

Abstract
Functional enzyme—nanoparticle bioconjugates are increasingly important in biomedical and biotechnology applications such as drug delivery and biosensing. Optimization of the function of such bioconjugates requires careful control and characterization of their structures and activity, but current methods are inadequate for this purpose. A key shortcoming of existing approaches is the lack of an accurate method for quantitating protein content of bioconjugates for low (monolayer) surface coverages. In this study, an integrated characterization methodology for protein—gold nanoparticle (AuNP) bioconjugates is developed, with a focus on site-specific attachment and surface coverage of protein on AuNPs. Single-cysteine-containing mutants of dihydrofolate reductase are covalently attached to AuNPs with diameters of 5, 15, and 30 nm, providing a range of surface curvature. Site-specific attachment to different regions of the protein surface is investigated, including attachment to a flexible loop versus a rigid α helix. Characterization methods include SDS-PAGE, UV—vis spectrophotometry, dynamic light scattering, and a novel fluorescence-based method for accurate determination of low protein concentration on AuNPs. An accurate determination of both protein and AuNP concentration in conjugate samples allows for the calculation of the surface coverage. We find that surface coverage is related to the surface curvature of the AuNP, with a higher surface coverage observed for higher surface curvature. The combination of these characterization methods is important for understanding the functionality of protein—AuNP bioconjugates, particularly enzyme activity.
INTRODUCTION
The conjugation of proteins to nanoparticles is an active area of research due to potential biomedical and nanotechnological applications and the challenges that remain in realizing this potential.1–4 More specifically, protein—gold nanoparticle (AuNP) bioconjugates have attracted significant interest as hybrid materials for biosensing,5–6 enzyme immobilization,7 drug delivery,8–10 and bioimaging.11,12 For example, AuNPs have been used as colorimetric biosensors in ligand—receptor chemistry to isolate viable cells from a population of cells, with possible diagnostic applications and cancer biology assays.13 Extending the fundamental research into clinical trials, a first- in-man trial has been completed, where silica coated AuNPs have been bioengineered into a transplantation patch that was implanted onto a cardiac artery in patients with atherosclerosis (ClinicalTrials.gov Identifier: NCT01270139).14 AuNPs are of particular interest because of their biocompatibility and their ability to be surface functionalized and used as a delivery vehicle. Further, AuNPs have useful optical properties due to their surface plasmon resonance (SPR), which is the collective oscillation of electrons on the nanoparticle surface.15,16 The SPR absorption of AuNPs depends on the size of the particle and the dielectric constant of the capping molecules on the AuNP surface. In 100 nm spherical AuNPs or smaller, the SPR absorption band is centered in the green region of the visible spectrum.
Proteins have been attached to AuNPs mainly using two methods: adsorption by electrostatic interactions or covalent binding via functional groups.17–20 With electrostatic attachment, the binding is via noncovalent interactions specific to the charge of the capping ligand on the AuNP and the charge on the protein surface. Other noncovalent forces may help stabilize protein attachment, such as hydrogen bonding, van der Waals, or hydrophobic interactions.21–23 This type of conjugation is nonspecific because the protein can associate to the AuNP surface in many different orientations, depending on protein structure and surface charge. In contrast, attachment of the protein through covalent binding occurs with a specific functional group that binds to either the gold itself or a capping ligand.19,20,24,25 A surface-exposed cysteine has been used as an attachment site on proteins, as the thiol will covalently bind to the gold. This covalent method of conjugation is beneficial because the attachment point on the protein is will covalently bind to the gold. This covalent method of conjugation is beneficial because the attachment point on the protein is site-specific. The cysteine can be selectively placed such that the protein binds to a specific area of interest in the protein.26−28 This ability to control the location of the nanoparticle in relation to the protein and to create a tighter bond between the protein and nanoparticle are key motivations for covalent bioconjugation.
There have been significant efforts to characterize protein— nanoparticle bioconjugates, and surface coverage is an important parameter in bioconjugate stability and protein functionality. The activity of an enzyme—AuNP bioconjugate is directly related to the number of proteins bound to the nanoparticle surface; higher surface coverage gives a greater number of catalytic sites per AuNP and usually increases the bioconjugate activity.29 At high surface coverage levels, however, protein crowding may become an issue.30 Protein crowding might restrict access of the substrate and cofactor to their respective binding sites on the enzyme, or it might hinder the dynamics necessary for the protein to function, such as loop motions. In addition, the protein structure itself might be stabilized or destabilized due to electrostatics and sterics of protein—protein interactions.31,32 These effects of crowding negatively influence the activity of the enzyme. In contrast, for enzyme cascades, having multiple enzymes in close proximity can enhance the overall efficiency by substrate channeling or by speeding up the diffusion process.33 Therefore, the design of functional bioconjugates should consider the balance between the number of available catalytic sites and the effects of protein crowding on the nanoparticle surface. Further, surface coverage can be affected by the method of attachment (covalent or noncovalent) and the size and surface curvature of the AuNP (larger AuNP means smaller curvature). All of these factors should be considered when designing bioconjugates for optimum function. Multiple approaches have been developed to determine surface coverage of biomolecules on nanoparticle surfaces, but these methods either require a significant amount of biomolecule—NP conjugates or rely on specialized instrumentation that is not generally available to researchers in this field.34–36 We present a simple, sample-conservative, fluorescence-based assay in conjunction with UV—vis absorption spectroscopy method to accurately determine the surface coverage of protein on AuNPs.
The most common methods for bioconjugate characterization employ UV—vis spectrophotometry and transmission electron microscopy (TEM).17–19 However, these methods alone do not account for unbound protein or the molar ratio of bound protein to nanoparticle. A method has been reported for measuring the concentration of protein bound to AuNPs using circular dichroism (CD) spectroscopy.37 The lack of sensitivity of CD and large scattering background from nanoparticles limit this approach to high total protein concentrations, and the method assumes the protein bound to the nanoparticle surface is folded in the same manner (and thus has the same spectrum) as free in solution. Further, there are no reported methods for accurate protein concentration determination for low protein concentration samples (ie. a monolayer of protein on AuNPs), as AuNPs scatter significantly in the near UV and UV region of the absorption spectrum. Clearly, a new approach is required to determine the ratio of bound protein to nanoparticles to characterize the surface coverage of bioconjugates.
In this study, E. coli dihydrofolate reductase (ecDHFR) is used as a model enzyme for bioconjugate characterization studies. DHFR catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) via the oxidation of the cofactor nicotinamide adenine dinucleotide phosphate (NADPH) to NADP+.38 The enzyme is covalently linked to the AuNPs via a site-specific cysteine engineered into the enzyme surface. An integrated suite of physical methods is used to isolate and characterize the bioconjugates, including a method to separate free protein from bioconjugates and account for the free protein that might still be present. A novel method to determine the protein concentration of bioconjugates directly and accurately is reported, which is used in conjunction with UV-vis absorption spectra of the AuNPs to accurately determine surface coverage of protein on the AuNPs. This novel approach for measuring the protein/AuNP ratio is combined with dynamic light scattering (DLS) to demonstrate that the surface coverage is monolayer or submonolayer. The effect of AuNP surface coverage on protein binding and surface coverage are assessed by varying the AuNP sizes over a range of diameters from 5 to 30 nm. The surface coverage of protein is found to be related to the curvature of the AuNP surface, with higher surface coverage on AuNPs of greater curvature. The combination of these characterization methods provides structural characterization of the protein-AuNP bioconjugates that is important for understanding their functionality, particularly enzyme activity.
