

Key Points
CD73-mediated adenosine production contributes to SCD pathogenesis by promoting erythrocyte 2,3-BPG production and sickling.
Specific inhibition of CD73 significantly attenuates disease severity of SCD mice and provides a novel therapeutic strategy to treat SCD.
Abstract
Although excessive plasma adenosine is detrimental in sickle cell disease (SCD), the molecular mechanism underlying elevated circulating adenosine remains unclear. Here we report that the activity of soluble CD73, an ectonucleotidase producing extracellular adenosine, was significantly elevated in a murine model of SCD and correlated with increased plasma adenosine. Mouse genetic studies demonstrated that CD73 activity contributes to excessive induction of plasma adenosine and thereby promotes sickling, hemolysis, multiorgan damage, and disease progression. Mechanistically, we showed that erythrocyte adenosine 5′-monophosphate-activated protein kinase (AMPK) was activated both in SCD patients and in the murine model of SCD. AMPK functions downstream of adenosine receptor ADORA2B signaling and contributes to sickling by regulating the production of erythrocyte 2,3-bisphosphoglycerate (2,3-BPG), a negative allosteric regulator of hemoglobin-O2 binding affinity. Preclinically, we reported that treatment of α,β-methylene adenosine 5′-diphosphate, a potent CD73 specific inhibitor, significantly decreased sickling, hemolysis, multiorgan damage, and disease progression in the murine model of SCD. Taken together, both human and mouse studies reveal a novel molecular mechanism contributing to the pathophysiology of SCD and identify potential therapeutic strategies to treat SCD.
Visual Abstract
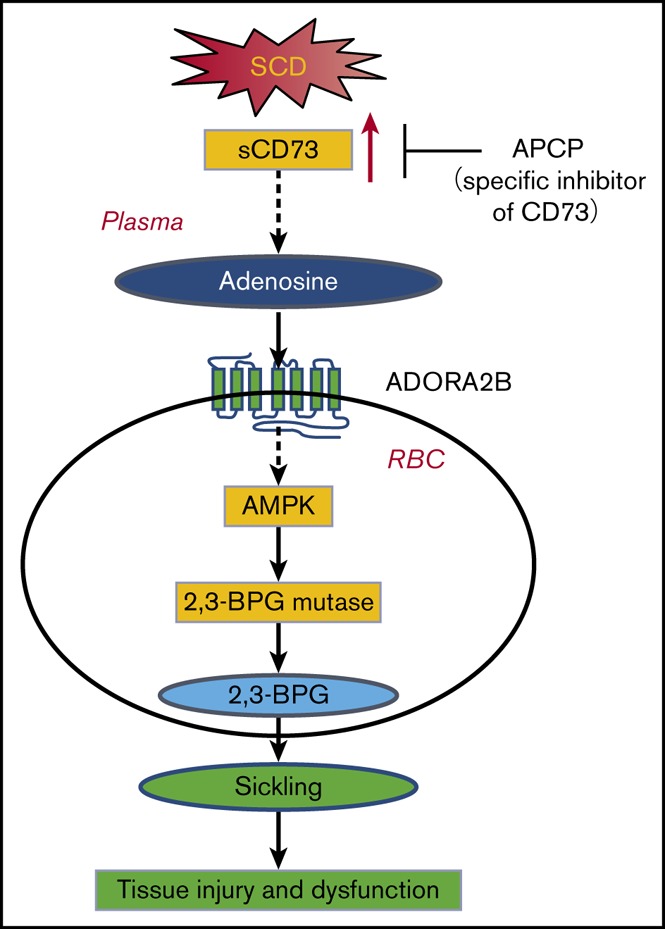
Introduction
Sickle cell disease (SCD) is a devastating genetic disease resulting from a point mutation in the β chain of hemoglobin (Hb), causing formation of an abnormal sickle Hb termed HbS.1 The deoxygenated HbS forms long insoluble polymers resulting in the red blood cells adopting a sickle shape that impairs blood flow through small blood vessels, leading to vaso-occlusion.2 As a result, SCD patients constantly face chronic hypoxia that is associated with high morbidity and mortality worldwide. Although this disease was identified more than a century ago, current treatment of this disease is still very limited. Earlier studies showed that excessive adenosine signaling via erythrocyte adenosine A2B receptor contributes to the pathophysiology of SCD by stimulating the production of erythrocyte 2,3-bisphosphoglycerate (2,3-BPG), a potent negative regulator of Hb-O2 binding affinity. The induction of deoxyHbS, and subsequent erythrocyte sickling, leads to chronic hemolysis, inflammation, and multiorgan damage.3-5
Adenosine functions as an important signaling molecule orchestrating the cellular response to hypoxia, energy depletion, and tissue damage by engagement of its G protein–coupled receptors on various cell types.6,7 Under stress conditions, adenosine triphosphate (ATP) is released from injured cells via transmembrane protein channels, including pannexins or connexins, and is subsequently converted to extracellular adenosine by the sequential action of 2 ectonucleotidases, CD39, which converts ATP to adenosine 5′-diphosphate (ADP)/adenosine 5′-monophosphate (AMP) and CD73, which converts AMP to adenosine.8-10 However, whether the CD73 activity is enhanced and contributes to excessive elevation of plasma adenosine and a detrimental role in SCD is unclear. Here we sought to investigate the molecular mechanism responsible for the increased plasma adenosine associated with SCD and to identify novel therapeutic possibilities for treating SCD.
