

Abstract
α-Ketoglutarate (αKG, also known as 2-oxoglutarate)-dependent mononuclear non-haem iron (αKG-NHFe) enzymes catalyze a wide range of biochemical reactions, including hydroxylation, ring fragmentation, C-C bond cleavage, epimerization, desaturation, endoperoxidation and heterocycle formation. These enzymes utilize iron (II) as the metallo-cofactor and αKG as the co-substrate. Herein, we summarize several novel αKG-NHFe enzymes involved in natural product biosyntheses discovered in recent years, including halogenation reactions, amino acid modifications and tailoring reactions in the biosynthesis of terpenes, lipids, fatty acids and phosphonates. We also conducted a survey of the currently available structures of αKG-NHFe enzymes, in which αKG binds to the metallo-centre bidentately through either a proximal- or distal-type binding mode. Future structure–function and structure–reactivity relationship investigations will provide crucial information regarding how activities in this large class of enzymes have been fine-tuned in nature.
Graphical Abstract
Proximal- and distal-type αKG binding to the Fe(II) centre might play a crucial role in fine-tuning the catalysis of αKG-dependent non-haem iron enzymes.
1 Introduction
In nature, α-ketoglutarate-dependent mononuclear non-haem iron (αKG-NHFe) enzymes catalyse a wide range of biochemical reactions. αKG-NHFe enzymes require iron (II) as a metallo-cofactor and αKG as a co-substrate.1–6 Throughout several decades, various aspects of this large enzyme superfamily have been summarised in many excellent reviews, including the crystal structures and mechanisms,6–13 reaction diversity and natural product biosynthetic pathways,1, 5, 6, 8, 14–24 mechanistic investigations using small molecular model systems6, 25 and their relevance to biological processes and human diseases.4–6, 26 Members of this superfamily are widely distributed across prokaryotes, eukaryotes and archaea. Among the pathways involving αKG-NHFe enzymes, those involved in antibiotic biosynthesis are some of the most extensively investigated areas, and several reviews have been devoted to penicillin, cephalosporin, cephamycin and clavam biosyntheses.27–30 Some pathways with pharmaceutical or agricultural relevance have also been summarised, including the biosynthesis of ethylene,31, 32 carnitine,33 collagen34 and coumarin.16 Some αKG-NHFe enzymes are known to be related to human diseases, including phytanoyl-CoA hydroxylase (PAHX) in Refsum disease,35 4-hydroxy-phenylpyruvate dioxygenase (4-HPPD) in tyrosinaemia type II and hawkinsinuria,36, 37 prolyl hydroxylase (P4H) in alcoholic liver cirrhosis38, 39 and lysyl hydroxylase (LH) in Ehlers–Danlos syndrome type VI.38 Knowledge gained from mechanistic characterisations of αKG-NHFe enzymes has been applied to guide inhibitor design and development, which was recently summarised by Schofield et al.26
αKG-NHFe enzymes catalyse reactions as wide as those catalysed by haem-containing enzymes. Besides mechanistic investigations on enzymatic systems, studies on small molecular model systems are also one of the key sources for our mechanistic understanding of the catalytic processes catalysed by αKG-NHFe enzymes.6, 25 Based on the initial mechanistic proposal by Hanauske-Abel and Günzler,40 together with experimental and computational data accumulated over the last few decades, a generic mechanism for αKG-mediated oxygen activation was proposed involving Fe(IV)=O species as one of the key intermediates (Fig. 1A).6–12, 41 Starting from Fe(IV)=O species, hydroxylation is the most common type of reaction (e.g. hydroxylation of taurine 1 catalysed by TauD, Fig. 1B) catalysed by αKG-NHFe enzymes.42–47 In the absence of sulfate under aerobic conditions, Escherichia coli can utilize aliphatic sulfonates as sulfur sources. TauD and FMNH2-dependent SsuD are the key enzymes in this process.48, 49 TauD oxidises taurine 1 to 1-hydroxy-2-aminoethanesulfonic acid 2, which then spontaneously decomposes to aminoacetaldehyde 3 and sulfite (Fig. 1B).42–47 In addition to taurine 1, TuaD can oxidise taurine analogues, including pentanesulfonic acid, 3-(N-morpholino)propanesulfonic acid and 1,3-dioxo-2-isoindolineethanesulfonic.50 Several lines of evidence, including a combination of stopped-flow UV-visible absorption spectroscopy,45, 46, 51 EXAFS and Mössbauer spectroscopy,45, 46, 51–56 and isotope labelling,45, 57 support the involvement of an Fe(IV)=O species (A-6, Fig. 1A) as a kinetically competent intermediate. In hydroxylation reactions catalysed by αKG-NHFe enzymes, the reactive Fe(IV)=O species abstracts a hydrogen atom from the substrate to generate a substrate-based radical, and simultaneously, the Fe(IV)=O intermediate is reduced to the Fe(III)-OH species (A-6 → A-7, Fig. 1A). A subsequent hydroxyl radical rebound completes the substrate hydroxylation reaction and the αKG-NHFe enzyme returns to its initial Fe(II) state (A-7 → A-8, Fig. 1A). The proposed Fe(IV)=O intermediate (species A-6, Fig. 1A) was first trapped and characterised in TauD studies (Fig. 1B).45, 51 Since then, this key intermediate has been trapped and characterised in a few other enzymatic and model systems.54, 58–63 In addition to hydroxylation reactions, in natural product biosyntheses, many other types of reactions have also been attributed to αKG-NHFe enzymes, including desaturation,64 ring formation,65 ring expansion,66 halogenation,67 endoperoxidation68 and carbon skeleton rearrangements.69 αKG-NHFe enzymes also participate in modifications and repairs of macromolecules (DNA, RNA and proteins).6
Fig. 1.

Hydroxylation mediated by αKG-dependent NHFe enzymes. (A) The generic mechanism of the αKG-NHFe enzyme-mediated hydroxylation reaction, involving an Fe(IV)=O species.45 (B) Hydroxylation of taurine catalysed by TauD.
In 2013, Hangasky et al. reported a survey of 25 αKG-NHFe structures deposited in the protein data bank (PDB) shown as part of Table 1.9 αKG-NHFe enzymes possess a double-stranded β-helical fold (DSBH fold), which has also been called a cupin or jelly-roll fold.70, 71 The DSBH fold (Fig. 2A) was first observed in the crystallographic studies of isopenicillin N synthase (IPNS). Intriguingly, IPNS-catalysis did not require αKG as a co-substrate, and all four electrons required for catalysis were shown to be from its substrate.72–74 αKG-NHFe enzymes share a conserved His-X-Asp/Glu-Xn-His (2-His-1-carboxylate) motif, in which two His residues and a Glu or an Asp residue serve as the ligands to the iron centre.70, 75 Exceptions to this 2-His-1-carboxylate facial triad have also been reported, e.g. αKG-NHFe halogenases.76 In most αKG-NHFe enzymes, the metallo-centres and their ligands form octahedral complexes.1, 70, 71, 75 αKG coordinates to the NHFe centre bidentately using its C2 keto oxygen and C1 carboxylate as the ligands replacing two water molecules. In most reported structures, in addition to its bidentate interactions with the metallo-centre, αKG also interacts with a basic residue in the active site (e.g. Arg or Lys) through electrostatic interactions using its C5 carboxylate, which facilitates the positioning of αKG in the active sites.6
Table 1.
Key enzymes relevant to this review and those summarized by Hangasky et al. 9
| No. | PDB ID | Abbreviation | Full name | Stereo-isomer | Reaction type | Biological functional |
|---|---|---|---|---|---|---|
| 1 | -- | JOX | Jasmonate-induced oxygenase | -- | Hydroxylation | Jasmonic acid hydroxylation |
| 2 | 3BUC84 | ABH2 | AlkB homologue 2 | Distal | Demethylation | Demethylation of DNA and RNA |
| 3 | 2IUW85 | ABH3 | AlkB homologue 3 | Distal | Demethylation | Demethylation of DNA and RNA |
| 4 | 3I3O86 | AlkB | AlkB | Distal | Demethylation | Demethylation of DNA and RNA |
| 5 | 3THT87 | ABH8 | AlkB homologue 8 | Distal | Demethylation | Demethylation of DNA and RNA |
| 6 | -- | AmbO5 | -- | -- | Halogenation | Ambiguine, fischerindole and hapalindole biosynthesis |
| 7 | 2BRT88 | ANS | Anthocyanidin synthase | Distal | Hydroxylation | Tricyclic flavonoid biosynthesis |
| 8 | 2OG789 | AsnO | Asparagine oxygenase | -- | Hydroxylation | Calcium-dependent antibiotics biosynthesis |
| 9 | 5DAP65 | AsqJ | -- | Distal | Desaturation and epoxidation | Aspoquinolones biosynthesis |
| 10 | 1OII90 | AtsK | Alkylsulfatase | Proximal | Oxygenolytic cleavage | Oxygenolytic cleavage of alkyl sulfate esters |
| 11 | 5YBL91 | AusE | -- | -- | Desaturation and spiro-ring forming rearrangement | Acetoxydehydroaustin biosynthesis |
| 12 | -- | BarB1 | -- | -- | Halogenation on protein-tethered substrates | Barbamide biosynthesis |
| 13 | -- | BarB2 | -- | -- | Halogenation on protein-tethered substrates | Barbamide biosynthesis |
| 14 | 3O2G92 | BBOX | γ-Butyrobetaine hydroxylase | Distal | Hydroxylation | Stereo-selective hydroxylation6 of γ-butyro-betaine in L-carnitine biosynthesis |
| 15 | -- | BcmB | -- | -- | Desaturation and epoxidation | Bicyclomycin biosynthesis |
| 16 | 1NX864 | CarC | Carbapenem synthase | Distal | Desaturation | Epimerization/desaturation of carbapenam |
| 17 | 1DS193 1GVG78 |
CAS | Clavaminate synthase | Proximal Distal |
Oxidation | Biosynthesis of clavulanic acid |
| 18 | 3N9N94 | ceKDM7A | Jumonji C-domain-containing histone demethylas | Proximal | Lys demethylation | Histone demethylation |
| 19 | -- | CmaB | -- | -- | Halogenation-initiated formation of cyclopropane | Coronatine biosynthesis |
| 20 | -- | Cpr19 | -- | -- | Hydroxylation | Uridine-5-carboxamide |
| 21 | -- | CrtW | -- | -- | Dehydrogenation | β-Carotene analogues biosynthesis |
| 22 | -- | CrtZ | -- | -- | Hydroxylation | β-Carotene analogues biosynthesis |
| 23 | 3NNF95 | CurA | CurA halogenase | Proximal | Halogenation-initiated formation of cyclopropane | Curacin biosynthesis |
| 24 | 3GJB96 | CytC3 | -- | Proximal | Halogenation on protein-tethered substrates | Dichloroaminobutyrate biosynthesis |
| 25 | 4YJD97 | DAO | Dioxygenase for auxin oxidation | -- | Oxidation | 2-Oxoindole-3-acetic acid biosynthesis |
| 26 | 1RXG66 | DAOCS | Deacetoxycephalosporin C synthase | Distal | Dehydrogenation and ring expansion | Penicillin G biosynthesis |
| 27 | -- | DhpA | -- | -- | Hydroxylation | Dehydrophos biosynthesis |
| 28 | -- | DhpJ | -- | -- | Hydroxylation | Dehydrophos biosynthesis |
| 29 | 4MHR98 4MHU98 |
EctD | Ectoine hydroxylase | -- | Hydroxylation | 5-Hydroxyectoine biosynthesis |
| 30 | 5LUN81 5V2Y79 5V2X79 |
EFE | Ethylene-forming enzymes | Distal Distal Monodentate |
Hydroxylation | L-Arg hydroxylation and ethylene formation |
| 31 | 1H2L99 | FIH | Factor inhibiting HIF-1α (asparagine hydroxylase) | Proximal | Hydroxylation | Asparagine hydroxylation |
| 32 | -- | Fr9P | -- | -- | Hydroxylation | Hemiketal FR901464 biosynthesis |
| 33 | 4Y5S68 | FtmOx1 | -- | Distal | Endoperoxide formation | Verruculogen biosynthesis |
| 34 | -- | FzmG | -- | -- | Hydroxylation | Fosfazinomycin A biosynthesis |
| 35 | -- | GLOXY4 | -- | -- | Dehydrogenation | Pnsumocandin A0 biosynthesis |
| 36 | -- | GRS1 | Glucoraphasatin synthase1 | -- | Hydroxylation | Glucoerucin biosynthesis |
| 37 | 2YU1100 | HJHDM1A | Jumonji C-domain-containing histone demethylase 1A | Distal | Lys demethylation | Histone demethylation |
| 38 | -- | HmaS | Hydroxymandelate synthase | N/A | Decarboxylation and hydroxylation | Vancomycin biosynthesis |
| 39 | 1SP9101 | HPPD | 4-Hydroxyphenylpyruvate dioxygenase | N/A | Decarboxylation and hydroxylation | Homogentisate biosynthesis |
| 40 | -- | HtcB | -- | -- | Halogenation on protein-tethered substrates | Hectochlorin biosynthesis |
| 41 | -- | IDO | L-isoleucine dioxygenase | -- | Hydroxylation | Isoleucine Hydroxylation |
| 42 | -- | JamE | -- | -- | Halogenation-initiated formation of cyclopropane | Jamaicamide biosynthesis |
| 43 | 2OQ7102 | JMJD2A | Jumonji C-domain-containing histone demethylase 2A | Proximal | Lys methylation | Histone demethylation |
| 44 | 3DXU103 | JMJD2D | Jumonji C-domain-containing histone demethylase 2D | Proximal | Lys methylation | Histone demethylation |
| 45 | 3LDB104 | JMJD6 | Jumonji C-domain-containing histone demethylase 6 | Distal | Lys 5-S-hydroxylation; Arg demethylation | Protein hydroxylation; Histone demethylation |
| 46 | 3KVB105 | KIAA1718 | Jumonji C-domain-containing histone demethylase 1D | Distal | Lys demethylation | Histone demethylation |
| 47 | -- | KthP | -- | -- | Halogenation on protein-tethered substrates | Kutzneride biosynthesis |
| 48 | -- | KtzD | -- | -- | Halogenation-initiated formation of cyclopropane | Kutzneride biosynthesis |
| 49 | -- | KtzO/KtzP | -- | -- | Hydroxylation | β-Hydroxylation of glutamic acid biosynthesis |
| 50 | -- | LpxO | -- | -- | Hydroxylation | Lipid A modification |
| 51 | -- | M2H | Melatonin-2-hydroxylase | -- | Hydroxylation | 2-Hydroxymelatonin biosynthesis |
| 52 | -- | MppO | -- | -- | Hydroxylation | Mannopeptimycin biosynthesis |
| 53 | -- | OkaE | -- | Azetidine ring formation and hydroxylation | Okaramine D biosynthesis | |
| 54 | 2A1X106 | PAHX | Phytanoyl-CoA 2-hydroxylase | Distal | Hydroxylation | 2-Hydroxy-phytanoyl-CoA biosynthesis |
| 55 | 4P7W107 | PH4 | Proline cis-4-hydroxylase | Proximal | Hydroxylation | Proline hydroxylation |
| 56 | 1E5R108 1E5S108 |
PH3 | Proline 3-hydroxylase (Type II) | -- | Hydroxylation | Proline hydroxylation |
| 57 | 3OUJ109 | PHD2 | Prolyl hydroxylase dioxygenase 2 | Distal | Hydroxylation | Hypoxia-inducible factor (HIF) transcription factor |
| 58 | -- | PhnY | -- | -- | Hydroxylation | 2-Amino-1-hydroxyethylphosphonic acid biosynthesis |
| 59 | -- | PoIL | -- | -- | Hydroxylation | CPOAA biosynthesis |
| 60 | 5YBN91 | PrhA | -- | Distal | Desaturation and cycloheptadiene formation | Paraherquonin biosynthesis |
| 61 | -- | PtlD | -- | -- | Desaturation and epoxidation | Pentalenolactone biosynthesis |
| 62 | 2RDS110 2RDQ110 |
PtlH | 1-Deoxypentalenic acid 11-β hydroxylase | Proximal | Hydroxylation | Pentalenolactone biosynthesis |
| 63 | -- | PlaO1 | -- | -- | Hydroxylation | Phenalinolactone biosynthesis |
| 64 | -- | RbtB | -- | -- | Hydroxylation | Rubratoxin A biosynthesis |
| 65 | 3OPT111 | RPH1 | Jumonji C-domain-containing histone demethylase 2A | Distal | Lys demethylation | Histone demethylation |
| 66 | 3W21112 | SadA | N-substituted L-amino acid dioxygenase | Proximal | Hydroxylation | β-Hydroxylating activity of N-succinyl L-leucine |
| 67 | 2FCT113 | SyrB2 | -- | Proximal | Halogenation on protein-tethered substrates | Syringomycin E biosynthesis |
| 68 | 1OS777 | TauD | Taurine/αKG dioxygenase | Proximal | Hydroxylation | Aminoacetaldehyde and sulfite formation |
| 69 | -- | TMLH | Trimethyllysine hydroxylase | -- | Hydroxylation | Carnitine biosynthesis |
| 70 | 3AL6114 | TYW5 | tRNA yW-synthesizing enzyme 5 | Proximal | Hydroxylation | tRNA modification |
| 71 | 3AVS115 | UTX/KDM6A | Lysine-specific demethylase 6A | Proximal | Demethylation | Histone H3 Lys 27 demethylation |
| 72 | 6ALM116 | VioC | L-Arg-3-hydroxylase | Proximal (shifted ~35°) | Hydroxylation | Viomycin biosynthesis |
| 73 | 5IQS117 | WelO5 | -- | Proximal | Halogenation on freestanding substrates | Hapalindole-type molecules biosynthesis |
Fig. 2.

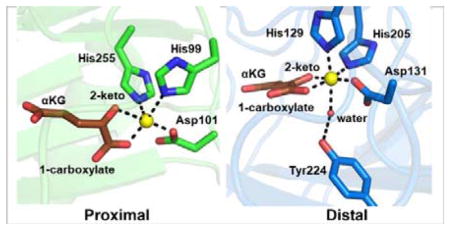
αKG-NHFe enzyme structural information. (A) The double-stranded helical fold (DSBH fold) first observed in IPNS structure.70 (B) A typical proximal αKG binding conformation represented by the TauD•Fe•αKG complex.77 (C) Distal-type αKG binding conformation represented by the FtmOx1•Fe•αKG complex.68 The proposed αKG conformational switch from the proximal (D) to the distal mode upon exposing the CAS•Fe•αKG•substrate complex to NO (E).78 (F) Schematic representation of αKG conformational switch between a proximal (F1)- and distal (F2)-type of conformation. (G) Another type of αKG binding conformation observed in the EFE•Fe•αKG binary complex where αKG binds to the Fe(II) centre monodentately using its C5 carboxylate.79 The iron centre is shown as yellow sphere, αKG is shown as brown sticks, water is shown as a red sphere, and NO is shown as sticks.
Two different αKG binding modes have been observed upon inspection of the structure in PDB: the proximal and distal types.9, 71 In the proximal-type αKG binding mode (e.g. TauD•Fe(II)• αKG binary complex in Fig. 2B), the αKG C1 carboxylate coordinates to the Fe(II) centre at a position trans to the first histidine (His99), while the αKG C2 keto is at a position opposite to the acidic ligand Asp101 of the 2-His-1-carboxylate facial triad. In Table 1, we labelled this type of αKG binding mode as the proximal type. Upon substrate binding, the remaining water dissociates from the Fe(II) centre opening up the site for oxygen binding and activation (A-3, Fig. 1A). In a typical hydroxylation reaction, this oxygen binding site is adjacent to the substrate binding site.77 The resulting Fe(IV)=O species (A-6, Fig. 1A) points towards the substrate, allowing direct oxidation of the substrate by Fe(IV)=O species.
Interestingly, in nearly ~50% of known αKG-NHFe structures (Table 1), αKG coordinates to the Fe(II) centre in a conformation different from the proximal type observed in TauD (Fig. 2B). For example, in the FtmOx1•Fe(II)• αKG binary complex shown in Fig. 2C, αKG displays a bidentate coordination to the Fe(II) centre and its C2 keto is at a position opposite to an acidic residue (Asp131 in Fig. 2C). However, in this structure, the C1 carboxylate of αKG coordinates to the Fe(II) centre at a position opposite to the distal histidine (His205) of the 2-His-1-carboxylate facial triad (Fig. 2C).68 This type of αKG binding mode is termed distal (Fig. 2C). Due to the change in αKG binding conformation relative to that of TauD, the remaining site for O2 binding and activation in FtmOx1 is not adjacent to the substrate binding pocket. Following the generic mechanistic model discussed in Fig. 1A for αKG-mediated oxygen activation, the Fe(IV)=O species formed (Fig. 2C) points away from the substrate binding site, making it inaccessible for subsequent chemical transformation.
Two scenarios have been proposed to explain the catalytic processes in distal-type αKG-NHFe enzymes. In the first scenario, as exemplified by the endoperoxidation reaction catalyzed by FtmOx1 (Figs. 2C),68 αKG coordinates to the Fe(II) centre using the distal binding mode and the oxygen binding site is not adjacent to the substrate verruculogen. However, a tyrosine residue (Y224) is next to the oxygen binding site and is crucial to the endoperoxidation reaction (see Section 3.6.6).68 Alternatively, αKG can undergo a conformational switch from the distal to proximal mode, re-orienting the oxygen binding and activation site towards the substrate. In clavaminic acid synthase (CAS), two αKG binding modes have been observed experimentally (Figs 2D & 2E).78 In the absence of NO, the C1 carboxylate of αKG coordinates to the Fe(II) centre from a site opposite to that of the proximal histidine (His144, Fig. 2D). Interestingly, upon the introduction of NO to the CAS•Fe(II)• αKG•substrate complex (Fig. 2E), the C1 carboxylate is now positioned opposite to the distal histidine (His279) of the His-X-Asp/Glu-Xn-His facial triad. Two αKG binding modes (distal and proximal) have also been observed for AlkB,80 which catalyzes oxidative DNA demethylation. The presence of two αKG binding modes in both CAS and AlkB has led to the proposed αKG conformational switch (Fig. 2F), which is necessary to properly orient the Fe(IV)=O species towards the substrate for catalysis.71
Early in 2017, two αKG binding modes were also observed for the ethylene-forming enzyme (EFE).79 In one structure, αKG displays a bidentate coordination to the Fe(II) centre in the distal-type binding mode.79, 81 Interestingly, in another EFE structure, αKG displays a monodentate coordination to the Fe(II) centre using its C5 carboxylate as the ligand (Fig. 2G).79 It is not yet known whether this binding mode is relevant to EFE catalysis.
In natural product biosynthesis, reactions catalyzed by αKG-NHFe enzymes are widely used to either produce biosynthetic precursors or to modify the natural product skeletons after assembly. In this review, we summarize recent examples of αKG-NHFe enzymes involved in the modification of amino acids, and biosynthesis of terpenes, lipids and phosphorous-containing secondary metabolites. The materials covered here complement many of the recent reviews in this area, especially a recent book on αKG-dependent oxygenases.6 Halogenases, which can install a halogen atom, Cl− or Br−, to an inactivated carbon centre are a subset of the αKG-NHFe enzyme superfamily and are covered in Section 2. Section 3 is devoted to αKG-NHFe enzymes involved in amino acid modifications, and are prevalent in the biosynthesis of several types of natural products. Terpenes are one the largest classes of natural products.82 After assembly of their skeleton, extensive modifications are introduced to produce the final products. Some of the required tailoring reactions are catalyzed by αKG-NHFe enzymes (Section 4). Phosphonates are C-P bond-containing natural products with great pharmaceutical potential due to their structural similarity to phosphates and carboxylates.83 Phosphonate biosynthesis involves many novel reactions, some of which are mediated by αKG-NHFe enzymes and are described Section 5. In addition, Sections 6 and 7 are devoted to αKG-NHFe enzymes involved in lipid and fatty acid modifications and nucleoside antibiotics biosynthesis, respectively.
In Table 1, we list all of the αKG-NHFe enzymes discussed in this review and an additional 25 enzymes covered by Hangasky et al. in their structural analysis of αKG-NHFe enzymes previously deposited in the PDB.9 For enzymes listed in Table 1, when structural information was available, they have been classified as either the proximal or distal type to indicate their αKG binding mode. Interestingly, the number of proximal- and distal-type αKG-NHFe enzymes are approximately equal, indicating that both binding modes are common in nature. Thus far, most of the mechanistic information on αKG-NHFe enzymes is based on the characterization of proximal-type αKG-NHFe enzymes. Distal-type αKG-NHFe enzymes have to change the binding conformation of αKG to properly orient the Fe(IV)=O species for catalysis, and this conformational switch may be a critical mechanistic feature. In cases where a conformational switch is not employed, it is highly possible that nature explores the uniqueness of the distal-type binding mode to mediate novel chemical transformations (e.g. FtmOx1 catalysis, Fig. 2C). Future mechanistic characterizations of more proximal-type αKG-NHFe enzymes will provide answers to these questions.
2 Halogenation
In this section, we summarize some recent examples of halogenation reactions catalyzed by αKG-NHFe enzymes. Unlike flavin-dependent halogenases, which catalyze halogenation at aromatic or electron-rich carbons,20, 67, 118, 119 αKG-dependent halogenases perform much more challenging chemical transformations, catalyzing halogenation reactions at aliphatic carbons.120, 121 Most of the halogenases characterized to date act on substrates tethered to the phosphopantetheinyl arm of carrier proteins (Sections 2.1 and 2.2).6 In recent years, halogenases using stand-alone small molecules as the substrates have also been discovered (Section 2.3).
Several crystal structures of αKG-dependent halogenases have been reported, including SyrB2, CytC3, CurA and WelO5 (Table 1). Interestingly, they do not have the typical 2-His-1-carboxylate “facial triad” as observed in other αKG-NHFe enzymes. Instead, the carboxylate ligand of the 2-His-1-carboxylate facial triad is replaced by a halide ligand, which together with some other active site interaction network modifications, is key to the selectivity of these halogenases.76, 95, 96, 113, 117
2.1 Halogenation on carrier protein-tethered substrates
SyrB2 catalyzes monochlorination of the methyl group of L-Thr-SyrB1 4 to 4-Cl-L-Thr-SyrB1 5, which is one of the steps in the biosynthesis of the phytotoxin syringomycin E 6 (Fig. 3A), using SyrB1 as the carrier protein.113, 120, 122 Wild-type SyrB2 can also catalyze aliphatic nitration or azidation reactions.123 Drennan et al. reported the first structure of the αKG-NHFe halogenase SyrB2 and showed that its Fe(II) centre has 2-His and 1-chloride ligand, instead of the classic 2-His-1-carboxylate facial triad.113
Fig. 3.