RESULTS AND DISCUSSION
The design of DHFR—AuNP conjugates for this study incorporates a site-specific, surface-exposed cysteine that is capable of forming a covalent bond with the gold atoms of the AuNP. These conjugates were purified from free, unbound enzyme (Figure 1) and then characterized with an integrated suite of biophysical methods. The concentrations of AuNPs were determined by UV-vis spectroscopy, and protein was determined by fluorescence after dissolution of AuNPs by KCN; these concentrations were used to calculate surface coverage of proteins on the AuNPs.
Figure 1.
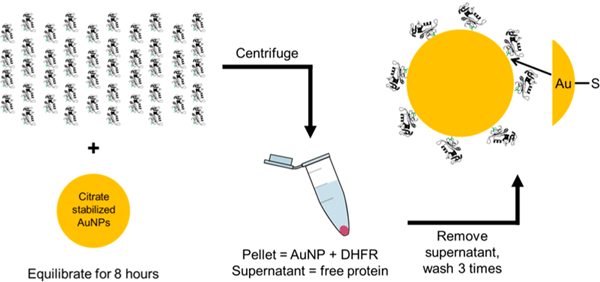
Schematic of protein−AuNP conjugation process. A large excess of DHFR is added to citrate stabilized AuNPs and allowed to equilibrate for no less than 8 h. The samples are centrifuged to pellet the conjugates, and the supernatant of free protein and citrate is removed by pipetting. Buffer is added to resuspend the pellet, and this process is repeated at least three times to remove all free protein from the conjugate solution
Conjugate Design.
Citrate, a weak capping ligand, was used to cap the AuNPs so that the protein thiolate S- would easily displace the citrate molecules from the AuNP surface. Wild-type (WT) DHFR contains two intrinsic cysteines at positions 85 and 152. When WT DHFR is folded, C85 is buried and not exposed to the surrounding solution, while C152 is surface exposed, located just off the GH loop of the protein. Because these cysteines might compete with the intended sites of attachment, both of them were replaced by site-specific mutation (ΔCys), C85 to an alanine (C85A) and C152 to a serine (C152S). Because the hexa-histidine tag added to the C-terminus of the protein for purification strongly associates to AuNPs,39 it was cleaved prior to formation of the bioconjugate to leave only the surface cysteine as the single binding point to the AuNPs. Two additional variants of DHFR were made to introduce non-native cysteines in surface- exposed positions as specific points of attachment, designated as the FG Loop mutant and Alpha Helix mutant (Figure 2a,b).
Figure 2.
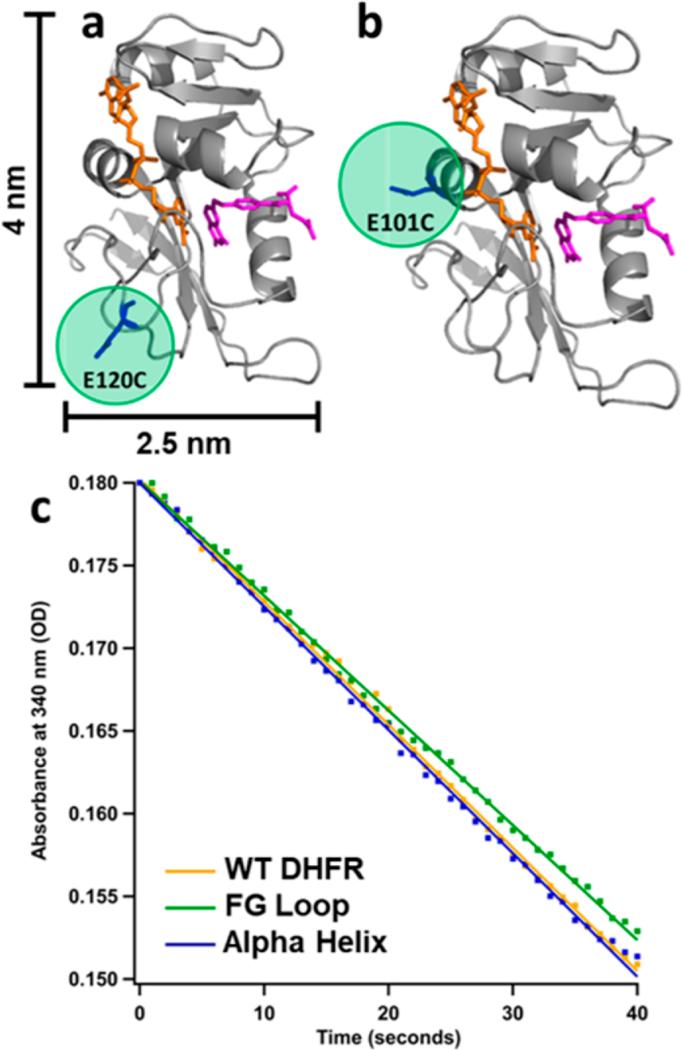
Location of cysteine mutation (highlighted in green) for (a) FG Loop mutant and (b) Alpha Helix mutant DHFR. Crystal structure from PDB 1RX2 with backbone shown in cartoon view (gray), substrate dihydrofolate in stick view (pink), and cofactor NADPH in stick view (orange). (c) Initial rate data for free protein when [DHFR] = 10 nM, [DHF] = 50 μM, and [NADPH] = 50 μM at 37°C. Yellow = WT DHFR. Green = FG Loop mutant. Blue = Alpha Helix mutant.
The FG Loop mutant (E120CΔCys, Figure 2a) has E120 on the FG loop mutated to a cysteine (E120C). The Alpha Helix mutant has E101 on a rigid α helix mutated to a cysteine (E101CΔCys, Figure 2b). These mutants are used to test the effect of the location of the point of attachment to the protein on the bioconjugate properties. The FG Loop mutant is expected to sterically hinder the substrate and cofactor from entering the active site, but the orientation on the AuNP should not completely prohibit the substrate and cofactor binding. The Alpha Helix mutant might sterically prevent the cofactor from having free access, but just as with the FG Loop mutant, it should not completely prevent binding. The focus of the present study is on the characterization of the bioconjugates formed with these enzyme constructs; the full evaluation of the reactivity of these bioconjugates will be published elsewhere.
Mutant Activity.
The enzyme activities of the free mutants were assayed in comparison to WT DHFR to ensure that the mutations did not greatly affect enzyme function. The steady- state kinetics of cofactor oxidation (NADPH absorbance at 340 nm) for the FG Loop mutant, Alpha Helix mutant, and WT DHFR in the presence of a large excess of substrate and cofactor are compared in Figure 2c. Since these kinetics measurements were obtained under identical conditions, they can be compared to assess the relative enzyme activity of each enzyme variant. The kinetics and thus the activity of each mutant is nearly identical to WT DHFR, so these mutations do not affect the ability of DHFR to catalyze the reduction of DHF.
AuNP Synthesis.
Citrate-stabilized AuNPs with a diameter of 15 nm were synthesized via the citrate reduction method.40 TEM was used to characterize the synthesized AuNPs, and the size of the AuNPs was analyzed with ImageJ (Figure 3 and Figure S1), giving an average particle size distribution of 14.1 ± 1.4 nm. Citrate-stabilized 5 and 30 nm AuNPs were purchased from Nanocomposix (San Diego, CA) and used without modification.
Figure 3.