Materials and methods
Human subjects
Individuals with SCD were identified by a hematologist on the faculty of the University of Texas Medical School at Houston. Subjects participating in this study had had no blood transfusion for at least 6 months before blood samples were collected. One-half of the individuals with SCD were treated with hydroxyurea. Control subjects were of African descent and were free of hematological disease. The research protocol, which included informed consent from the subjects, was approved by the University of Texas Health Science Center at Houston Committee for the Protection of Human Subjects.3
Mouse experiments
Eight- to 10-week-old C57BL/6 wild-type (WT) mice were purchased from Harlan Laboratories (Indianapolis, IN). Berkley SCD transgenic mice (SCD Tg) expressing exclusively human HbS were purchased from The Jackson Laboratory. CD73-deficient (Cd73−/−) mice with C57BL/6 background were generated and genotyped as described previously.11,12 All protocols involving animal studies were reviewed and approved by the Institutional Animal Welfare Committee of the University of Texas Health Science Center at Houston.
Irradiation and BMT
WT mice and CD73−/− mice (12 to 14 weeks of age) were treated with neomycin at 2 μg/mL in drinking water the day before irradiation, as described previously.13 The next day, mice were exposed to 5 Gy body irradiation with a RS X-ray irradiator (Rad Source Technologies, Suwanee, GA). Four hours later, the mice were exposed to the same dose of irradiation. Bone marrow cells (BMCs) isolated from WT mice were transplanted to lethally irradiated WT recipients for generation of WT bone marrow transplantation (BMT) controls (1 × 106 BMC per mouse IV). Similarly, BMC from SCD mice were transplanted to lethally irradiated WT recipients or CD73-deficient recipients, respectively, to create a SCD phenotype in WT (SCD BMT) or CD73-deficient (SCD/CD73−/− BMT) mice. After BMT, the mice were injected intraperitoneally with 1 μg/kg body weight erythropoietin per day for 3 days and treated with 2 μg/mL neomycin in drinking water for 2 weeks. Mice were sacrificed 16 weeks later for experiments.5,13
Chimerism measurement in SCD mouse BMT
Sixteen weeks post-BMT, flow cytometry was used to examine chimeras by quantifying blood CD45.1 (from recipient mice) and CD45.2 (from donor mice) positive cells, as in our previous publications.2,14
Blood collection and preparation
Human and mouse blood was collected and stored as described earlier. Approximately 4 mL human blood was collected with heparin as an anticoagulant for 2,3-BPG measurement with a commercial assay (Roche) as previously described.3,11,13,15 For plasma adenosine assay, 1 mL EDTA-collected blood was divided into aliquots in tubes containing 10 μmol/L dipyridamole (an inhibitor of equilibrative nucleoside transporters) and 10 μmol/L deoxycoformycin (an inhibitor of adenosine deaminase). Approximately 1 mL of mouse blood was collected and used for 2,3-BPG and plasma adenosine measurement, similar to human blood described previously.11
Plasma adenosine measurement
Plasma adenosine concentration was measured by reverse phase high-performance liquid chromatography (HPLC), as previously described.3,11,16 Briefly, 500 μL plasma was used to isolate adenosine by sequentially adding 0.6 mol/L cold perchloric acid, 3 mol/L KHCO3/3.6N KOH, 1.8 mol/L ammonium dihydrogen phosphate (pH 5.1), and phosphoric acid (30%). Next, the sample was centrifuged at 20 000g for 5 minutes, and the supernatant was transferred to a new tube for reverse phase HPLC analysis. Adenosine content was normalized to volume and expressed as a concentration.
ELISA measurement of erythrocyte phosphorylation of AMPKα at Thr172
Erythrocyte protein extract from human and mouse blood was collected as described previously.11 Next, phosphorylation levels of erythrocyte AMPKα at Thr172 were quantitatively measured by using commercially available enzyme-linked immunosorbent assay (ELISA) kits (A Solid Phase Sandwich ELISA, Cell Signaling).
BALF, total cell counts, albumin concentration, and IL-6 measurement
Bronchoalveolar lavage fluid (BALF) of mouse was collected as previously described.17 Briefly, lungs of anesthetized mice were lavaged 4 times with 0.3 mL phosphate-buffered saline (PBS). The collected BALF was separated for cell count quantification with a hemocytometer. The rest of the BALF was centrifuged and the supernatant was stored for further analysis. The albumin concentration in the BALF was measured by a mouse ELISA (Immunology Consultants Laboratory, Inc); the interleukin 6 (IL-6) concentration of BALF was detected by a mouse IL-6 ELISA according to the manufacturer’s instructions (BD Biosciences, US).11,17
MPO activity measurement in the lung, spleen, and liver
The myeloperoxidase (MPO) activity was measured by a commercial colorimetric activity assay kit (Sigma-Aldrich, St. Louis, MO). Briefly, harvested kidneys, lungs, and hearts were rapidly homogenized with MPO assay buffer, and centrifuged at 13 000g for 10 minutes at 4°C. Supernatants were used for MPO activity measurement according to manufacturer’s instructions.11
Isolation of total erythrocytes and treatment of human and mouse erythrocytes in vitro
Both human and mouse blood samples were collected with heparin as an anticoagulant, and erythrocytes were isolated by centrifugation at 240g for 10 minutes at room temperature. Packed erythrocytes were washed 3 times with culture media (F-10 nutrients mix, Life Technology) and resuspended to 4% hematocrit. One milliliter of erythrocytes was added to each well of a 12-well plate.3,11 Cultured erythrocytes were treated with BAY 60-6583 at 10 μM, AICAR (Tocris, Minneapolis, MN) at 1 mM, or BAY 60-6583 at 10 μM cotreated with Compound C (Tocris) at 20 μM for 4 hours under hypoxic conditions.3,11 At the end of experiments, 2,3-BPG levels were measured as mentioned previously (Roche, Nutley, NJ).
