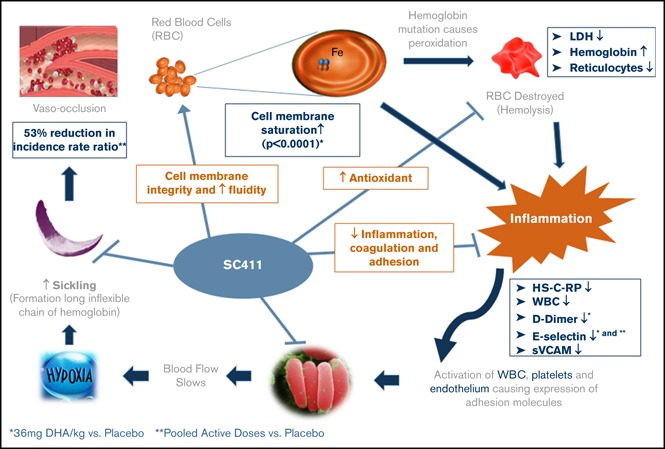
Introduction
Sickle cell disease (SCD) is a multisystem disorder caused by a mutation in the β-globin gene resulting in the sickle hemoglobin (HbS).1 Globally, approximately 300 000 infants are born annually with homozygous SCD, and that number could rise to 400 000 by 2050.2 In the United States, population estimates suggest that approximately 100 000 Americans have the disease.3 In Africa, mortality rates among children were reported to be as high as 90%.4,5 SCD is characterized by chronic hemolysis, painful crises, multiorgan dysfunction, and death. Sickle cell crisis (SCC) is the main cause of hospitalization, a negative effect on quality of life, and high risk for death.6
Although HbS polymerization is central to the pathophysiology of the disease,7 recent advances in SCD research suggest a more complex process.8-10 Instability of HbS induces impairments in erythrocyte membrane structure, elasticity, viscosity, and biochemical properties.10,11 In addition, oxidative stress, inflammation, enhanced adhesiveness of neutrophils, and hypercoagulability are increasingly recognized to play a primary role in vaso-occlusion, tissue ischemia, and organ damage.1,12,13
Given the complex nature of SCD pathophysiology, a multimodal therapy or a single drug with multiple mechanisms of action may be necessary to achieve clinical effectiveness.14,15 The multiple biological and pleiotropic effects of the ω-3 fatty acid docosahexaenoic acid (DHA, 22:6ω3) that include, but are not limited to, its antioxidant, anti-inflammatory, antiadhesion, anti-aggregation properties and its positive effects on erythrocyte rheology and vasodilatation16-25 all offer potential therapeutic benefits in SCD. In addition, resolvins, endogenous lipid mediators generated from DHA and eicosapentaenoic acid (EPA; 20:5ω3), have been shown to increase nitric oxide availability and reduce inflammatory pain.25,26
Interestingly, blood cells and plasma of patients with SCD show decreased levels of DHA and EPA, with increased levels of the pro-inflammatory ω-6 fatty acid arachidonic acid (20:4ω6).20,27-29 Treatment of patients with SCD with the ω-3 fatty acids DHA and EPA has been shown to correct cell membrane fatty acid balance and confer protection against vaso-occlusive pain episodes, hemolysis, oxidative stress, and inflammation.16-21 Treatment of mouse models of SCD with DHA and EPA also improves erythrocyte flexibility and reduces vascular endothelial activation and sickle cell-related end-organ injury.30,31
SC411 is a novel DHA ethyl ester formulation with a proprietary delivery platform (Advanced Lipid Technology, or ALT) that enhances DHA bioavailability and overcomes the variable absorption related to dietary fat (“food effect”)32,33 To investigate the safety and efficacy of 3 dose levels of SC411 in children with SCD, the phase 2 SCOT (Sickle Cell Omega-3 Treatment) trial (NCT02973360) was conducted at 11 sites in the United States.
Methods
Subjects
Eligible subjects were aged from at least 5 years to 17 or fewer years, with SCD (homozygous hemoglobin SS, hemoglobin SC, and hemoglobin S/β0-thalassemia documented by high-performance liquid chromatography or electrophoresis), and having had 2 to 10 documented clinical sickle cell crises (cSCC) within the 12 months before screening. cSCC were defined as painful crisis or acute chest syndrome. A painful crisis was defined as new onset of pain lasting 2 or more hours for which there is no explanation other than vaso-occlusion and that requires therapy with oral or parenteral opioids, nonsteroidal anti-inflammatories, or other analgesics prescribed by a health care provider in a medical setting (eg, hospital, clinic, or emergency room visit) or documented telephone management.14,34 Acute chest syndrome was defined as acute illness characterized by a new segmental pulmonary infiltrate on a chest X-ray, excluding atelectasis, and fever (≥38.5°C) or respiratory symptoms such as hypoxia, chest pain, tachypnea, wheezing, or cough.34
Subjects treated with hydroxyurea (HU) before screening were eligible if they were receiving treatment for a minimum of 6 months before screening and intended to continue at the same weight-based dose for the duration of the study. Subjects who were not receiving HU at screening were not allowed to initiate HU during the blinded part of the study.
Exclusion criteria included documented abnormal or “high conditional” transcranial Doppler ultrasonography within 12 months of screening, history of known cerebrovascular disease, blood transfusion within 3 months of screening, and chronic daily use of opioid analgesia for any reason (>30 consecutive days during the 6 months before screening) (for full list of the inclusion and exclusion criteria, see supplemental Figure 1).
Participation required written informed consent from the subject’s parent or legal representative and documented assent from participants aged 7 years and older. Ethical approval was obtained from either the local site Institutional Review Board or the central Western Institutional Review Board.
Study design
The SCOT trial was a phase 2, randomized, double-blind, placebo-controlled, parallel-group, dose-finding study to evaluate the safety, tolerability, and efficacy of 3 weight-based dose levels of SC411 (20 [range: 12-26]; 36 [range: 26-48], or 60 [range: 51-72] mg DHA/kg per day) in children with SCD. The trial was conducted in 2 parts, including a randomized, double-blind, placebo-controlled, parallel-group, dose-finding study (part A) and an ongoing optional 19-month open-label extension (part B) for subjects who completed part A. Part A consisted of a screening period of up to 2 weeks, an 8-week treatment period, and a 4-week safety follow-up period for those who opted not to join the open-label extension (OLE) part, for a maximum total of 14 weeks. Participation in both the double-blind part A and OLE part B could result in up to a maximum of 22.5 months of therapy. Only data from part A are reported in this manuscript.