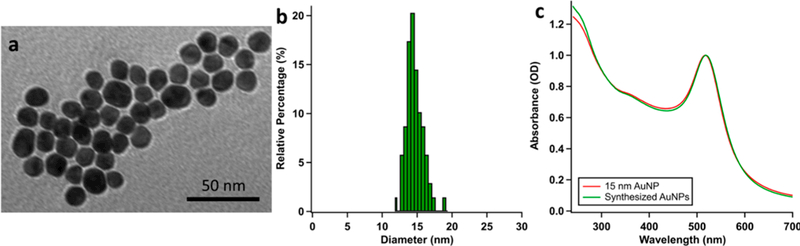
Characterization of 15 nm AuNPs. (a) TEM image of synthesized AuNPs. The scale bar is 50 nm. (b) Particle size distribution. The TEM images were analyzed with ImageJ. Sizes of 75 AuNPs were measured, and the size distribution was plotted in 0.5 nm increments. The average diameter of the AuNPs is 14.1 ± 1.4 nm. (c) UV-vis absorption spectra of synthesized AuNPs (green) and 15 nm AuNPs bought from Nanocomposix (red).
Protein-AuNP Conjugation and Isolation.
DHFR was conjugated to AuNPs through covalent binding with the surface cysteine as shown in Figure 1. A large excess of protein was added to AuNPs in the binding step, which means most of protein was not bound to the AuNPs. The free protein was separated from the conjugates via centrifugation, which pellets the conjugates and leaves the remaining unbound protein and displaced citrate in the supernatant. Multiple rounds of centrifugation and washing with 0.005% Tween 20, 10 mM sodium phosphate, pH 7 buffer removed the free protein. The conjugates were then stored in the same buffer at 4°C until characterized.
There are three lines of evidence that support covalent attachment rather than nonspecific chemisorption of DHFR to AuNPs. First, we have introduced surface Cys into the DHFR structure in positions that are highly exposed. Furthermore, we have tested whether these Cys are chemically active by labeling these positions with a thiol reactive dye, Badan (6-Bromoacetyl-2-dimethylaminonaphthalene). Attachment efficiency of the Badan dye to the E120CΔCys and E101CΔCys mutants is >90%, which indicates the surface Cys is highly reactive in both cases. Second, reactive thiols (thiolates) invariably form strong covalent bonds with a gold surface. Covalent attachment of proteins to Au surfaces through reactive Cys has been established for many proteins, including cytochrome c, glucose oxidase and Protein G.26−28 Therefore, it is highly likely that since our DHFR constructs have reactive surface Cys they will form covalent attachments with a gold surface. Finally, we conducted a series of control experiments to verify covalent attachment (Figure S2). Protein that is covalently bound on a gold surface cannot be removed by denaturing the protein or by the introduction of high salt concentration, whereas such treatment often removes chemisorbed protein. Furthermore, introduction of high salt concentration to AuNPs that are not stabilized by bound protein causes the AuNPs to aggregate and crash out of solution, a process that is easily detected by a color change from pink to blue. Capping the AuNP binding sites and exposed thiols on the protein with an excess of 2-mercaptoethanol (βME) blocks covalent attachment of the protein. These properties of the protein−AuNP conjugates can be used to test the mode of attachment. When covalent attachment of DHFR is blocked with βME, addition of a high concentration of salt causes the nanoparticles to immediately aggregate, with an associated color change (Figure S2a). Without βME, addition of the same large excess of salt does not crash out the AuNPs because the covalently bound DHFR stabilizes them in this case (Figure S2a). This same effect is observed when the protein is denatured in preparation for an SDS-PAGE gel. The βME containing conjugate samples aggregate upon denaturation of DHFR, whereas this is not the case for the samples without βME, due to covalent attachment of the protein (Figure S2b). SDS-PAGE of these samples shows that only the DHFR−AuNP conjugates and DHFR−AuNP + NaCl samples are stable (Figure S2c). After staining the gel, free protein can be seen in the lanes containing DHFR−AuNP + βME and DHFR−AuNP + βME + NaCl, indicating that the protein is not bound to the AuNPs in the presence of βME (Figure S2d). These results demonstrate that the DHFR−AuNP conjugates contain protein that is covalently bound to the AuNPs.
Removal of Free Protein from Conjugates.
SDS-PAGE of the conjugates was used to determine the efficiency of removal of the free protein from conjugates. The unstained gel (Figure S3) shows that DHFR—AuNP conjugates run on the gel (lanes 2—7) and are pink in color, while unbound AuNPs aggregate in the well (lanes 8—10) and are black in color. The free AuNPs actually aggregate immediate upon addition to loading buffer, as they are weakly capped with citrate and salt easily destabilizes them. The absence of aggregation in the case of the conjugates indicates that they are stabilized relative to citrate-capped AuNPs. The 5 nm AuNP-DHFR conjugates run the fastest and 30 nm AuNP-DHFR conjugates run the slowest, as expected based on size. Coomassie Blue stain was used to detect the presence of protein. In the stained gel (Figure 4), the pink conjugates turn purple, which indicates the presence of protein directly on the AuNPs. The free protein bands run at approximately 21 kDa in lanes 11 — 12, and these free protein bands are not present in any of the conjugate lanes, indicating that there is no (within the detection limit) free protein in the conjugate samples after separation and washing. The detection limit of a stainedprotein band on the gel is approximately 3% of the AuNP bound protein concentration.
Figure 4.
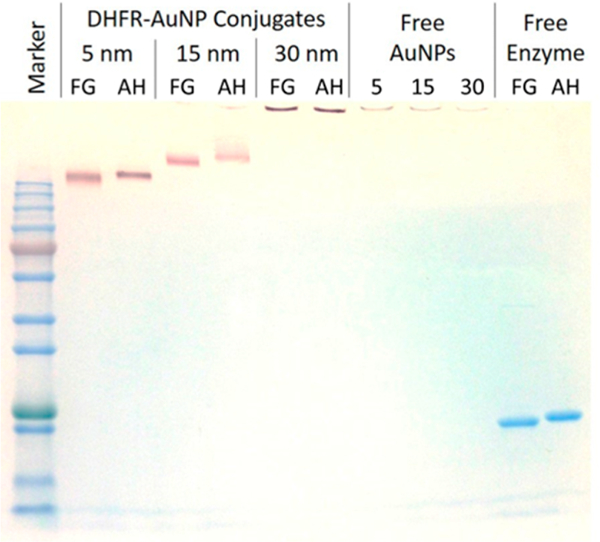
Stained SDS-PAGE gel of DHFR−AuNP conjugates and free DHFR. Lane 1 is protein marker. Lanes 2−3 are 5 nm AuNP- DHFR conjugates. Lanes 4−5 are 15 nm AuNP-DHFR conjugates. Lanes 6−7 are 30 nm AuNP-DHFR conjugates. Lanes 8−10 are free AuNPs: 5, 15, and 30 nm. Lanes 11 −12 are free protein. FG = FG Loop mutant. AH = Alpha Helix mutant. AuNPs: 5, 15, and 30 nm. Lanes 11 −12 are free protein. FG = FG Loop mutant. AH = Alpha Helix mutant.
UV-vis Absorption Spectra of Conjugates.
UV-vis spectrophotometry was used to verify the replacement of the citrate capping ligands with protein on the AuNP surface. For free citrate-stabilized 5 nm AuNPs, the SPR absorbance maximum is at 515 nm. When protein has replaced citrate, the SPR band redshifts due to the change in dielectric constant of the molecules on the surface of the AuNP. This redshift is approximately 6 nm, to 521 nm (Figure S4a). The SPR shift for the 15 nm AuNP conjugates is from 518 to 523 nm (Figure S4b), and for 30 nm AuNP conjugates, the shift is from 520 to 524 nm (Figure S4c). These SPR redshifts from free AuNPs to DHFR−AuNP conjugates are consistent for both the FG Loop mutant and Alpha Helix mutant conjugates, which indicates that the protein is indeed bound to the AuNP surfaces.
Dynamic Light Scattering.