Measurement of plasma CD73 activity
Plasma CD73 enzyme activity was measured by quantifying the rate of the adenosine(E-ADO) production as described previously.12,18 In brief, isolated plasma were used for plasma CD73-specific activity measurement. First, 20 μL of plasma was preincubated at room temperature with 200 nM deoxycoformycin (an inhibitor of AMP-deaminase) in 0.1 M N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.4), with 50 μM MgCl, with or without α,β-methylene ADP, a specific inhibitor of CD73 (APCP, Sigma-Aldrich). Then, samples were incubated at 37°C for 30 minutes in the presence of 100 μM E-AMP. Plasma CD73 activity was measured by determining E-ADO concentrations with reversed phase HPLC as described previously. Absorbance was measured at 260 nm. Plasma CD73 activity was expressed as the rate of E-ADO produced from E-AMP that is inhibited by APCP.
Measurement of lifespan of erythrocytes in SCD mice
Berkley SCD Tg mice were treated with or without APCP for 8 weeks. At 6-week treatment, erythrocytes were labeled in vivo by using N-hydroxysuccinimide biotin and the lifespan of circulating red blood cells was measured as described previously.5 Specifically, 50 mg/kg−1 of N-hydroxysuccinimide biotin was injected into the retroorbital plexus of SCD Tg mice (prepared in 100 μL sterile saline just before injection; initially dissolved at 50 mg/mL in N,-N,-dimethylacetamide). Blood samples (only 5 μL) were collected after first day of biotin injection from tail veins by venipuncture to determine the percentage of erythrocytes labeled with biotin. Subsequently, 5 μL of blood was obtained from tail veins every 3 days for measurement of biotinylated erythrocytes until the 10th day. The percentage of biotinylated erythrocytes was calculated by determining the fraction of peripheral blood cells labeled with Ter-119 (to identify erythrocytes) that were also labeled with a streptavidin-conjugated fluorochrome by flow cytometry.13
Mouse organ isolation and histological analysis
Mice were euthanized and body weight was determined. Organs were isolated and weighed. Half of each organ was quickly frozen in liquid nitrogen and then stored at –80°C for heme content measurements as described in the following section. The remaining half of each organ was fixed with 4% paraformaldehyde in PBS overnight at 4°C. Fixed tissues were rinsed in PBS, dehydrated through graded ethanol washes, and embedded in paraffin. Five-micrometer sections were collected on slides and stained with hematoxylin and eosin (H&E). Semiquantification of histological changes was analyzed by Image-Plus, Pro software.3
Measurement of erythrocyte 2,3-BPG and 2,3-BPG mutase activity
Erythrocyte 2,3-BPG mutase was isolated as described previously and measured by a commercially available assay (Roche). Frozen erythrocyte pellets were used for protein extraction following the protocol as mentioned previously. Erythrocyte protein extract was subsequently used for measurement of 2,3-BPG mutase activity, as described previously with modification.11,19 Briefly, erythrocyte protein extract was incubated in 100 μL prepared reaction mixture (100 mM triethanolamine pH 7.6, 1 mM MgSO4, 4 mM ATP, 3 mM 3-phosphoglycerate, and 10 U phosphoglycerate kinase) for 20 minutes at 300°C. Then, the reaction was terminated by adding 25 μL of trichloroacetic acid and subsequently centrifuged at 10 000g for 5 minutes. One hundred microliters of the supernatant was mixed with 17 μL of 1.8 M Tris base; the 2,3-BPG level of the mixture was measured by using a commercial assay (Roche) as previous.10
Hb-O2 P50 measurement
Blood was collected as described previously. Ten microliters of whole blood was aliquoted and mixed with 4.5 hemox buffer (TCS Scientific Corporation, New Hope, PA), 10 μL anti-foaming reagent (TCS Scientific Corporation), and 20 μL 22% BSA in PBS; the mixture was then injected in the Hemox Analyzer (TCS Scientific Corporation) for measurement of O2 equilibrium curve at the temperature of 37°C to monitor Hb-O2 binding affinity (P50).
Morphology study of erythrocytes
Blood smears were made using 1% glutaraldehyde fixed cultured human or tail blood from bone marrow transplanted mice. Blood smears were stained by WG16-500ML kit (Sigma-Aldrich) for sickle cell. Blood smears stained by these procedures were observed using the 40× objective of an Olympus BX60 microscope. Areas where red blood cells did not overlap were randomly picked, at least 10 fields were observed, and 1000 red blood cells including sickle cells were counted. The percentages of sickle cells in red blood cells were calculated.