Using an Interactive Response Technology system, subjects were randomly assigned 1:1:1 to 1 of the 3 dose levels. Then, patients at each dose level were randomly assigned 3:1 to SC411 and placebo. Randomization was stratified by baseline HU use. The subjects, investigators, and assessors were blinded to the treatment assignment. SC411 and a matching placebo were supplied as soft gel mini capsules. SC411 capsules contain 383 mg DHA, and matching placebo capsules contained approximately 410 mg soybean oil. Capsules were administered once daily, with or without food. Adherence was monitored by pill counts and a handheld, mobile electronic patient-reported outcome device (eDiary).
Subjects who participated in the blinded part A only underwent safety follow-up evaluation 4 weeks after the last dose. The subjects who consented to participate in both part A and part B, are receiving the lowest of the 3 active dose levels during the part B OLE for up to 19 more months (“Timeline for Clinical Study Visits”; supplemental Figure 2).
Efficacy endpoints
The primary efficacy endpoint was the percentage change from baseline in the total blood cell membrane DHA and EPA concentration after 4 weeks of treatment. EPA was also measured, in addition to DHA, because a modest increase in EPA was expected to occur as a result of enzymatic retroconversion of DHA to EPA.35 SC411 uses a proprietary Advanced Lipid Technology drug delivery platform that enhances DHA bioavailability.33 This may result in saturation of blood cell membrane with DHA by all 3 SC411 doses within 8 weeks of treatment. Hence, to identify the optimal SC411 dose capable of the greatest DHA increase within the shortest period of time, the primary endpoint analysis was determined to be performed at 4 weeks of exposure to SC411, instead of 8 weeks.
During part A, total blood cell membrane DHA and EPA concentration was determined by using a validated liquid chromatography–tandem mass spectrometry bioanalytical method (Medpace Bioanalytical Laboratories, Cincinnati, OH). The laboratory-based secondary and exploratory endpoints included plasma lactate dehydrogenase, D-dimer, reticulocyte count, white blood cell count, platelets, indirect bilirubin, high-sensitivity C-reactive protein, soluble vascular cell adhesion molecule-1, soluble E-selectin, and Hb. The secondary and exploratory laboratory-based endpoints were assessed as the change from baseline at 2, 4, and 8 weeks. The exploratory clinical endpoints included the rate of visits to a medical facility (hospital, clinic, or emergency room) for cSCC or other SCD-related complications, the number of hospitalization days for SCC, and number of blood transfusions (acute simple blood transfusions and exchange blood transfusions). Endpoints recorded using the eDiary included rate of painful crises, intensity of painful crises, frequency of analgesic use at home, and school attendance. An eDiary-recorded painful crisis was defined as a new onset of sickle cell pain accompanied by analgesic use, as recorded in the subject’s daily eDiary. The eDiary recorded pain crises must have been separated by at least 48 hours to be considered as a distinct painful crisis.
Safety assessment
Safety endpoints included the incidence of adverse events (AE), severe AEs (SAEs), treatment-emergent AEs, and AEs of special interest. Safety assessments were conducted at screening; at weeks 0, 2, 4, and 8 (part A and end of treatment); and 4 weeks after the last dose. Safety assessment included physical examination, vital signs (blood pressure, respiratory rate, pulse rate, oxygen saturation, and temperature), bleeding questionnaire, laboratory tests (complete blood count, chemistry panel, urinalysis, coagulation profile), serum (β-HCG) pregnancy test, and 12-lead electrocardiogram. Reported AEs were coded using the Medical Dictionary for Regulatory Activities (version 19.1) for medical history and AEs, and World Health Organization Drug Dictionary for prior and concomitant medications. An independent data and safety monitoring committee reviewed safety data during the study.
Trial oversight
Sancilio Pharmaceuticals Company, Inc. was responsible for study design, protocol development, and study implementation. Data were collected and analyzed by Cato Research (Durham, NC) data management and statistical departments. An independent Data and Safety Monitoring Committee monitored safety and overall implementation of the study at prespecified points during the study.
Statistical analysis
The median percentage of DHA and EPA erythrocyte membranes of the healthy pediatric population in the United States was estimated to be around 4%.36 Sickle cell patients have a lower DHA and EPA level in blood cell membranes. 20,27,28 Therefore, the sample size was based on the assumption that after 4 weeks of treatment, the mean blood cell membrane DHA and EPA percentage in at least 1 of the 3 SC411 dose levels would be increased by 100% compared with placebo treatment (approximately 7% for a SC411 dose and 3.5% for placebo). The standard deviation in each group was expected to be 3%. Under these assumptions, at least 15 subjects per group (total of 60 subjects) would provide approximately 87% power to test each dose vs placebo and would provide 97% power to test the combined doses vs placebo at the 0.05 α level. To account for a 10% expected attrition, a total of 67 subjects were randomized. The analysis of the primary, secondary, and exploratory endpoints were conducted on the intention-to-treat population. Compliance to the study medication was assessed by calculating the percentage of capsules taken ([capsules taken/capsules scheduled to be taken] × 100).
The primary endpoint, the percentage change in total blood cell membrane concentration of the sum of DHA and EPA after 4 weeks of intervention, was compared between the 3 dose groups and placebo, using a mixed-model repeated-measures analysis with baseline sum of DHA and EPA in total blood cell, baseline disease severity (based on the number of crises during the year before enrollment, dichotomized as 2-4 or 5-10 cSCC), age group, HU use, visit, treatment, and visit by treatment interaction as fixed effects, and subject as a random effect. The laboratory and biomarker-based secondary and exploratory endpoint variables were analyzed using the statistical model described in the analysis of the primary endpoint, but included data through week 8 of treatment.
The rate of cSCC, acute SCC, visits to a medical facility for SCC or SCD-related complications, rate of hospitalization, rate of blood transfusions, rate of eDiary-recorded painful crises, and rate of eDiary-recorded analgesic use were analyzed using a Poisson regression model with log-link, including terms for treatment, baseline disease severity, age at baseline, and prior HU use. A robust variance estimation was used to account for potential extra-Poisson variability (overdispersion) The log of the total number of days in the study from randomization was included in the model as the offset variable to provide the basis for the rate estimate. The rate estimate was multiplied by 365 to get the annualized rate. The annualized rate estimate by treatment, 95% confidence interval (CI) of the annualized rate, and the P value were calculated for comparisons between each treatment regimen, along with the average active-treatment effect across treatment regimens, and the placebo. The odds of school absenteeism because of sickle cell pain was analyzed using a logistic regression with treatment, baseline disease severity, age group, and HU use as covariates. Multiple imputation was used to account for missing data of the primary endpoint and cSCC.