Halogenation on protein-tethered substrates. (A) Halogenation reaction catalyzed by SyrB2 on L-Thr tethered on SyrB1 4 in Syringomycin E biosynthesis.113 (B) The proposed mechanism for SyrB2 reaction begins with oxygen activation, similarly to other enzymes in this family, to form the Fe(IV)=O species. Notably, the carboxylate ligand is replaced with a chloride ligand at the iron centre allowing the halogen atom to react with a substrate-based radical to give the halogenated product.113
SyrB2 undergoes oxygen activation similar to other αKG-NHFe enzymes (Fig. 1A), generating an Fe(IV)=O species (Fig. 3B).113 The Fe(IV)=O species (B-4, Fig. 3B) then abstracts a hydrogen atom from the substrate to create a substrate-based radical (B-5, Fig. 3B). Subsequently, the chlorine atom combines with the substrate-based radical, instead of going through a hydroxy-rebound to form a hydroxylation product, resulting in the formation of a chlorinated product (B-5 → B-1, Fig. 3B).60
The biosynthesis of barbamide 11 (Fig. 4A) involves two αKG-NHFe halogenases, BarB1 and BarB2, which work in tandem to trichlorinate the C5 methyl group of L-Leu-S-BarA 7. In these chlorination reactions, BarA is the carrier protein (Fig. 4A), and BarB2 chlorinates either L-Leu-S-BarA 7 or monochloro-Leu-S-BarA 8 to dichloro-leu-S-BarA 9. Interestingly, BarB1 can convert both mono- and di-chlorinated L-Leu-S-BarA (8 and 9) to (2S,4S)-5,5,5-trichloro-Leu-S-BarA 10 (Fig. 4A).124 CytC3 catalyzes the chlorination of aminobutyryl-S-CytC2 12 during biosynthesis of the Streptomycete antibiotic dichloroaminobutyrate 15, and functions in a similar manner to SyrB2, BarB1 and BarB2. Both γ-chloro- and γ, γ-dichloroaminobutyryl-S-CytC2 (13 and 14, Fig. 4B) are products of CytC3 catalysis.125
Fig. 4.

Halogenation on protein-tethered substrates. (A) In barbamide 11 biosynthesis, BarB2 works along with BarB1 to yield a trichloro-Leu 10, which is further incorporated into the final product 11.124 (B) A similar chlorination strategy is observed in CytC3 reaction in the biosynthesis of dichloroaminobutyrate 15.125 (C) HtcB-mediated chlorination reactions in hectochlorin 20 biosynthesis.126 (D) Chlorination reaction using a piperazyl-group tethered to a carrier protein as the substrate has been observed in KthP catalysis in the biosynthesis of kutzneride 2.127
In addition to the aforementioned cases where chlorinase is part of the non-ribosomal peptide synthetase (NRPS) machinery, chlorination has also been reported as a tailoring reaction for the biosynthesis of other natural products. HtcB is a fatty acyl halogenase involved in the biosynthesis of hectochlorin 20. The HtcB protein from Lyngbya majuscule contains three domains: an N-terminal catalytic αKG-NHFe halogenase domain, an acyl coenzyme A binding domain and an acyl carrier protein (ACP) domain.126 When ACP-tethered hexanoyl 16 was used as a substrate, 5-oxo- 17, 4-ene-5-chloro- 18 and 5,5-dichloro-haxanoyl-S-ACP 19 were all observed as products in HctB catalysis (Fig. 4C).128 After this tailoring reaction, 5,5-dichloro-haxanoyl-S-ACP 19 was utilized as one of the building blocks for the biosynthesis of hectochlorin 20.
In the above cases, amino acids tethered to the phosphopantetheinyl arm of carrier proteins or an acyl group tethered to an ACP domain were used as substrates by αKG-NHFe halogenases. KthP is an αKG-NHFe enzyme involved in the biosynthesis of kutzneride 2 (23). For the chlorination step in the production of kutzneride 2 (23), a piperazyl functional group was tethered to the thiolation protein KtzC first (21, Fig. 4D), and KthP then stereo- and regio-selectively chlorinated the tethered piperazyl ring to generate (3S,5S)-5-chloropiperazate-S-KtzC 22 (Fig. 4D).127
2.2 Halogenation-initiated formation of cyclopropanes
Chlorination is employed by several natural product biosynthetic pathways to activate C-H bonds and the resulting halides are then used for the construction of other functional groups, e.g. cyclopropane. During biosynthesis of the Pseudomonas syringae phytotoxin coronatine 28 (Fig. 5A), CmaB, an αKG-NHFe halogenase, chlorinates L-allo-isoleucine-S-CmaD 24 to γ-chloro-L-allo-isoleucine-S-CmaD 25 (Fig. 5A). Subsequent intramolecular γ-elimination is mediated by a zinc-dependent enzyme, CmaC, resulting in the formation of coronamic acid-S-CmaD 26 (Fig. 5A). Coronamic acid (CMA, 27) is then released from CmaD and used as a building block for the biosynthesis of coronatine 28 (Fig. 5A).121
Fig. 5.

Halogenation-initiated formation of cyclopropane. (A) Halogenation reactions catalyzed by CmaB on CmaD-tethered Ile 24 in the coronatine 28 biosynthetic pathway.121 (B) KtzD chlorinates KtzC-tethered L-Ile 29 and the chlorinated product is further cyclized by KtzA-catalysis.129
An analogous pathway of coronatine biosynthesis is present in the biosynthesis of kutzneride 2 (23, Fig. 5B). In this process, KtzD, an αKG-NHFe enzyme, chlorinates the γ position of L-Ile-S-KtzC 29 tethered to a KtzC carrier protein to generate γ chloro-L-allo-Ile-S-KtzC 30. Subsequent cyclopropyl ring formation mediated by the flavoprotein KtzA affords (1S, 2R)-allo-CMA 31, which is in contrast to the zinc-dependent protein in the coronatine biosynthesis (Fig. 5B).129 It is worth mentioning that, the cyclopropyl group present in the (1S, 2R)-allo-CMA-S-KtzC intermediate 31 (Fig. 5B)129 is structurally distinct from the 2-(2-methylcyclopropyl)glycine moiety in kutznerides, e.g. kutzneride 2 (23, Fig. 5B).130
CurA and JamE are two polyketide synthase (PKS) megasynthases involved in the Lyngbya majuscule curacin 37131 and janmaicamide 39132 biosynthetic pathways, respectively (Fig. 6A). An αKG-NHFe halogenase (Hal) domain is embedded in the CurA megasynthase, and plays a similar role to that of CmaB and KtzD in the construction of a cyclopropyl group. The Hal domain of CurA chlorinates (S)-3-hydroxy-3-methylglutaryl-ACP ((S)-HMG-ACP, 32) to produce 33. Then, a dehydratase (ECH1) domain embedded in CurE catalyzes the dehydration of 33 to yield 3-methylglutaconyl-ACP 34. Subsequent decarboxylation mediated by a decarboxylase (ECH2) domain embedded in CurF affords an α, β-enoyl thioester, 3-methylcrotonyl-ACP 35. Finally, an enoyl reductase (ER) domain embedded in CurF catalyzes the cyclopropanation of 35 to 36, which then serves as a building block in the biosynthesis of curacin A 37 (Fig. 6A).133
Fig. 6.

Halogenation-initiated formation of cyclopropane. (A) In the biosynthesis of curacin 37 and jamaicamide 39, two homologous megasynthases, namely CurA and JamE, catalyze the chlorination of (S)-3-hydroxy-3-methylglutaryl-ACP 32.133 In the curacin pathway, the ECH2 domain catalyzes the decarboxylation to give an α,β-enoyl thioester 35, while the ECH2 domain in the jamaicamide pathway catalyzes the formation of the vinyl chloride moiety 38.132 (B) Structure of CurA halogenase in a ligand-free open form. (C) CurA halogenase•Fe•αKG• structural complex showing that αKG (brown stick) and chloride ion (blue sphere) binding trigger a conformation change to a closed form, which allows substrate binding.
A similar biosynthetic route is present in the jamaicamide A pathway. Here, the halogenase domain embedded in JamE catalyzes the same chlorination step on 32 as that of CurA in curacin A biosynthesis (Fig. 6A). However, in contrast to the α, β-enoyl thioester-based product 35 from the CurF ECH2 domain, the JamJ ECH2 domain decarboxylates 34 to produce the β, γ-enoyl thioester intermediate 38 in the biosynthesis of jamaicamide 39 (Fig. 6A).132, 134
Several crystal structures of the CurA halogenase domain (Hal) in different ligand states have been reported.95 Two conformation states exist, namely an open and a closed state (Figs 6B & 6C), and the transition between the two states is triggered by αKG binding. A large lid covers the active site in the closed form, which is disordered in the open form. Upon αKG binding, CurA Hal undergoes a conformational change from the open to closed form, facilitating the substrate (S)-HMG-ACP binding (Figs 6B & 6C). Additionally, CurA Hal exhibits a high degree of substrate specificity, requiring both the C3 S-hydroxyl and C5 carboxylate on (S)-HMG-ACP 32 for recognition. It has been suggested that the halogenation vs hydroxylation outcome in these halogenase reactions depends on the substrate positioning. The conformational switch triggered by αKG binding may allow the precise positioning of the substrate in the active site, thereby minimizing the competing hydroxylation reaction.95
2.3 Halogenation on freestanding substrates
In most early descriptions of αKG-NHFe halogenase reactions (sections 2.1 & 2.2), their substrates were covalently tethered to carrier proteins. These halogenases do not recognize stand-alone small molecule as a substrate. New information regarding their substrate specificity was discovered recently. WelO5, an enzyme involved in welwitindolinone biosynthesis, is the first reported αKG-NHFe chlorinase that utilizes a stand-alone small molecule as its substrate.117, 135, 136 WelO5 stereo-specifically chlorinates hapalindole-type molecules (40 → 41, Fig. 7A).117, 135, 136 In addition to chloride, WelO5 can use other halides, including bromide, as alternative halogenation agents.137
Fig. 7.

Halogenation versatility of WelO5 and AmbO5. (A) WelO5 chlorinates hapalindole-type molecules, while AmbO5 exhibits a higher substrate tolerance chlorinating ambiguine, fisherindole and hapalindole alkaloids.138 (B) Structure of WelO5•Fe•αKG•substrate117 shows a chloride ligand (blue sphere) at the iron centre (yellow sphere). A second-coordination shell Ser189 was proposed to be involved in controlling the rearrangement of αKG (brown sticks) binding conformation to re-orient the chloride group in the halo-oxo-iron(IV) intermediate towards the substrate for the chlorination reaction.
Recently, the structure of the WelO5•αKG•substrate complex was reported (Fig. 7B).117 In this structure, after αKG and substrate binding, the vacant ligand site in the Fe(II) centre is directly facing the substrate. If this is the site for O2 binding and activation and no αKG rearrangement is involved, then the oxo group, instead of the chlorine group of the halo-oxo-iron(IV) intermediate, faces the substrate. To explain the chlorination activity, it was thus suggested that a switch in αKG conformation was required. A second-coordination shell residue, Ser189, was suggested to play a key role in controlling such an αKG conformational switch (Fig. 7B). This hypothesis is supported by results from the WelO5 S189A mutant, which produces an equal amount of halogenation and hydroxylation products.117
AmbO5 is another recent example of such an αKG-NHFe halogenase (Fig. 7A). AmbO5 has a high degree of substrate flexibility and catalyzes the chlorination of ambiguine (44, 46, 50), fischerindole (40, 42) and hapalindole 52 alkaloids (Fig. 7A).138 Comparative analysis of AmbO5 and WelO5 revealed that a fragment of the C-terminal portion of WelO5 might be important for substrate selection and specificity.138 Indeed, replacing a fragment of 18 residues in the WelO5 C-terminus with the corresponding AmbO5 sequence expanded the substrate scope of WelO5 catalysis.138
Many halogenated natural products exhibit biological activities. For example, salinisporamide shows anti-cancer activity,139 while syringomycin functions as an anti-fungal agent.140 Halogenation is critical for the biological activities of these compounds, and as a result, developing new halogenation strategies continues to be a key area of interest.20, 67 In the last few decades, while many investigations have focused on the structural and mechanistic characterizations of these halogenases, some efforts have been devoted to developing new halogenases. Several groups have attempted the conversion of a hydroxylase to a halogenase and vice versa.113, 117 One of the successful examples is the WelO5 S189A mutant, which has exhibited a relaxed selectivity relative to wild-type WelO5, producing an equal proportion of hydroxyl and chlorine-modified products.117 Recently, Boal et al. also engineered an N-acyl amino acid hydroxylase, SadA, into a halogenase.141
3 Amino Acid Modifications
The presence of non-proteinogenic amino acids in alkaloids and non-ribosomal peptides is very common and, in many cases, these unnatural amino acids are supplied through dedicated biosynthetic pathways. Alternatively, after natural product skeleton assembly using 21 proteinogenic amino acids, additional structural diversity is then introduced through extensive modifications by tailoring enzymes. αKG-NHFe enzymes are one of the most common types of tailoring enzymes. These αKG-NHFe enzymes often show some degree of substrate promiscuity, readily incorporating substrate analogues into natural products through pathway engineering or by in vitro biocatalytic processes. For some valuable compounds, these αKG-NHFe enzyme-mediated biocatalytic transformations may have advantages relative to their synthetic organic pathways. In this section, some recent amino acid tailoring reactions are summarized.
3.1 αKG-NHFe enzymes in carnitine biosynthesis
L-Carnitine 59 plays a key role in fatty acid metabolism in all animals and in some prokaryotes. As a result, the enzymes involved in carnitine biosynthesis have been explored for therapeutic purposes.146, 147 Carnitine biosynthesis148–150 (Fig. 8A) begins with trimethylated lysine 55, and involves the following reactions: Nε-trimethyllysine hydroxylase (TMLH), 3-hydroxy-Nε-trimethyllysine aldolase (HTML aldolase), 4-N-trimethylaminobutyraldehyde dehydrogenase (TMABA dehydrogenase) and γ-butyrobetaine hydroxylase (BBOX). Both TMLH and BBOX are αKG-NHFe enzymes. TMLH catalyzes the stereo-selective conversion of (2S)-Nε-trimethyllysine 55 to (2S,3S)-3-hydroxy-Nε-trimethyllysine 56 (Fig. 8A),142, 151, 152 one of the key reactions in carnitine biosynthesis.148–150 HTML aldolase catalyzes the cleavage of 56 into 4-N-trimethylaminobutyraldehyde 57 and glycine using pyridoxal phosphate (PLP) as a cofactor. Then, TMABA dehydrogenase, an NAD+-dependent enzyme, oxidizes 4-N-trimethylaminobutyraldehyde 57 to γ-butyrobetaine (γ-BB) 58. The last step of this pathway is the BBOX-catalyzed hydroxylation of 58 to L-carnitine 59.
Fig. 8.

Carnitine biosynthesis. (A) L-carnitine 59 biosynthesis involves two αKG-NHFes: TMLH and BBOX.142 (B) The biocatalytic versatility of TMLH-mediated hydroxylation on trimethyl-Lys analogues.143 (C) BBOX also oxidizes THP 68 as the substrate through a Stevens-type rearrangement reaction.144 (D) Structure of BBOX in complex with zinc (orange sphere), N-oxalylglycine (NOG, green sticks) andγBB substrate (magenta sticks) showing a distal-type αKG binding mode. (E) The studies of PsBBOX show that the positively charged trimethylammonium group on the substrate is crucial for recognition.145
TMLH has some degree of substrate flexibility and can accept several trimethyllysine analogues as alternative substrates (60–63, Fig. 8B), catalyzing the production of their corresponding hydroxylation products (64–67, Fig. 8B).143 TMLH’s substrate flexibility is reflected in at least two aspects: the chain length of the amino acid side-chain and the alkyl group on the lysine ε-nitrogen. Thus far, TMLH’s crystal structure has not been reported. However, the substrate promiscuity of TMLH implies that its active site pocket for the lysine side-chain binding is relatively flexible, and could be explored for the stereo-selective hydroxylation of substrate analogues for synthetic purposes. Recently, many αKG-NHFe enzymes involved in histone demethylation (e.g. the demethylation of methylated lysine residues) have been discovered and discussed in depth in a book edited by Schofield and Hausinger.6 Future structural work may also provide information on how this class of enzymes control regioselectivity.
BBOX is an αKG-NHFe enzyme involved in the last step of L-carnitine biosynthesis, hydroxylating γ-butyrobetaine (γ-BB, 58) to L-carnitine 59 (Fig. 8A).153, 154 Because of the importance of L-carnitine in fatty acid metabolism, BBOX has been explored as a target to develop treatments for myocardial infarction.147 Interestingly, BBOX catalyzes the oxidation of its inhibitor 3-(2,2,2-trimethylhydrazinium)propionate (THP) 68 and produces multiple products, including 68g–68j, and 3-amino-4-(methylamino)butanoic acid (AMBA) 69 (Fig. 8C).144 The oxidation of THP 68 involves N–N bond cleavage and C–C bond formation, which is likely achieved via a process related to a Stevens rearrangement (Stevens [1,2]-shift).155 The Fe(IV)=O intermediate abstracts a hydrogen atom from THP 68 to generate a radical intermediate 68a, which is followed by N–N bond cleavage and a [1,2]-shift to produce 68d (Fig. 8C). Two potential pathways (Pathways I and II) have been proposed for the subsequent steps (Fig. 8C). In pathway I, N-methyl hydroxylation followed by spontaneous decomposition of the hydroxylation product 68f affords the formaldehyde 68h. On the other hand, in pathway II, the methylene radical reacts with the imine 68b and the subsequent [1,5] H-shift results in a radical intermediate 68l. The hydroxyl radical rebounds to the intermediate 68l, generating the hydroxylation product 70 (acyclic aminal), which might be in equilibrium with its cyclic aminal form 71. Compound 70 is further converted to the final product AMBA 69 after eliminating the formaldehyde moiety (Fig. 8C).144
The structure of BBOX in complex with an αKG analogue, N-oxalylglycine (NOG) and γ-BB has been reported (Fig. 8D).92 In this structure, the αKG analogue coordinates to the iron centre in the distal-type binding mode, which implies that a conformational switch of αKG upon binding might be necessary in order to properly orient the Fe(IV)=O species towards the substrate for the hydroxylation reaction.92, 156 BBOX’s structural information also explains its substrate flexibility. When the nitrogen atom in the trimethylammonium group is replaced by either phosphorous or arsenic, the corresponding analogues (72, 74, 76, 78, 80 and 82 Fig. 8E) are also recognized as BBOX substrates, as demonstrated in a study of Pseudomonas sp. AK1 BBOX (PsBBOX). The BBOX active site possesses an aromatic cage that forms the binding pocket for the positively charged trimethylammonium group of γ-BB 58. Kinetic analyses have shown that cation–π interactions between BBOX and its substrate/substrate analogues are crucial for their recognition. Intriguingly, BBOX does not recognize an uncharged analogue of γ-BB 58 as an alternative substrate,145 providing an additional line of evidence supporting the importance of π-cation interactions in BBOX catalysis.145
3.2 Ethylene-forming enzymes (EFE)
Ethylene is one of the most widely used raw materials in the chemical industry, and has been widely adopted in the manufacture of plastics, textiles and solvents. The polymerization of ethylene is also used to produce hydrocarbons in the C5–C10 range. Currently, ethylene usage has approached ~150 million metric tons per year.157 To meet industrial needs, ethylene is primarily produced in massive quantities by the steam cracking of fossil fuels or from the dehydrogenation of ethane, representing one of the largest CO2-emitting processes in the chemical industry. Despite the usage of modern technology, approximately 2 MJ of energy are required per pound of ethylene produced. Given the size of the ethylene industry, this product alone accounts for at least 1.5% of United States’ carbon footprint.157, 158
In recent years, alternative routes for ethylene production have been explored, with one of these directions involving biocatalytic production processes. Ethylene is a plant hormone that plays a crucial role in plant growth and development.159 Plants use aminocyclopropane-1-carboxylic acid (ACC) oxidase to convert ACC, an intermediate generated from S-adenosyl-L-methionine, to ethylene, CO2 and HCN.6 ACC oxidase has been intensively investigated, with work in this area summarized in a recent book;6 however, this production route is not suitable for industrial-scale adaptation due to the toxicity of the HCN by-products.
More recently, a completely different ethylene-production system was discovered. It was recently found that some bacteria utilize an enzyme that catalyzes ethylene formation using αKG-based oxidative fragmentation, which is dependent on the presence of oxygen, Fe(II) and L-Arg.163, 164 Isotope labelling studies indicated that the ethylene precursor is αKG, and consequently, this class of enzyme was named as ethylene-forming enzymes (EFEs).165
EFEs are αKG-NHFe enzymes and have been found in several species, including Pseudomonas syringae,166 Ralstonia solanacearu166 and Penicillium digitatum.167 Biochemical characterizations have indicated that EFEs catalyze two reactions simultaneously (Fig. 9A).168 The first reaction is the hydroxylation of L-Arg 84 at its C5 position at the expense of αKG 88 to produce succinate and CO2. This is a very typical αKG-NHFe enzyme-catalyzed hydroxylation reaction. The second reaction in EFE catalysis is the fragmentation of αKG 88 to produce ethylene 89 and three molecules of CO2. The L-Arg hydroxylation product, 5-hydroxyl-L-arginine 85, is not stable and spontaneously decomposes to guanidine 86 and L-Δ-1-pyrroline-5-carboxylate (P5C) 87. The EFE-catalyzed production of ethylene through αKG fragmentation has not been observed in any other enzymes.
Fig. 9.