Hemolytic analysis
The Hb, haptoglobin, and total bilirubin in mouse plasma were quantified by ELISA kits following instructions provided by the vendor (BioAssay Systems, Hayward, CA).3
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM). Data were analyzed for statistical significance with GraphPad Prism 5 software. Nonparametric-equivalent Wilcoxon rank-sum tests were applied in 2-group analysis. Differences between the means of multiple groups were compared by 1-way analysis of variance, followed by a Tukey multiple-comparisons test. Comparison of the data obtained at different time points was analyzed by 2-way repeated measures, followed by the Tukey post hoc test. P < .05 was considered significant.
Results
Plasma CD73 activity is increased in SCD Tg mice and is essential for the induction of plasma adenosine, hemolysis, and decreased erythrocyte lifespan
The concentration of plasma adenosine was significantly increased in SCD Tg mice as compared with WT mice (Figure 1A). Because CD73 is the key enzyme to generate adenosine extracellularly,7 we hypothesized that CD73 contributes to excessive elevation of plasma adenosine in the SCD Tg mice. Indeed, we found that plasma CD73 activity was enhanced, and that the level of plasma CD73 activity was significantly correlated with the concentration of plasma adenosine in SCD Tg mice (Figure 1B-C). To further assess the functional role of CD73 in the SCD, we used a genetic approach coupled with BMT to create a SCD phenotype in WT (SCD BMT) or CD73-deficient (SCD/CD73−/− BMT) mice. Sixteen weeks after transplantation, we confirmed the success of our BMT by showing that the chimerisms in SCD BMT WT or CD73−/− mice were very high, up to 95% (supplemental Figure 1). Moreover, plasma CD73 activity was confirmed to be suppressed in the SCD/CD73−/− BMT mice compared with SCD BMT or WT BMT mice (Figure 1D). In addition, plasma adenosine was significantly reduced in the SCD/CD73−/− BMT mice compared with SCD BMT and WT BMT mice, thereby resulting in reduced sickled erythrocytes, improved hemolysis indicated by decreased plasma Hb and total bilirubin, and prolonged erythrocyte lifespan (Figure 1E-I).
Figure 1.

Genetic deletion of CD73 in SCD mice decreases hemolysis and increases erythrocyte lifespan by attenuating plasma CD73 activity and plasma adenosine level. (A-B) Plasma adenosine level and plasma CD73 activity level in WT and SCD mice. (C) Correlation of plasma CD73 activity and plasma adenosine level in SCD mice. (D) Plasma CD73 activity and (E) plasma adenosine level in WT BMT mice, SCD BMT mice, and SCD/CD73−/− BMT mice. (F) Representative image of sickled erythrocytes and the percentage of sickled erythrocytes in SCD BMT mice and SCD/CD73−/− BMT mice (magnification ×400). (G) Lifespan of erythrocytes in SCD BMT and SCD/CD73−/−BMT mice. (H) Plasma total hemoglobin and (I) plasma bilirubin in WT BMT mice, SCD BMT mice, and SCD/CD73−/− BMT mice. Data are expressed as mean ± SEM. n = 5 for each group; *P < .05 vs WT, **P < .05 vs SCD, Student t test.
CD73 contributes to multiorgan damage in SCD BMT mice
Histological analysis showed that tissue injury in multiple sites of SCD BMT mice including lung, spleen, and liver was substantially reduced in the SCD/CD73−/− BMT mice (Figure 2A-D).Because SCD patients are characterized with marked pulmonary dysfunction, we investigated the parameters of lung function in the SCD/CD73−/− BMT mice and SCD BMT mice. BALF was collected, and BALF cell count, BALF albumin concentration, and BALF IL-6 concentration were quantified in the WT BMT, SCD/CD73−/− BMT, and SCD BMT mice.
Figure 2.

Genetic deletion of CD73 in SCD mice decreases multiorgan damage including lung, spleen, and liver. (A) H&E staining of lungs, spleens, and livers of WT BMT, SCD BMT, and SCD/CD73−/− BMT mice (magnification ×200). (B-D) Semiquantitative analysis of H&E-stained sections show decreased lung congestion, spleen necrosis, and liver necrosis in SCD/CD73−/− BMT mice compared with SCD BMT mice. (E-G) MPO activity in multiple organs including lung, spleen, and liver in SCD/CD73−/− BMT mice compared with SCD BMT mice. Data are expressed as mean ± SEM. n = 5 for each group; *P < .05 vs WT, **P < .05 vs SCD, Student t test.
Consistent with the hemolysis and sickling data, the BALF cell count and BALF albumin concentration were significantly induced in the SCD BMT mice compared with WT BMT controls, indicating increased pulmonary vascular leakage (supplemental Figure 2A-B). Similarly, increased pulmonary inflammation was detected in the SCD BMT mice, including enhanced BALF IL-6, compared with WT BMT controls (supplemental Figure 2C). We further quantified neutrophils in multiple tissues by measuring the activity of MPO, a specific enzyme expressed in neutrophils. We found MPO activity was significantly induced in lung, spleen, and liver in the SCD BMT mice compared with WT BMT controls, whereas these pathological parameters were significantly attenuated in the SCD/CD73−/− BMT mice (Figure 2E-G).