The number of hospitalization days, number of days out of school, and diary-recorded pain intensity during the treatment period were analyzed using the analysis of variance method, with factors for disease severity and HU use included in the model. Safety variables were analyzed descriptively. SAS (version 9.4) statistical software was used for statistical analyses.
Results
Enrollment and follow-up
A total of 75 subjects were screened, and 67 subjects met the eligibility criteria and were enrolled in the study at 11 US sites between March and July 2017 (Figure 1). The enrolled subjects were randomly assigned to 1 of the 3 dose levels. Within each dose level, the subjects were randomly assigned to receive either SC411 or placebo in a 3:1 ratio. Sixteen subjects received 20 mg/kg DHA, 18 subjects received 36 mg/kg, 16 subjects received SC411 60 mg/kg, and 17 subjects received placebo. None of the patients who were receiving HU at enrollment stopped treatment during the blinded part of the study. A total of 62 subjects completed the double-blinded part A of the study (8 weeks postrandomization). Of the 5 subjects who did not complete the double-blinded part A, 3 subjects were lost to follow-up (one 20-mg DHA/kg and 2 placebo), 1 subject discontinued the assigned regimen (20 mg DHA/kg) because of drug-related abdominal pain, and 1 subject (placebo) was prematurely terminated because of noncompliance. Forty-one subjects (66.1%) enrolled in the 19 months OLE part B. All 21 subjects (33.9%) who decided not to enroll in the OLE part B underwent a safety follow-up visit 4 weeks after the end of part A treatment.
Figure 1.

Flowchart of patient enrollment, randomization assignments and follow-up.
Assessment of compliance during the blinded part of the study showed that the mean percentage of capsules taken were higher among those who received 36 and 60 mg DHA/kg (94.9% and 95.5%, respectively) and relatively lower among the patients who received 20 mg/kg (83.9%) or placebo (86.4%).
Baseline characteristics of the participants
The demographic and clinical baseline characteristics of the subjects were similar in the 4 groups (Table 1). Distribution of the subjects according to baseline disease severity, moderate (2-4 cSCC during the year before enrollment) vs severe (5-10 cSCC during the year before enrollment), was similar in 3 SC411 treatment groups; however, the placebo groups had relatively more subjects (n = 13, 76.5%) classified as moderate vs severe (n = 4, 23.5%).
Table 1.
Demographic and baseline characteristics of the patients in the intention-to-treat population
| Statistic/category | SC411 | Placebo (n = 17) | |||
|---|---|---|---|---|---|
| Dose level 1 (n=16) | Dose level 2 (n = 18) | Dose level 3 (n = 16) | Pooled (n = 50) | ||
| Age, mean (SD), y | 12.3 (2.3) | 11.3 (2.7) | 11.9 (2.8) | 11.8 (2.6) | 12.6 (3.7) |
| Age groups, y | |||||
| 5-11, n (%) | 5 (31.3) | 10 (55.6) | 8 (50) | 23 (46) | 7 (41.2) |
| 12-17, n (%) | 11 (68.8) | 8 (44.4) | 8 (50) | 27 (54) | 10 (58.8) |
| Sex, n (%) | |||||
| Male | 8 (50.0) | 9 (50.0) | 6 (37.5) | 23 (46.0) | 10 (58.8) |
| Female | 8 (50.0) | 9 (50.0) | 10 (62.5) | 27 (54.0) | 7 (41.2) |
| BMI, mean (SD) | 19 (4.5) | 18.5 (3.4) | 19.3 (3.6) | 18.9 (3.8) | 19.1 (4.6) |
| Race, n (%)* | |||||
| Black | 16 (100) | 18 (100) | 15 (93.8) | 49 (98) | 17 (100) |
| White | 0 (0) | 0 (0) | 1 (6.2) | 1 (2) | 0 (0) |
| Genotyping, n (%) | |||||
| Hemoglobin SS | 13 (81.3) | 14 (77.8) | 11 (68.8) | 38 (76) | 15 (88.2) |
| Hemoglobin SC | 2 (12.5) | 2 (11.1) | 4 (25) | 8 (16) | 2 (11.8) |
| Hemoglobin S/β0-thalassemia | 1 (6.3) | 2 (11.1) | 1 (6.3) | 4 (8) | 0 |
| Disease severity, n (%)† | |||||
| Moderate | 8 (50) | 11 (61.1) | 7 (43.8) | 26 (52) | 13 (76.5) |
| Severe | 8 (50) | 7 (38.9) | 9 (56.3) | 24 (48) | 4 (23.5) |
| HU use, n (%) | 13 (81.3) | 12 (66.7) | 12 (75.0) | 37 (74.0) | 14 (82.4) |
| Hematocrit, mean (SD), % | 28.6 (3.5) | 27.2 (4.8) | 29.7 (4.5) | 28.6 (4.3) | 27.5 (3.2) |
| Hemoglobin, mean (SD), g/dL | 9.19 (1.4) | 8.8 (1.4) | 9.7 (1.5) | 9.3 (1.4) | 9.0 (0.9) |
| Mean corpuscular hemoglobin, mean (SD), pg/cell | 29.0 (3.7) | 29.6 (4.4) | 30.4 (3.8) | 29.8 (3.9) | 30.8 (5.4) |
| Mean corpuscular volume, mean (SD), fL | 90.2 (9.7) | 90.3 (12.0) | 93.0 (11.1) | 91.3 (10.8) | 93.9 (14.1) |
| Platelets, mean (SD), ×109/L | 286.6 (143.2) | 334.9 (153.0) | 341.1 (112.1) | 325.7 (134.9) | 399.1 (178.9) |
| Erythrocytes, mean (SD), ×1012/L | 3.2 (0.6) | 3.1 (0.9) | 3.3 (0.8) | 3.2 (0.8) | 2.9 (0.8) |
| Reticulocytes, mean (SD), % | 6.6 (2.8) | 7.8 (4.3) | 6.3 (2.8) | 6.8 (3.3) | 6.7 (2.9) |
| Leukocytes, mean (SD), ×109/L | 8.2 (3.2) | 11.7 (5.4) | 8.8 (2.6) | 9.6 (4.0) | 10.6 (3.3) |
SD, standard deviation
Race was self-reported.