Amino acid modifications by αKG-NHFe enzymes. (A) In EFE catalysis, L-Arg 84 hydroxylation and αKG fragmentation to ethylene 89 are the two reactions. (B) EFE•Fe•αKG•L-Arg complex shows that αKG (brown stick) binds to the Fe(II) centre bidentately.79 (C) Hydroxylation on L-Ile mediated by IDO. (D) BtIOD not only catalyzes hydroxylation on a wide range of substrates, but also catalyzes reactions other than hydroxylations.160 (E) In the biosynthesis of glucoraphasatin, GRS1 catalyzes the desaturation of the side chain of compound 107 to form the aliphatic glucoraphasatin 108.161 (F) SadA-mediated β-hydroxylation of N-succinyl-L-Ile 109.162
Recently, EFE has been structurally and biochemically characterized.79, 81, 168 Initially, αKG was proposed to become conjugated to L-Arg through the formation of a Schiff base, and the two reactions in EFE catalysis were believed to be tightly coupled through a dual-circuit reaction mechanism.169 The results from these recent biochemical characterizations indicated that EFE also accepts some L-Arg analogues as alternative substrates, and in some cases, the ratio of these two reactions is different from that of wild-type EFE reactions, which provides some initial evidence against the proposed dual-circuit mechanism. Structural information reported in 2017 by Schofield, Hausinger and their co-workers79, 81 clearly indicated that αKG does not form a Schiff base with L-Arg; instead, αKG coordinates to the Fe(II) centre bidentately in a distal-type binding conformation (Fig. 9B). Therefore, an αKG conformational switch was proposed as an essential step for L-Arg hydroxylation in EFE.79 Interestingly, αKG in EFE also adopts another binding mode (Fig. 2G) that is completely different from all other reported structures (Figss 2B & 2C, and Table 1). In this EFE structure (Fig. 2G), αKG is a monodendate ligand and coordinates to the Fe(II) centre through its C5 carboxylate. To date, the mechanistic details of EFE remain to be elucidated, and the importance of this new type of αKG binding mode in EFE catalysis remains enigmatic.79
Beyond these efforts towards structural and mechanistic characterizations, the production of ethylene through the heterologous expression of EFE has also been demonstrated in several model organisms, including E. coli, Saccharomyces cerevisiae and cyanobacteria.170–172 Although native EFE-producing hosts are less amenable to genetic manipulation, ethylene is produced in much higher yields compared to those achieved in heterologous hosts.173 One of key goals in EFE engineering is to de-couple L-Arg hydroxylation from ethylene production so that a more efficient ethylene-production process can be achieved.
3.3 L-Isoleucine hydroxylase
L-Isoleucine hydroxylase, also known as L-isoleucine dioxygenase (IDO), is an αKG-NHFe enzyme that coverts L-Ile 91 to (2S,3R,4S)-4-hydroxyisoleucine 92 (Fig. 9C).174 (2S,3R,4S)-4-hydroxyisoleucine 92 shows anti-diabetic and anti-obesity activities, and has been explored as one of the components in functional foods.175, 176 IDO from Bacillus thuringiensis (BtIDO) has been characterized. After oxidizing L-Ile 91 to 92, BtIDO can further oxidize 92 to (2S,3R)-2-amino-3-methyl-4-ketopentanoate 93 (Fig. 9D).160 BtIDO can also tolerate some degree of variations in the L-Ile side-chain (Fig. 9D). The use of substrate analogues alters both the position and stereo-chemistry of the hydroxylation reactions, suggesting that the L-Ile side-chain may have some degree of flexibility within the binding pocket.
BtIDO shows considerable substrate promiscuity, recognizing a wide range of L-Ile analogues as alternative substrates.160 Intriguingly, the reactions in BtIDO-catalysis are not limited to hydroxylation (Fig. 9C).160 Using L-Met 103 or its analogue 105 as the substrate, BtIDO catalyzes the sulfoxidation of these sulfur-containing L-amino acids, producing L-methionine sulfoxide 104 and L-ethionine sulfoxide 106, respectively (Fig. 9D).160 Furthermore, some efforts have also been devoted to applying IDO-catalysis to the fermentation-based production of compound 92. In αKG-NHFe catalysis, along with the formation of Fe(IV)=O species, the co-substrate αKG is oxidized to succinate. The conversion of αKG to succinate is one of the key steps in the tricarboxylic acid cycle, which is catalyzed by αKG dehydrogenase. In an engineered E. coli strain, involving the replacement of αKG dehydrogenase with BtIDO to couple BtIDO-catalysis to the tricarboxylic acid cycle, L-Ile 91 was readily converted to 92 in a high yield.177
In BtIDO-catalysis, L-Met 103 is oxidized to the sulfoxide 104. However, in glucoraphasatin synthase 1 (GRS1), a different type of chemistry was observed in an αKG-NHFe enzyme involved in glucoraphasatin 108 biosynthesis.178 After the conversion of L-Met 103 to 4-methylthiobutyl glucoerucin 107, GRS1 catalyzes the desaturation of 107, introducing a double bond into the product 108 (Fig. 9E).161 Thus far, no structural information is available for GRS1. Future comparative studies between BtIDO and GRS1 might reveal the factors governing sulfoxidation vs. desaturation in these two enzymatic systems.
SadA is an αKG-NHFe enzyme that catalyzes the β-hydroxylation of several N-substituted L-amino acids, especially N-succinyl L-leucine 109.162 In vitro characterizations of SadA indicated that when N-succinyl L-Leu 109 was used as the substrate, N-succinyl L-threo-β-hydroxyleucine 110 could be obtained in an over 99% diastereomeric excess (Fig. 9F).179 The crystal structures of SadA•Zn(II) and SadA•Zn(II)•αKG complexes were reported,112 and showed a bidentate coordination of the αKG molecule to the Zn(II) metal ion. In addition, N-succinyl-L-leucine and N-succinyl-L-phenylalanine were modelled into the active site of SadA and revealed that the binding pocket of the N-succinyl group is located in an electropositive-rich cavity formed by the side chains of Arg83, Arg163 and Arg203. This structural feature accounts for SadA’s high selectivity towards N-succinyl-L-amino acids relative to other N-substituted amino acids.112, 162
As discussed in Section 2.3, SadA represents an elegant example of a hydroxylase that has been engineered to function as a halogenase. In the SadA D157G mutant, one of the active site Fe(II) ligands, Asp157, is replaced by a Gly residue. In the reaction using the SadA D157G mutant in the presence of NaCl or NaBr, the formation of chlorine- and bromine-substituted products were observed.141 In the previous section, we described how BtIDO can accept L-Leu 98 as a substrate, catalyzing a γ-hydroxylation reaction (98 → 99 conversion, Fig. 9D). Taken together, IDO and SadA are another pair of αKG-NHFe enzymes that are suitable for comparative studies to elucidate the structure–function relationship.
3.4 Lysyl hydroxylase
Post-translational modifications of proteins are one of the key strategies in signal transduction pathways and in the epigenetic regulation of biological processes, e.g. histone modifications.6 The post-translational modification of proteins is not only key to tuning the functions of structural proteins, such as collagen by introducing intra- and inter-molecular cross-linking, but also provide attachment sites for other modifications, e.g. the glycosylation of hydroxylysine residues in collagen.180 An in-depth discussion of protein hydroxylation, and histone and nucleic acid demethylation reactions can be found in a recently published book on αKG-NHFe enzymes.6 Herein, we briefly touch upon this topic.
In collagen post-translational modification processes, in addition to the prolyl hydroxylase discussed in the next section (Section 3.5), another important αKG-NHFe enzyme in this pathway is lysyl hydroxylase (LH).181, 182 Three isoforms of lysyl hydroxylase (LH1, LH2 and LH3) were isolated from human and mouse tissues and were shown to mediate the hydroxylation of lysyl residues in collagen polypeptide chains 111 (Fig. 10A).183 LH1 is associated with the genetically inherited disorder Ehlers–Danlos syndrome type VI (Kyphoscoliotic form).184, 185 Recent characterizations suggest that LH2 lysyl hydroxylase has two alternatively spliced forms (LH2a and LH2b).186, 187 In contrast to LH1 and LH2, LH3 is a multi-functional enzyme. In addition to a hydroxylation reaction to produce hydroxylysyl (Hyl, 112), LH3 is responsible for further glycosylations at the hydroxylation site, resulting in the addition of galactosylhydroxylysyl (Gal-Hyl, 113) and glucosylgalactosyl hydroxylysyl (Glyc-Gal-Hyl, 114) glycans (Fig. 10A).188, 189 Thus far, the structural information on LH1, LH2 and LH3 has not been available. However, the crystal structure has been solved for JMJD6, another αKG-NHFe lysyl hydroxylase. JMJD6 belongs to the JmjC subfamily. JmjC enzymes are responsible for the demethylation of Nε-methylated lysine residues of histones.190 Unlike many other enzymes in the JmjC subfamily, JMJD6 mediates both the lysyl hydroxylation and arginyl demethylation of various protein substrates.191 For example, JMJD6 hydroxylases the lysyl residues of the arginine-serine-rich domains of U2AF65, an RNA-splicing-related protein.192, 193 Structural analysis of JMJD6 revealed how lysyl residues of the protein peptide substrate 115 bind to JMJD6 in an orientation that promotes C5 hydroxylation to produce 116 (Fig. 10B) rather than Nε-demethylation, which is a typical reaction of many JMJD6-class histone demethylases.6 Another example is JMJD4, which mediates lysyl C4 hydroxylation to produce 117 (Fig. 10B).194 Recently, Schofield et al. summarized the structure–function relationship of human JmjC oxygenases and highlighted the key differences between hydroxylases and demethylases.195 For interested readers, another recent review summarizes the various biological processes that JMJD6 proteins are involved in.191
Fig. 10.

Hydroxylations of lysyl residues. (A) Collagen polypeptide lysyl 111 hydroxylation mediated by LH1/LH2/LH3.196 In contrast to LH1 and LH2, LH3 can further modify hydroxylysyl 112 to galactosyl hydroxylysyl 113 and glucosylgalactosyl hydroxylysyl 114.188 (B) Reactions catalyzed by JMJD6/JMJD4 on lysyl residue in post-translational modifications of proteins.193, 194
3.5 Proline hydrolase (PH)
Hydroxylprolines (Fig. 11) have been identified as components in both small molecular natural products (e.g. actinomycin I, etamycin and echinocandins) and in proteins.197–199 L-Pro 118 hydroxylases (PHs) are αKG-NHFe enzymes.198, 200–204 Both 3-hydroxy-L-proline and 4-hydroxy-L-proline have been identified as proline hydroxylation products. Early studies led to the discovery of cis-3- and trans-4-L-proline hydroxylases (cis-3-PH and trans-4-PH), which have been applied in the industrial production of cis-3- and trans-4-hydroxy-L-proline (cis-3-L-Hyp 119a and trans-4-L-Hyp 119d, Fig. 11A), respectively. Over the years, many more PHs have been discovered, and all four isomers of monohydroxy-L-proline can be produced enzymatically using stereo- and regio-specific PHs (Fig. 11A).199, 205, 206
Fig. 11.

L-Pro modifications catalyzed by PHs. (A) Four different isomers of monohydroxyl-L- Pro (119a–d) produced by stereo- and regio-specific PHs. (B) Stereospecific epoxidation reactions catalyzed by SgP4H. (C) SrPH can hydroxylate both L-Pro 118 and L-pipcolinic acid 122.207 (D) In pneumocandin (126 and 127) biosynthesis, Gloxy4 catalyzes the oxidative cyclization of L-Leu 97 to methyl-Pro 124, which is further hydroxylated by GloF to produce hydroxyproline as one of the building blocks for pneumocandin biosynthesis.208
Furthermore, many PHs can accept proline analogues as alternative substrates to carry out reactions that are distinct from hydroxylation. For example, a 4-proline hydrolase from Streptomyces griseoviridus P8648 (SgP4H) mediated the stereospecific epoxidation of 3,4-dehydro-L-pro 120 to trans-3,4-epoxy-L-Pro 121 (Fig. 11B).197 The reaction was stereospecific and cis-3,4-epoxy-L-proline was not detected. SrPH, another αKG-NHFe enzyme from Streptosporangium roseum NBRC 3776T, can accept both L-Pro 118 and L-pipecolinic acid 122 as substrates. Using L-Pro 118 as a substrate, SrPH catalyzes the formation of both cis-3-hydroxy-L-proline 119a and cis-4-hydroxy-L-proline 119c (Fig. 11C). Similarly, SrPH catalyzes the hydroxylation of L-pipecolinic 122 to cis-3-hydroxy-L-pipecolinic acid 123a and cis-5-hydroxy-L-pipecolinic acid 123b (Fig. 11C).207 GloF, an αKG-dependent proline hydroxylase from the pneumocandin pathway (Fig. 11D), accepts both proline 118 and trans-4-methyl-L-proline 124 as substrates. When proline 118 is the substrate, GloF catalyzes the formation of both trans-4- and trans-3-hydroxy-L-proline (119d and 119b) at a ratio of 8:1 (Fig. 11D). When trans-4-methyl-L-proline 124 is utilized as a substrate, GloF-catalyzed hydroxylation leads to the production of (3S,4S)-4-methyl-3-hydroxyl-L-proline 125 (Fig. 11D).199 All three hydroxyprolines (119b, 119d and 125) are building blocks required for the biosynthesis of pneumocandins (126 & 127).
Interestingly, in pneumocandin biosynthesis (Fig. 11D), there is another αKG-NHFe enzyme, GLOXY4, which catalyzes the oxidative cyclization of L-Leu 97 to produce trans-4-methylproline 124. The anti-fungal agent pneumocandin possesses 4S-methyl-L-proline as part of its hexapeptide core (the R group in 126 vs 127).209 Results from gene deletion studies (e.g. the disruption of GLOXY4 from the pneumocandin gene cluster of the fungus Glarea lozoyensis) suggested that GLOXY4 is responsible for the production of 124 (Fig. 11D).209 The inactivation of GLOXY4 abolishes the production of both 124 and pneumocandin A0 126; however, the GLOXY4 deletion strain remains capable of producing pneumocandin B0 127 as the exclusive product.209 Pneumocandin B0 127 still maintains the anti-fungal activity of pnuemocandin A0, but with reduced toxicity. For this reason, pneumocandin B0 127 was chosen for use as a semisynthetic precursor of the clinical anti-fungal drug, caspofungin acetate.210 Therefore, the inactivation of GLOXY4 provides a route for the industrial production of pneumocandin B0 127 to increase its production titre. The mechanistic details for GLOXY4-catalyzed oxidative cyclization remain to be explored.
In addition to being key components of small molecular metabolites, hydroxylprolines also exist in proteins as a result of protein post-translational modification.6
3.6 αKG-NHFe oxidases associated with non-ribosomal peptide synthetase (NRPS) systems
Non-ribosomal peptides are assembled by NRPS systems, and the diversity of this group of natural products can be achieved through a few different approaches, including the selective incorporation of a variety of precursors, the modification of incorporated building blocks on carrier proteins or by the modification of the non-ribosomal peptide skeletons after they are released from biosynthetic machinery.211, 212 Some halogenases that play a role in these pathways were discussed in Section 2. However, many other types of reactions catalyzed by αKG-NHFe enzymes also contribute to the structural and functional diversity of this large class of natural products. In this section, we summarize the results from a few key cases reported recently.
3.6.1 L-Arginine related hydroxylase
Viomycin 130 (Fig. 12A) belongs to the tuberactinomycin family of non-ribosomal peptide antibiotics.213 Its skeleton is assembled by NRPS and one of the building blocks is a non-proteinogenic amino acid (2S,3R)-capreomycidine 129 derived from L-Arg. (2S,3R)-Capreomycidine 129 is produced by a combination of reactions mediated by VioC and VioD using L-Arg as the substrate (Fig. 12A). The αKG-NHFe enzyme VioC catalyzes the C3 hydroxylation of L-Arg 84 to 3S-hydroxyl-L-Arg 128 (Fig. 12A),214, 215 which is then further converted to (2S,3R)-capreomycidine 129 in a reaction catalyzed by VioD (Fig. 12A).216 In the final product viomycin 130, there is an additional C5 hydroxylation catalyzed by VioQ, a Rieske-type of NHFe enzyme.217
Fig. 12.

L-Arg hydroxylation reactions catalyzed by αKG-NHFe oxygenases. (A) VioC hydroxylates L-Arg 84 and VioD further hydroxylates the product 128 to (2S,3R)-capreomycidine 129, which serves as a building block for viomycin biosynthesis. (B) In mannopeptimycin β 135, L-Arg 84 hydroxylation is mediated by MppO to produce a hydroxyenduracididine 134 moiety in the final product.218 (C) Crystal structure of VioC•Fe•αKG complex.116
Mannopeptimycins (MPPs) have exceptional in vitro and in vivo antibacterial activities against methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci and penicillin-resistant Streptococcus pneumoniae.219 In the biosynthesis of mannopeptimycin β 135, addition of the β-hydroxyenduracididine moiety 134 (Fig. 12B) involves reactions similar to those discussed in viomycin biosynthesis.218 Mannopeptimycin β 135 contains both D- and L-forms of β-hydroxyenduracididine, which are produced by hydroxylation of the unnatural amino acid L-enduracididine 133. Enduracididine 133 has a unique five-membered cyclic guanidine moiety.220 The first step in enduracidine 133 biosynthesis is the hydroxylation and deamination of L-Arg 84 catalyzed by MppP (a PLP-dependent hydroxylase), producing 2-oxo-4-hydroxy-5-guanidinovaleric acid 131.221 Subsequently, the pyruvate aldose, MppR, catalyzes the dehydration/cyclization of 2-oxo-4-hydroxy-5-guanidinovaleric acid 131 to produce a cyclic guanidine intermediate 132,222 followed by transamination catalyzed by MppQ to produce L-enduracidine 133.220 MppO, an αKG-NHFe enzyme, then hydroxylates the β-carbon of L-enduracididine 133, resulting in β-hydroxy-enduracididine 134 (Fig. 12B).218
L-Arg, a substrate for both VioC (Fig. 12A) and EFE (Fig. 9A), and L-enduracidine 133, a substrate of MppO, shares the guanidine moiety. In EFE (Fig. 9A), αKG fragments to produce ethylene and three molecules of CO2. However, αKG fragmentation activity has not been reported for VioC and MppO.
The structural information of MppO is not yet available, but the structures of both VioC and EFE have been reported,79, 81, 116, 215 including the structures of VioC•αKG•L-Arg, VioC•αKG•3-OH-L-Arg, VioC•L-Arg•peroxysuccinate, VioC•3-OH-L-Arg•succinate and VioC•L-Arg•succinate•photoreduced vanadyl ion complexes.116 VioC binds αKG binding in a proximal-type conformation (Table 1), which slightly differs from the typical proximal mode of binding. In the VioC•αKG•L-Arg complex, the Fe(II) centre exhibits a distorted 5-coordinate geometry in which the C1 carboxylate of αKG is ~35° out of the equatorial plane defined by H168, E170 and H316 (Fig. 12C).116 In addition, structural information from the Fe(II)-peroxysuccinate complex and vanadium(IV)-oxo species in VioC revealed coordinated motions of the active site residues, which may properly orient the Fe(IV)=O species for catalysis.
A structural and biochemical comparison between EFE (Fig. 2G & Fig. 9B) and VioC (Fig. 12C) revealed several key differences.79, 81, 116 First, the L-Arg hydroxylation positions are different. VioC and EFE hydroxylate L-Arg at the C3 and C5 positions, respectively. Second, in the absence of L-Arg, αKG binds to EFE in a monodentate fashion, while upon L-Arg binding, αKG shifts to the bidentate fashion. Third, in EFE, due to the distal-type αKG binding, a conformational switch is required to re-orient the Fe(IV)=O species for hydroxylation. Fourth, a phenylalanine residue (F283) was proposed to play a key role in controlling the αKG conformational switch to re-orient Fe(IV)=O, which might be important for determining the ratio between ethylene formation and L-Arg hydroxylation. Thus far, there have been no compelling mechanistic results obtained to explain αKG fragmentation in EFE, while other homologues (e.g. VioC) do not show such activity.
3.6.2 L-Asparagine hydroxylase
Calcium-dependent antibiotics (CDA) (138, Fig. 13A) are acidic lipopeptides that are promising candidates for the development of new antibiotics.224 CDA are synthesized through NRPS, with one of the building blocks being L-β-hydroxy-asparagine 137. The hydroxylation of L-Asn 136 to L-β-hydroxy-Asn 137 is catalyzed by an αKG-NHFe enzyme: AsnO (Fig. 13A).89 The crystal structures of AsnO•Fe(II) and AsnO•Fe(II)•2S,3S-3-hydroxy-Asn•succinate have been reported. AsnO displays an overall DSBH fold, where H155, E157 and H287 form the 2-His-1-carboxylate facial triad.89 The AsnO active site closes upon substrate binding via a lid-like region. Biochemical data revealed that AsnO uses stand-alone L-Asn as the substrate instead of L-Asn tethered to the carrier domain or the peptide released from the NRPS.225
Fig. 13.