Dynamic light scattering (DLS) was used to determine the hydrodynamic diameter of the AuNPs and conjugates. DLS measures the rate of diffusion of particles through a confocal observation volume that is defined by imaging the scattered light through a pinhole. This diffusion rate is then used to calculate the hydrodynamic diameter of the particles. It is difficult to measure the smallest particle size (5 nm) with conventional DLS instruments that employ a red light-source, so we applied this technique to characterize all samples except free 5 nm AuNPs. The time decay of the autocorrelation function (ACF) of the scattering signal is plotted for free AuNPs and conjugates in Figure 5a-c. The ACF decay is slower for the conjugates than for free AuNPs, as expected for larger, slower diffusing particles. The plot of the intensity distributions (inset of Figure 5a-c) shows narrow size distributions for free AuNPs, centered at 18 and 35 nm for the 15 and 30 nm AuNPs, respectively. The DLS measurement determines hydrodynamic diameters from diffusion rates, which are slightly higher than the physical diameter determined by TEM. The intensity distributions of 5 nm AuNP-FG Loop and Alpha Helix conjugates (Figure 5a) are centered at 16.5 and 19.4 nm, respectively, where the slight difference could be due to the higher concentration of conjugates required to obtain DLS data with the smaller particles. Both 15 nm AuNP-DHFR conjugates have intensity distributions centered at 27 nm and are slightly broader than the free AuNP distribution. A similar increase in size to 48 nm is observed for 30 nm AuNP-FG Loop conjugates and to 46 nm for 30 nm AuNP-Alpha Helix conjugates. The roughly 912 nm increase in hydrodynamic diameter for the DHFR- AuNP conjugates is consistent with the known structural properties of DHFR. The physical diameter of DHFR is at maximum 3.5 nm,7 and the hydrodynamic diameter is somewhat larger (4.5 nm); therefore, the measured hydrodynamic diameters of the conjugates indicate a monolayer or slightly submonolayer of protein bound to the AuNP.
Figure 5.
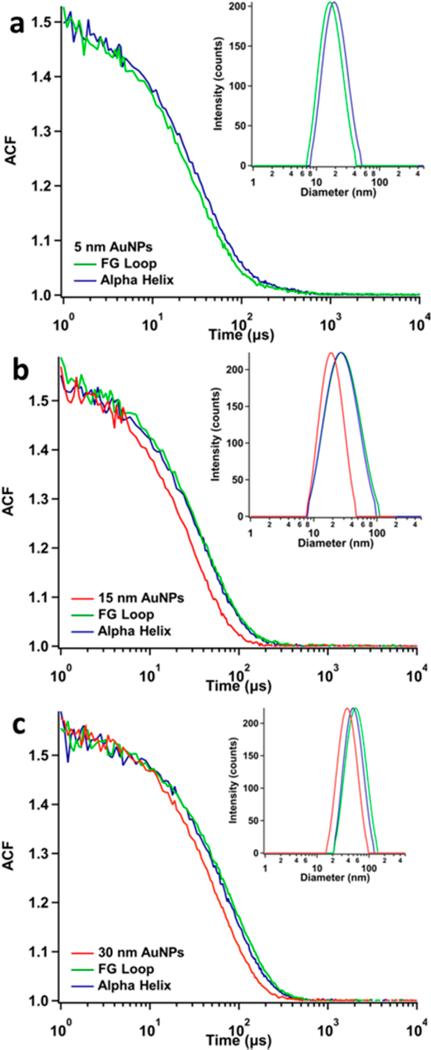
DLS of DHFR−AuNP conjugates. (a) 5 nm AuNP conjugates: (green) FG loop conjugates (16.5 nm), (blue) Alpha Helix conjugates (19.4 nm). Free AuNPs are too small to obtain accurate DLS measurements. (b) 15 nm AuNP conjugates: (red) free 15 nm AuNPs (18 nm), both AuNP-DHFR conjugates (27 nm). (c) 30 nm AuNP conjugates: free 30 nm AuNPs (35 nm), FG loop conjugates (48 nm), Alpha Helix conjugates (46 nm).
Protein Concentration Determination of Conjugates.
Currently, there is very little information in the literature regarding the determination of protein concentration on AuNPs at low surface coverage; however, this information is crucial to understanding bioconjugate function and stability. The most common methods for quantifying protein concentration are spectrophotometric, using the intrinsic UV absorption of the protein at 280 nm or the visible absorption at 595 nm in a Bradford assay. The strong absorption and scattering background of AuNPs introduce large errors into these methods, however, making them unsuitable for application to protein−AuNP conjugates. The method we have developed for protein concentration determination in protein−AuNP conjugates is a fluorescence assay (Figure 6), following the dissolution of AuNPs with potassium cyanide (KCN) (Figure 6a).41 When KCN is added to a sample of conjugates, KCN and Au atoms form a complex, dissolving the AuNPs and leaving the protein free in solution. The dissolution of the AuNPs removes the absorption and scattering spectral contributions due to the particles.41 The protein in this state (for this sample) is too dilute to measure via standard UV−vis absorption, so the much more sensitive fluorescence detection is used. WT DHFR has five intrinsic tryptophan residues, so there is a strong native fluorescence signal that can be used to quantify the amount of protein. Additionally, fluorescence spectroscopy intrinsically has zero background, so it is much more sensitive than absorption spectroscopy. Known concentrations of WT DHFR samples were used to create a calibration curve, prepared in the same manner as the conjugate samples, including identical additions of TCEP, Tween 20, and KCN (Figure 6b). The fluorescence spectra are taken with 280 nm excitation to excite the tryptophan residues, and the tryptophan emission peak is centered at approximately 340 nm. The area under the emission curve is plotted versus the respective protein concentration. The linear fit of this calibration curve is used to determine the protein concentration in the conjugate samples, which were also excited at 280 nm and analyzed in the same manner as the WT DHFR standards (Figure 6c).
Figure 6.
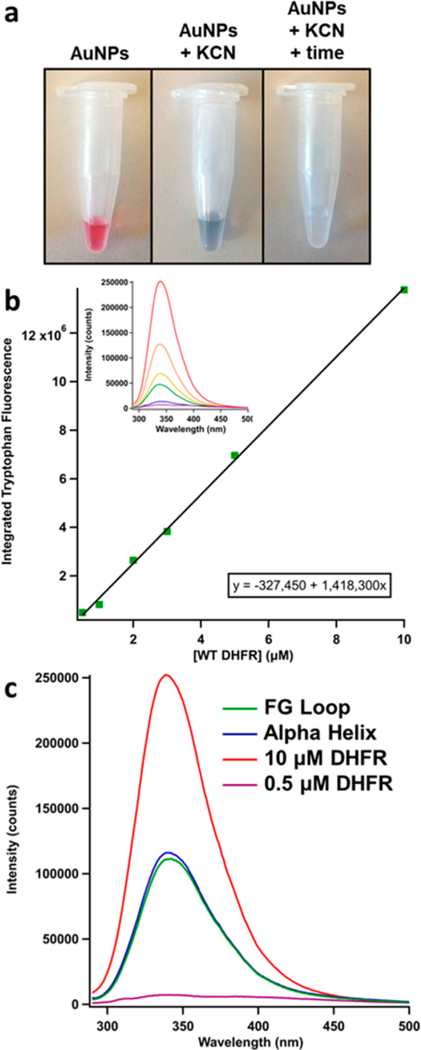
Fluorescence assay for protein concentration determination on the AuNPs. (a) Left: DHFR−AuNP conjugates are stable, as indicated by their pink color. Middle: Immediately after the addition of KCN, the AuNPs start to aggregate, indicated by the deep purple/ blue color. Right: After reacting for 1 h, the AuNPs are dissolved and are now Au−KCN complexes, showing no visible color. (b) Standard curve for WT DHFR protein samples under identical conditions as the unknown conjugate samples (same KCN and Tween 20 concentrations). The inset shows the fluorescence spectra obtained with 280 nm excitation. The area under each curve integrated from 300 to 385 nm is plotted versus the respective concentration to form the standard curve. (c) The tryptophan fluorescence spectra for the conjugate samples at the upper and lower bounds of the calibration curve [WT DHFR] and at intermediate concentrations of the mutants (green = FG loop conjugates. blue = alpha helix conjugates). The area under each conjugate spectrum is used in conjunction with the standard curve equation to determine the concentration of protein in the AuNP samples.