Patients who experienced 2 to 4 SCD crises during the year before randomization are classified as moderate. Those who experienced ≥5 to 10 SCD crises during the year before randomization are classified as severe.
Primary endpoint
The analysis of the primary endpoint showed a significant mean percentage increase from baseline in the sum of total blood cell DHA and EPA concentration of 109.0 (CI, 46.7-171.3), 163.8 (CI, 108.3-219.2), 170.8 (CI, 90.25-251.4), and 28.6 (CI, −50.1 to 107.3) in doses of 20, 36, 60 mg DHA/kg and placebo, respectively, after 4 weeks of treatment (Table 2; Figure 2); 36 and 60 mg DHA/kg showed a significant increase in the sum of DHA and EPA vs placebo.
Table 2.
Primary endpoint and selected secondary and exploratory biochemical endpoints in the intention-to-treat population
| Statistic/ category | SC411 | ||||
|---|---|---|---|---|---|
| Dose level 1 (n = 16) | Dose level 2 (n = 18) | Dose level 3 (n = 16) | Pooled (n = 50) | Placebo (n = 17) | |
| Primary endpoint: DHA + EPA blood cell membrane levels at week 4 | |||||
| Baseline (SD), μg/mL | 36.32 (11.74) | 38.59 (11.92) | 36.66 (14.70) | 37.28 (12.56) | 44.64 (18.30) |
| Week 4 (SD), μg/mL | 74.71 (20.63) | 108.40 (43.38) | 99.38 (24.90) | 96.01 (34.66) | 43.74 (18.82) |
| Change vs baseline (95% CI), %* | 109.00 (46.67-171.33) | 163.78 (108.32-219.24) | 170.84 (90.25-251.42) | 147.87 (105.54-190.21) | 28.61 (−50.07-107.29) |
| P value vs baseline | .0007 | <.0001 | <.0001 | <.0001 | .4730 |
| P value vs placebo | .0814 | .0037 | .0096 | .0043 | |
| Biomarkers associated with SCD pathophysiology at week 8 | |||||
| Hemoglobin | |||||
| Baseline (SD), μg/mL | 9.06 (1.23) | 8.84 (1.34) | 9.66 (1.47) | 9.18 (1.37) | 8.96 (0.89) |
| Week 8 (SD), μg/mL | 10.29 (1.16) | 9.58 (1.21) | 9.63 (1.33) | 9.79 (1.25) | 9.43 (0.96) |
| Change vs baseline (95% CI), g/dL† | 0.97 (0.51-1.42) | 0.70 (0.31-1.09) | 0.01 (−0.37 to 0.39) | 0.56 (0.31-0.81) | 0.33 (−0.11 to 0.78) |
| P value vs baseline | <.0001 | .0006 | .9402 | <.0001 | .1389 |
| P value vs placebo | .0389 | .2010 | .2667 | .3488 | |
| Reticulocytes | |||||
| Baseline (SD), % | 6.65 (2.82) | 7.52 (3.90) | 6.25 (2.83) | 6.83 (3.23) | 6.66 (2.91) |
| Week 8 (SD), % | 5.02 (1.55) | 5.85 (2.71) | 5.53 (2.81) | 5.51 (2.46) | 5.76 (3.02) |
| Change vs baseline (95% CI), %† | −0.98 (−2.3 to 0.3) | −0.86 (−1.9 to 0.2) | −0.57 (−1.7 to 0.5) | −0.80 (−1.5 to −0.1) | −0.07 (−1.3 to 1.2) |
| P value vs baseline | .1292 | .1165 | .2981 | .0266 | .9051 |
| P value vs placebo | .2786 | .3227 | .5320 | .2744 | |
| Exploratory endpoints: biomarkers associated with SCD at week 8 | |||||
| E-selectin | |||||
| Baseline (SD), ng/mL | 57.86 (19.32) | 72.10 (41.63) | 62.48 (24.56) | 64.74 (30.97) | 53.34 (19.77) |
| Week 8 (SD), ng/mL | 57.17 (17.78) | 60.77 (33.40) | 59.30 (23.38) | 59.23 (28.84) | 53.06 (19.88) |
| Change vs baseline (95% CI), ng/mL† | −2.89 (−10.8 to 5.0) | −8.95 (−15.1 to −2.8) | −1.60 (−8.1 to 5.0) | −4.48 (−8.8 to −0.2) | 1.74 (−5.4 to 8.9) |
| P value vs baseline | .4682 | .0052 | .6265 | .0429 | .6281 |
| P value vs placebo | .3435 | .0219 | .4691 | .1068 | |
| D-dimer | |||||
| Baseline (SD), μg/mL | 1.66 (2.87) | 1.18 (1.0) | 1.16 (0.73) | 1.33 (1.74) | 1.36 (0.69) |
| Week 8 (SD), μg/mL | 1.12 (0.75) | 0.71 (0.36) | 1.17 (1.00) | 0.99 (0.76) | 2.58 (3.02) |
| D-dimer change vs baseline (95% CI), μg/mL | −0.31 (−1.16 to 0.54) | −0.63 (−1.33 to 0.06) | −0.02 (−0.81 to 0.77) | −0.32 (−0.81 to 0.17) | 0.54 (−0.27 to 1.34) |
| P value vs baseline | .4683 | .0728 | .9541 | .1926 | .1845 |
| P value vs placebo | .1216 | .0250 | .3009 | .0521 | |
LS Means and LS mean differences are from a mixed-model repeated-measures analysis with baseline blood cell DHA+EPA, HU use, age group, baseline disease severity, and treatment as covariates. The model uses subject as a repeated measure with a compound symmetry covariance structure and includes a visit by treatment interaction. Contrasts, dose trend, and P values are produced inside the model.
LS means and LS mean differences are from a mixed-model repeated-measures analysis with each parameter's respective baseline, HU use, age group, baseline disease severity, visit, and treatment as covariates, and subject as a random variable. The model uses subject as a repeated measure with an unstructured covariance structure and includes a visit by treatment interaction. Contrasts and P values are produced inside the model.
Figure 2.

Percentage change of DHA+EPA blood cell membrane levels from baseline at week 4. After 4 weeks of treatment, blood cell membrane DHA and EPA levels were significantly increased in all SC411 doses (P < .001) vs baseline; 36 mg/kg, 60 mg/kg, and pooled treatments were also significantly increased vs placebo (P < .01).