AMPK underlies hypoxia-induced CD73-dependent erythrocyte sickling by regulation of 2,3-BPG mutase activity and 2,3-BPG level in SCD mice
Recent studies showed that AMPK, an energy sensor functioning downstream of ADORA2B, underlies adenosine-mediated activation of 2,3-BPG mutase, which leads to increased production of 2,3-BPG, a potent regulator of Hb deoxygenation in normal erythrocytes.11 However, the functional role of this newly identified pathway in sickling remains unclear. To investigate whether CD73-mediated elevation of adenosine is capable of inducing AMPK activation, subsequent activation of BPG mutase and increased 2,3-BPG production, we measured AMPK activity, 2,3-BPG mutase activity, and 2,3-BPG levels in WT BMT controls, SCD BMT mice, and SCD/CD73−/− BMT mice. The results show that AMPK activity, 2,3-BPG mutase activity, 2,3-BPG levels, and Hb-O2 P50 were significantly increased in erythrocytes of SCD BMT mice compared with WT BMT controls, but were reduced in SCD/CD73−/− BMT mice, which were associated with reduced plasma CD73 activity and plasma adenosine levels (Figure 3A-D).
Figure 3.

CD73 is important for elevation of plasma adenosine, erythrocyte p-AMPK, erythrocyte 2,3-BPG mutase activity, and erythrocyte 2,3-BPG in both SCD mice and SCD patients. (A) Erythrocyte p-AMPK, (B) erythrocyte 2,3-BPG mutase activity, (C) erythrocyte 2,3-BPG level, and (D) hemoglobin oxygen release capacity (P50) in WT BMT, SCD BMT, and SCD/CD73−/− BMT mice. (E) Plasma CD73 activity, (F) plasma adenosine level, (G) erythrocyte p-AMPK levels quantified by ELISA, (H) erythrocyte 2,3-BPG mutase activity, and (I) 2,3-BPG level in normal individuals and sickle cell disease patients. Data are expressed as mean ± SEM. n = 5 for each group; *P < .05 vs control, **P < .05 vs SCD, Student t test.
Next, to test the functional role of AMPK in sickle erythrocytes in response to hypoxia, we conducted in vitro studies of cultured erythrocytes isolated from SCD Tg mice. We found that phosphorylation of AMPK, 2,3-BPG mutase activity, 2,3-BPG levels, and sickling were significantly induced in response to hypoxia exposure. ADORA2B activation by Bay Compound, a specific ADORA2B agonist, further induced erythrocyte phosphorylation of AMPK, 2,3-BPG mutase activity, 2,3-BPG levels, and sickling compared with hypoxia treatment alone. However, inhibition of AMPK by Compound C, a specific AMPK antagonist, attenuated 2,3-BPG mutase activity, 2,3-BPG level, and sickling under hypoxia, indicating the importance of AMPK activation in erythrocytes of SCD Tg mice under hypoxia (supplemental Figure 3A-C).
Plasma CD73 activity, plasma adenosine, erythrocyte AMPK phosphorylation, 2,3 BPG mutase activity, and 2,3 BPG levels are increased in the SCD patients
To investigate the importance of our newly identified signaling components in SCD patients and translate our findings from mice to humans, we measured plasma CD73 activity and plasma adenosine levels in SCD patients and normal individuals. Plasma CD73 activity and plasma adenosine were significantly increased in the SCD patients compared with normal individuals. Meanwhile, phosphorylation of AMPK, 2,3-BPG mutase activity, and 2,3-BPG levels were induced in the SCD patients compared with normal individuals (Figure 3E-I). To translate our SCD mouse findings on the pathophysiological significance of CD73 and adenosine to SCD patients, we further analyzed the correlation of plasma CD73 activity and plasma adenosine levels with hemolysis parameters including plasma Hb levels and plasma bilirubin levels. Intriguingly, the plasma CD73 activity and plasma adenosine levels were positively correlated with hemolysis parameters including plasma Hb levels and plasma bilirubin levels in the SCD patients, respectively (Figure 4A-D). These results indicate that plasma CD73 activity and plasma adenosine are potential pathological biomarkers that can be used to predict the clinical severity of SCD patients.
Figure 4.

AMPK underlies the detrimental role of CD73-mediated adenosine signaling in erythrocytes in SCD patients. Plasma adenosine levels were correlated to (A) plasma hemoglobin and (B) bilirubin. Plasma CD73 activity was correlated to plasma (C) hemoglobin and (D) bilirubin. (E-H) Changes in erythrocyte-phosphorylated AMPK level, 2,3-BPG mutase activity, 2,3-BPG concentrations and percentage of sickled cells in erythrocytes isolated from SCD patients after incubation under hypoxic conditions in the absence or presence of Bay Compound (ADORA2B agonist), AICAR (AMPK agonist), Bay Compound + Compound C (AMPK antagonist), Compound C, respectively. *P < .05 vs normoxic condition; **P < .05 vs untreated samples under hypoxic condition; #P < .05 versus Bay Compound–treated samples under hypoxic condition. ##P < .05 vs untreated samples under hypoxic condition.