The pelleting and washing of the protein Au−NPs is necessary to remove free protein from the bioconjugates. However, some conjugates are lost in the process of removing the supernatant, as it is performed by pipetting the supernatant away from the pellet, so there is a balance between efficient removal of free protein and loss of conjugates. To limit loss of conjugates, narrow pipet tips can be used during supernatant removal when the level is close to the pellet. Three centrifugation steps are sufficient for washing the conjugates without losing much sample, as the free protein is below the detection limit in the SDS-PAGE gel (Figure 4). Consistent protein concentrations, typically in the range of 3−5 yμM, are obtained with this method for many repetitions of the conjugate preparation.
The first step in characterizing the surface coverage of the protein—AuNP conjugates is to determine the molar ratio of bound protein to AuNP. The number of moles of protein is determined from the concentration assay described above. The UV−is absorption spectrum of the conjugates provides the AuNP concentration and therefore moles of AuNPs (ε521 nm = 1.10 × 107 M−1 cm−1 for 5 nm AuNPs, e523 nm = 3.67 × 108 M−1 cm−1 for 15 nm AuNPs, e524 nm = 3.36 × 109 M−1 cm−1 for 30 nm AuNPs). The molar ratio of protein to AuNPs is summarized for each of the conjugates in Table 1. This method has been tested on many different conjugate preparations, and the ratio of DHFR to AuNPs is reproducible, with standard deviations equal to or less than 10% of the protein:AuNP ratio.
Table 1.
Surface Coverage of DHFR−AuNP Conjugates
| proteins per AuNPa surface coverage percentage | |||
|---|---|---|---|
| mutant | 5 nm AuNPs | 15 nm AuNPs | 30 nm AuNPs |
| FG loop conjugates | 7 ± 1 | 35 ± 4 | 180 ± 10 |
| 89% | 50% | 63% | |
| alpha helix conjugates | 7 ± 1 | 34 ± 4 | 183 ± 11 |
| 89% | 48% | 65% | |
aThe uncertainty is determined from the standard deviation of at least three separate conjugate preparations.
Surface area calculations are an important test of whether the experimentally determined ratio of protein to AuNP is physically reasonable. The surface area of the protein is taken to be a rectangle of 2.5 nm by 4 nm based on the binding geometry (Figure 2), which is 10 nm2. The surface area of the AuNPs is calculated to be 78 nm2 for 5 nm AuNPs, 707 nm2 for 15 nm AuNPs, and 2827 nm2 for 30 nm AuNPs. The maximum number of proteins that can fit on the AuNPs is calculated by dividing the surface area of the AuNP by the surface area of the protein. With this simplifying assumption, the maximum possible number of bound DHFR molecules is 8 for the 5 nm AuNPs, 70 for the 15 nm AuNPs, and 282 for the 30 nm AuNPs. These numbers represent an upper bound because the calculation assumes no empty space in the packing of the protein molecules on the surface, which is of course not possible. In addition, the actual contact area of the protein with AuNP surface is probably less than the rectangular cross section used in the calculation. All of the experimentally determined ratios of protein to AuNPs are less than the upper bound determined from this simplified model, consistent with a monolayer or submonolayer of protein on the surface, in agreement with the DLS results.
The surface coverage of the protein on the AuNPs (Table 1) is determined by dividing the experimentally determined number of proteins per AuNP by the maximum number of proteins that can bind based on the surface area. The average surface coverage is 89% for DHFR-5 nm AuNP conjugates, 49% for DHFR-15 nm AuNP conjugates, and 64% for DHFR- 30 nm AuNP conjugates. The DLS results indicate that there is at most a monolayer of protein on each AuNP, consistent with these calculated surface coverages. Furthermore, the protein surface coverage appears to be related to the size of the AuNP. We postulate that the surface coverage is correlated to the curvature of the surface (which depends on the size of the nanoparticle) because of protein—protein steric effects. The greater the surface curvature, the more volume is available to pack proteins on the nanoparticle surface. Thus, we attribute the nearly 100% surface coverage for 5 nm AuNP conjugates to its high curvature (relative to the size of the protein) that minimizes steric clash of bound proteins. When the radius of curvature decreases with the increasing AuNP size, there is more steric interaction of bound proteins, restricting the number that can bind, which is demonstrated by the 50—60% surface coverage in 15 and 30 nm AuNP conjugates. As the nanoparticle becomes large compared with the size of the protein, this effect is expected to saturate, which could explain the minimal difference between surface coverage in the larger conjugates. A similar protein crowding effect is observed for proteins bound on membrane vesicle surfaces; at high protein surface coverage, the steric pressure is sufficient to drive nanotubule formation, which relieves the steric pressure by creating a much greater surface curvature of the mem- brane.42,43 Thus, it is important to consider how curvature impacts surface coverage in bioconjugate systems, and the methods presented herein allow for the accurate quantification of parameters to determine surface coverage of protein−AuNP conjugates.
CONCLUSIONS
In this study, we present a thorough protein−AuNP bioconjugate characterization methodology using SDS-PAGE, UV−vis absorption, DLS and most importantly, an accurate method for surface coverage determination. The characterization scheme was applied to DHFR−AuNP conjugates of three different AuNP sizes: 5, 15, and 30 nm, as a means of varying the surface curvature and surface coverage. We show that the protein−AuNP conjugates were successfully synthesized, only a monolayer of protein is bound to the nanoparticles, and free protein was removed in the washing steps. We also present a novel methodology for accurately determining the protein concentration on AuNPs using KCN dissolution and a fluorescence assay. The conjugate synthesis is reproducible, with a consistent average surface coverage per AuNP. Further, the surface coverage of proteins on the AuNPs seems to be related to the curvature of the AuNP. There is higher surface coverage with smaller AuNPs, which have greater curvature and allow for higher loading density due to sterics. For larger AuNPs (smaller surface curvature), steric crowding of the attached proteins increases, causing a decrease in surface coverage on the AuNP.
In summary, a thorough characterization of the surface coverage in protein—nanoparticle conjugates is key to understanding their functionality, particularly enzyme activity. The integrated characterization methodology developed in this work to determine surface coverage can be applied to many other types of protein—nanoparticle conjugates. The methods of detection are UV—vis absorption and fluorescence for these AuNPs, which can be used not only for protein detection but also for DNA detection. Further, the developed protocols could be broadly applied to other systems, such as silver nanoparticles and quantum dots, where just the KCN step for concentration determination would need to be modified.
MATERIALS AND METHODS
Materials.
All chemicals were used as purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. DHF was synthesized using the dithionite reduction method,44 lyophilized, and stored at −20°C with desiccant.
Protein Expression and Purification.