Secondary and exploratory endpoints
After 8 weeks of treatment, significant changes against placebo were observed in D-dimer (P = .025) and soluble E-selectin (P = .0219) only in 36-mg DHA/kg-treated patients. Significant increase in Hb vs placebo was observed only in the 20-mg DHA/kg group (P = .039; Table 2; Figure 3) Directional positive trends were observed on plasma high-sensitivity C-reactive protein, lactate dehydrogenase, soluble vascular cell adhesion molecule, and white blood cell count, but these were not statistically significant (supplemental Figure 3).
Figure 3.

Effect of SC411 on selected biomarkers of adhesion, coagulation, and hemolysis. After 8 weeks of treatment, statistically significant reductions were observed in soluble E-selectin (A) (P = .0219), and D-dimer (B) (P = .025) in patients exposed to the 36 mg DHA/kg dose of SC411 vs placebo. Hemoglobin was significantly increased compared with baseline at doses 20 and 36 mg/kg and against placebo at 20 mg DHA/kg vs placebo (C) (P < .001). No significant difference was observed lactate dehydrogenase (D). *Significance vs placebo.
A total of 24 cSCC occurred during the double-blinded part of the study. Nine of these cSCCs occurred among 6 subjects receiving placebo and 15 among 14 subjects in the SC411 treatment groups. A reduction in the rate of cSCC was observed in the pooled treatment groups vs the placebo; however, it did not reach statistical significance (rate ratio, 0.45; 95% CI, 0.19-1.07; P = .07; Figure 4A).
Figure 4.

Ratio of the clinical SCC. The clinical sickle cell crisis rate ratio was calculated in all patients (A), patients receiving HU treatment at baseline (B), and patients not receiving HU treatment at baseline (C). The analysis of the sum of events for each subject was performed using Poisson regression (Proc Genmod). The model statement included covariates for either treatment (all doses vs placebo) or for the 4 dose levels vs placebo. Each analysis was adjusted for disease severity at baseline, the subject’s age group, and the subject’s use of HU at the time of the baseline evaluation. The model was standardized to an annual rate, using a SAS offset variable. Each table shows the estimate, its standard error, the risk ratio, and the 95% CI for the risk ratio, which was done by exponentiation of the lower and upper CIs for the treatment β coefficients.
Significant reduction in eDiary-recorded SCC, analgesic use, opioid use, and school absences resulting from SCD pain were observed on 36 and 60 mg/kg pooled SC411 subjects compared with placebo (Figure 4).
Reduction in hospitalization rate, hospitalization days, visits to medical facility for cSCC or sickle cell-related complications, eDiary-recorded sickle cell pain intensity, and number of school day absences resulting from SCD were observed in active groups compared with placebo; however, these were not statistically significant (supplemental Figure 4; Figure 5).
Figure 5.

eDiary-recorded endpoints. The rate ratio (active vs placebo) of pain crises (A), analgesic use at home (B), and opioid analgesic use at home (C) were calculated using a Poisson regression model with a log-link and with treatment, baseline disease severity, age group, and HU use as covariates. The model used the log of (days in the study/365) as the offset variable to produce the annualized rates. P values were testing to see whether the rate ratio is different from placebo. The odds of absence from school because of SCD pain (D) were calculated as the number of days missed (eDiary Q7 response of “Yes”) divided by the number of days available to be missed (eDiary Q7 response of “Yes,” “No, went to school,” or “No, other”). Least squares proportion estimates, CIs, and comparison P values are from a logistic regression with treatment, baseline disease severity, age group, and HU use as covariates. P values are testing to see whether the odds ratio is different from 1.
Subgroup analysis
There appeared to be a greater reduction in the rate ratio of cSCC for those receiving only SC411 treatment compared with the subjects receiving both HU and SC411; however, neither was statistically significant (supplemental Figure 4; Figure 4).
Safety
SAEs were reported in 9 placebo subjects (52.9%), 5 subjects (31.3%) at active 20 mg/kg dose, 6 subjects (33.3%) at active 36 mg/kg dose, and 5 subjects at active 60 mg/kg dose (31.3%; supplemental Figure 5). The most frequent SAE (sickle cell crisis) occurred in 8 placebo subjects (47.1%) and 11 subjects treated with 1 of the 3 active doses (22.0%). The percentage of subjects who experienced severe SAEs was higher in the placebo group (52.9%) than in the group randomly assigned to active doses 20 mg/kg (31.3%), 36 mg/kg (33.3%), and 60 mg/kg (31.3%).
There were only 2 AEs determined to be related to SC411: abdominal pain and nausea, experienced by 1 subject. AEs reported in more than 5% of the subjects who received 1 of the 3 active doses were abdominal pain, nausea, pyrexia, and headache.
Discussion
SCD was first described in the Western medical literature more than 100 years ago37; however, affected patients still have very limited disease-modifying treatment options. Until recently, HU was the only US Food and Drug Administration–approved medication in SCD with the adult indication to reduce the frequency of painful crises and the need for blood transfusions. Nevertheless, HU is not effective in all patients and may not prevent the accumulation of end organ damage. 38,39 Recently, the US Food and Drug Administration also approved l-glutamine treatment, although little is known about its clinical effectiveness.40 Therefore, there is still an urgent unmet need for new therapeutic options for children with SCD.
The SCOT trial showed that all 3 dose levels of SC411 significantly increased blood cell membrane concentration of the sum of DHA and EPA after 4 weeks of treatment. In addition, 36 and 60 mg/kg doses achieved relatively higher blood cell membrane concentration of DHA and EPA vs 20 mg/kg. Remarkably, doses 36 and 60 mg/kg achieved a comparable level of the DHA and EPA concentration in blood cell membrane, suggesting that 36 mg DHA/kg is the lowest effective dose to achieve the highest DHA and EPA concentration after 4 weeks of treatment.