Asparagine and aspartate hydroxylation reactions mediated by αKG-NHFe enzymes. (A) L-Asn 136 hydroxylation is catalyzed by AsnO, to generate the building block 137 for CDA 138 biosynthesis.89 (B) Biosynthetic pathway of ectoine 144. The hydroxylation of ectoine 144 to hydroxylectoine 145 is mediated by an αKG-dependent EctD.223
3.6.3 Ectoine hydroxylase
Ectoine 144 and hydroxyectoine 145 are zwitterionic small molecules produced by many halophilic and halotolerant bacteria (Fig. 13B);223 however, they are not part of the NRPS biosynthetic machinery. We have included them in this section only for comparison to reactions covered in Sections 3.6.1 and 3.6.2. Ectoine 144 and hydroxyectoine 145 are biologically inert and do not interfere with overall cellular functions, even when present at high concentrations in the cytoplasm. The proposed function of ectoine 144 and hydroxyectoine 145 is to cope with osmotic stress at high external salinity. In addition, ectoine 144 and hydroxyectoine 145 may also function as effective stabilizers of proteins226 and cellular membranes.227 Due to these unique properties, ectoine and hydroxyectoine biosynthetic gene clusters have been identified, and their biosynthetic pathways have been biochemically characterized.223 Ectoine biosynthesis in initiated by the Ask-catalyzed activation of L-Asp 139 to β-aspartylphosphate 140, which is then reduced to β-semialdehyde 141 by Asd. Subsequently, a PLP-dependent transaminase, EctB, converts 141 to L-diaminobutyric acid 142. The EtcA acetyltransferase catalyzes the acetylation of the side-chain amino group in 142 to Nβ-acetyl-diaminobutyric acid 143. The final step of ectoine biosynthesis is the EctC-catalyzed condensation between the α-amino group and the keto of the Nγ-acetyl group of compound 143. The further conversion of ectoine 144 to hydroxylectoine 145 is mediated by an αKG-NHFe enzyme: ectoine hydroxylase EctD (Fig. 12B). Interestingly, compound 145 is superior to its precursor 144 in protecting microorganisms against environmentally imposed stresses and in preserving the functionality of macromolecules and cells.228, 229 Therefore, EctD could be important for the industrial production of hydroxylectoine 145. Structures of the apo form of EctD have been reported,98, 230 but no substrate complex structure is available at this time.
3.6.4 Glutamate hydroxylase
The biosynthesis of kutzneride 2 (23, Fig. 4D) involves an αKG-NHFe chlorinase (e.g. KtzD in Section 2.2). This pathway also includes two more αKG-NHFe enzymes, KtzO and KtzP, which catalyze the stereospecific hydroxylation of L-glutamate tethered to the carrier domains (L-glutamate-S-PCP 146, Fig. 14). The catalysis of KtzO and KtzP produces threo 147 and erythro 148 isomers (Fig. 14). Both threo and erythro isomers can be found in different kutznerides.231 Thus far, no structural information is available for these enzymes and their stereo-selectivity remains to be addressed.
Fig. 14.

Hydroxylation of glutamate tethered to a carrier protein mediated by KtzO/KtzP in kutzneride 2 biosynthesis, which results in the production of threo 147 and erytho 148.231
3.6.5 4-Hydroxyphenylpyruvate oxygenase
Vancomycin 153 biosynthesis involves several tyrosine-derived building blocks.232 In this biosynthetic pathway, the 4-hydroxyphenylpyruvate (4-HPPA) 149 to L-4-hydroxymandelate 150 reaction catalyzed by hydroxymandelate synthase (HmaS) is unique (Fig. 15A). Although HmaS is an NHFe enzyme, it does not require αKG for its catalytic activity. Instead, the α-keto-carboxylate moiety of αKG has been incorporated as part of the substrate 4-HPPA 149. As a result, the α-keto-carboxylate moiety of the substrate 4-HPPA 149 plays a similar role to that of αKG during the oxidative decarboxylation reaction.6 In HmaS catalysis, the α-keto-carboxylate moiety of 4-HPPA 149 is most likely coordinated to the iron centre in a fashion similar to that of αKG. After O2 activation and decarboxylation, Fe(IV)=O species hydroxylates the decarboxylation product from 4-HPPA 149, resulting in the production of L-4-hydroxymandelate 150 (Fig. 15A).232–234 The hydroxyl group in compound 150 is then oxidized by Hmo-catalysis to produce 151, with subsequent transamination catalyzed by HpgT leading to 4-hydroxyphenylglycine 152 (Fig. 15A),232 which is one of the unnatural amino acids used in vancomycin 153 biosynthesis.232
Fig. 15.

Reactions of 4-hydroxyphenylpyruvate oxygenases. (A) In vancomycin biosynthesis, HmaS-mediated hydroxylation affords L-4-hydroxymandelate 150. (B) HPPD hydroxylates aromatic carbon to yield homogentisate. (C) In the reaction catalyzed by the HPPD F337I variant, both 150 and 154 are produced.236
Another similar example is 4-hydroxyphenylpyruvate dioxygenase (HPPD), which mediates the oxidative decarboxylation of 4-HPPA 149, followed by aromatic ring hydroxylation (Fig. 15B).6, 235 HPPD plays a crucial role in tyrosine metabolism and has a high sequence homology to HmaS (Fig. 15A).
These two enzymes share the common substrate 149, and accomplish their catalytic chemistry without requiring αKG, which is supplied from the carboxylate moiety of the substrate.232–234 The decarboxylation half-reaction of these enzymes is similar, while the other half-reactions differ in regioselectivity and complexity. A point mutant of HPPD (F337I) was shown to produce a mixture of 154 and 150 (Fig. 15C).236 The crystal structures of HPPD from Zea mays, Arabidopsis and Streptomyces avermitilis have been reported,101, 237, 238 and it was suggested that the relative orientation of the substrate aromatic ring relative to the Fe(IV)=O species is responsible for the two activities in HPPD and HMS.239
3.6.6 Tryptophan or indole hydroxylation
Tryptophan or indole-derived natural products are also widely distributed in nature and αKG-NHFe enzymes play a role in many biosynthetic pathways. In the following section, a few of these examples are discussed. Indole-3-acetic acid (IAA, 155) is an natural auxin in plants, regulating many aspects of growth and development.241 In vitro, a rice αKG-NHFe enzyme, DAO, hydroxylates IAA 155 to a biologically inactive molecule 2-oxoindole-3-acetic acid (OxIAA, 156, Fig. 16A). This observation suggests that DAO might play a crucial role in IAA catabolism to maintain IAA homeostasis during a plant’s reproductive development.241
Fig. 16.

Hydroxylation of tryptophan derivatives. (A) DAO-catalyzed production of the natural plant auxin IAA 155. (B) M2H-catalyzed melatonin 157 hydroxylation. (C) FqzB-catalyzed rearrangement in the biosynthesis of spirotryprostatin A 162.240
The crystal structure of DAO has not been reported yet. A similar example is the metabolism of melatonin, which is mediated by melatonin-2-hydroxylase M2H (Fig. 16B). Four αKG-NHFe enzymes (M2Hs) from rice have been shown to hydroxylate melatonin 157 to 2-hydroxy-melatonin 158.242 It is not yet known whether DAO and M2H-catalysis involves an epoxide intermediate, which is a key species in the biosynthesis of some spiro-indole alkaloids.240 For example, the generation of the spiro-carbon moiety during the biosynthesis of spirotryprostatin A 162 was suggested to involve an expoxide intermediate. After the epoxide is incorporated by FqzB-catalysis (160a, Fig. 16C), the electron-donation property of the methoxy oxygen lone pair initiates the opening of a 2,3-epoxide ring in 160a. A subsequent semipinacol-like rearrangement in 160b produces a 2-indolone 161 via the migration of its C2-substitute to the C3 position (Fig. 16C).240
FtmOx1 is another excellent example of αKG-NHFe enzyme functional diversity in indole-alkaloid biosynthesis. Both gene disruption114 and biochemical characterizations114, 243 have clearly demonstrated that FtmOx1 is responsible for endoperoxidation in the biosynthesis of verruculogen 164 (Fig. 17A), a tremorgenic mycotoxin found in various Aspergillus244 and Penicillium species.245, 246 In earlier studies, both αKG and ascorbate were reported to be required for the endoperoxidation reaction, while ascorbate was proposed to be essential for catalysis (Fig. 17A).114, 243 Our recent characterisations revealed that under single turnover conditions, ascorbate is not needed, and the oxidation of the C13-hydroxyl of verruculogen into a keto group was also observed (163 → 165, Fig. 17A).68 When the FtmOx1 reaction was conducted under a mixed 18O2/16O2 atmosphere, analysis of the products suggested that dioxygen gas is incorporated into the endoperoxide moiety of verruculogen without O-O bond cleavage (Fig. 17A).243 The structure of FtmOx1 was recently reported and showed αKG coordinates to the Fe(II) centre through a distal-type binding mode (Fig. 2C).68 Most importantly, immediately adjacent to the remaining site for O2 binding and activation, there is a tyrosine residue (Y224), which plays a key role in the endoperoxidation reaction based on our recent biochemical and spectroscopic characterization of FtmOx1 (Fig. 17B).68 In reactions catalyzed by FtmOx1 Y224 variants, the dominant products are N1 deprenylation reaction products (163 → 166, Fig. 17A), which are decomposition products from hydroxylation instead of endoperoxidation reactions.68
Fig. 17.

Endoperoxide formation in verroculogen biosynthesis. (A) FtmOx1 reaction with ascorbate affords verruculogen 164 as the dominant product, while in the absence of ascorbate, compound 165 is the dominant product. The reactions of FtmOx1 Y224 variants produce the N-1 deprenylation 166 as the major product. (B) Proposed FtmOx1 catalysis involves a tyrosyl radical species, which is key to the endoperoxidation reaction.68
The carbon skeleton of 4′-methoxyviridicatin 170 (Fig. 18A) was constructed by an NRPS, which is a similar strategy to that used in the biosynthesis of verruculogen. The 6,6-bicyclic scaffold of compound 170 is also found in many other bioactive compounds.248, 249 In the biosynthetic pathway of 4′-methoxyviridicatin 170, the condensation between anthranilic acid and methyltyrosine affords 4′-methoxycyclopeptin 167, which is further converted to 6,6-bicyclic quinolone 170 by AsqJ-mediated sequential dehydrogenation and epoxidation reactions.247 AsqJ first catalyzes the dehydrogenation of 167 to incorporate a double bond into the 6,7-bicyclic intermediate 168, and then a subsequent AsqJ-mediated reaction results in the incorporation of an epoxide into the product (168 ® 169, Fig. 18A).62, 247
Fig. 18.

AsqJ catalysis. (A) AsqJ-catalyzed dehydrogenation and epoxidation.247 (B) Probes used in the AsqJ-dehydrogenation reaction mechanistic study.62
An Fe(IV)=O species has been trapped in AsqJ catalysis and characterized spectroscopically.62 In addition, crystallographic studies of A. nidulans AsqJ revealed that it makes use of a 2-His-1-carboxylate facial triad ligand environment (His34, Asp136 and His211).65 In this structure, αKG coordinates to the iron centre bidentately in the distal-type binding mode. The AsqJ•substrate binary structure is also available.65 Similar to other distal-type αKG-NHFe enzymes, the substrate is not adjacent to the remaining site for O2 binding and activation. A plausible explanation for AsqJ catalysis is that an βKG conformational switch is required to properly re-orient the Fe(IV)=O species towards the substrate for the dehydrogenation and epoxidation reactions.
AsqJ catalysis has been examined recently using quantum mechanics and molecular mechanics calculations (QM/MM).250 In this study, the desaturation reaction mediated by AsqJ was proposed to proceed through two consecutive hydrogen atom transfer processes (Pathway I, Fig. 18A). In this mechanistic model, the Fe(IV)=O species abstracts a hydrogen atom from the C3 or C10 position of the substrate. Subsequently, a second hydrogen atom abstraction from the substrate affords a di-radical intermediate 167b, which recombines to yield the desaturated product 168. The findings from this QM/MM study contradict the recently reported AsqJ study by Chang and co-worker.251 In this study, cyclopeptin 171, an analogue of compound 169, and its C3 epimer 172 were employed to decipher the desaturation mechanism of AsqJ (Fig. 18B).251 Both isomers functioned as efficient AsqJ substrates, and a pre-steady state characterization of these two reactions indicated that they proceeded with similar kinetic parameters. These results highly suggested that the AsqJ-catalyzed desaturation reaction does not go through a hydrogen abstraction step at the C3 position of the substrate, as was proposed in the QM/MM study. Instead, the combined kinetic and spectroscopic results using substrate analogues suggested that the desaturation process was initiated from a hydrogen atom abstraction by the Fe(IV)-oxo species from the C10 position of the substrate. Most likely, the desaturation reaction either employs a hydroxylated intermediate 167c or a carbocation intermediate 167d, for the AsqJ-catalyzed desaturation reaction (Pathway II or III, Fig. 18A).251 Additional mechanistic studies are required to decipher this AsqJ-mediated desaturation reaction.
Interestingly, after AsqJ-catalyzed desaturation and epoxidation, the resulting product 169 underwent a non-enzymatic arrangement converting the 6,7-bicyclic benzodiazepinedione core in 169 to the 6,6-bicyclic quinolone scaffold present in 170 (Fig. 18A).247 The proposed mechanism for this rearrangement step begins with ring opening of the epoxide 169 to 169a, triggered by the electron-donation property of the methoxyl group. A two-step rearrangement of the 6,7-bicyclic moiety in 169a results in the formation of 6,6-bicyclic quinolone in 170 through elimination of a methyl isocyanate unit from 169b (Fig. 18A).247
3.6.7 Amino modification associated with cyclodipeptide synthase (CDPS)
Diketopiperazines (DKPs) serve as a scaffold in many alkaloids. Bicyclomycin (BCM, 183) is a DKP-type alkaloid isolated from Streptomyces,253, 254 and exhibits activity against a broad spectrum of Gram-negative bacteria.255 The BCM biosynthetic gene cluster was identified256 and the functions of the proposed enzymes have also been validated in vitro (Fig. 19).252 The biosynthetic pathway starts with BcmA-catalyzed condensation using Leu-tRNALeu 173 and Ile-tRNAIle 174 as substrates, producing a DKP scaffold.256 After the construction of the DKP scaffold in 175, the subsequent tailoring reactions involve one cytochrome P450 (BcmD) and five αKG-NHFe enzymes (BcmB, BcmC, BcmE, BcmF and BcmG, Fig. 19). BcmC, BcmE and BcmG are hydroxylases, which incorporate the three hydroxyl groups (C2′, C3 and C3′ positions, see the labels at compound 176) on the Leu and Ile side-chains to produce 181. Similar to AsqJ discussed in Section 3.6.6, BcmB is a bi-functional enzyme that desaturates the C1 and C1′ positions to introduce the double bond present in 179. BcmB then uses 179 as a substrate to incorporate an epoxide moiety into the C1, C1′ positions of 180. The C3-OH group undergoes intramolecular attacks to open the epoxide, resulting in the O-bridged bicycle-[4,2,2]piperazinedione ring in 181. BcmD is a P450 monooxygenase, and in the presence of spinach ferredoxin, ferredoxin reductase and NADH, BcmD successfully hydroxylates 181 at the C6 position to produce 182. Finally, BcmF, another αKG-NHFe enzyme, introduces a double bond between the C5 and C5a positions. The bicyclomycin biosynthetic pathway represents an excellent system for demonstrating the catalytic diversities of αKG-NHFe enzymes (BcmB, BcmC, BcmE, BcmF and BcmG, Fig. 19). The activities of all of these enzymes have been demonstrated in vitro; however, mechanistic information is not yet available for them.
Fig. 19.

The biosynthetic pathway of bicyclomycin involves five αKG-NHFe enzymes in the tailoring reactions to produce the final product bicyclomycin 183.252
3.6.8 Aspartate modification associated with ribosomally synthesized and post-translationally modified peptides (RiPPS) systems
In the above sections, we discussed modifications in NRPS catalysis. Very similar cases have been observed in RiPPS systems, e.g. a 19-amino acid antibiotic cinnamycin 184. In cinnamycin, there are nine post-translational modifications that are critical for the biological activities of cinnamycin 184.257 One of these nine post-translational modifications is the β-hydroxylation of Asp15 in the precursor peptide CinA, which is catalyzed by the αKG-NHFe enzyme CinX (Fig. 20).257, 258 In vitro studies confirmed that CinX accepts the precursor peptide CinA as a substrate, and that the leader sequence at the N-terminus of CinA is not required for its activity.257 Further heterologous co-expression experiments combining CinA, CinX and other tailoring enzymes in E. Coli led to the production of cinnamycin with full antibacterial activity.257
Fig. 20.

β-Hydroxylation of Asp15 in the precursor peptide CinA catalyzed by an αKG-NHFe enzyme CinX in the cinnamycin 184 biosynthetic pathway.257
4 Terpene Biosynthesis
Terpenes are one of the largest classes of natural products.82 All terpenes are derived from two building blocks: isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Condensation between IPP and DMAPP leads to the production of prenyldiphophates, which are used by terpene cyclases or prenyltransferases to create the skeletons of terpenes.259 After the terpene skeletons are assembled, many of them go through extensively modifications, with αKG-NHFe enzymes being among the most frequently used tailoring enzymes. In this section, we discuss a few cases where αKG-NHFe enzymes are key to their structural diversities.
Astaxanthin 189 is one of the most commonly found carotenoid pigments in marine animals.260 Two αKG-NHFe enzymes, CtrZ and CrtW, are responsible for multiple oxidative tailoring reactions in astaxanthin biosynthesis. CrtZ hydroxylates either the 3 or 3′ position of the β-ionone ring, while CrtW oxidizes methylene to keto groups at the 4 or 4′ position of the β-ionone ring (Fig. 21).261–265 CrtZ sequentially hydroxylates β-carotene 185 to β-cryptoxanthin 186 and then to zeaxanthin 187. CrtW then introduces keto groups into the 4- and 4′-positions of 187 to produce astaxanthin 189 (187 → 188 → 189, Fig. 21).266 Alternatively, CrtW can sequentially oxidize β-carotene 185 to echinenone 190 and then to canthaxanthin 191. CrtZ hydroxylates 191 to phoenicoxanthin 192. Another round of CrtZ-catalyzed hydroxylation on 192 yields astaxanthin 189 (Fig. 21). Thus far, no structural information is available for either CrtZ or CrtW.
Fig. 21.

Multiple oxidative modifications in the astaxanthin 189 biosynthetic pathway involving two αKG-dependent NHFe enzymes: CtrZ and CrtW. CrtZ hydroxylates either the 3 or 3′ position of the β-ionone ring, while CrtW oxidizes methylene to keto groups at the 4 or 4′ position of the β-ionone ring.263
Pentalenolactone 203 (Fig. 22A) is a sesquiterpenoid antibiotic isolated from more than 30 species of Streptomyces.267, 268 The electrophilic epoxylactone moiety of pentalenolactone allows it to alkylate an active site cysteine residue of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase.269–272 Over the years, three pentalenolactone biosynthetic gene clusters have been identified, namely pen, pnt and ptl.273–275
Fig. 22.

Pentalenolactone and neopentalenolactone biosynthesis. (A) The biosynthetic pathways of pentalenolactone 203 and neopentalenolactone 206. PenH/PntH/PtlH catalyzes the hydroxylation of 1-deoxypentalenic acid to 11-β-hydroxy-1-deoxypentalenic acid 198.275 (B) Crystal structure of PtlH•Fe•αKG showing a proximal-type αKG (brown sticks) coordination to the iron centre (yellow sphere). (C) Notably, the structure of PtlH with the substrate analogue ent-1-deoxypentalenic acid (green sticks) reveals a conformation change of an active site Y142 relative to that in (B), where there is no substrate.
Pentalenolactone biosynthesis is initiated by the cyclization of farnesyl diphosphate (FPP, 193) to the tricyclic hydrocarbon pentalenene 194.272, 276 After the pentalenene skeleton is assembled, it is extensively modified by six redox enzymes.273–275 An αKG-NHFe enzyme (PenH/PntH/PtlH)275, 277 catalyzes the hydroxylation of 1-deoxypentalenic acid 197 to the 11-β-hydroxy-1-deoxypentalenic acid 198. In the pentalenolactone pathway, another αKG-NHFe enzyme (PenD/PntD)273 catalyzes the epoxidation reaction (200 → 202).273 In the neopentalenoketolactone 206 pathway, PtlD catalyzes the conversion of 204 to 205. However, the epoxide 205 is not stable and spontaneously re-arranges forming 206 as the final product (Fig. 21A). In this biosynthetic pathway, PtlD, PenD and PntD are multi-functional enzymes, catalyzing both dehydrogenation and epoxidation reactions, similar to the AsqJ catalysis discussed in Fig. 18.
A crystal structure of the PtlH tertiary complex (PtlH•αKG•substrate analogue) has been reported.110 PtlH has the common DSBH fold with a typical 2-His-1-carboxylate facial triad ligand environment (His137, Asp139 and His226). The other three ligands are water ligands. Upon αKG binding, it replaces two of the water ligands by coordinating to the Fe(II) centre in a proximal-type conformation.110 The structure of the PtlH tertiary complex has been reported,110 in which an inactive substrate analogue ent-1-deoxypentalenic acid was used to replace the substrate 197. In the absence of the substrate analogue ent-1-deoxypentalenic acid, the active site tyrosine residue (Y142) adopts two different conformations (Figs 22B and C). However, upon the binding of ent-1-deoxypentalenic acid, Y142 is locked into one conformation by forming a H-bond with the remaining H2O ligand of the iron centre (Fig. 22C).110 The role of this active site tyrosine residue in PtlH-catalysis remains to be addressed.
Phenalinolactone A (PL A, 214) is a terpene glycoside that shows antibacterial activity (Fig. 23). PL A possesses a tricyclic backbone conjugated to a γ-hydroxybutyrolactone. After oxidative modification of the phenalinolactione scaffold, these sites are further glycosylated and acylated by both acetyl and 5-methylpyrrole-2-carboxylic groups to produce the final product PL A (214, Fig. 23).278 The gene cluster encoding pathway enzymes for phenalinolactones biosynthesis from Streptomyces sp. Tü6071 have been identified (pla biosynthetic gene cluster). Many biosynthetic genes have been tentatively assigned functions based on results from heterologous expression and biochemical analyses, by the characterization of gene deletion mutants, or via sequence homology with other known genes.278, 279 The pla biosynthetic gene cluster encodes 1-deoxy-D-xylulose-5-phosphate synthase (DXP) and 1-hydroxy-2-methyl-2(E)-butenyl-4-diphosphate synthase (HMBPP synthase), which implies that the precursors (IPP and DMAPP) for phenalinolactone biosynthesis are most likely provided by the non-mevalonate biosynthetic pathway.82 The pla biosynthetic pathway has five oxygenases (one αKG-NHFe enzyme and four P450s).278, 279 Inactivation of the gene plaO1, which encodes an αKG-NHFe enzyme, results in disruption of the formation of the γ-butyrolactone moiety, and accumulates PL CD6 207.279 When PL CD6 207 was incubated with recombinant PlaO1 in vitro in the presence of αKG and ascorbate, a new compound with a UV-absorption spectrum, mass spectrum and HPLC retention time identical to that of PL HS6 208 was identified (Fig. 23).278 This result suggested that PlaO1 is most likely responsible for the complicated 207 → 208 conversion (Fig. 23). A mechanism for PlaO1 catalysis has been proposed.279 In this reaction, after the hydroxylation mediated by PlaO1, intramolecular C-C bond migration might go through a cyclopropanone intermediate 207b. The subsequent ring opening of this leads to an aldehyde and an α-keto acid 207c, which then cyclizes to produce PL HS6 208 (Fig. 23). More detailed characterizations are needed to uncover the mechanistic details for this complex chemical transformation.
Fig. 23.