A detailed protein expression and purification protocol has been previously described.45 Briefly, C-terminal hexa-histidine tagged E. coli DHFR was cloned and expressed in BL21(DE3). WT DHFR had only the hexa-histidine tag modification. The FG Loop and Alpha Helix DHFR mutants had a TEV cleavage site (Glu-Asn- Leu-Tyr-Phe-Gln-Gly) inserted between the protein and the hexa-Histidine tag, where Gly was inserted directly on the protein. Luria—Bertani (LB) medium containing ampicillin was used. The growth of cells was stopped by addition of isopropyl β-D-thiogalactopyranoside (IPTG). The culture was grown overnight, and the cells were harvested. The protein was purified via a nickel HisPrep affinity column on a GE Healthcare AKTA FPLC system (Pittsburgh, PA).
TEV Cleavage of Histidine Tag.
DHFR is purified via a hexa-Histidine tag on a nickel column. The histidine tag has a strong association to the AuNP (1−5 nM dissociation constant),39 which would cause two binding sites of the protein on AuNPs, the cysteine and the His tag (Figure S5). A tobacco etched virus (TEV) protease cleavage site of Glu-Asn- Leu-Tyr-Phe-Gln-Gly is inserted between the protein and the Histidine tag, and TEV protease is then used to cleave the Histidine tag from the rest of the protein after initial purification. Briefly, a minimum of 1:20 molar ratio of TEV:DHFR was allowed to react for at least 24 h at 4°C. The sample was then purified via a Ni-NTA (nitrilotriacetic acid) column, as the uncleaved His tag containing DHFR and His tag containing TEV protease bind to the nickel while the cleaved DHFR products flows through the column. The flow through from the column was buffer exchanged into 50 mM sodium phosphate, pH 7 buffer with Centricon filters for six rounds. The buffer exchanged sample was diluted to 50 μM, aliquoted into tubes, lyophilized, and stored at −20°C. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) is used to further confirm the cleavage (Figure S6). The denaturing gel shows that cleaved protein travels faster than uncleaved protein, and the mass difference is approximately 2 kDa (cleaved: 21 kDa, uncleaved: 23 kDa), verifying successful cleavage of the His tag. CD spectroscopy was performed on the mutants at 5 μM concentration in 10 mM sodium phosphate buffer on a Jasco J-810 spectropo- larimeter (Easton, MD) to confirm the folded state of the mutated enzyme in comparison to WT DHFR (Figure S7). The FG Loop mutant and Alpha Helix mutant have similar CD spectra to WT DHFR, indicating that the mutants fold properly. The cleavage has approximately a 90% yield when run for either 25 h with 1:10 TEV:DHFR or 40 h with 0.5:10 TEV:DHFR.
Free Enzyme Activity Assays.
Activity assays were performed by monitoring the decrease in 340 nm absorbance on an Ocean Optics QE65000 spectrometer (Winterpark, FL) with a xenon lamp source, which monitors the oxidation of NADPH to NADP+. 50 mM sodium phosphate, pH 7 buffer, NADPH (5×10–8 moles), and DHFR (1×10–11 moles) were added into a cuvette and allowed to equilibrate at 37°C in an Ocean Optics QPOD temperature controlled cuvette stage (Winterpark, FL) for 5 min. DHF (5×10–8 moles) was added to initiate the reaction, and the 340 nm absorbance was measured over 5 min. All reactions were run in at least triplicate. Enzyme turnover was calculated by the method of initial rates, using a linear fit to the first 40 s of the reaction. The linear fits were used with the integrated extinction coefficient to determine the initial rate. Free protein used in each sample were 1×10–11 moles. The concentration of protein was used with the initial rate to determine enzyme turnover rate (s-1). The assumption in this method is that there is a fast-equilibrium step with the substrate, producing pseudo first order kinetics. This is a reasonable assumption, as the Km for DHFR affinity to the substrate DHF is known to be 1.2 0μM,46 the enzyme concentration is very low (1 μM per assay), and the substrate concentration is in large excess (2.5 mM per assay).
Synthesis and Characterization of 15 nm AuNPs.
Citrate-stabilized 15 nm AuNPs were synthesized via citrate reduction method.40 Briefly, 0.1969 g of hydrogen tetrachlor- oaurate (III) trihydrate was combined with 500 mL of DI water in a 1 L two neck round-bottom flask. The reaction mixture was stirred, heated, and under reflux. Once vigorously boiling (refluxing at 1 drip per second), 0.5704 g sodium citrate dihydrate in 50 mL water was quickly poured in. The reaction mixture was allowed to reflux for 15 min, and the color changed from yellow to clear to black to purple and finally to red. The heat and water for the condenser were turned off, and the mixture was allowed to cool overnight. The mixture of synthesized AuNPs was filtered with a 0.2 μm filter to get rid of the larger aggregates of particles. AuNPs are stored in a glass bottle covered with aluminum foil at 4°C.
The synthesized AuNPs were dispersed and dried on 200 mesh copper TEM grids, and TEM images were taken on a Hitachi H7000 TEM (Hitachi High-Technologies America, Inc., Pleasanton, CA) with an accelerating voltage of 80 kHz. The program ImageJ was used for analysis of AuNP size (Figure S1). The AuNPs were determined to be 14 ± 1.4 nm. UV-vis absorption spectra of the synthesized AuNPs were measured on a PerkinElmer Lambda 35 spectrophotometer (Waltham, MA) and were comparable to spectra of NanoXact citrate stabilized 0.05 mg/mL 15 nm diameter AuNPs from Nanocomposix (San Diego, CA) (Figure 3c).
Functionalizing AuNPs with Enzyme.
DHFR (1.2×10–8 moles) was added to AuNPs at 1500 times excess (8.0×10–12 moles), and the solution was incubated at 4°C for at least 8 h to ensure complete binding of protein to AuNPs. The DHFR-15 nm AuNP conjugates were then centrifuged at 8000 rpm for 90 min on an Eppendorf 5415 D centrifuge (Hauppauge, NY). The supernatant (containing free protein and citrate) was removed, and the pellet (containing the conjugates) was resuspended in 0.005% Tween 20, 10 mM sodium phosphate, pH 7 buffer. The resulting solution was centrifuged for 40 min and resuspended in Tween 20 buffer twice more to wash away as much free protein as possible from the conjugates. The conjugates were stored in 0.005% Tween 20, 10 mM sodium phosphate pH 7 buffer in an 8 times dilution from the combined pellets. For the 5 nm AuNP conjugates, centrifugation cycles are at 13 200 rpm for 90 min during all cycles. For the 30 nm AuNP conjugates, centrifugation cycles are at 8000 rpm for 10 min during all cycles. Since the AuNPs from Nanocomposix are more dilute than the synthesized 15 nm AuNPs, the final dilution for these conjugates is 2 times rather than 8 times. The low salt concentration and surfactant help to stabilize the AuNPs, keeping them from sticking to tubes and pipet tips. The stability of the conjugates can be visually seen via colorimetric inspection, as pink represents stable conjugates and purple/ blue represents aggregated AuNPs (Figure 6a for visualization), where the SPR band drastically broadens and redshifts when aggregated..
Characterization of the Conjugates.
SDS-PAGE.
A Biorad Mini-PROTEAN tetra vertical electrophoresis cell (Hercules, CA) was used to run SDS-PAGE gels. The denaturing gels were run with Tris-Glycine-SDS running buffer. Laemmli sample loading buffer was added such that the final concentration of free protein was 10 μM, and the final dilution of 5 nm conjugates was 4X, 15 nm conjugates was 20X, and 30 nm conjugates was 4X. Ten microliters of color prestained protein standard, broad range (11–245 kDa) was added to ladder wells, and 15 gL protein or bioconjugate samples was added to each well. The gel was run at 200 V for 35 min. A 0.1% Coomassie R-250, 50% methanol, 7% acetic acid solution was used to stain the gel for 30 min. A 5% methanol, 7% acetic acid solution was used to destain the gel and was replaced several times for several days before imaging.