Although SCOT was not designed or powered to provide definitive conclusions regarding reduction in cSCC, a nonstatistically significant reduction of at least 45% in the rate of cSCC was reported in all SC411 dose levels vs placebo during the double-blind part A. We hypothesized that SC411 would affect SCD pathophysiology through a distinct mechanism of action from HU that may result in additive effects. However, we observed a greater reduction (68%) in cSCC in the HU-naive subjects compared with 38% lower among those also receiving HU (Figure 4). There were also significant reductions in eDiary-reported sickle cell pain crisis, analgesic use at home to treat SCD pain, and opioid analgesic use to treat SCD pain in subjects who received either 36 or 60 mg DHA/kg. These beneficial effects were also reflected in a significant reduction in school absence resulting from sickle cell pain in SC411 treatment groups compared with placebo. The observed improvements in the clinical endpoints, despite not being statistically significant because of the limited study sample size, may suggest a beneficial therapeutic effect of SC411 on SCD children and supports progressing to a pivotal trial.41
Consistent with previously described mechanisms of action of ω-3 FAs in SCD,19,20,31 this study shows positive trends on markers of inflammation, adhesion, coagulation, and hemolysis among subjects who received 20 and 60 mg DHA/kg. The positive effects on the tested biomarkers were more pronounced in patients treated with 36 mg DHA/kg vs placebo, as statistically significant reduction was observed in biomarkers of endothelial activation (E-selectin) and coagulation (D-dimer).42,43 Intriguingly, the Hb concentration was significantly increased in doses 20 and 36 mg/kg against baseline, and in 20 mg/kg against placebo, but not in patients treated with 60 mg/kg to either baseline or placebo. Taking into account the relatively smaller changes in reticulocyte and lactate dehydrogenase in 60 mg/kg compared with baseline, the observed unchanged level of Hb seem not to be a result of increased hemolysis.
In addition to the putative mechanisms of action, some reports indicate that membrane DHA enrichment induces modifications in the cell membrane molecular organization, phospholipid composition, flip-flop dynamics, and functions.44-47 Dysfunctional lipid bilayer in sickle cell membrane is characterized by externalization of serine phosphoglycerides.48,49 It has been proposed that external serine phosphoglycerides contributes to thrombogenesis,50 enhanced adhesion with other blood cells and endothelium,51-53 shortened red blood cell lifespan in SCD,54,55 the provision of a cellular substrate for secretory phospholipase A256,57, and ultimately, vaso-occlusion and chronic hemolysis.58 Therefore, the observed therapeutic effects of DHA in patients with SCD could partially be attributable to its possible effects on cell membrane structure and function. Moreover, the potential antioxidant activity of DHA may also play a role in improving cell membrane integrity and functionality.15,19,59
The observed beneficial biochemical and clinical effects of SC411 in SCD children in the SCOT trial agree with previous studies of high-dose DHA or combination DHA/EPA treatment in patients with SCD for periods of 6 months or more.16,18,21 However, this is the first study that used multiple doses of a highly purified DHA during a relatively short period and in children who were also receiving concomitant HU treatment.
Pure DHA was used to develop SC411, as it has been conjectured that DHA could be a more efficacious and safer therapy for patients with SCD than others, similar to a combination of DHA and EPA or pure EPA.16 The reduction in ω-3 fatty acids levels observed in blood cell membrane of patients with SCD is more pronounced for DHA than EPA.29,60 In addition, it has been shown in several cell types that DHA has a greater influence on cell membrane deformability and fluidity as compared with EPA because of its higher unsaturation index,61,62 which could produce a beneficial improvement also in sickle erythrocyte rheology. In addition, DHA also can be enzymatically retroconverted to EPA to correct any EPA deficiency.35
The AEs, SAEs, and AEs of special interest did not differ significantly between the 3 active dose groups. It is important to note that higher frequency of SAEs occurred in the placebo group compared with the active groups. SC411 was well tolerated, as indicated by high retention rate, adherence to study medication, and high percentage of subjects opting to participate on the OLE part of the study.
In conclusion, the results from this 8-week study of multiple doses of a highly purified DHA in children with SCD, many of whom were still symptomatic despite receiving concomitant HU treatment, supports the progression to a larger phase 3 trial of SC411 in children with SCD.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and families who participated in the study, as well as the study coordinators and support staff. Clinical trial operational support was provided by George Steinfeld and Rebecca Avila; both are employees of Sancilio Pharmaceuticals Company, Inc. (SCI). Special thanks to Miguel Lopez-Toledano for his contribution in editing the manuscript.
This study was funded by SCI.
Authorship
Contribution: A.A.D. and A.L.R. conceived and designed the study and analyzed and interpreted data; A.A.D. wrote the first draft of the manuscript; C.D.D., A.L.R., and M.M.H. revised the manuscript; C.D.D., B.F., J.K., O.A.A., L.V.B., M.A.M., M.U.C., A.G., L.N., L.M.H., and M.M.H. performed the research (contributed patients to the study); and all authors thoroughly reviewed and approved the manuscript.
Conflict-of-interest disclosure: A.A.D. and A.L.R. are employees of SCI. O.A.A. has been an advisory board member for Novartis. C.D.D. has been an advisory board member for Novartis, Pfizer, and SCI; worked as a consultant for Novartis, Pfizer, GBT, Epizyme, and Prolong Therapeutics, and Data and Safety Monitoring Committee chair for Ironwood; and obtained research funding for Pfizer, SCI, Lilly, Novartis, Katz Foundation, and the National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development. J.K. has worked as a consultant for Bluebird Bio, Novartis, CRISPR, and AstraZeneca (steering committee), as well as participated in the National Heart, Lung, and Blood Institute Sickle Cell Advisory Committee. L.V.B. has worked as a consultant for Prolong Pharmaceuticals and Mast Therapeutics; been a paid member of the American Society of Pediatric Hematology/Oncology; participated in the advisory committee of Bioverativ; and obtained research funding from the National Institutes of Health, Pfizer, and Mast Therapeutics. M.U.C. participated in the Advisory Board of Shire, Octapharma, Grifols, Pfizer, Bayer, Roche, Bioverativ, and Hema; has been part of the Speakers Bureau of Shire, Roche/Genentech, Bayer, and Novo Nordisk; has obtained research support from Shire and Pfizer; has participated as site investigator for Pfizer, Roche/Genentech, Novo Nordisk, Global Blood Therapeutics, SCI, and Amgen; and holds stocks in Alnylum Pharmaceuticals. L.N. worked as a consultant for Emmaus, Bayer, CTD Holdings, and Pfizer; is currently on a Data and Safety Monitoring Committee for ApoPharma; is a site principal investigator for Pfizer, Sancilio, and PCORI; is a co-investigator for ApoPharma, Novartis, Bluebird Bio, Sangamo Therapeutics, Global Blood Therapeutics, Silarus, Celgene, Terumo, La Jolla Pharmaceuticals, and Imara; and also is an investigator on National Heart, Lung, and Blood Institute, Agency for Healthcare Research and Quality, US Food and Drug Administration, Health Resources and Services Administration, Centers for Disease Control and Prevention, Doris Duke, the State of California, University of California Office of the President, and Seattle Children's Research grants. F.S. has worked as a consultant for Clearway Global, LLC; and is a member of the Board of Directors of SCI; and participated in the Advisory Committees of Noble Financial Company. M.M.H. has worked as a consultant for SCI and AstraZeneca; is part of the Advisory Committees of SCI and Novartis; and received research support from SCI, Pfizer, and Intrinsic Life Sciences. The remaining authors declare no competing financial interests.