Modification reactions in terpene biosynthesis. In the biosynthesis of phenalinolactone A, PlaO1 is responsible for converting PL CD6 207 to a γ-butyrolactone moiety formation of PL HS6 208 through a proposed cyclopropanone intermediate.278
Paraherquonin (218, Fig. 24A) biosynthesis280 is another example where an αKG-NHFe enzyme mediates multiple reactions. In the biosynthesis of the fungal meroterpenoid paraherquonin 218, an αKG-NHFe enzyme PrhA catalyzes multiple chemical reactions (215 → 216 → 217, Fig. 24A). The proposed mechanism for PrhA-catalyzed reactions is shown in Fig. 24A.280 The PrhA-catalysis starts with a dehydrogenation reaction at the C5 position of preaustinoid A1 215 to produce bekeleyone B 216. A second round of oxidation creates a radical species at the C1 position in species 216a, which initiates the subsequent carbon skeleton rearrangements, as proposed in Fig. 24A. The proposed conversions from 216a to 216c are known as the homoallyl-homoallyl radical rearrangement.281
Fig. 24.

Multiple chemical transformations catalyzed by αKG-NHFe enzymes in the paraherquonin and acetoxydehydroaustin pathways. (A) PrhA mediates the dehydrogenation of 215, followed by oxidation to yield paraherquonin as the final product.280 AusE acts on the same substrate 215. Unlike PrhA, AusE-catalysis goes through a dehydrogenation reaction followed by rearrangements to form the spiro-ring.69 (B) Crystal structure of AusE•Mn•αKG with substrate 215 modelled into the active site. (C) Crystal structure of PrhA•Fe•αKG•substrate preaustinoid A1 215 (cyan sticks). The residues involved in the structure–function studies are labelled in red.
Another αKG-NHFe enzyme (AusE) from P. brasilianum MG11 shares 92% sequence identity with PrhA. AusE catalyzes a complicated skeleton rearrangement in acetoxydehydroaustin 221 biosynthesis (Fig. 24A).280 Despite its high sequence identity to PrhA, AusE exhibits a completely different activity. AusE catalyzes the conversion of preaustinoid A1 215 to preaustinoid A3 220 (Fig. 24A) by desaturation and an unusual spiro-ring forming rearrangement reaction (Fig. 24A).280 AusE-catalysis is initiated by hydrogen abstraction of the C1 position of the substrate, resulting in a net dehydrogenation reaction to generate preaustinoid A2 219, which is an isomer of 216 produced from the PrhA reaction. In a second round of AusE-catalysis, the Fe(IV)=O species abstracts a hydrogen atom from the C5 position of 219, leading to the formation of a radical species 219a. Subsequent hydroxyl rebound leads to hydroxylation at C5 (219b). Finally, the skeleton rearrangements in 219b generate the spiro-lactone moiety in 220, which is a key intermediate in acetoxydehydroaustin 221 biosynthesis.69 In another AusE-mechanistic model, a cyclopropyl intermediate similar to the one in PrhA-catalysis (216b, Fig. 24A) has been suggested.91
Structural characterization of both AusE and PrhA indicated that three active site residues may play a key role in determining the outcome of the reactions.91 The active sites of AusE and PrhA are highly similar and share a typical 2-His-1-carboxylate as the Fe(II) centre ligands (His130, Asp132 and His214). AusE and PrhA make use of the same substrate, but differ in their reaction outcomes (Fig. 24A). Recently, Abe and co-workers solved the crystal structures of AusE•Mn•αKG and PrhA•Fe•αKG•substrate where the substrate is preaustinoid A1 215 (Figs 24B and 24C). They then focused on the interactions between the substrate and enzymes in the regions next to the A and B rings of the substrate because these are the reaction sites. In AusE, V150 and A232 interact with the A ring of the substrate, while in PhrA, an identical interaction is formed by L150 and S232. Based on these differences, Abe and co-workers conducted mutagenesis studies guided by the information gained from these protein-substrate complexes (Figs 24B & 24C). By mutating the residues in AusE (V150 and A232) to the corresponding residues in PrhA (L150 and S232), they successfully tuned the activities of AusE into that of PrhA. For example, the AusE-S232A mutant produced a mixture of 217 and 220, while the AusE-L150V/S232A double mutant produced 217 as the exclusive product, instead of producing 220 in the wild-type AusE enzyme. Similarly, PhrA-V150L/A232S completely lost its wild-type PhrA activity and produced compound 220 as the sole product.
The PrhA and AusE reactions are great examples demonstrating how αKG-NHFe enzymes can fine-tune their activities by subtle changes in the active site residues. Interestingly, in the PrhA-V150L/A232S double mutant, when a third mutation M241V was introduced, several products different from those produced by wild-type enzymes were observed. The results from this study nicely demonstrated that these multi-functional αKG-NHFe enzymes may be amenable to engineering efforts for the biocatalytic generation of natural product derivatives.91
Okaramines (Fig. 25) are complex indole alkaloids with potent insecticidal activity.282 Among all the members of the okaramine family of indole alkaloids, okaramine B is the most potent insecticide.283 Compounds in this family selectively activate glutamate-gated chloride channels (GluCls) in invertebrates.284 Okaramine biosynthetic gene clusters (oka) have been identified from the P. simplicissimum strain ATCC 90288 (AK-40) and A. aculeatus strain ATCC 16872, respectively.285 Okaramine C 223, one of the key intermediates for this biosynthetic pathway, was proposed to be assembled by a three-step process involving an NRPS-catalyzed condensation between two tryptophan molecules to form the diketopiperazine ring, followed by a prenyltransferase-catalyzed transfer of the dimethylallyl moiety, and finally an epoxidation reaction mediated by a flavin-dependent enzyme that catalyzes the ring closure. Subsequent modification reactions are mediated by a P450 enzyme (OkaD) and an αKG-NHFe (OkaE) (Fig. 25). The function of OkaD was proposed based on the results from characterizations of the okaE and okaD deletion mutants. The okaE deletion mutant accumulates okaramine C 223, while the okaD deletion mutant accumulates both compounds 223 and 225. It was thus proposed that the P450 OkaD catalyzes the four-electron-oxidation of okaramine C 223 to form Okaramine A 225. However, the biochemical details for this proposed biocatalytic transformation have not yet been reported.
Fig. 25.

The biosynthetic pathway of okaramine D. Okaramine A 225 can be converted to 12-deshydroxyl okaramine E 226 and okaramine 227 by OkaE-catalysis via radical intermediates.
OkaE, an αKG-NHFe enzyme, was proposed to catalyze the formation of the azetidine ring in 227. The inactivation of okaE leads to an accumulation of okaramine C 223 and okaramine A 225. Okaramine A 225 could be converted to 12-deshydroxyl okaramine E 226 and okaramine E 227 using OkaE expressed in Saccharomyces cerevisiae BJ5464-NpgA. In the presence of FeSO4, αKG and ascorbate, OkaE converts 225 to 227 in vitro. However, in the presence of reductants (e.g. β-mercaptoethanol), αKG and O2, using okaramine A 225 as the substrate, OkaE-catalysis produces compound 226 as the major product (Fig. 25). OkaE can also hydroxylate compound 226 to produce okaramine 227 in vitro. OkaE-catalysis was proposed to start with the abstraction of a hydrogen atom from the C8a position of compound 225, followed by an exo-cyclization to forge the azetidine ring (Fig. 25).285 The mechanistic details for 225a → 227 or 225b →226 remain to be characterized (Fig. 24A). From okaramine 227, a hydroxylation mediated by OkaG, a P450 enzyme, followed by a methylation catalyzed by a methyltransferase (OkaF) leads to the production of okaramine D 228.
Rubratoxin A 242 is a fungal polyketide, comprised of cyclononane ring and two fused maleic anhydrides forming a 5/9/5 core ring system (Fig. 26). Rubratoxin A is a potent inhibitor of protein phosphatase 2 (PP2A) and is a lead compound for the development of anti-cancer drugs.286 The biosynthetic information for the formation of the 5/9/5 core ring system of rubratoxin A was drawn upon genetic analysis of byssochlamic acid and the agenstadride A biosynthetic gene cluster. These two compounds also have medium-sized carbocycles fused with maleic anhydride moieties.287 After the formation of polyketide-derived monomers, a methylcitrate dehydratase, RbtK, and an alkylcitrate synthase, RbtL, catalyze the formation of maleidride-type building block 231 and 232 (Fig. 26). To assemble the nonadride core in 234, one ketosteroid isomerase (RbtR) and two putative phosphatidylethanolamine binding proteins (RbtM and RbtO) work together to mediate the dimerization of two maleidride-type precursors via the intermediate 233.287 The mechanistic details for this dimerization process remain to be characterized. It was proposed that the core of rubratoxin A is assembled through a similar process, followed by extensive oxidative modifications to convert the nonadride 234 to 242.288 Genetic analysis of the rubratoxin A biosynthetic gene cluster indicated that it encodes four αKG-dependent NHFe enzymes (RbtB, RbtG, RbtE and RbtU), one P450 monooxygenase (RbtI) and one flavin-dependent monooxygenase (RbtA). Results from characterization of the ΔrbtI deletion mutant suggested that it might be involved in the hydroxylation reaction in the production of maleidride monomers. Genetic and biochemical characterizations have suggested that the four αKG-dependent NHFe enzymes (RbtB, RbtG, RbtE and RbtU) are hydroxylases, catalyzing four hydroxylations at the C5, C6, C11 and C7′-position, respectively (Fig. 26).288 RbtA then catalyzes the oxidation of 238 to an aldehyde 239. RbtB is a bi-functional enzyme, which further oxidizes 239 to generate a carboxylate 240.288
Fig. 26.

Proposed biosynthetic pathway of rubratoxin A. Four αKG-NHFe enzymes RbtB, RbtG, RbtE and RbtU catalyze the sequential hydroxylations, converting 234 to 238.
5 Biosynthesis and Metabolism of Phosphorous Products
Over the last sixty years, ~40 natural phosphonates have been isolated.289 Due to their structural mimicry to carboxylates and phosphates,83, 290, 291 phosphonates have great pharmaceutical potential.290, 292 Some representative phosphonates and their corresponding enzyme substrate/transition states are shown in Fig. 27. Fosfomycin 243 has long been used in the treatment of urinary tract infection293, 294 and, in conjunction with other antibiotics, it has also been used for the treatment of multidrug resistant bacteria.295 Fosfomycin inactivates UDP-GlcNAc enoylpyruvyl transferase (MurA), which is the first committed step in bacterial peptidoglycan biosynthesis.296 Phosphinothricin 245 (part of phosphinothricin-tripeptide, PTT 256) is unique due to its C-P-C functionality.83, 290, 297–299 Phosphinothricin 245 binds slowly to glutamine synthetase and is subsequently phosphorylated by ATP to produce a phosphorylated product 246, which irreversibly inhibits glutamine synthetase (Fig. 27A).300, 301 As a result, PTT 256 is widely used as the herbicide commercially known as Glufosinate.302, 303 Dehydrophos 247 was isolated as a tripeptide, and once this tripeptide is taken up by a cell, it is hydrolyzed into an analogue of dehydroalanine, a mimic of pyruvate 248, and inhibits alanine racemase activity.304–306 Dehydrophos 247 has been used as a broad-spectrum antibiotic.307 Fosmidomycin 249 and related compounds were isolated from a few Streptomyces strains.308–310 It is an inhibitor of 1-deoxy-D-xylulose 5-phosphate (DXP, 250) synthase, which is one of the key steps in the methyl erythritol phosphate pathway, an essential pathway for providing isoprenoid biosynthetic precursors in bacteria.82 Fosmidomycin 249 is currently being evaluated for the treatment of malaria.311 (Z)-L-2-amino-5-phosphono-3-pentenoic acid (APPA, 251) is a key component of several natural products (e.g. rhizocticins A-D, plumbermycins A-B, and phosacetamycin)312–314 and exhibits anti-fungal activities (Fig. 27A).315 K-4 and K-26 (252 & 253, Fig. 27B) contain a phosphonate analogue of tyrosine, (R)-1-amino-2-(4-hydroxyphenyl) ethylphosphonic acid (AHEP) 260 (Fig. 27C).316, 317 K-4 and K-26 are inhibitors of the angiotensin-converting enzyme (ACE) and are anti-hypertensive drug-candidates.318
Fig. 27.

Natural phosphonates. (A) Phosphonates and their corresponding enzymatic substrates or transition state analogues. (B) Four categories of phosphonates are represented by K-26: 253, 2-aminoethylphosphonic acid (AEP) 254, N-acetyl demethylphos-phinothricin (AcDMPT) 255 and PTT 256, based on how their C-P bonds are constructed. (C) Key steps of the biosynthesis of PnAA 258, PTT 256 and K26 253.
Based on how their C-P bonds are constructed, these natural phosphonates can be roughly divided into four categories,83, 290 as represented by K-26 253, 2-aminoethylphosphonic acid (AEP, 254), N-acetyl demethylphos-phinothricin (AcDMPT, 255) and PTT 256 (Fig. 27B). Phosphoenolpyruvate mutase (Ppm) is still the only well-characterized enzyme involved in C-P bond formation.319–321 Ppm catalyzes the isomerization of PEP 244 to phosphonopyruvate (PnPy 257). Equilibrium favours PEP by a factor of more than 500-fold.319 As a result, in all Ppm-involving biosynthetic pathways, the immediate next step is an irreversible reaction,290 and the decarboxylation of PnPy 257 to phosphonoacetaldehyde PnAA 258 (Fig. 27C) is one of these reactions that drives the equilibrium between PnPy and PnAA towards PnAA during Ppm-catalysis. Analysis of the publicly-available actinomycete genomes has indicated that ~5% of the sequenced actinomycetes have Ppm genes,289 implying that many more Ppm-involving phosphonate biosynthetic pathways might be uncovered in the future. PTT (256, Fig. 27) has a C-P-C functionality and involves phosphinate (e.g. AcDMPT, 255) as a key intermediate. Its second C-P bond formation is catalyzed by a P-methylase (255 → 259 conversion, Fig. 27C),297, 322–326 which is a member of the radical SAM enzyme superfamily (>114,000 members).327, 328 K-4 and K-26 (252 & 253, Fig. 27B) contain a phosphonate analogue of tyrosine, AHEP. The gene cluster for K-26 has not yet been discovered and the results from feeding studies suggest that K-26 biosynthesis does not involve the Ppm gene for its C-P bond construction, and instead an unknown type of C-P bond formation chemistry is likely involved.316, 317
Thus far, a few phosphonate biosynthetic pathways have been biochemically characterized. As with other types of natural products, extensive modifications are part of the phosphonate biosynthetic pathways, with many of them being unprecedented chemical reactions, suggesting that biosynthetic studies of natural phosphonates production are a gold mine for the identification of novel chemistries.83 Here, we briefly summarize some αKG-NHFe enzymes involved in several phosphonate biosynthetic pathways. Two αKG-NHFe enzymes play a role in the biosynthesis of O-methylated dehydroaminophosphonate (266, Fig. 28),305 the precursor for the phosphorous antibiotic dehydrophos 247. The first enzyme, DhpA, hydroxylates 2-hydroxyethyl-phosphonate (2-HEP, 261) to generate a 1,2-dihydroxyethylphosphonate (1,2-DHEP, 262) during the early stage of the biosynthesis of 266 (Fig. 28A).329 Another αKG-NHFe enzyme, DhpJ, is active in the later stage of this pathway, where it converts 264 to 265.330 Subsequent glycine addition mediated by DhpK, a Gly-tRNAGly-dependent peptidyl transferase, affords 266 as the final product (Fig. 28A).330 DhpJ can also catalyzes the hydroxylation of 263 to compound 267 (Fig. 28A).330 However, the 263 → 267 conversion is not as efficient as the 264 → 265 conversion, suggesting that compound 264 is probably the native substrate for DhpJ.
Fig. 28.

Phosphonate modifications mediated by αKG-NHFe enzymes. (A) Reactions catalyzed by DhpA and DhpJ in the O-methylated dehydroamino phosphonate 266 biosynthetic pathway.329 (B) FzmG mediates multiple hydroxylation reactions in the biosynthesis of fosfazinomycin A 271.331 (C) The organophosphate metabolism involves the PhnY-mediated hydroxylation of 254 to yield 2-amino-1-hydroxyethylphosphonic acid 272.332
FzmG catalyzes multiple hydroxylation reactions in biosynthesis of fosfazinomycin A 271, including the oxidation of PnAA 258 to PnA 268 and a stereospecific hydroxylation of Me-PnA 269 to compound 270 (Fig. 28B).331 The subsequent steps of fosfazinomycin A biosynthesis, especially the N-N and N-P bond construction steps (270 → 271), have not yet been biochemically characterized.
AEP 254 is a prevalent organophosphonate in many organisms and is frequently used to complement inorganic phosphate in phosphate-limited environments.290, 333 The αKG-NHFe enzyme PhnY plays an important role in this process by mediating the hydroxylation of the α-carbon of 254 to produce 2-amino-1-hydroxyethylphosphonic acid (2-AHEP, 272).332 The C-P bond of 272 is then cleaved to generate an inorganic phosphate and glycine (PhnZ-catalysis, Fig. 28C).332
Thus far, structural information for these αKG-NHFe enzymes is not yet available. With the discovery of new phosphonate biosynthetic pathways, many more αKG-NHFe enzymes are highly likely to be uncovered in the future.
6 Lipid and fatty acid modifications
Jasmonic acid (JA, 273, Fig. 29A) is a hormone synthesized by plants for the activation of self-defence mechanisms against attacks by pathogens and herbivores.334 Four αKG-NHFe enzymes, named jasmonate-induced oxygenases 1–4 (JOXs 1–4), play a crucial role in balancing the JA level during plant growth by mediating the hydroxylation of JA 273 to yield the inactive 12-OH-JA 274, which prevents the inhibitory effects of high JA levels on plant growth and development (Fig. 29A).335
Fig. 29.

Lipid and fatty acid modification reactions. (A) Hydroxylation of jasmonic acid 273 mediated by JOXs 1–4.335 (B) LpxO and KdoO-catalyzed hydroxylations of Kdo2-lipid A 275.336, 337 (C) Hydroxylation of phytanoyl-CoA 278 catalyzed by PhyH in the phytanic acid metabolism.338
Lipid A is the hydrophobic membrane anchor for lipopolysaccharides, which are the principal constituents of the outer membrane of Gram-negative bacteria.339 Some lipid A molecules contain hydroxylated acyl chains.340 Two αKG-NHFe enzymes, LpxO336 and KdoO,337 catalyze the hydroxylation of Kdo2-lipid A 275 (Fig. 29B). When LpxO was heterologeously expressed in E. coli K-12, the lipid A produced contained 2-hydroxymyristate, providing evidence supporting the proposed function of LpxO as a lipid hydroxylase.341 Recently, a LpxO homologue KdoO was identified from both Burkholderia ambifaria and Yersinia pestis.337 KdoO also makes use of the Kdo2-lipid A 275 as a substrate. However, the KdoO enzyme from B. ambifaria and Y. pestis hydroxylates the deoxysugar moiety of Kdo2-lipid A 275 to produce 277 (Fig. 29B).337 Therefore, LpxO and KdoO have different regioselectivity.
Similar to the α-hydroxylation reaction observed in LpxO catalysis, PhyH, an αKG-NHFe enzyme, hydroxylates phytanoyl-CoA 278 to 2-hydroxyphytanoyl-CoA 279, which is a fatty acid α-hydroxylation reaction (Fig. 29C). Phytanic acid is a compound found in common dietary sources. Impaired PhyH activity is responsible for 90% of cases of the neurological condition called Refsum disease,338 although the pathological mechanisms of this disease are not yet well understood. No cure has been found for this disease, but strict dietary restriction can slow the pathological progress. The mapping of clinically relevant mutations in the published structure of PhyH strongly support the conclusion that a loss of PhyH enzymatic activity is the main cause of Refsum disease.338
The reported structure of PhyH indicates that αKG coordinates the Fe(II) centre in a distal-type binding mode.106 As a result, the O2 binding and activation site is away from the substrate. One likely scenario is that αKG goes through a conformational change to re-orient the Fe(IV)=O species towards the substrate, enabling a direct hydrogen atom abstraction and subsequent hydroxyl radical rebound to form the hydroxylation product 279 (Fig. 29C).
7 Examples of nucleoside antibiotics
Many nucleoside-derived natural products exhibit biological activity.342 Their biosyntheses employ materials from primary metabolism (e.g. nucleic acids, proteins and glycans). Like other classes of natural products summarized in previous sections, extensive modifications are common in this class of natural products, and in this section, some examples of αKG-NHFe enzymes are briefly discussed.
5′-C-glycyluridine (GlyU) 282 (Fig. 30), a component of the nucleoside antibiotic capuramycin 283,343 is synthesized from uridine monophosphate (UMP) 280 and L-Thr by sequential reactions catalyzed by the αKG-NHFe enzyme Cpr19 and a transaldolase Cpr25 (Fig. 30).344 In vitro characterization of Cpr19 indicated that it catalyzes the hydroxylation at the C5 position of 280 to produce a germinal hydroxyl-phosphoester intermediate 280a, which is followed by phosphate elimination to yield uridine-5-aldehyde 281 (Fig. 30).344–346
Fig. 30.

Biosynthesis of capuramycin 283. The αKG-NHFe Cpr19 catalyzes the conversion of UMP 280 to uridine-5-aldehyde 281 through a germinal hydroxyl-phosphoester intermediate 280a, followed by phosphate elimination.345
Polyoxin 292 (Fig. 31), a nucleoside-derived natural product, can inhibit fungal cell wall biosynthesis by targeting chitin synthetase. Polyoxin is also an efficient agricultural fungicide.347 Structurally, polyoxin is constructed from three building blocks: a nucleoside and two amino acids (L-Ile and L-Glu). Many of the biosynthetic details of polyoxin remain enigmatic. The polyoxin biosynthetic gene cluster has been identified from Streptomyces cacaoi.348 Biosynthesis of the nucleoside portion of polyoxin is initiated by the condensation of UMP 280 with PEP 244 by PolA, followed by a rearrangement, which is proposed to be catalyzed by PolJ, to form octofuranuloseuronic acid 288. In the subsequent steps, oxidative elimination of the two terminal carbon atoms and the introduction of an amino group at the C5′ position were achieved by PolH, PolD, PolI and PolK catalysis to form the nucleoside skeleton 291.
Fig. 31.