ADORA2B-induced AMPK activation underlies hypoxia-induced erythrocyte 2,3-BPG mutase activity, 2,3-BPG levels, and subsequent sickling in both SCD patients and SCD Tg mice
To investigate the pathophysiological significance of the ADORA2B-mediated AMPK signaling pathway in SCD, we conducted both human and mouse in vitro studies by culturing isolated erythrocytes from SCD patients and SCD Tg mice. First, we showed that hypoxia exposure induced phosphorylation of AMPK, 2,3-BPG mutase activity, 2,3-BPG levels, and sickling in cultured erythrocytes (Figure 4E-H; supplemental Figure 3A-D). Next, we found that activation of ADORA2B by a specific receptor agonist, Bay 60-5683, induced a greater elevation of phosphorylation of AMPK, 2,3-BPG mutase activity, 2,3-BPG levels, and sickling (Figure 4E-H; supplemental Figure 3A-D). Furthermore, by treating the cultured erythrocytes from SCD patients with AICAR, a specific AMPK agonist, under hypoxic conditions, we observed a similar elevation of phosphorylation of AMPK, 2,3-BPG mutase activity, 2,3-BPG levels, and sickling as the effect of Bay Compound treatment. However, inhibition of AMPK by Compound C significantly attenuated the hypoxia-induced effect, indicating that ADORA2B-mediated AMPK activation contributes to hypoxia-induced signaling by regulation of erythrocyte 2,3-BPG mutase activity and 2,3-BPG levels (Figure 4E-H; supplemental Figure 3A-D).
In vivo beneficial effect of CD73 inhibition in SCD mice
To investigate the therapeutic possibility of CD73 inhibition in the pathophysiology of SCD, we treated the SCD mice with APCP, a potent CD73-specific inhibitor. Consistent with the genetic evidence, pharmacological inhibition of CD73 significantly attenuated plasma CD73 activity and plasma adenosine, thereby decreasing erythrocyte phosphorylation of AMPK, 2,3-BPG mutase activity, 2,3-BPG levels, and P50 (Figure 5A-F). Moreover, reduced hemolysis, indicated by reduced plasma Hb and plasma bilirubin levels, as well as prolonged erythrocyte lifespan, were detected in the SCD mice with APCP treatment compared with vehicle controls (Figure 5G-I). Histological analysis showed that improved tissue injury including lung, spleen, and liver was observed in the SCD mice with APCP treatment (Figure 6A-D). Reduced pulmonary vascular leakage reflected by BALF cell count, BALF albumin concentration, and improved pulmonary inflammation reflected by reduced BALF IL-6 levels was seen in the SCD mice after APCP treatment (Figure 6E-G). MPO activity assay showed that reduced tissue inflammation in lung, spleen, and liver were observed in the SCD mice with APCP treatment compared with vehicle controls (Figure 6H). Therefore, our study showed that specific inhibition of CD73 provides potential therapeutic possibility to treat SCD.
Figure 5.

APCP treatment decreases plasma CD73 activity, plasma adenosine level, erythrocyte phosphorylation of AMPK, erythrocyte 2,3-BPG mutase activity, and erythrocyte 2,3-BPG level. (A-B) Plasma CD73 activity and plasma adenosine level in SCD mice with or without APCP treatment. (C-F) Erythrocyte-phosphorylated AMPK level, 2,3-BPG mutase activity, 2,3-BPG concentrations, and P50 in SCD mice with or without APCP treatment. (G-I) Plasma hemoglobin, plasma total bilirubin, and erythrocyte lifespan in SCD mice with or without APCP treatment. Data are expressed as mean ± SEM. n = 5 for each group; *P < .05 vs SCD, Student t test.
Figure 6.

APCP treatment in SCD mice decreases multiorgan damage, including lung, spleen, and liver. (A) H&E staining of lungs, spleens, and livers of SCD mice with or without APCP treatment (magnification ×200). (B-D) Semiquantitative analysis of H&E-stained sections showed decreased lung congestion, spleen necrosis, and liver necrosis in SCD mice following APCP treatment. (E) BALF total cell count, (F) BALF albumin concentration, and (G) BALF IL-6 concentration in SCD mice with or without APCP treatment. (H) MPO activity in multiple organs, including lung, spleen, and liver in SCD mice with or without APCP treatment. Data are expressed as mean ± SEM. *P < .05 vs SCD, Student t test. (I) Working model: CD73 is essential for induction of plasma adenosine in SCD. Elevated plasma adenosine contributes to erythrocyte sickling, tissue injury, and dysfunction by engagement of ADORA2B on erythrocytes to induce 2,3-BPG mutase activity and 2,3-BPG production. AMPK is a key enzyme that functions downstream of ADORA2B to activate 2,3-BPG mutase and promote 2,3-BPG production and subsequent erythrocyte sickling. Preclinically, inhibition of CD73 is a promising therapeutic strategy to treat SCD or prevent hypoxia-induced tissue damage. APCP treatment decreases multiorgan damage, including lung, spleen, and liver in SCD mice.