UV-vis Absorption.
UV-vis absorption spectra of conjugates were taken on a Thermo Scientific Nanodrop 2000 spectrophotometer (Waltham, MA). Appropriate dilutions were used for the conjugate samples, and the spectra were normalized at the peak maximum: 521 nm for 5 nm conjugates, 523 nm for 15 nm conjugates, 524 nm for 30 nm conjugates, 515 nm for free 5 nm AuNPs, 518 nm for free 15 nm AuNPs, and 520 nm for free 30 nm AuNPs.
Dynamic Light Scattering (DLS).
A Micromeritics Instrument Corporation NanoPlus DLS Nano Particle Size Analyzer instrument (Norcross, GA) was used for DLS measurements. Conjugate samples were diluted by 5–10 times, and all samples were filtered with a 0.2 μm filter before running, as dust or larger can greatly affect the DLS data.
Determining Protein Concentration on AuNPs.
The amount of protein bound to AuNPs was determined through fluorescence assays after dissolving AuNPs with KCN. Sixteen gL saturated KCN was added to 32 gL washed conjugates. The sample was sonicated in a Fisher Scientific Sonic Dissemble model 500 (Pittsburgh, PA) until the AuNPs were completely dissolved, and 52 gL of 10 mM sodium phosphate buffer was added for the final dilution. The dissolved AuNP samples were stored at room temperature until use. Stock solutions for a calibration were made in 0.005% Tween 20, 10 mM sodium phosphate buffer to keep the amount of Tween 20 consistent among all samples. The stock solutions were WT DHFR at 10, 5, 3, 2, 1, and 0.5 03bcM. The samples were run on a Horiba Scientific Dual-FL fluorometer (Edison, NJ) using a quartz fluorometer cuvette. The excitation wavelength was 280 nm with a 2 pixel increment. The tryptophan emission peak is highly concentration dependent and is used as the calibration factor for the standards. The integration (peak area) of the emission peakfrom 300 to 385 nm was plotted versus the concentration of protein in the standard to generate the standard curve. A linear fit to the data was used to determine the concentration of the unknown samples. There is a minor effect of KCN on the emission spectrum, so the WT stock samples were prepared with an equivalent amount of KCN.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. This work was also supported by National Institutes of Health grant GM068036 (R.B.D.) Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
ABBREVIATIONS
- DHFR
dihydrofolate reductase
- WT
wildtype
- AuNP
gold nanoparticle
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- DHF
dihydrofolate
- TEV
tobacco etched virus
- TEM
transmission electron microscopy
- DLS
dynamic light scattering
- PDB
Protein Data Bank
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconj-chem.8b00366.
TEM images and ImageJ analysis of synthesized AuNPs; Evidence for covalent binding of protein to AuNPs; Unstained SDS-PAGE conjugate gel; UV-vis absorption spectra of conjugates; Stability of AuNPs pictures; DLS of His tag containing DHFR-AuNP conjugates; SDS- PAGE of TEV cleaved protein; CD spectra of proteins (PDF)
REFERENCES
- (1).Ghosh P, Han G, De M, Kim CK, and Rotello VM (2008) Gold nanoparticles in delivery applications. Adv. Drug Delivery Rev. 60 (11), 1307–1315. [DOI] [PubMed] [Google Scholar]
- (2).Medintz IL, Uyeda HT, Goldman ER, and Mattoussi H (2005) Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 4, 435–446. [DOI] [PubMed] [Google Scholar]
- (3).Grzelczak MP, Danks SP, Klipp RC, Belic D, Zaulet A, Kunstmann-Olsen C, Bradley DF, Tsukuda T, Vmas C, Teixidor F, et al. (2017) Ion Transport across Biological Membranes by Carborane-Capped Gold Nanoparticles. ACS Nano 11 (12), 12492–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jain PK, Lee KS, El-Sayed IH, and El-Sayed MA (2006) Calculated Absorption and Scattering Properties of Gold Nanoparticles of Different Size, Shape, and Composition: Applications in Biological Imaging and Biomedicine. J. Phys. Chem. B 110 (14), 7238–7248. [DOI] [PubMed] [Google Scholar]
- (5).Hutter E, and Maysinger D (2013) Gold-nanoparticle-based biosensors for detection of enzyme activity. Trends Pharmacol. Sci. 34 (9), 497–507. [DOI] [PubMed] [Google Scholar]
- (6).Zong J, Cobb SL, and Cameron NR (2017) Peptide- functionalized gold nanoparticles: versatile biomaterials for diagnostic and therapeutic applications. Biomater. Sci. 5, 872–886. [DOI] [PubMed] [Google Scholar]
- (7).Ditzler LR, Sen A, Gannon MJ, Kohen A, and Tivanski AV (2011) Self-Assembled Enzymatic Monolayer Directly Bound to a Gold Surface: Activity and Molecular Recognition Force Spectroscopy Studies. J. Am. Chem. Soc. 133 (34), 13284–13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dreaden EC, Austin LA, Mackey MA, and El-Sayed MA (2012) Size matters: gold nanoparticles in targeted cancer drug delivery. Ther. Delivery 3 (4), 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhou J, Patel TR, Sirianni RW, Strohbehn G, Zheng MQ, Duong N, Schafbauer T, Huttner AJ, Huang Y, Carson RE, et al. (2013) Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc. Natl. Acad. Sci. U. S. A. 110 (29), 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Alkilany AM, Thompson LB, Boulos SP, Sisco PN, and Murphy CJ (2012) Gold nanorods: Their potential for photothermal therapeutics and drug delivery, tempered by the complexity of their biological interactions. Adv. Drug Delivery Rev.64 (2), 190–199. [DOI] [PubMed] [Google Scholar]
- (11).Sharma P, Brown S, Walter G, Santra S, and Moudgil B (2006) Nanoparticles for bioimaging. Adv. Colloid Interface Sci. 123–126, 471–485. [DOI] [PubMed] [Google Scholar]
- (12).Dreaden EC, Alkilany AM, Huang X, Murphy CJ, and El-Sayed MA (2012) The golden age: gold nanoparticles for biomedicine. Chem. Soc. Rev. 41 (7), 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rauta PR, Hallur PM, and Chaubey A (2018) Gold nanoparticle-based rapid detection and isolation of cells using ligand- receptor chemistry. Sci. Rep. 8 (1), 2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kharlamov AN, Feinstein JA, Cramer JA, Boothroyd JA, Shishkina EV, and Shur V (2017) Plasmonic photothermal therapy of atherosclerosis with nanoparticles: long-term outcomes and safety in NANOM-FIM trial. Future Cardiol. 13 (4), 345–363. [DOI] [PubMed] [Google Scholar]
- (15).Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E, Stewart MH, and Medintz IL (2013) Functionalizing Nanoparticles with Biological Molecules: Developing Chemistries that Facilitate Nanotechnology. Chem. Rev. 113 (3), 1904–2074. [DOI] [PubMed] [Google Scholar]
- (16).Govorov AO, and Richardson HH (2007) Generating heat with metal nanoparticles. Nano Today 2 (1), 30–38. [Google Scholar]
- (17).Piella J, Basffis NG, and Puntes V (2017) Size-Dependent Protein-Nanoparticle Interactions in Citrate-Stabilized Gold Nanoparticles: The Emergence of the Protein Corona. Bioconjugate Chem. 28, 88–97. [DOI] [PubMed] [Google Scholar]