The biosynthetic pathway of polyoxin 292. The reactions catalyzed by PolL introduce two hydroxyl groups onto the structure of CPOAA 303, which is one of the modules used in the biosynthesis of polyoxin.348
Homologues of these enzymes have also been identified in the nikkomycin biosynthetic pathway.349 The polyoxamic acid (295, POIA) moiety was derived from L-Ile 90 through the introduction of a β,γ-double bond followed by a cyclization reaction. These chemical transformations were catalyzed by PolC, PolE and PolF. The genes responsible for biosynthesis of the carbamoylpolyoxamic acid moiety (303, CPOAA) were assigned based on the results from biochemical characterizations (Fig. 31).350 L-Glu is first acetylated by a bi-functional N-aceytyltransferase, PolN, followed by PolP-mediated phosphorylation resulting in 298. Subsequently, an NADPH-dependent PolM sequentially reduces the acyl-phosphate to an aldehyde moiety in 299, and to an alcohol moiety in 300.350 The subsequent deacetylation of 300 is also mediated by the aforementioned N-aceytyltransferase, PolN, resulting in α-amino-δ-hydroxyvaleric acid (AHV, 301). Transcarbamoylation-mediated PolO affords α-amino-δ-carbamoylhydroxyvaleric acid (ACV, 302). In the last two steps of CPOAA 303 biosynthesis, two hydroxyl groups are introduced by an αKG-NHFe enzyme, PolL. The three moieties, namely the nucleoside skeleton 291, POIA 295 and CPOAA 303, are then assembled into polyoxin by PolG (Fig. 31). In this biosynthetic pathway, the mechanistic details for several novel reactions, including the PolL catalysis, still await further exploration.350
8 Conclusions
NHFe enzymes catalyze reactions as diverse as haem-containing enzymes,1, 12, 25, 41, 71, 351–353 including hydroxylation, ring fragmentation, C-C bond cleavage, epimerization, desaturation and heterocycle formation via either C-N, C-O or C-S bond formation. Recently, some unique transformations were reported as additional examples of NHFe enzyme functional versatility, including oxidative dehydrogenation in epoxidation, chlorination, epimerization and endoperoxidation. αKG-NHFe enzymes are one of the main sub-groups of NHFe enzymes. The examples covered in this article are some recent examples; and additional examples of this enzyme family in fungal biosynthetic pathways can be found in a recent review by Abe et a.l354 and a recent edited book on αKG-NHFe enzymes.6
Given that more and more structural information is becoming available for these enzymes, and that many novel reactions are constantly being discovered, increased structure–function and structure–reactivity relationship information will further enrich our knowledge on this large class of enzymes. One of the interesting discoveries from their structural characterizations is that both the proximal- and distal-type αKG binding modes are equally distributed among the structures in PDB, which suggests that a switching of αKG binding conformation is most likely a common phenomenon in cases where the corresponding enzyme adopts the distal-type αKG binding conformation. It is also possible that nature specifically exploits the distal-type αKG binding conformation to catalyze novel chemical transformations, as we recently observed in FtmOx1 catalysis. Given the prevalence of distal-type αKG-NHFe enzymes, many more examples of this type are likely to be uncovered in the future.
Acknowledgments
Some of the work covered here is supported in part by grants from the National Institutes of Health (R01 GM093903) and the National Science Foundation (CHE-1309148) to P.L., and by a grant from the National Natural Science Foundation of China (81573341 and 31720103901) to X.L. X.L. is supported by a fellowship from Chinese Scholarship Council.
Biographies

Shu-Shan Gao
Shu-Shan Gao received his Ph.D. in Marine Pharmacology in 2011 from the Institute of Oceanology, Chinese Academy of Sciences, under the direction of Professor Bingui Wang. He spent three years performing first postdoctoral research with Professor Tom Simpson at the School of Chemistry, University of Bristol, UK, where he studied the biosynthesis of trans-AT polyketide biosynthesis (2011-2014). Then, he moved to the University of California, Los Angeles, USA, under the direction of Prof. Yi Tang as a postdoctoral researcher to study the biosynthesis of fungal polyketides. He is now a professor at the Institute of Microbiology, Chinese Academy of Sciences, since February 2018. His research focuses on the biosynthesis and biosynthetic engineering of fungal natural products.

Nathchar Naowarojna
Nathchar Naowarojna was born in Thailand and received a BA in Chemistry with Biochemistry concentration (2015) from Boston University, USA. Currently, she is a doctoral candidate in the Chemistry programme of the Chemistry Department, Boston University, under the supervision of Professor Pinghua Liu. Her research interests lie in the mechanistic studies of enzymes and biosynthesis redesign for the production of sulfur-containing natural products.

Ronghai Cheng
Ronghai Cheng obtained his B.S degree in Biology in 2016 from the Southern University of Science and Technology, China. He is currently studying for a Ph.D. degree in Chemistry from Boston University, supervised by Prof. Pinghua Liu. His current research interests are in bioinformatics and biochemical characterization of sulfur-containing natural products.

Xueting Liu
Xueting Liu received her Ph.D. in 2006 from China Pharmaceutical University under the direction of Professor Zhida Min, where she studied Natural Product Chemistry. After a year postdoctoral research with Professor Biao Yu at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, she moved to the University of Wisconsin, La Crosse, WI, USA, to work with Professor Aaron Monte on microbial natural products. She worked at the Institute of Microbiology, Chinese Academy of Sciences, as an associate professor in 2010–2016 and then was appointed as professor in 2016 at the East China University of Technology and Sciences, Shanghai. Her research interests mostly focus on natural products biosynthesis based on genome-mining and synthetic biology approaches.