Discussion
Although CD73 is known to play important roles in various systems,20,21 no studies have reported that plasma CD73 activity was induced in SCD, and that the induction of plasma CD73 activity contributes to the pathogenesis and progression of SCD before our current studies. Here we report that plasma CD73 activity and plasma adenosine levels were significantly increased in the SCD Tg mice compared with WT mice. Genetic evidence showed that SCD/CD73−/− BMT mice displayed attenuated severity of disease including reduced hemolysis, sickling, pulmonary dysfunction, tissue inflammation, and multiple tissue injury. Mechanistically, we identified that erythrocyte AMPK was activated both in the SCD patients and in the murine models of SCD and functions downstream of adenosine ADORA2B signaling to contribute to sickling by regulation of 2,3-BPG mutase and 2,3-BPG production. Significantly, we found that plasma CD73 activity and plasma adenosine were positively correlated with the clinical pathological characteristics of SCD, including elevated plasma Hb and plasma bilirubin. Preclinically, we report that APCP treatment significantly decreased sickling, hemolysis, multiorgan damage, and disease progression in the murine models of SCD by interfering with CD73-mediated adenosine signaling in erythrocytes (Figure 6I). Therefore, both human and mouse studies reveal a novel molecular mechanism that contributes to the pathophysiology of SCD and identify potential therapeutic strategies to treat SCD.
As the central pathophysiology of SCD, molecular basis underlying hypoxia-mediated sickling has been intensively investigated. For example, early studies showed that increased reactive oxygen species with excessive oxidization of lipids and auto-oxidization of HbS coupled with an insufficient antioxidant defense system contribute to the sickling process.22,23 Further studies identified that impaired Lands’ cycle plays a detrimental role in SCD.11 More recent studies demonstrated that sphingosine kinase 1, the major enzyme to produce S1P in erythrocytes, significantly attenuated sickling by reducing S1P in the erythrocytes of SCD mice.5,13,14 Additional studies have identified that adenosine signaling is detrimental to SCD by induction of erythrocyte 2,3-BPG.24 However, the molecular basis responsible for the induction of plasma adenosine in SCD remained unknown. Here we revealed that plasma CD73 activity is increased in both SCD mice and humans. Genetic deletion of peripheral CD73 attenuates elevation of plasma adenosine and thus sickling and hemolysis in SCD BMT mice. As such, we observed significant reduction of tissue inflammation and damage in SCD/CD73−/− BMT mice. We provided strong evidence that elevated CD73 activity is detrimental in SCD by inducing elevation of plasma adenosine and subsequent sickling.
Adenosine-mediated biological function is mainly dependent on activation of adenosine receptors. Early studies showed that elevated adenosine signaling via ADORA2B contributes to sickling in SCD mice and cultured human sickle erythrocytes.24 Here we further demonstrated that CD73-dependent production of plasma adenosine signaling via ADORA2B induces 2,3-BPG production, deoxygenated Hb, and sickling by activating BPG mutase in a AMPK-dependent manner in both in vivo SCD mice and cultured human sickle erythrocytes. As such, inhibition of CD73 or AMPK activation reduced BPG mutase, 2,3-BPG production, deoxygenated Hb, and thus sickling, a central pathogenesis of SCD. In contrast to the detrimental effects of elevated adenosine in sickling, early studies showed that inhibition of invariant natural killer T (iNKT) cells through activation of ADORA2A reduces pulmonary dysfunction in the NY1DD mouse model of SCD.25 In view of this finding, a recent randomized phase 2 clinical trial26 reported that low-dose infusion of regadenoson (a specific ADORA2A agonist) intended to reduce the activity of iNKT cells is not sufficient to produce a statistically significant clinical efficacy. Thus, a potentially better therapeutic choice for adenosine-based therapy in SCD is a combination therapy including an ADORA2B antagonist-mediated antisickling effect in SCD erythrocytes and an ADORA2A agonist-mediated activation of iNKT cells, which may be a better therapeutic choice than CD73 inhibitor, because specific inhibition of ADORA2B will not prevent the potentially beneficial effects of adenosine in activating other types of adenosine receptors on cell types other than erythrocytes.
Adenosine and AMPK are 2 important hypoxic sensors regulating multicellular functions under physiological and pathological conditions.7,27,28 Recent human high-altitude studies and mouse genetic work have demonstrated that CD73-dependent elevation of plasma adenosine is beneficial to induce 2,3-BPG production in normal erythrocytes to counteract tissue hypoxia by activating AMPK.24 In contrast, here we provide both patient and mouse in vitro evidence that this newly identified pathway is detrimental for SCD by inducing 2,3-BPG production, deoxygenated HbS, sickling, hemolysis, shortened lifespan, and multiple tissue damage. Thus, our findings dissect the differential role of CD73-mediated elevation of adenosine and AMPK-induced 2,3-BPG production in normal and sickle erythrocytes.
Taken together, our findings support a working model that elevated plasma CD73 activity leads to increased adenosine and increased adenosine signaling via ADORA2B induces sickling and hemolysis by promoting mutase activity, 2,3-BPG production, and deoxygenated HbS via activation of AMPK activity. As such, plasma ATP and ADP are elevated from lysed sickle erythrocytes. Thus, increased plasma CD73 coupled with elevated plasma ATP and ADP work together as a malicious cycle to induce elevation of plasma adenosine, sickling, and hemolysis. Without inference, this cycle will progress to multiple tissue damage. Significantly, our findings immediately indicate that enhancing CD73-adenosine-AMPK signaling cascade-mediated BPG mutase activation can be beneficial for patients with normal HbA under hypoxia, whereas this approach can be detrimental for SCD patients with HbS. As such, our findings support the concept that precision medicine is critical for an effective treatment of individuals with SCD.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL114457, HL113574, HL137990, and HL136969) (Y.X.).