- (18).Colangelo E, Comenge J, Paramelle D, Volk M, Chen Q, and Levy R (2017) Characterizing Self-Assembled Monolayers on Gold Nanoparticles. Bioconjugate Chem. 28, 11–22. [DOI] [PubMed] [Google Scholar]
- (19).Lata JP, Gao L, Mukai C, Cohen R, Nelson JL, Anguish L, Coonrod S, and Travis AJ (2015) Effects of Nanoparticle Size on Multilayer Formation and Kinetics of Tethered Enzymes. Bioconjugate Chem. 26 (9), 1931–1938. [DOI] [PubMed] [Google Scholar]
- (20).Hondred JA, Breger J, Garland N, Oh E, Susumu K, Walper S, Medintz I, and Claussen JC (2017) Enhanced enzymatic activity from phosphotriesterase trimer gold nanoparticle bioconjugates for pesticide detection. Analyst 142, 3261–3271. [DOI] [PubMed] [Google Scholar]
- (21).Vilanova O, Mittag JJ, Kelly PM, Milani S, Dawson KA, Radler JO, and Franzese G (2016) Understanding the Kinetics of Protein-Nanoparticle Corona Formation. ACS Nano 10 (12), 10842–10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang J, Jensen UB, Jensen GV, Shipovskov S, Balakrishnan VS, Otzen D, Pedersen JS, Besenbacher F, and Sutherland DS (2011) Soft Interactions at Nanoparticles Alter Protein Function and Conformation in a Size Dependent Manner. Nano Lett. 11 (11), 4985–4991. [DOI] [PubMed] [Google Scholar]
- (23).Cui M, Liu R, Deng Z, Ge G, Liu Y, and Xie L (2014) Quantitative study of protein coronas on gold nanoparticles with different surface modifications. Nano Res. 7 (3), 345–352. [Google Scholar]
- (24).Schade M, Moretto A, Donaldson PM, Toniolo C, and Hamm P (2010) Vibrational Energy Transport through a Capping Layer of Appropriately Designed Peptide Helices over Gold Nanoparticles. Nano Lett. 10 (8), 3057–3061. [DOI] [PubMed] [Google Scholar]
- (25).Hassan S, Schade M, Shaw CP, Levy R, and Hamm P (2014) Response of Villin Headpiece-Capped Gold Nanoparticles to Ultrafast Laser Heating. J. Phys. Chem. B 118 (28), 7954–7962. [DOI] [PubMed] [Google Scholar]
- (26).Aubin-Tam M-E, Hwang W, and Hamad-Schifferli K (2009) Site-Directed Nanoparticle Labeling of Cytochrome C. Proc. Natl. Acad. Sci. U. S.A. 106 (11), 4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rodriguez-Quijada C, Sanchez-Purra M, de Puig H, and Hamad-Schifferli K (2018) Physical Properties of Biomolecules at the Nanomaterial Interface. J. Phys. Chem. B 122 (11), 2827–2840. [DOI] [PubMed] [Google Scholar]
- (28).Lee JM, Park HK, Jung Y, Kim JK, Jung SO, and Chung BH (2007) Direct Immobilization of Protein G Variants with Various Numbers of Cysteine Residues on a Gold Surface. Anal. Chem. 79 (7), 2680–2687. [DOI] [PubMed] [Google Scholar]
- (29).Liu F, Wang L, Wang H, Yuan L, Li J, Brash JL, and Chen H (2015) Modulating the Activity of Protein Conjugated to Gold Nanoparticles by Site-Directed Orientation and Surface Density of Bound Protein. ACS Appl. Mater. Interfaces 7 (6), 3717–3724. [DOI] [PubMed] [Google Scholar]
- (30).Kuznetsova IM, Turoverov KK, and Uversky VN (2014) What Macromolecular Crowding Can Do to a Protein. Int. J. Mol. Sci. 15 (12), 23090–23140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wieczorek G, and Zielenkiewicz P (2008) Influence of Macromolecular Crowding on Protein-Protein Association Rates—a Brownian Dynamics Study. Biophys. J. 95 (11), 5030–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Phillip Y, Sherman E, Haran G, and Schreiber G (2009) Common Crowding Agents Have Only a Small Effect on Protein- Protein Interactions. Biophys. J. 97 (3), 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Keighron JD, and Keating CD (2010) Enzyme:Nano- particle Bioconjugates with Two Sequential Enzymes: Stoichiometry and Activity of Malate Dehydrogenase and Citrate Synthase on Au Nanoparticles. Langmuir 26 (24), 18992–19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liu S, Horak J, Holdrich M, and Lammerhofer M (2017) Accurate and reliable quantification of the protein surface coverage on protein-functionalized nanoparticles. Anal. Chim. Acta 989, 29–37. [DOI] [PubMed] [Google Scholar]
- (35).Schneck NA, Phinney KW, Lee SB, and Lowenthal MS (2016) Quantification of antibody coupled to magnetic particles by targeted mass spectrometry. Anal. Bioanal Chem. 408 (29), 8325–8332. [DOI] [PubMed] [Google Scholar]
- (36).Grell TAJ, Paredes E, Das SR, and Aslan K (2010) Quantitative Comparison of Protein Surface Coverage on Glass Slides and Silver Island Films in Metal-Enhanced Fluorescence-based Biosensing Applications. Nano Biomed. Eng. 2 (3), 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li S, Peng Z, and Leblanc RM (2015) Method To Determine Protein Concentration in the Protein-Nanoparticle Conjugates Aqueous Solution Using Circular Dichroism Spectroscopy. Anal. Chem. 87 (13), 6455–6459. [DOI] [PubMed] [Google Scholar]
- (38).Fierke CA, Johnson KA, and Benkovic SJ (1987) Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 26 (13), 4085–92. [DOI] [PubMed] [Google Scholar]
- (39).Aldeek F, Safi M, Zhan N, Palui G, and Mattoussi H (2013) Understanding the Self-Assembly of Proteins onto Gold Nanoparticles and Quantum Dots Driven by Metal-Histidine Coordination. ACS Nano 7 (11), 10197–10210. [DOI] [PubMed] [Google Scholar]
- (40).Hill HD, and Mirkin CA (2006) The bio-barcode assay for the detection of protein and nucleic acid targets using DTT-induced ligand exchange. Nat. Protoc. 1 (1), 324–336. [DOI] [PubMed] [Google Scholar]
- (41).Yehl K, Joshi JP, Greene BL, Dyer R B., Nahta, R., and Salaita, K. (2012) Catalytic Deoxyribozyme-Modified Nanoparticles for RNAi-Independent Gene Regulation. ACS Nano 6 (10), 9150–9157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Stachowiak JC, Hayden CC, and Sasaki DY (2010) Steric confinement of proteins on lipid membranes can drive curvature and tubulation. Proc. Natl. Acad. Sci. U. S. A. 107 (17), 7781–7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Stachowiak JC, Schmid EM, Ryan CJ, Ann HS, Sasaki DY, Sherman MB, Geissler PL, Fletcher DA, and Hayden CC (2012) Membrane bending by protein-protein crowding. Nat. Cell Biol. 14, 944–949. [DOI] [PubMed] [Google Scholar]
- (44).Blakley R L. (1960) Crystalline dihydropteroylglutamic acid. Nature 188, 231–232. [Google Scholar]
- (45).Reddish MJ, Vaughn MB, Fu R, and Dyer R B. (2016) Ligand-Dependent Conformational Dynamics of Dihydrofolate Reductase. Biochemistry 55 (10), 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gekko K, Kunori Y, Takeuchi H, Ichihara S, and Kodama M (1994) Point mutations at glycine-121 of Escherichia coli dihydrofolate reductase: important roles of a flexible loop in the stability and function. J. Biochem. 116 (1), 34–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.