Correspondence: Matthew M. Heeney, Boston Children's Hospital, 300 Longwood Ave, Boston, MA 02115; e-mail: matthew.heeney@childrens.harvard.edu.
References
- 1.Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561-1573. [DOI] [PubMed] [Google Scholar]
- 2.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512-S521. [DOI] [PubMed] [Google Scholar]
- 4.Rahimy MC, Gangbo A, Ahouignan G, et al. Effect of a comprehensive clinical care program on disease course in severely ill children with sickle cell anemia in a sub-Saharan African setting. Blood. 2003;102(3):834-838. [DOI] [PubMed] [Google Scholar]
- 5.Molineaux L, Fleming AF, Cornille-Brøgger R, Kagan I, Storey J. Abnormal haemoglobins in the Sudan savanna of Nigeria. III. Malaria, immunoglobulins and antimalarial antibodies in sickle cell disease. Ann Trop Med Parasitol. 1979;73(4):301-310. [DOI] [PubMed] [Google Scholar]
- 6.Darbari DS, Wang Z, Kwak M, et al. Severe painful vaso-occlusive crises and mortality in a contemporary adult sickle cell anemia cohort study. PLoS One. 2013;8(11):e79923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Platt OS. Sickle cell anemia as an inflammatory disease. J Clin Invest. 2000;106(3):337-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation. 2009;16(1):97-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Embury SH. The not-so-simple process of sickle cell vasoocclusion. Microcirculation. 2004;11(2):101-113. [DOI] [PubMed] [Google Scholar]
- 10.Hebbel RP. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood. 1991;77(2):214-237. [PubMed] [Google Scholar]
- 11.Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64(1):72-80. [DOI] [PubMed] [Google Scholar]
- 12.Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9(2):101-106. [DOI] [PubMed] [Google Scholar]
- 13.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018-2031. [DOI] [PubMed] [Google Scholar]
- 14.Heeney MM, Hoppe CC, Abboud MR, et al. ; DOVE Investigators. A multinational trial of prasugrel for sickle cell vaso-occlusive events. N Engl J Med. 2016;374(7):625-635. [DOI] [PubMed] [Google Scholar]
- 15.Jagadeeswaran R, Rivers A. Evolving treatment paradigms in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2017;2017:440-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daak AA, Ghebremeskel K, Hassan Z, et al. Effect of omega-3 (n-3) fatty acid supplementation in patients with sickle cell anemia: randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2013;97(1):37-44. [DOI] [PubMed] [Google Scholar]
- 17.Okpala I, Ezenwosu O, Ikefuna A, et al. Addition of multimodal therapy to standard management of steady state sickle cell disease. ISRN Hematol. 2013;2013:236374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomer A, Kasey S, Connor WE, Clark S, Harker LA, Eckman JR. Reduction of pain episodes and prothrombotic activity in sickle cell disease by dietary n-3 fatty acids. Thromb Haemost. 2001;85(6):966-974. [PubMed] [Google Scholar]
- 19.Daak AA, Ghebremeskel K, Mariniello K, Attallah B, Clough P, Elbashir MI. Docosahexaenoic and eicosapentaenoic acid supplementation does not exacerbate oxidative stress or intravascular haemolysis in homozygous sickle cell patients. Prostaglandins Leukot Essent Fatty Acids. 2013;89(5):305-311. [DOI] [PubMed] [Google Scholar]
- 20.Daak AA, Elderdery AY, Elbashir LM, et al. Omega 3 (n-3) fatty acids down-regulate nuclear factor-kappa B (NF-κB) gene and blood cell adhesion molecule expression in patients with homozygous sickle cell disease. Blood Cells Mol Dis. 2015;55(1):48-55. [DOI] [PubMed] [Google Scholar]
- 21.Okpala I, Ibegbulam O, Duru A, et al. Pilot study of omega-3 fatty acid supplements in sickle cell disease. APMIS. 2011;119(7):442-448. [DOI] [PubMed] [Google Scholar]
- 22.Awoda S, Daak AA, Husain NE, Ghebremeskel K, Elbashir MI. Coagulation profile of Sudanese children with homozygous sickle cell disease and the effect of treatment with omega-3 fatty acid on the coagulation parameters. BMC Hematol. 2017;17(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruxton CH, Calder PC, Reed SC, Simpson MJ. The impact of long-chain n-3 polyunsaturated fatty acids on human health. Nutr Res Rev. 2005;18(1):113-129. [DOI] [PubMed] [Google Scholar]
- 24.Calder PC. Omega-3 polyunsaturated fatty acids and inflammatory processes: nutrition or pharmacology? Br J Clin Pharmacol. 2013;75(3):645-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011;34(11):599-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aslan M, Celmeli G, Özcan F, Kupesiz A. LC-MS/MS analysis of plasma polyunsaturated fatty acids in patients with homozygous sickle cell disease. Clin Exp Med. 2015;15(3):397-403. [DOI] [PubMed] [Google Scholar]
- 28.Daak AA, Ghebremeskel K, Elbashir MI, Bakhita A, Hassan Z, Crawford MA. Hydroxyurea therapy mobilises arachidonic Acid from inner cell membrane aminophospholipids in patients with homozygous sickle cell disease. J Lipids 2011;2011:718014. [DOI] [PMC free article] [PubMed]
- 29.Ren H, Okpala I, Ghebremeskel K, Ugochukwu CC, Ibegbulam O, Crawford M. Blood mononuclear cells and platelets have abnormal fatty acid composition in homozygous sickle cell disease. Ann Hematol. 2005;84(9):578-583. [DOI] [PubMed] [Google Scholar]
- 30.Wandersee NJ, Maciaszek JL, Giger KM, et al. Dietary supplementation with docosahexanoic acid (DHA) increases red blood cell membrane flexibility in mice with sickle cell disease. Blood Cells Mol Dis. 2015;54(2):183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalish BT, Matte A, Andolfo I, et al. Dietary ω-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica. 2015;100(7):870-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez-Toledano MA, Thorsteinsson T, Daak AA, et al. Minimal food effect for eicosapentaenoic acid and docosahexaenoic acid bioavailability from omega-3-acid ethyl esters with an Advanced Lipid TechnologiesTM (ALT®)-based formulation. J Clin Lipidol. 2017;11(2):394-405. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Toledano MA, Thorsteinsson T, Daak A, et al. A novel ω-3 acid ethyl ester formulation incorporating Advanced Lipid TechnologiesTM (ALT®) improves docosahexaenoic acid and eicosapentaenoic acid bioavailability compared with Lovaza®. Clin Ther. 2017;39(3):581-591. [DOI] [PubMed] [Google Scholar]