Authorship
Contribution: H.L. designed and conducted in vitro and in vivo experiments, analyzed all the experimental data, drew the figures, and wrote the manuscript; R.R.L., M.A., and J.M. conducted bone marrow transplantation and helped maintain SCD mice; Y.E.W. and A.Q.W. helped with mouse phenotype examination; A.S., T.W., and J.K. helped measure the soluble CD73 activity and plasma adenosine concentration; R.E.K., H.K.E., and M.R.B. provided expertise in adenosine signaling and proofread the manuscript; H.S.J. and M.I. provided expertise in sickle cell disease; and Y.X. oversaw the design of experiments and interpretation of results, the writing and organization of the manuscript, and did final editing of manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yang Xia, UT Health Science Center at Houston, 6431 Fannin, MSB 6.202, Houston, TX 77030; e-mail: yang.xia@uth.tmc.edu.
References
- 1.Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia a molecular disease. Science. 1949;110(2865):543-548. [DOI] [PubMed] [Google Scholar]
- 2.Hu X, Adebiyi MG, Luo J, et al. Sustained elevated adenosine via ADORA2B promotes chronic pain through neuro-immune interaction. Cell Reports. 2016;16(1):106-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Dai Y, Wen J, et al. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17(1):79-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Xia Y. Adenosine signaling in normal and sickle erythrocytes and beyond. Microbes Infect. 2012;14(10):863-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun K, Zhang Y, Bogdanov MV, et al. Elevated adenosine signaling via adenosine A2B receptor induces normal and sickle erythrocyte sphingosine kinase 1 activity. Blood. 2015;125(10):1643-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karmouty-Quintana H, Xia Y, Blackburn MR. Adenosine signaling during acute and chronic disease states. J Mol Med (Berl). 2013;91(2):173-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu H, Xia Y. Beneficial and detrimental role of adenosine signaling in diseases and therapy. J Appl Physiol (1985). 2015;119(10):1173-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eltzschig HK, Eckle T, Mager A, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99(10):1100-1108. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X, Teng B, Tilley S, Mustafa SJ. A1 adenosine receptor negatively modulates coronary reactive hyperemia via counteracting A2A-mediated H2O2 production and KATP opening in isolated mouse hearts. Am J Physiol Heart Circ Physiol. 2013;305(11):H1668-H1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson LF, Eltzschig HK, Ibla JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200(11):1395-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, Bogdanov M, Zhang Y, et al. Hypoxia-mediated impaired erythrocyte Lands’ Cycle is pathogenic for sickle cell disease. Sci Rep. 2016;6(1):29637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang W, Zhang Y, Wang W, et al. Elevated ecto-5′-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res. 2013;112(11):1466-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Berka V, Song A, et al. Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J Clin Invest. 2014;124(6):2750-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun K, D’Alessandro A, Ahmed MH, et al. Structural and functional insight of sphingosine 1-phosphate-mediated pathogenic metabolic reprogramming in sickle cell disease. Sci Rep. 2017;7(1):15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun K, Xia Y. New insights into sickle cell disease: a disease of hypoxia. Curr Opin Hematol. 2013;20(3):215-221. [DOI] [PubMed] [Google Scholar]
- 16.Song A, Zhang Y, Han L, et al. Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat Commun. 2017;8:14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo F, Le NB, Mills T, et al. Extracellular adenosine levels are associated with the progression and exacerbation of pulmonary fibrosis. FASEB J. 2016;30(2):874-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iriyama T, Sun K, Parchim NF, et al. Elevated placental adenosine signaling contributes to the pathogenesis of preeclampsia. Circulation. 2015;131(8):730-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallego C, Graña X, Carreras J. Increase of 2,3-bisphosphoglycerate synthase/phosphatase during maturation of reticulocytes with high 2,3-bisphosphoglycerate content. Mol Cell Biochem. 1991;102(2):183-188. [DOI] [PubMed] [Google Scholar]
- 20.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. 2006;2(2):351-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Fan J, Thompson LF, et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest. 2011;121(6):2371-2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silva DGH, Belini Junior E, de Almeida EA, Bonini-Domingos CR. Oxidative stress in sickle cell disease: an overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic Biol Med. 2013;65:1101-1109. [DOI] [PubMed] [Google Scholar]
- 23.Rogers SC, Ross JG, d’Avignon A, et al. Sickle hemoglobin disturbs normal coupling among erythrocyte O2 content, glycolysis, and antioxidant capacity. Blood. 2013;121(9):1651-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun K, Liu H, Song A, et al. Erythrocyte purinergic signaling components underlie hypoxia adaptation. J Appl Physiol. 2017;123(4):951-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood. 2010;116(23):5010-5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Field JJ, Majerus E, Gordeuk VR, et al. Randomized phase 2 trial of regadenoson for treatment of acute vaso-occlusive crises in sickle cell disease. Blood Adv. 2017;1(20):1645-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116(7):1776-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.