Pinghua Liu
After receiving his BA in Chemistry from Nankai Univeristy, China, Pinghua Liu joined the Dalian Institute of Chemical Physics, Chna, and received his Master’s degree in Natural Gas Chemical Engineering. He obtained his Ph.D. in Bioorganic Chemistry from the University of Minnesota, USA, with Professor Hung-wen Liu on natural product biosynthetic studies. In his last year of graduate studies, after Professor Hung-wen Liu moved to the University of Texas at Austin, he stayed at Minnesota for an additional year working under the guidance of Professor Hung-wen Liu and Professors John Lipscomb and Lawrence Que to continue his metallo-enzyme mechanistic studies using biophysical approaches. He joined MIT as a postdoctoral scientist with Professor JoAnne Stubbe studying transcriptional regulations in the production of biodegradable polymers. Currently, he is an associate professor at the Department of Chemistry of Boston University, USA. His research interests are developing new tools for natural product biosynthetic studies, mechanistic enzymology with a special emphasis on novel transformations catalyzed by metallo-enzymes, and the application of natural products as tools in studying biological processes (e.g. ageing and ageing-associated diseases).
Footnotes
9 Conflicts of Interest
There are no conflicts to declare.
References
- 1.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 2.Simmons JM, Müller TA, Hausinger RP. Dalton Trans. 2008:5132–5142. doi: 10.1039/b803512a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Purpero V, Moran GR. J Biol Inorg Chem. 2007;12:587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]
- 4.Loenarz C, Schofield CJ. Nat Chem Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 5.Loenarz C, Schofield CJ. Trends Biochem Sci. 2011;36:7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Schofield CJ, Hausinger RP, editors. 2-Oxoglutarate-dependent oxygenases. Royal Society of Chemistry; 2015. [Google Scholar]
- 7.Huang X, Groves JT. J Biol Inorg Chem. 2017;22:185–207. doi: 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kal S, Que L. J Biol Inorg Chem. 2017;22:339–365. doi: 10.1007/s00775-016-1431-2. [DOI] [PubMed] [Google Scholar]
- 9.Hangasky JA, Taabazuing CY, Valliere MA, Knapp MJ. Metallomics. 2013;5:287–301. doi: 10.1039/c3mt20153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lundberg M, Borowski T. Coord Chem Rev. 2013;257:277–289. [Google Scholar]
- 11.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Curr Opin Struct Biol. 2010;20:659–672. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Krebs C, Galonic Fujimori D, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koehntop KD, Emerson JP, Que L. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 14.Wu LF, Meng S, Tang GL. Biochim Biophys Acta, Proteins Proteomics. 2016;1864:453–470. doi: 10.1016/j.bbapap.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Araújo WL, Martins AO, Fernie AR, Tohge T. Front Plant Sci. 2014;5:552. doi: 10.3389/fpls.2014.00552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu B-I. Front Plant Sci. 2014;5:549. doi: 10.3389/fpls.2014.00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai Y, Ono E, Mizutani M. Plant J. 2014;78:328–343. doi: 10.1111/tpj.12479. [DOI] [PubMed] [Google Scholar]
- 18.Cochrane RV, Vederas JC. Acc Chem Res. 2014;47:3148–3161. doi: 10.1021/ar500242c. [DOI] [PubMed] [Google Scholar]
- 19.Smith DR, Grüschow S, Goss RJ. Curr Opin Chem Biol. 2013;17:276–283. doi: 10.1016/j.cbpa.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Agarwal V, Miles ZD, Winter JM, Eustaquio AS, El Gamal AA, Moore BS. Chem Rev. 2017;117:5619–5674. doi: 10.1021/acs.chemrev.6b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagel JM, Facchini PJ. Front Physiol. 2010;1:14. doi: 10.3389/fphys.2010.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou WM, Kutchan TM. Plant J. 1998;15:289–300. doi: 10.1046/j.1365-313x.1998.00220.x. [DOI] [PubMed] [Google Scholar]
- 23.Hedden P. Physiol Plant. 1997;101:709–719. [Google Scholar]
- 24.Prescott AG, Lloyd MD. Nat Prod Rep. 2000;17:367–383. doi: 10.1039/a902197c. [DOI] [PubMed] [Google Scholar]
- 25.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 26.Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 27.Baldwin J, Abraham E. Nat Prod Rep. 1988;5:129–145. doi: 10.1039/np9880500129. [DOI] [PubMed] [Google Scholar]
- 28.Baldwin J. J Heterocycl Chem. 1990;27:71–78. [Google Scholar]
- 29.Hamed RB, Gomez-Castellanos JR, Henry L, Ducho C, McDonough MA, Schofield CJ. Nat Prod Rep. 2013;30:21–107. doi: 10.1039/c2np20065a. [DOI] [PubMed] [Google Scholar]
- 30.Baggaley KH, Brown AG, Schofield CJ. Nat Prod Rep. 1997;14:309–333. doi: 10.1039/np9971400309. [DOI] [PubMed] [Google Scholar]
- 31.Fukuda H, Ogawa T, Tanase S. Adv Microb Physiol. 1993;35:275–306. doi: 10.1016/s0065-2911(08)60101-0. [DOI] [PubMed] [Google Scholar]
- 32.John P. Physiol Plant. 1997;100:583–592. [Google Scholar]
- 33.Rebouche CJ. Am J Clin Nutr. 1991;54:1147S–1152S. doi: 10.1093/ajcn/54.6.1147s. [DOI] [PubMed] [Google Scholar]
- 34.Kivirikko KI, Pihlajaniemi T. Adv Enzymol Relat Areas Mol Biol. 2009;72:325–398. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 35.Verhoeven N, Wanders R, Saudubray J-M, Jakobs C. J Inherit Metab Dis. 1998;21:697–728. doi: 10.1023/a:1005476631419. [DOI] [PubMed] [Google Scholar]
- 36.Hocart CH, Halpern B, Hick LA, Wong CO, Hammond JW, Wilcken B. J Chromatogr B: Biomed Sci Appl. 1983;275:237–243. doi: 10.1016/s0378-4347(00)84371-6. [DOI] [PubMed] [Google Scholar]
- 37.Niederwieser A, Wadman S, Danks D. Clin Chim Acta. 1978;90:195–200. doi: 10.1016/0009-8981(78)90522-3. [DOI] [PubMed] [Google Scholar]
- 38.Kivirikko KI. Ann Med. 1993;25:113–126. doi: 10.3109/07853899309164153. [DOI] [PubMed] [Google Scholar]
- 39.Eeva-Riitta K-S, Juha R, Miettinen TA, Kivirikko K. Eur J Clin Invest. 1979;9:89–95. doi: 10.1111/j.1365-2362.1979.tb01672.x. [DOI] [PubMed] [Google Scholar]
- 40.Hanauske-Abel H, Günzler V. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 41.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 42.Proshlyakov DA, Hausinger RP. Iron-containing enzymes: versatile catalysts of hydroxylation reactions in nature. ch. 3. Royal Society of Chemistry; Cambridge, UK: 2011. pp. 67–87. [Google Scholar]
- 43.Bollinger JM, Price JC, Hoffart LM, Barr EW, Krebs C. Eur J Inorg Chem. 2005;2005:4245–4254. [Google Scholar]
- 44.Grzyska PK, Appelman EH, Hausinger RP, Proshlyakov DA. Proc Natl Acad Sci U S A. 2010;107:3982–3987. doi: 10.1073/pnas.0911565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM. J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 46.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 47.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 48.Uria-Nickelsen MR, Leadbetter ER, Godchaux W. Arch Microbiol. 1994;161:434–438. doi: 10.1007/BF00288955. [DOI] [PubMed] [Google Scholar]
- 49.van der Ploeg JR, Eichhorn E, Leisinger T. Arch Microbiol. 2001;176:1–8. doi: 10.1007/s002030100298. [DOI] [PubMed] [Google Scholar]
- 50.Eichhorn E, van der Ploeg JR, Kertesz MA, Leisinger T. J Biol Chem. 1997;272:23031–23036. doi: 10.1074/jbc.272.37.23031. [DOI] [PubMed] [Google Scholar]
- 51.Price JC, Barr EW, Tirupati B, Bollinger JM, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 52.Krebs C, Price JC, Baldwin J, Saleh L, Green MT, Bollinger JM. Inorg Chem. 2005;44:742–757. doi: 10.1021/ic048523l. [DOI] [PubMed] [Google Scholar]
- 53.Lim MH, Rohde J-U, Stubna A, Bukowski MR, Costas M, Ho RY, Münck E, Nam W, Que L. Proc Natl Acad Sci U S A. 2003;100:3665–3670. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 55.Grapperhaus CA, Mienert B, Bill E, Weyhermüller T, Wieghardt K. Inorg Chem. 2000;39:5306–5317. doi: 10.1021/ic0005238. [DOI] [PubMed] [Google Scholar]
- 56.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Krebs C. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 57.Bollinger JM, Krebs C. J Inorg Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 58.Galonić DP, Barr EW, Walsh CT, Bollinger JM, Krebs C. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 59.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM. Proc Natl Acad Sci U S A. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang W-C, Guo Y, Wang C, Butch SE, Rosenzweig AC, Boal AK, Krebs C, Bollinger JM. Science. 2014;343:1140–1144. doi: 10.1126/science.1248000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang W-C, Li J, Lee JL, Cronican AA, Guo Y. J Am Chem Soc. 2016;138:10390–10393. doi: 10.1021/jacs.6b05400. [DOI] [PubMed] [Google Scholar]
- 63.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Krebs C. Proc Natl Acad Sci U S A. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clifton IJ, Doan LX, Sleeman MC, Topf M, Suzuki H, Wilmouth RC, Schofield CJ. J Biol Chem. 2003;278:20843–20850. doi: 10.1074/jbc.M213054200. [DOI] [PubMed] [Google Scholar]
- 65.Bräuer A, Beck P, Hintermann L, Groll M. Angew Chem Int Ed. 2016;55:422–426. doi: 10.1002/anie.201507835. [DOI] [PubMed] [Google Scholar]
- 66.Valegård K, van Scheltinga ACT, Lloyd MD, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee H-J, Baldwin JE, Schofield CJ. Nature. 1998;394:805–809. doi: 10.1038/29575. [DOI] [PubMed] [Google Scholar]
- 67.Weichold V, Milbredt D, van Pée KH. Angew Chem Int Ed. 2016;55:6374–6389. doi: 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- 68.Yan W, Song H, Song F, Guo Y, Wu C-H, Sae Her A, Pu Y, Wang S, Naowarojna N, Weitz A, Hendrich MP, Costello CE, Zhang L, Liu P, Jessie Zhang Y. Nature. 2015;527:539. doi: 10.1038/nature15519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Matsuda Y, Awakawa T, Wakimoto T, Abe I. J Am Chem Soc. 2013;135:10962–10965. doi: 10.1021/ja405518u. [DOI] [PubMed] [Google Scholar]
- 70.Aik WS, McDonough MA, Thalhammer A, Chowdhury R, Schofield CJ. Curr Opin Struct Biol. 2012;22:691–700. doi: 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 71.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. J Inorg Biochem. 2006;100:644–669. doi: 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 72.Roach PL, Clifton IJ, Fulop V, Harlos K. Nature. 1995;375:700. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 73.Roach PL, Clifton IJ, Hensgens CM, Shibata N. Nature. 1997;387:827. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- 74.Clifton IJ, Ge W, Adlington RM, Baldwin JE, Rutledge PJ. Arch Biochem Biophys. 2011;516:103–107. doi: 10.1016/j.abb.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 75.Hegg EL, Que L. FEBS J. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 76.Blasiak LC, Drennan CL. Acc Chem Res. 2008;42:147–155. doi: 10.1021/ar800088r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Brien JR, Schuller DJ, Yang VS, Dillard BD, Lanzilotta WN. Biochemistry. 2003;42:5547–5554. doi: 10.1021/bi0341096. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Z, Ren J-S, Harlos K, McKinnon CH, Clifton IJ, Schofield CJ. FEBS Lett. 2002;517:7–12. doi: 10.1016/s0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- 79.Martinez S, Fellner M, Herr CQ, Ritchie A, Hu J, Hausinger RP. J Am Chem Soc. 2017;139:11980–11988. doi: 10.1021/jacs.7b06186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu B, Edstrom WC, Benach J, Hamuro Y, Weber PC, Gibney BR, Hunt JF. Nature. 2006;439:879–884. doi: 10.1038/nature04561. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Z, Smart TJ, Choi H, Hardy F, Lohans CT, Abboud MI, Richardson MS, Paton RS, McDonough MA, Schofield CJ. Proc Natl Acad Sci U S A. 2017;114:4667–4672. doi: 10.1073/pnas.1617760114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao LS, Chang WC, Xiao YL, Liu HW, Liu PH. Annu Rev Biochem. 2013;82:497–530. doi: 10.1146/annurev-biochem-052010-100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horsman GP, Zechel DL. Chem Rev. 2016;117:5704–5783. doi: 10.1021/acs.chemrev.6b00536. [DOI] [PubMed] [Google Scholar]
- 84.Yang C-G, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sundheim O, Vågbø CB, Bjørås M, Sousa MM, Talstad V, Aas PA, Drabløs F, Krokan HE, Tainer JA, Slupphaug G. EMBO J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu B, Hunt JF. Proc Natl Acad Sci U S A. 2009;106:14315–14320. doi: 10.1073/pnas.0812938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pastore C, Topalidou I, Forouhar F, Yan AC, Levy M, Hunt JF. J Biol Chem. 2012;287:2130–2143. doi: 10.1074/jbc.M111.286187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Welford RW, Clifton IJ, Turnbull JJ, Wilson SC, Schofield CJ. Org Biomol Chem. 2005;3:3117–3126. doi: 10.1039/b507153d. [DOI] [PubMed] [Google Scholar]
- 89.Strieker M, Kopp F, Mahlert C, Essen L-O, Marahiel MA. ACS Chem Biol. 2007;2:187–196. doi: 10.1021/cb700012y. [DOI] [PubMed] [Google Scholar]
- 90.Müller I, Kahnert A, Pape T, Sheldrick GM, Meyer-Klaucke W, Dierks T, Kertesz M, Usón I. Biochemistry. 2004;43:3075–3088. doi: 10.1021/bi035752v. [DOI] [PubMed] [Google Scholar]
- 91.Nakashima Y, Mori T, Nakamura H, Awakawa T, Hoshino S, Senda M, Senda T, Abe I. Nat Commun. 2018;9 doi: 10.1038/s41467-017-02371-w. Article number: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leung IK, Krojer TJ, Kochan GT, Henry L, von Delft F, Claridge TD, Oppermann U, McDonough MA, Schofield CJ. Chem Biol. 2010;17:1316–1324. doi: 10.1016/j.chembiol.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Z, Ren J, Stammers DK, Baldwin JE, Harlos K, Schofield CJ. Nat Struct Mol Biol. 2000;7:127–133. doi: 10.1038/72398. [DOI] [PubMed] [Google Scholar]
- 94.Yang Y, Hu L, Wang P, Hou H, Lin Y, Liu Y, Li Z, Gong R, Feng X, Zhou L. Cell Res. 2010;20:886–898. doi: 10.1038/cr.2010.86. [DOI] [PubMed] [Google Scholar]
- 95.Khare D, Wang B, Gu L, Razelun J, Sherman DH, Gerwick WH, Håkansson K, Smith JL. Proc Natl Acad Sci U S A. 2010;107:14099–14104. doi: 10.1073/pnas.1006738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wong C, Fujimori DG, Walsh CT, Drennan CL. J Am Chem Soc. 2009;131:4872. doi: 10.1021/ja8097355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakano S, Yasukawa K, Kawahara N, Ishitsubo E, Tokiwa H, Asano Y. PDB accession number: 4YJD. 2015 doi: 10.2210/pdb4yjd/pdb. [DOI] [Google Scholar]
- 98.Höppner A, Widderich N, Lenders M, Bremer E, Smits SH. J Biol Chem. 2014;289:29570–29583. doi: 10.1074/jbc.M114.576769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 100.Han Z, Liu P, Gu L, Zhang Y, Li H, Chen S, Chai J. Frontier Sci. 2007;1:52–61. [Google Scholar]
- 101.Fritze IM, Linden L, Freigang J, Auerbach G, Huber R, Steinbacher S. Plant Physiol. 2004;134:1388–1400. doi: 10.1104/pp.103.034082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, Von Delft F. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 103.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hong X, Zang J, White J, Wang C, Pan C-H, Zhao R, Murphy RC, Dai S, Henson P, Kappler JW. Proc Natl Acad Sci U S A. 2010;107:14568–14572. doi: 10.1073/pnas.1008832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X. Nat Struct Mol Biol. 2010;17:38–43. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McDonough MA, Kavanagh KL, Butler D, Searls T, Oppermann U, Schofield CJ. J Biol Chem. 2005;280:41101–41110. doi: 10.1074/jbc.M507528200. [DOI] [PubMed] [Google Scholar]
- 107.Koketsu K, Shomura Y, Moriwaki K, Hayashi M, Mitsuhashi S, Hara R, Kino K, Higuchi Y. ACS Syn Biol. 2015;4:383–392. doi: 10.1021/sb500247a. [DOI] [PubMed] [Google Scholar]
- 108.Clifton IJ, Hsueh LC, Baldwin JE, Harlos K, Schofield CJ. FEBS J. 2001;268:6625–6636. doi: 10.1046/j.0014-2956.2001.02617.x. [DOI] [PubMed] [Google Scholar]
- 109.Rosen MD, Venkatesan H, Peltier HM, Bembenek SD, Kanelakis KC, Zhao LX, Leonard BE, Hocutt FM, Wu X, Palomino HL. ACS Med Chem Lett. 2010;1:526. doi: 10.1021/ml100198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.You Z, Omura S, Ikeda H, Cane DE, Jogl G. J Biol Chem. 2007;282:36552–36560. doi: 10.1074/jbc.M706358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chang Y, Wu J, Tong X-J, Zhou J-Q, Ding J. Biochem J. 2011;433:295–302. doi: 10.1042/BJ20101418. [DOI] [PubMed] [Google Scholar]
- 112.Qin H-M, Miyakawa T, Jia MZ, Nakamura A, Ohtsuka J, Xue Y-L, Kawashima T, Kasahara T, Hibi M, Ogawa J, Tanokura M. PLoS One. 2013;8:e63996. doi: 10.1371/journal.pone.0063996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 114.Kato N, Suzuki H, Takagi H, Uramoto M, Takahashi S, Osada H. ChemBioChem. 2011;12:711–714. doi: 10.1002/cbic.201000562. [DOI] [PubMed] [Google Scholar]
- 115.Sengoku T, Yokoyama S. Genes Dev. 2011;25:2266–2277. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mitchell AJ, Dunham NP, Martinie RJ, Bergman JA, Pollock CJ, Hu K, Allen BD, Chang W-C, Silakov A, Bollinger JM, Krebs C, Boal AK. J Am Chem Soc. 2017;139:13830–13836. doi: 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, Boal AK. Nat Chem Biol. 2016;12:636–640. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dong C, Flecks S, Unversucht S, Haupt C, van Pée K-H, Naismith JH. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neumann CS, Fujimori DG, Walsh CT. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 120.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 122.Guenzi E, Galli G, Grgurina I, Gross DC, Grandi G. J Biol Chem. 1998;273:32857–32863. doi: 10.1074/jbc.273.49.32857. [DOI] [PubMed] [Google Scholar]
- 123.Matthews ML, Chang W-C, Layne AP, Miles LA, Krebs C, Bollinger JM., Jr Nat Chem Biol. 2014;10:209–215. doi: 10.1038/nchembio.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Galonić DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 125.Ueki M, Galonić DP, Vaillancourt FH, Garneau-Tsodikova S, Yeh E, Vosburg DA, Schroeder FC, Osada H, Walsh CT. Chem Biol. 2006;13:1183–1191. doi: 10.1016/j.chembiol.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 126.Ramaswamy AV, Sorrels CM, Gerwick WH. J Nat Prod. 2007;70:1977–1986. doi: 10.1021/np0704250. [DOI] [PubMed] [Google Scholar]
- 127.Jiang W, Heemstra JR, Forseth RR, Neumann CS, Manaviazar S, Schroeder FC, Hale KJ, Walsh CT. Biochemistry. 2011;50:6063–6072. doi: 10.1021/bi200656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pratter SM, Ivkovic J, Birner-Gruenberger R, Breinbauer R, Zangger K, Straganz GD. ChemBioChem. 2014;15:567–574. doi: 10.1002/cbic.201300345. [DOI] [PubMed] [Google Scholar]
- 129.Neumann CS, Walsh CT. J Am Chem Soc. 2008;130:14022–14023. doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Proc Natl Acad Sci U S A. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 132.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 133.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jones AC, Monroe EA, Eisman EB, Gerwick L, Sherman DH, Gerwick WH. Nat Prod Rep. 2010;27:1048–1065. doi: 10.1039/c000535e. [DOI] [PubMed] [Google Scholar]
- 135.Hillwig ML, Liu X. Nat Chem Biol. 2014;10:921–923. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 136.Zhu Q, Liu X. Beilstein J Org Chem. 2017;13:1168–1173. doi: 10.3762/bjoc.13.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhu Q, Hillwig ML, Doi Y, Liu X. ChemBioChem. 2016;17:466–470. doi: 10.1002/cbic.201500674. [DOI] [PubMed] [Google Scholar]
- 138.Hillwig ML, Zhu Q, Ittiamornkul K, Liu X. Angew Chem Int Ed. 2016;55:5780–5784. doi: 10.1002/anie.201601447. [DOI] [PubMed] [Google Scholar]
- 139.Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Biorg Med Chem. 2009;17:2175–2180. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.De Lucca A, Jacks T, Takemoto J, Vinyard B, Peter J, Navarro E, Walsh T. Antimicrob Agents Chemother. 1999;43:371–373. doi: 10.1128/aac.43.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mitchell AJ, Dunham NP, Bergman JA, Wang B, Zhu Q, Chang W-c, Liu X, Boal AK. Biochemistry. 2017;56:441–444. doi: 10.1021/acs.biochem.6b01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Reddy YV, Al Temimi AHK, White PB, Mecinović J. Org Lett. 2017;19:400–403. doi: 10.1021/acs.orglett.6b03608. [DOI] [PubMed] [Google Scholar]
- 143.Al Temimi AHK, Pieters BJGE, Reddy YV, White PB, Mecinovic J. Chem Commun. 2016;52:12849–12852. doi: 10.1039/c6cc07845a. [DOI] [PubMed] [Google Scholar]
- 144.Henry L, Leung IKH, Claridge TDW, Schofield CJ. Bioorg Med Chem Lett. 2012;22:4975–4978. doi: 10.1016/j.bmcl.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 145.Kamps JJAG, Khan A, Choi H, Lesniak RK, Brem J, Rydzik AM, McDonough MA, Schofield CJ, Claridge TDW, Mecinović J. Chem Eur J. 2016;22:1270–1276. doi: 10.1002/chem.201503761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liepinsh E, Makrecka-Kuka M, Kuka J, Vilskersts R, Makarova E, Cirule H, Loza E, Lola D, Grinberga S, Pugovics O, Kalvins I, Dambrova M. Br J Pharmacol. 2015;172:1319–1332. doi: 10.1111/bph.13004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tars K, Leitans J, Kazaks A, Zelencova D, Liepinsh E, Kuka J, Makrecka M, Lola D, Andrianovs V, Gustina D, Grinberga S, Liepinsh E, Kalvinsh I, Dambrova M, Loza E, Pugovics O. J Med Chem. 2014;57:2213–2236. doi: 10.1021/jm401603e. [DOI] [PubMed] [Google Scholar]
- 148.Steiber A, Kerner J, Hoppel CL. Mol Aspects Med. 2004;25:455–473. doi: 10.1016/j.mam.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 149.Vaz FM, Wanders RJA. Biochem J. 2002;361:417–429. doi: 10.1042/0264-6021:3610417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Strijbis K, Vaz FM, Distel B. IUBMB life. 2010;62:357–362. doi: 10.1002/iub.323. [DOI] [PubMed] [Google Scholar]
- 151.Hulse JD, Ellis SR, Henderson LM. J Biol Chem. 1978;253:1654–1659. [PubMed] [Google Scholar]
- 152.Swiegers JH, Vaz FM, Pretorius IS, Wanders RJA, Bauer FF. FEMS Microbiol Lett. 2002;210:19–23. doi: 10.1111/j.1574-6968.2002.tb11154.x. [DOI] [PubMed] [Google Scholar]
- 153.RÜEtschi U, Nordin I, OdelhÖG B, JÖRnvall H, Lindstedt S. Eur J Biochem. 1993;213:1075–1080. doi: 10.1111/j.1432-1033.1993.tb17855.x. [DOI] [PubMed] [Google Scholar]
- 154.Vaz FM, van Gool S, Ofman R, Ijlst L, Wanders RJA. Biochem Biophys Res Commun. 1998;250:506–510. doi: 10.1006/bbrc.1998.9343. [DOI] [PubMed] [Google Scholar]
- 155.Vanecko JA, Wan H, West FG. Tetrahedron. 2006;62:1043–1062. [Google Scholar]
- 156.Tars K, Rumnieks J, Zeltins A, Kazaks A, Kotelovica S, Leonciks A, Sharipo J, Viksna A, Kuka J, Liepinsh E, Dambrova M. Biochem Biophys Res Commun. 2010;398:634–639. doi: 10.1016/j.bbrc.2010.06.121. [DOI] [PubMed] [Google Scholar]
- 157.Eckert C, Xu W, Xiong W, Lynch S, Ungerer J, Tao L, Gill R, Maness P-C, Yu J. Biotechnol Biofuels. 2014;7:33. doi: 10.1186/1754-6834-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Worrell E, Phylipsen D, Einstein D, Martin N. Energy use and energy intensity of the US chemical industry. Lawrence Berkeley National Laboratory; 2000. [Google Scholar]
- 159.Bleecker AB, Kende H. Annu Rev Cell Dev Biol. 2000;16:1–18. doi: 10.1146/annurev.cellbio.16.1.1. [DOI] [PubMed] [Google Scholar]
- 160.Hibi M, Kawashima T, Kodera T, Smirnov SV, Sokolov PM, Sugiyama M, Shimizu S, Yokozeki K, Ogawa J. Appl Environ Microbiol. 2011;77:6926–6930. doi: 10.1128/AEM.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kakizaki T, Kitashiba H, Zou Z, Li F, Fukino N, Ohara T, Nishio T, Ishida M. Plant Physiol. 2017;173:1583–1593. doi: 10.1104/pp.16.01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hibi M, Kawashima T, Kasahara T, Sokolov PM, Smirnov SV, Kodera T, Sugiyama M, Shimizu S, Yokozeki K, Ogawa J. Lett Appl Microbiol. 2012;55:414–419. doi: 10.1111/j.1472-765X.2012.03308.x. [DOI] [PubMed] [Google Scholar]
- 163.Hottiger T, Boller T. Arch Microbiol. 1991;157:18–22. [Google Scholar]
- 164.Fukuda H, Fujii T, Ogawa T. Agric Biol Chem. 1986;50:977–981. [Google Scholar]
- 165.Chou T, Yang S. Arch Biochem Biophys. 1973;157:73–82. doi: 10.1016/0003-9861(73)90391-3. [DOI] [PubMed] [Google Scholar]
- 166.Weingart H, Völksch B, Ullrich MS. Phytopathology. 1999;89:360–365. doi: 10.1094/PHYTO.1999.89.5.360. [DOI] [PubMed] [Google Scholar]
- 167.Fukuda H, Kitajima H, Fujii T, Tazaki M, Ogawa T. FEMS Microbiol Lett. 1989;59:1–5. doi: 10.1111/j.1574-6968.1989.tb03428.x. [DOI] [PubMed] [Google Scholar]
- 168.Martinez S, Hausinger RP. Biochemistry. 2016;55:5989–5999. doi: 10.1021/acs.biochem.6b00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fukuda H, Ogawa T, Tazaki M, Nagahama K, Fujiil T, Tanase S, Morino Y. Biochem Biophys Res Commun. 1992;188:483–489. doi: 10.1016/0006-291x(92)91081-z. [DOI] [PubMed] [Google Scholar]
- 170.Xiong W, Morgan JA, Ungerer J, Wang B, Maness P-C, Yu J. Nat Plants. 2015;1:15053. [Google Scholar]
- 171.Lynch S, Eckert C, Yu J, Gill R, Maness P-C. Biotechnol Biofuels. 2016;9:3. doi: 10.1186/s13068-015-0413-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pirkov I, Albers E, Norbeck J, Larsson C. Metab Eng. 2008;10:276–280. doi: 10.1016/j.ymben.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 173.Guerrero F, Carbonell V, Cossu M, Correddu D, Jones PR. PLoS One. 2012;7:e50470. doi: 10.1371/journal.pone.0050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Haefelé C, Bonfils C, Sauvaire Y. Phytochemistry. 1997;44:563–566. doi: 10.1016/s0031-9422(96)00620-6. [DOI] [PubMed] [Google Scholar]
- 175.Broca C, Gross R, Petit P, Sauvaire Y, Manteghetti M, Tournier M, Masiello P, Gomis R, Ribes G. Am J Physiol Endocrinol Metab. 1999;277:E617–E623. doi: 10.1152/ajpendo.1999.277.4.E617. [DOI] [PubMed] [Google Scholar]
- 176.Handa T, Yamaguchi K, Sono Y, Yazawa K. Biosci, Biotechnol, Biochem. 2005;69:1186–1188. doi: 10.1271/bbb.69.1186. [DOI] [PubMed] [Google Scholar]
- 177.Smirnov SV, Kodera T, Samsonova NN, Kotlyarova VA, Rushkevich NY, Kivero AD, Sokolov PM, Hibi M, Ogawa J, Shimizu S. Appl Microbiol Biotechnol. 2010;88:719–726. doi: 10.1007/s00253-010-2772-3. [DOI] [PubMed] [Google Scholar]
- 178.Visentin M, Tava A, Iori R, Palmieri S. J Agric Food Chem. 1992;40:1687–1691. [Google Scholar]
- 179.Hibi M, Kasahara T, Kawashima T, Yajima H, Kozono S, Smirnov SV, Kodera T, Sugiyama M, Shimizu S, Yokozeki K, Ogawa J. Adv Synth Catal. 2015;357:767–774. [Google Scholar]
- 180.Vogel W, Gish GD, Alves F, Pawson T. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 181.Hautala T, Byers MG, Eddy RL, Shows TB, Kivirikko KI, Myllyla R. Genomics. 1992;13:62–69. doi: 10.1016/0888-7543(92)90202-4. [DOI] [PubMed] [Google Scholar]