- 34.Ballas SK, Lieff S, Benjamin LJ, et al. ; Investigators, Comprehensive Sickle Cell Centers. Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol. 2010;85(1):6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J Clin Nutr. 2006;83(6 Suppl):1467S-1476S. [DOI] [PubMed] [Google Scholar]
- 36.Harris WS, Pottala JV, Varvel SA, Borowski JJ, Ward JN, McConnell JP. Erythrocyte omega-3 fatty acids increase and linoleic acid decreases with age: observations from 160,000 patients. Prostaglandins Leukot Essent Fatty Acids. 2013;88(4):257-263. [DOI] [PubMed] [Google Scholar]
- 37.Savitt TL, Goldberg MF. Herrick’s 1910 case report of sickle cell anemia. The rest of the story. JAMA. 1989;261(2):266-271. [PubMed] [Google Scholar]
- 38.Wang WC, Ware RE, Miller ST, et al. ; BABY HUG investigators. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raghupathy R, Billett HH. Promising therapies in sickle cell disease. Cardiovasc Hematol Disord Drug Targets. 2009;9(1):1-8. [DOI] [PubMed] [Google Scholar]
- 40.L-glutamine (Endari) for sickle cell disease. Med Lett Drugs Ther 2018;60(1539):21-22. [PubMed]
- 41.Kaul S, Diamond GA. Trial and error. How to avoid commonly encountered limitations of published clinical trials. J Am Coll Cardiol. 2010;55(5):415-427. [DOI] [PubMed] [Google Scholar]
- 42.Telen MJ. Biomarkers and recent advances in the management and therapy of sickle cell disease. F1000 Res. 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br J Haematol. 2013;162(1):3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akbar M, Calderon F, Wen Z, Kim HY. Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci USA. 2005;102(31):10858-10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams EE, May BD, Stillwell W, Jenski LJ. Docosahexaenoic acid (DHA) alters the phospholipid molecular species composition of membranous vesicles exfoliated from the surface of a murine leukemia cell line. Biochim Biophys Acta. 1999;1418(1):185-196. [DOI] [PubMed] [Google Scholar]
- 46.Shaikh SR, Kinnun JJ, Leng X, Williams JA, Wassall SR. How polyunsaturated fatty acids modify molecular organization in membranes: insight from NMR studies of model systems. Biochim Biophys Acta. 2015;1848(1 1 Pt B):211-219. [DOI] [PubMed] [Google Scholar]
- 47.Ibarguren M, López DJ, Escribá PV. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim Biophys Acta. 2014;1838(6):1518-1528. [DOI] [PubMed] [Google Scholar]
- 48.Middelkoop E, Lubin BH, Bevers EM, et al. Studies on sickled erythrocytes provide evidence that the asymmetric distribution of phosphatidylserine in the red cell membrane is maintained by both ATP-dependent translocation and interaction with membrane skeletal proteins. Biochim Biophys Acta. 1988;937(2):281-288. [DOI] [PubMed] [Google Scholar]
- 49.Kuypers FA, Lewis RA, Hua M, et al. Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled annexin V. Blood. 1996;87(3):1179-1187. [PubMed] [Google Scholar]
- 50.Atichartakarn V, Angchaisuksiri P, Aryurachai K, et al. Relationship between hypercoagulable state and erythrocyte phosphatidylserine exposure in splenectomized haemoglobin E/beta-thalassaemic patients. Br J Haematol. 2002;118(3):893-898. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz RS, Tanaka Y, Fidler IJ, Chiu DT, Lubin B, Schroit AJ. Increased adherence of sickled and phosphatidylserine-enriched human erythrocytes to cultured human peripheral blood monocytes. J Clin Invest. 1985;75(6):1965-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manodori AB, Barabino GA, Lubin BH, Kuypers FA. Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood. 2000;95(4):1293-1300. [PubMed] [Google Scholar]
- 53.Setty BN, Kulkarni S, Stuart MJ. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood. 2002;99(5):1564-1571. [DOI] [PubMed] [Google Scholar]
- 54.Bratosin D, Estaquier J, Petit F, et al. Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 2001;8(12):1143-1156. [DOI] [PubMed] [Google Scholar]
- 55.Lang KS, Lang PA, Bauer C, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15(5):195-202. [DOI] [PubMed] [Google Scholar]
- 56.Neidlinger NA, Larkin SK, Bhagat A, Victorino GP, Kuypers FA. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J Biol Chem. 2006;281(2):775-781. [DOI] [PubMed] [Google Scholar]
- 57.Fourcade O, Simon MF, Viodé C, et al. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell. 1995;80(6):919-927. [DOI] [PubMed] [Google Scholar]
- 58.Boas FE, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci USA. 1998;95(6):3077-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okuyama H, Orikasa Y, Nishida T. Significance of antioxidative functions of eicosapentaenoic and docosahexaenoic acids in marine microorganisms. Appl Environ Microbiol. 2008;74(3):570-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Connor WE, Lin DS, Thomas G, Ey F, DeLoughery T, Zhu N. Abnormal phospholipid molecular species of erythrocytes in sickle cell anemia. J Lipid Res. 1997;38(12):2516-2528. [PubMed] [Google Scholar]
- 61.Hashimoto M, Hossain S, Yamasaki H, Yazawa K, Masumura S. Effects of eicosapentaenoic acid and docosahexaenoic acid on plasma membrane fluidity of aortic endothelial cells. Lipids. 1999;34(12):1297-1304. [DOI] [PubMed] [Google Scholar]
- 62.Hashimoto M, Hossain S, Shido O. Docosahexaenoic acid but not eicosapentaenoic acid withstands dietary cholesterol-induced decreases in platelet membrane fluidity. Mol Cell Biochem. 2006;293(1-2):1-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.