- 182.Passoja K, Rautavuoma K, Ala-Kokko L, Kosonen T, Kivirikko KI. Proc Natl Acad Sci U S A. 1998;95:10482–10486. doi: 10.1073/pnas.95.18.10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mercer DK, Nicol PF, Kimbembe C, Robins SP. Biochem Biophys Res Commun. 2003;307:803–809. doi: 10.1016/s0006-291x(03)01262-2. [DOI] [PubMed] [Google Scholar]
- 184.Eyre D, Shao P, Ann Weis M, Steinmann B. Mol Genet Metab. 2002;76:211–216. doi: 10.1016/s1096-7192(02)00036-7. [DOI] [PubMed] [Google Scholar]
- 185.Hyland J, Ala-Kokko L, Royce P, Steinmann B, Kivirikko KI, Myllylä R. Nat Genet. 1992;2:228. doi: 10.1038/ng1192-228. [DOI] [PubMed] [Google Scholar]
- 186.Yeowell HN, Walker LC. Matrix Biol. 1999;18:179–187. doi: 10.1016/s0945-053x(99)00013-x. [DOI] [PubMed] [Google Scholar]
- 187.Ruotsalainen H, Vanhatupa S, Tampio M, Sipilä L, Valtavaara M, Myllylä R. Matrix Biol. 2001;20:137–146. doi: 10.1016/s0945-053x(01)00130-5. [DOI] [PubMed] [Google Scholar]
- 188.Heikkinen J, Risteli M, Wang C, Latvala J, Rossi M, Valtavaara M, Myllylä R. J Biol Chem. 2000;275:36158–36163. doi: 10.1074/jbc.M006203200. [DOI] [PubMed] [Google Scholar]
- 189.Wang C, Luosujärvi H, Heikkinen J, Risteli M, Uitto L, Myllylä R. Matrix Biol. 2002;21:559–566. doi: 10.1016/s0945-053x(02)00071-9. [DOI] [PubMed] [Google Scholar]
- 190.Klose RJ, Kallin EM, Zhang Y. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 191.Kwok J, O’Shea M, Hume DA, Lengeling A. Front Genet. 2017;8:32. doi: 10.3389/fgene.2017.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mantri M, Krojer T, Bagg EA, Webby CJ, Butler DS, Kochan G, Kavanagh KL, Oppermann U, McDonough MA, Schofield CJ. J Mol Biol. 2010;401:211–222. [PubMed] [Google Scholar]
- 193.Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR. Science. 2009;325:90–93. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 194.Feng T, Yamamoto A, Wilkins SE, Sokolova E, Yates LA, Münzel M, Singh P, Hopkinson RJ, Fischer R, Cockman ME. Mol Cell. 2014;53:645–654. doi: 10.1016/j.molcel.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Markolovic S, Leissing TM, Chowdhury R, Wilkins SE, Lu X, Schofield CJ. Curr Opin Struct Biol. 2016;41:62–72. doi: 10.1016/j.sbi.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 196.Anastasia L, Rota P, Anastasia M, Allevi P. Org Biomol Chem. 2013;11:5747–5771. doi: 10.1039/c3ob40945g. [DOI] [PubMed] [Google Scholar]
- 197.Baldwin JE, Field RA, Lawrence CC, Lee V, Robinson JK, Schofield CJ. Tetrahedron Lett. 1994;35:4649–4652. [Google Scholar]
- 198.Lawrence CC, Sobey WJ, Field RA, Baldwin JE, Schofield CJ. Biochem J. 1996;313:185–191. doi: 10.1042/bj3130185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Houwaart S, Youssar L, Hüttel W. ChemBioChem. 2014;15:2365–2369. doi: 10.1002/cbic.201402175. [DOI] [PubMed] [Google Scholar]
- 200.Mori H, Shibasaki T, Uozaki Y, Ochiai K, Ozaki A. Appl Environ Microbiol. 1996;62:1903–1907. doi: 10.1128/aem.62.6.1903-1907.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mori H, Shibasaki T, Yano K, Ozaki A. J Bacteriol. 1997;179:5677–5683. doi: 10.1128/jb.179.18.5677-5683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shibasaki T, Mori H, Chiba S, Ozaki A. Appl Environ Microbiol. 1999;65:4028–4031. doi: 10.1128/aem.65.9.4028-4031.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shibasaki T, Mori H, Ozaki A. Biosci, Biotechnol, Biochem. 2000;64:746–750. doi: 10.1271/bbb.64.746. [DOI] [PubMed] [Google Scholar]
- 204.Shibasaki T, Mori H, Ozaki A. Biotechnol Lett. 2000;22:1967–1973. [Google Scholar]
- 205.Hara R, Kino K. Biochem Biophys Res Commun. 2009;379:882–886. doi: 10.1016/j.bbrc.2008.12.158. [DOI] [PubMed] [Google Scholar]
- 206.Petersen L, Olewinski R, Salmon P, Connors N. Appl Microbiol Biotechnol. 2003;62:263–267. doi: 10.1007/s00253-003-1264-0. [DOI] [PubMed] [Google Scholar]
- 207.Hara R, Uchiumi N, Kino K. J Biotechnol. 2014;172:55–58. doi: 10.1016/j.jbiotec.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 208.Denning DW. Lancet. 2003;362:1142–1151. doi: 10.1016/S0140-6736(03)14472-8. [DOI] [PubMed] [Google Scholar]
- 209.Chen L, Yue Q, Li Y, Niu X, Xiang M, Wang W, Bills GF, Liu X, An Z. Appl Environ Microbiol. 2015;81:1550–1558. doi: 10.1128/AEM.03256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Balkovec JM, Hughes DL, Masurekar PS, Sable CA, Schwartz RE, Singh SB. Nat Prod Rep. 2014;31:15–34. doi: 10.1039/c3np70070d. [DOI] [PubMed] [Google Scholar]
- 211.Walsh CT, O’Brien RV, Khosla C. Angew Chem Int Ed. 2013;52:7098–7124. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Finking R, Marahiel MA. Annu Rev Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 213.Thomas MG, Chan YA, Ozanick SG. Antimicrob Agents Chemother. 2003;47:2823–2830. doi: 10.1128/AAC.47.9.2823-2830.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yin X, Zabriskie TM. ChemBioChem. 2004;5:1274–1277. doi: 10.1002/cbic.200400082. [DOI] [PubMed] [Google Scholar]
- 215.Helmetag V, Samel SA, Thomas MG, Marahiel MA, Essen L-O. FASEB J. 2009;276:3669–3682. doi: 10.1111/j.1742-4658.2009.07085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yin XH, McPhail KL, Kim KJ, Zabriskie TM. ChemBioChem. 2004;5:1278–1281. doi: 10.1002/cbic.200400187. [DOI] [PubMed] [Google Scholar]
- 217.Fei X, Yin X, Zhang L, Zabriskie TM. J Nat Prod. 2007;70:618–622. doi: 10.1021/np060605u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Haltli B, Tan Y, Magarvey NA, Wagenaar M, Yin X, Greenstein M, Hucul JA, Zabriskie TM. Chem Biol. 2005;12:1163–1168. doi: 10.1016/j.chembiol.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 219.Singh MP, Petersen PJ, Weiss WJ, Janso JE, Luckman SW, Lenoy EB, Bradford PA, Testa RT, Greenstein M. Antimicrob Agents Chemother. 2003;47:62–69. doi: 10.1128/AAC.47.1.62-69.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Atkinson DJ, Naysmith BJ, Furkert DP, Brimble MA. Beilstein J Org Chem. 2016;12:2325–2342. doi: 10.3762/bjoc.12.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Han L, Schwabacher AW, Moran GR, Silvaggi NR. Biochemistry. 2015;54:7029–7040. doi: 10.1021/acs.biochem.5b01016. [DOI] [PubMed] [Google Scholar]
- 222.Burroughs AM, Hoppe RW, Goebel NC, Sayyed BH, Voegtline TJ, Schwabacher AW, Zabriskie TM, Silvaggi NR. Biochemistry. 2013;52:4492–4506. doi: 10.1021/bi400397k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Khmelenina VN, Mustakhimov II, Reshetnikov AS, Kalyuzhnaya M, Trotsenko YA. Am J Agric Biol Sci. 2010;5:446–458. [Google Scholar]
- 224.Baltz RH, Miao V, Wrigley SK. Nat Prod Rep. 2005;22:717–741. doi: 10.1039/b416648p. [DOI] [PubMed] [Google Scholar]
- 225.Hojati Z, Milne C, Harvey B, Gordon L, Borg M, Flett F, Wilkinson B, Sidebottom PJ, Rudd BAM, Hayes MA, Smith CP, Micklefield J. Chem Biol. 2002;9:1175–1187. doi: 10.1016/s1074-5521(02)00252-1. [DOI] [PubMed] [Google Scholar]
- 226.Lippert K, Galinski EA. Appl Microbiol Biotechnol. 1992;37:61–65. [Google Scholar]
- 227.Graf R, Anzali S, Buenger J, Pfluecker F, Driller H. Clin Dermatol. 2008;26:326–333. doi: 10.1016/j.clindermatol.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 228.Bursy J, Kuhlmann AU, Pittelkow M, Hartmann H, Jebbar M, Pierik AJ, Bremer E. Appl Environ Microbiol. 2008;74:7286–7296. doi: 10.1128/AEM.00768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Manzanera M, García de Castro A, Tøndervik A, Rayner-Brandes M, Strøm AR, Tunnacliffe A. Appl Environ Microbiol. 2002;68:4328–4333. doi: 10.1128/AEM.68.9.4328-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Reuter K, Pittelkow M, Bursy J, Heine A, Craan T, Bremer E. PLoS One. 2010;5:e10647. doi: 10.1371/journal.pone.0010647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Strieker M, Nolan EM, Walsh CT, Marahiel MA. J Am Chem Soc. 2009;131:13523–13530. doi: 10.1021/ja9054417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hubbard BK, Thomas MG, Walsh CT. Chem Biol. 2000;7:931–942. doi: 10.1016/s1074-5521(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 233.Choroba OW, Williams DH, Spencer JB. J Am Chem Soc. 2000;122:5389–5390. [Google Scholar]
- 234.Di Giuro CML, Konstantinovics C, Rinner U, Nowikow C, Leitner E, Straganz GD. PLoS One. 2013;8:e68932. doi: 10.1371/journal.pone.0068932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lindblad B, Lindstedt G, Lindstedt S, Rundgren M. J Biol Chem. 1977;252:5073–5084. [PubMed] [Google Scholar]
- 236.Gunsior M, Ravel J, Challis GL, Townsend CA. Biochemistry. 2004;43:663–674. doi: 10.1021/bi035762w. [DOI] [PubMed] [Google Scholar]
- 237.Brownlee JM, Johnson-Winters K, Harrison DHT, Moran GR. Biochemistry. 2004;43:6370–6377. doi: 10.1021/bi049317s. [DOI] [PubMed] [Google Scholar]
- 238.Serre L, Sailland A, Sy D, Boudec P, Rolland A, Pebay-Peyroula E, Cohen-Addad C. Structure. 1999;7:977–988. doi: 10.1016/s0969-2126(99)80124-5. [DOI] [PubMed] [Google Scholar]
- 239.Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, Spencer JB, Solomon EI. Proc Natl Acad Sci U S A. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Tsunematsu Y, Ishikawa N, Wakana D, Goda Y, Noguchi H, Moriya H, Hotta K, Watanabe K. Nat Chem Biol. 2013;9:818–825. doi: 10.1038/nchembio.1366. [DOI] [PubMed] [Google Scholar]
- 241.Zhao Z, Zhang Y, Liu X, Zhang X, Liu S, Yu X, Ren Y, Zheng X, Zhou K, Jiang L, Guo X, Gai Y, Wu C, Zhai H, Wang H, Wan J. Dev Cell. 2013;27:113–122. doi: 10.1016/j.devcel.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 242.Byeon Y, Back K. J Pineal Res. 2015;58:343–351. doi: 10.1111/jpi.12220. [DOI] [PubMed] [Google Scholar]
- 243.Steffan N, Grundmann A, Afiyatullov S, Ruan H, Li S-M. Org Biomol Chem. 2009;7:4082–4087. doi: 10.1039/b908392h. [DOI] [PubMed] [Google Scholar]
- 244.Patterson D, Shreeve B, Roberts B, MacDonald S. Appl Environ Microbiol. 1981;42:916–917. doi: 10.1128/aem.42.5.916-917.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Fill TP, Asenha HBR, Marques AS, Ferreira AG, Rodrigues-Fo E. Nat Prod Res. 2013;27:967–974. doi: 10.1080/14786419.2012.701210. [DOI] [PubMed] [Google Scholar]
- 246.Cole R, Kirksey J, Moore J, Blankenship B, Diener U, Davis N. Appl Microbiol. 1972;24:248–250. doi: 10.1128/am.24.2.248-250.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ishikawa N, Tanaka H, Koyama F, Noguchi H, Wang CCC, Hotta K, Watanabe K. Angew Chem Int Ed. 2014;53:12880–12884. doi: 10.1002/anie.201407920. [DOI] [PubMed] [Google Scholar]
- 248.Daneshtalab M, Ahmed A. J Pharm Pharm Sci. 2011;15:52–72. doi: 10.18433/j3302n. [DOI] [PubMed] [Google Scholar]
- 249.Cushnie TT, Cushnie B, Lamb AJ. Int J Antimicrob Agents. 2014;44:377–386. doi: 10.1016/j.ijantimicag.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 250.Su H, Sheng X, Zhu W, Ma G, Liu Y. ACS Catal. 2017;7:5534–5543. [Google Scholar]
- 251.Liao HJ, Li JK, Huang JL, Davidson M, Kurnikov I, Lin TS, Lee JL, Kurnikova M, Guo YS, Chan NL, Chang WC. Angew Chem Int Ed. 2018;57:1831–1835. doi: 10.1002/anie.201710567. [DOI] [PubMed] [Google Scholar]
- 252.Meng S, Han W, Zhao J, Jian XH, Pan HX, Tang GL. Angew Chem Int Ed. 2018;130:727–731. doi: 10.1002/anie.201710529. [DOI] [PubMed] [Google Scholar]
- 253.Miyoshi T, Miyairi N, Aoki H, Kohsaka M, Sakai H-I, Imanaka H. J Antibiot. 1972;25:569–575. doi: 10.7164/antibiotics.25.569. [DOI] [PubMed] [Google Scholar]
- 254.Miyamura S, Ogasawara N, Otsuka H, Niwayama S, Tanaka H, Take T, Uchiyama T, Ochiai H, Abe K, Koizumi K. J Antibiot. 1972;25:610–612. doi: 10.7164/antibiotics.25.610. [DOI] [PubMed] [Google Scholar]
- 255.Kohn H, Widger W. Curr Drug Targets Infect Disord. 2005;5:273–295. doi: 10.2174/1568005054880136. [DOI] [PubMed] [Google Scholar]
- 256.Patteson JB, Cai W, Johnson RA, Santa Maria KC, Li B. Biochemistry. 2017;57:61–65. doi: 10.1021/acs.biochem.7b00943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ökesli A, Cooper LE, Fogle EJ, van der Donk WA. J Am Chem Soc. 2011;133:13753–13760. doi: 10.1021/ja205783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Widdick DA, Dodd HM, Barraille P, White J, Stein TH, Chater KF, Gasson MJ, Bibb MJ. Proc Natl Acad Sci U S A. 2003;100:4316–4321. doi: 10.1073/pnas.0230516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 260.Matsuno T. In: Carotenoids: Chemistry and Biology. Krinsky NI, Mathews-Roth MM, Taylor RF, editors. Springer US; 1989. pp. 59–74. [Google Scholar]
- 261.Misawa N, Satomi Y, Kondo K, Yokoyama A, Kajiwara S, Saito T, Ohtani T, Miki W. J Bacteriol. 1995;177:6575–6584. doi: 10.1128/jb.177.22.6575-6584.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Misawa N, Nakagawa M, Kobayashi K, Yamano S, Izawa Y, Nakamura K, Harashima K. J Bacteriol. 1990;172:6704–6712. doi: 10.1128/jb.172.12.6704-6712.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tian L, DellaPenna D. Plant Mol Biol. 2001;47:379–388. doi: 10.1023/a:1011623907959. [DOI] [PubMed] [Google Scholar]
- 264.Breitenbach J, Misawa N, Kajiwara S, Sandmann G. FEMS Microbiol Lett. 1996;140:241–246. doi: 10.1016/0378-1097(96)00187-5. [DOI] [PubMed] [Google Scholar]
- 265.Kajiwara S, Kakizono T, Saito T, Kondo K, Ohtani T, Nishio N, Nagai S, Misawa N. Plant Mol Biol. 1995;29:343–352. doi: 10.1007/BF00043657. [DOI] [PubMed] [Google Scholar]
- 266.Fraser PD, Miura Y, Misawa N. J Biol Chem. 1997;272:6128–6135. doi: 10.1074/jbc.272.10.6128. [DOI] [PubMed] [Google Scholar]
- 267.Martin D, Slomp G, Mizsak S, Duchamp D, Chidester C. Tetrahedron Lett. 1970;11:4901–4904. doi: 10.1016/s0040-4039(00)99739-9. [DOI] [PubMed] [Google Scholar]
- 268.Takeuchi S, Ogawa Y, Yonehara H. Tetrahedron Lett. 1969:2737–2740. doi: 10.1016/s0040-4039(01)88256-3. [DOI] [PubMed] [Google Scholar]
- 269.Duszenko M, Balla H, Mecke D. Biochim Biophys Acta, Gen Subj. 1982;714:344–350. doi: 10.1016/0304-4165(82)90343-9. [DOI] [PubMed] [Google Scholar]
- 270.Hartmann S, Neeff J, Heer U, Mecke D. FEBS Lett. 1978;93:339–342. doi: 10.1016/0014-5793(78)81135-1. [DOI] [PubMed] [Google Scholar]
- 271.Cane DE, Sohng J-K. Arch Biochem Biophys. 1989;270:50–61. doi: 10.1016/0003-9861(89)90006-4. [DOI] [PubMed] [Google Scholar]
- 272.Cane DE, Sohng J-K. Biochemistry. 1994;33:6524–6530. doi: 10.1021/bi00187a020. [DOI] [PubMed] [Google Scholar]
- 273.Seo M-J, Zhu D, Endo S, Ikeda H, Cane DE. Biochemistry. 2011;50:1739–1754. doi: 10.1021/bi1019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jiang J, Tetzlaff CN, Takamatsu S, Iwatsuki M, Komatsu M, Ikeda H, Cane DE. Biochemistry. 2009;48:6431–6440. doi: 10.1021/bi900766w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. Biochemistry. 2006;45:6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cane DE, Abell C, Harrison PH, Hubbard BR, Kane CT, Lattman R, Oliver JS, Weiner SW. Philos Trans R Soc, B. 1991;332:123–129. doi: 10.1098/rstb.1991.0040. [DOI] [PubMed] [Google Scholar]
- 277.You Z, Omura S, Ikeda H, Cane DE. J Am Chem Soc. 2006;128:6566–6567. doi: 10.1021/ja061469i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Daum M, Schnell H-J, Herrmann S, Günther A, Murillo R, Müller R, Bisel P, Müller M, Bechthold A. ChemBioChem. 2010;11:1383–1391. doi: 10.1002/cbic.201000117. [DOI] [PubMed] [Google Scholar]
- 279.Dürr C, Schnell H-J, Luzhetskyy A, Murillo R, Weber M, Welzel K, Vente A, Bechthold A. Chem Biol. 2006;13:365–377. doi: 10.1016/j.chembiol.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 280.Matsuda Y, Iwabuchi T, Fujimoto T, Awakawa T, Nakashima Y, Mori T, Zhang H, Hayashi F, Abe I. J Am Chem Soc. 2016;138:12671–12677. doi: 10.1021/jacs.6b08424. [DOI] [PubMed] [Google Scholar]
- 281.Toyota M, Wada T, Fukumoto K, Ihara M. J Am Chem Soc. 1998;120:4916–4925. [Google Scholar]
- 282.Shiono Y, Akiyama K, Hayashi H. Biosci, Biotechnol, Biochem. 2000;64:1519–1521. doi: 10.1271/bbb.64.1519. [DOI] [PubMed] [Google Scholar]
- 283.Hayashi H, Takiuchi K, Murao S, Arai M. Agric Biol Chem. 1989;53:461–469. [Google Scholar]
- 284.Furutani S, Ihara M, Kai K, Tanaka K, Sattelle DB, Hayashi H, Matsuda K. Neurotoxicology. 2017;60:240–244. doi: 10.1016/j.neuro.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 285.Lai C-Y, Lin H-C, Lo I-W, Hewage RT, Chen Y-C, Chen C-T, Lee C-F, Lin S, Tang M-C. Angew Chem Int Ed. 2017;56:9478–9482. doi: 10.1002/anie.201705501. [DOI] [PubMed] [Google Scholar]
- 286.Wada Si, Usami I, Umezawa Y, Inoue H, Ohba Si, Someno T, Kawada M, Ikeda D. Cancer Sci. 2010;101:743–750. doi: 10.1111/j.1349-7006.2009.01438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Williams K, Szwalbe AJ, Mulholland NP, Vincent JL, Bailey AM, Willis CL, Simpson TJ, Cox RJ. Angew Chem Int Ed. 2016;55:6784–6788. doi: 10.1002/anie.201511882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bai J, Yan D, Zhang T, Guo Y, Liu Y, Zou Y, Tang M, Liu B, Wu Q, Yu S. Angew Chem Int Ed. 2017;56:4782–4786. doi: 10.1002/anie.201701547. [DOI] [PubMed] [Google Scholar]
- 289.Ju KS, Gao JT, Doroghazi JR, Wang KKA, Thibodeaux CJ, Li S, Metzger E, Fudala J, Su J, Zhang JK, Lee J, Cioni JP, Evans BS, Hirota R, Labeda DP, van der Donk WA, Metcalf WW. Proc Natl Acad Sci U S A. 2015;112:12175–12180. doi: 10.1073/pnas.1500873112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Metcalf WW, vd Donk WA. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.White AK, Metcalf WW. Annu Rev Microbiol. 2007;61:379–400. doi: 10.1146/annurev.micro.61.080706.093357. [DOI] [PubMed] [Google Scholar]
- 292.Ju K-S, Doroghazi JR, Metcalf WW. J Ind Microbiol Biotechnol. 2014;41:345–356. doi: 10.1007/s10295-013-1375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hendlin D, Stapley E, Jackson M, Wallick H, Miller A, Wolf F, Miller T, Chaiet L, Kahan F, Foltz E. Science. 1969;166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 294.Keating GM. Drugs. 2013;73:1951–1966. doi: 10.1007/s40265-013-0143-y. [DOI] [PubMed] [Google Scholar]
- 295.Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Clin Infect Dis. 2008;46:1069–1077. doi: 10.1086/527442. [DOI] [PubMed] [Google Scholar]
- 296.Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure. 1996;4:1465–1474. doi: 10.1016/s0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- 297.Schwartz D, Berger S, Heinzelmann E, Muschko K, Welzel K, Wohlleben W. Appl Environ Microbiol. 2004;70:7093–7102. doi: 10.1128/AEM.70.12.7093-7102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Blodgett JA, Thomas PM, Li G, Velasquez JE, Van Der Donk WA, Kelleher NL, Metcalf WW. Nat Chem Biol. 2007;3:480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Seto H, Kuzuyama T. Nat Prod Rep. 1999;16:589–596. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- 300.Abell LM, Villafranca JJ. Biochemistry. 1991;30:6135–6141. doi: 10.1021/bi00239a008. [DOI] [PubMed] [Google Scholar]
- 301.Gill HS, Eisenberg D. Biochemistry. 2001;40:1903–1912. doi: 10.1021/bi002438h. [DOI] [PubMed] [Google Scholar]
- 302.Bayer vE, Gugel K, Hägele K, Hagenmaier H, Jessipow S, König W, Zähner H. Helv Chim Acta. 1972;55:224–239. doi: 10.1002/hlca.19720550126. [DOI] [PubMed] [Google Scholar]
- 303.Omura S, Hinotozawa K, Imamura N, Murata M. J Antibiot. 1984;37:939–940. doi: 10.7164/antibiotics.37.939. [DOI] [PubMed] [Google Scholar]
- 304.Hunt AH, Elzey TK. J Antibiot. 1988;41:802–802. doi: 10.7164/antibiotics.41.802. [DOI] [PubMed] [Google Scholar]
- 305.Whitteck JT, Ni W, Griffin BM, Eliot AC, Thomas PM, Kelleher NL, Metcalf WW, van der Donk WA. Angew Chem Int Ed. 2007;46:9089–9092. doi: 10.1002/anie.200703810. [DOI] [PubMed] [Google Scholar]
- 306.Conti P, Tamborini L, Pinto A, Blondel A, Minoprio P, Mozzarelli A, De Micheli C. Chem Rev. 2011;111:6919–6946. doi: 10.1021/cr2000702. [DOI] [PubMed] [Google Scholar]
- 307.Kuemin M, Van Der Donk WA. Chem Commun. 2010;46:7694–7696. doi: 10.1039/c0cc02958k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Masakuni Okuhara YK, Goto Toshio, Okamoto Masanori, Terano Hiroshi, Kohsaka Masanobu, Aoki Hatsuo, Imanaka Hiroshi. J Antibiot. 1980;33:13–17. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- 309.Kuroda YOM, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot. 1980;33:29–35. doi: 10.7164/antibiotics.33.29. [DOI] [PubMed] [Google Scholar]
- 310.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki HIH. J Antibiot. 1980;33:24–28. doi: 10.7164/antibiotics.33.24. [DOI] [PubMed] [Google Scholar]
- 311.Fernandes JF, Lell B, Agnandji ST, Obiang RM, Bassat Q, Kremsner PG, Mordmüller B, Grobusch MP. Future Microbiol. 2015;10:1375–1390. doi: 10.2217/FMB.15.60. [DOI] [PubMed] [Google Scholar]
- 312.Park BK, Hirota A, Sakai H. Agric Biol Chem. 1977;41:573–579. [Google Scholar]
- 313.Fredenhagen A, Angst C, Peter HH. J Antibiot. 1995;48:1043–1045. doi: 10.7164/antibiotics.48.1043. [DOI] [PubMed] [Google Scholar]
- 314.Evans BS, Zhao C, Gao J, Evans CM, Ju K-S, Doroghazi JR, Van Der Donk WA, Kelleher NL, Metcalf WW. ACS Chem Biol. 2013;8:908–913. doi: 10.1021/cb400102t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Ntai I, Phelan VV, Bachmann BO. Chem Commun. 2006:4518–4520. doi: 10.1039/b611768f. [DOI] [PubMed] [Google Scholar]
- 317.Ntai I, Manier ML, Hachey DL, Bachmann BO. Org Lett. 2005;7:2763–2765. doi: 10.1021/ol051091d. [DOI] [PubMed] [Google Scholar]
- 318.Yamato M, Koguchi T, Okachi R, Yamada K, Nakayama K, Kase H, Karasawa A, Shuto K. J Antibiot. 1986;39:44–52. doi: 10.7164/antibiotics.39.44. [DOI] [PubMed] [Google Scholar]
- 319.Bowman E, McQueney M, Barry RJ, Dunaway-Mariano D. J Am Chem Soc. 1988;110:5575–5576. [Google Scholar]
- 320.Seidel HM, Freeman S, Seto H, Knowles JR. Nature. 1988;335:457–458. doi: 10.1038/335457a0. [DOI] [PubMed] [Google Scholar]
- 321.Hidaka T, Mori M, Imai S, Hara O, Nagaoka K, Seto H. J Antibiot. 1989;42:491–494. doi: 10.7164/antibiotics.42.491. [DOI] [PubMed] [Google Scholar]
- 322.Kamigiri K, Hidaka T, Imai S, Murakami T, Seto H. J Antibiot. 1992;45:781–787. doi: 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]
- 323.Hidaka T, Hidaka M, Kuzuyama T, Seto H. Gene. 1995;158:149–150. doi: 10.1016/0378-1119(95)00101-b. [DOI] [PubMed] [Google Scholar]
- 324.Blodgett JA, Zhang JK, Metcalf WW. Antimicrob Agents Chemother. 2005;49:230–240. doi: 10.1128/AAC.49.1.230-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Werner WJ, Allen KD, Hu K, Helms GL, Chen BS, Wang SC. Biochemistry. 2011;50:8986–8988. doi: 10.1021/bi201220r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Allen KD, Wang SC. Biochim Biophys Acta. 2014;1844:2135–2144. doi: 10.1016/j.bbapap.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Landgraf BJ, McCarthy EL, Booker SJ. Annu Rev Biochem. 2016;85:485–514. doi: 10.1146/annurev-biochem-060713-035504. [DOI] [PubMed] [Google Scholar]
- 329.Circello BT, Eliot AC, Lee J-H, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Bougioukou DJ, Mukherjee S, van der Donk WA. Proc Natl Acad Sci U S A. 2013;110:10952–10957. doi: 10.1073/pnas.1303568110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Huang Z, Wang K-KA, Lee J, van der Donk WA. Chem Sci. 2015;6:1282–1287. doi: 10.1039/c4sc03095h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.McSorley FR, Wyatt PB, Martinez A, DeLong EF, Hove-Jensen B, Zechel DL. J Am Chem Soc. 2012;134:8364–8367. doi: 10.1021/ja302072f. [DOI] [PubMed] [Google Scholar]
- 333.Van Mooy BAS, Fredricks HF, Pedler BE, Dyhrman ST, Karl DM, Koblizek M, Lomas MW, Mincer TJ, Moore LR, Moutin T, Rappe MS, Webb EA. Nature. 2009;458:69–72. doi: 10.1038/nature07659. [DOI] [PubMed] [Google Scholar]
- 334.Campos ML, Kang J-H, Howe GA. J Chem Ecol. 2014;40:657–675. doi: 10.1007/s10886-014-0468-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Caarls L, Elberse J, Awwanah M, Ludwig NR, de Vries M, Zeilmaker T, Van Wees SCM, Schuurink RC, Van den Ackerveken G. Proc Natl Acad Sci U S A. 2017;114:6388–6393. doi: 10.1073/pnas.1701101114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Gibbons HS, Reynolds CM, Guan Z, Raetz CR. Biochemistry. 2008;47:2814–2825. doi: 10.1021/bi702457c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Chung HS, Raetz CR. Proc Natl Acad Sci U S A. 2011;108:510–515. doi: 10.1073/pnas.1016462108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Mihalik SJ, Morrell JC, Kim D, Sacksteder KA, Watkins PA, Gould SJ. Nat Genet. 1997;17:185–189. doi: 10.1038/ng1097-185. [DOI] [PubMed] [Google Scholar]
- 339.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 340.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. Science. 1997;276:250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 341.Gibbons HS, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2000;275:32940–32949. doi: 10.1074/jbc.M005779200. [DOI] [PubMed] [Google Scholar]
- 342.Ichikawa S, Matsuda A. Expert Opin Ther Pat. 2007;17:487–498. [Google Scholar]
- 343.Murakami R, Fujita Y, Kizuka M, Kagawa T, Muramatsu Y, Miyakoshi S, Takatsu T, Inukai M. J Antibiot. 2007;60:690–695. doi: 10.1038/ja.2007.88. [DOI] [PubMed] [Google Scholar]
- 344.Cai W, Goswami A, Yang Z, Liu X, Green KD, Barnard-Britson S, Baba S, Funabashi M, Nonaka K, Sunkara M, Morris AJ, Spork AP, Ducho C, Garneau-Tsodikova S, Thorson JS, Van Lanen SG. J Biol Chem. 2015;290:13710–13724. doi: 10.1074/jbc.M115.646414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Goswami A, Liu X, Cai W, Wyche TP, Bugni TS, Meurillon M, Peyrottes S, Perigaud C, Nonaka K, Rohr J, Van Lanen SG. FEBS Lett. 2017;591:468–478. doi: 10.1002/1873-3468.12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Yang Z, Chi X, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen JM, Elliott GI, Rohr J, Van Lanen SG. J Biol Chem. 2011;286:7885–7892. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hori MK, Eguchi J, Kakiki K, Misato T. J Antibiot. 1974;27:260–266. doi: 10.7164/antibiotics.27.260. [DOI] [PubMed] [Google Scholar]
- 348.Chen W, Huang T, He X, Meng Q, You D, Bai L, Li J, Wu M, Li R, Xie Z, Zhou H, Zhou X, Tan H, Deng Z. J Biol Chem. 2009;284:10627–10638. doi: 10.1074/jbc.M807534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lauer B, Russwurm R, Schwarz W, Kalmanczhelyi A, Bruntner C, Rosemeier A, Bormann C. Mol Gen Genet. 2001;264:662–673. doi: 10.1007/s004380000352. [DOI] [PubMed] [Google Scholar]
- 350.Qi J, Wan D, Ma H, Liu Y, Gong R, Qu X, Sun Y, Deng Z, Chen W. Cell Chem Biol. 2016;23:935–944. doi: 10.1016/j.chembiol.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 351.Buongiorno D, Straganz GD. Coord Chem Rev. 2013;257:541–563. doi: 10.1016/j.ccr.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Naowarojna N, Cheng R, Chen L, Quill M, Xu M, Zhao C, Liu P. Biochemistry. 2018 doi: 10.1021/acs.biochem.8b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Nakamura H, Matsuda Y, Abe I. Nat Prod Rep. 2018 doi: 10.1039/c7np00055c. [DOI] [PubMed] [Google Scholar]