

Abstract
SLC30 proteins belong to the cation diffusion facilitator (CDF) superfamily of metal transporters. SLC30A10 mediates manganese efflux, while other SLC30 members transport zinc. Metal specificity of CDFs may be conferred by amino acids that form a transmembrane metal binding site (Site A). Site A of zinc-transporting CDFs, such as SLC30A1/ZnT1, have a HXXXD motif, but manganese transporters, such as SLC30A10, harbor a NXXXD motif. This critical histidine-to-asparagine substitution, at residue 43, was proposed to underlie manganese transport specificity of SLC30A10. However, we recently discovered that asparagine-43 was dispensable for manganese efflux in HeLa cells; instead, glutamate-25, aspartate-40, asparagine-127, and aspartate-248 were required. In contrast, another group reported that asparagine-43 was required in a chicken cell line. The goal of this study was to resolve the divergent results about the requirement of the crucial asparagine-43 residue. For this, we compared the manganese efflux activity of four cell types that stably over-expressed SLC30A10WT, SLC30A10N43A or SLC30A10E25A: physiologically-relevant hepatic HepG2 and neuronal AF5 cells, HEK cells, and embryonic fibroblasts from Slc30a10−/− mice. In all cell types, manganese efflux activity of SLC30A10N43A was comparable to WT, while SLC30A10E25A lacked activity. Importantly, unlike SLC30A10, the histidine residue of the HXXXD motif of SLC30A1/ZnT1 was required for zinc transport. These results imply that the mechanisms of ion coordination within the transmembrane domain of SLC30A10 substantially differ from previously-studied CDFs, suggest that factors beyond Site A residues may confer metal specificity to CDFs, and improve understanding of the pathobiology of manganese toxicity due to mutations in SLC30A10.
Keywords: manganese, zinc, transporter, metal homeostasis, structure-function, cation diffusion facilitator, efflux
Table of Contents Entry
Mechanism by which the cation diffusion facilitator SLC30A10 transports manganese is fundamentally different from that of previously-studied proteins in this superfamily.
Introduction
Cation diffusion facilitators (CDFs) are a ubiquitous superfamily of metal transporters present in bacteria, archaea and eukaryotes 1–4. Proteins of the SLC30 family are the mammalian representatives of this superfamily 1, 2, 4. Humans express ten SLC30 proteins – SLC30A1-A10 1, 4. SLC30A1-A8 (also called ZnT1-8) transports zinc from the cytosol to the exterior of cells or into the lumen of intracellular organelles (i.e. mediate zinc efflux) 1. Classification of SLC30A9 as a transporter is likely incorrect; this protein functions as a nuclear receptor coactivator and is now called GAC63 1. In contrast, it is now well established that SLC30A10 functions as a cell surface-localized manganese efflux transporter that reduces cellular manganese levels and protects cells and organisms against manganese toxicity 5–9. Importantly, SLC30A10 lacks zinc efflux activity 5–10. (Note that we include the ZnT name whenever we refer to any zinc-transporting SLC30 protein so as to distinguish these proteins from SLC30A10, which lacks zinc transport activity). Understanding the mechanisms that confer manganese transport specificity to SLC30A10 should provide insights into the principles that guide metal selectivity in the CDF superfamily itself. Additionally, manganese is an essential metal, but elevated levels are toxic and induce an incurable parkinsonian syndrome 11. Loss-of-function mutations in SLC30A10 cause an inherited form of manganese toxicity in humans 12–18. Slc30a10 knockout mice also develop severe manganese toxicity 5, 7. Knowledge about the mechanisms by which SLC30A10 mediates manganese transport and regulates manganese homeostasis should improve understanding of the pathobiology of manganese toxicity and may also aid in the generation of new therapeutic strategies for the management of this devastating disease.
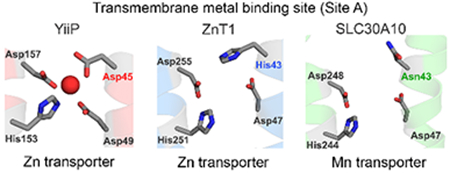
Thus far, the crystal structure of only one CDF, YiiP, a bacterial zinc/cadmium transporter 19, 20, has been solved 21, 22. Consequently, YiiP has become the prototypical CDF. YiiP has one transmembrane domain, with six membrane spanning helices, and a cytoplasmic C-terminal domain (refs.21, 22; Fig.1A). Within the transmembrane domain, side chain oxygen or nitrogen atoms of four residues, Asp-45 and Asp-49 from the second and His-153 and Asp-157 from the fifth transmembrane segments, directly coordinate the zinc ion that is transported (refs.21, 22; Fig.1A). Bioinformatic and experimental studies suggest that, in other cation diffusion facilitators, residues that correspond to site A of YiiP are crucial for metal coordination and that the identities of these amino acids may play an important role in determining metal specificity 1, 3, 19, 23–26. In most zinc transporting-CDFs, a HXXXD motif in the second transmembrane segment contributes to site A 3, 26. This HXXXD motif is present in zinc-transporting SLC30 proteins and in ZRC1 (Fig.1B), which is the yeast homolog of SLC30A10 and which, unlike the mammalian counterpart, is a zinc transporter 1. Similarly, in bacterial YiiP, a DXXXD motif from the second transmembrane segment contributes to site A 21, 22; mutation of this motif to HXXXD enhances zinc transport specificity of YiiP 19. In contrast to zinc transporters, in manganese-transporting CDFs, a NXXXD motif from the second transmembrane segment often contributes to site A 23, 26. The critical histidine-to-asparagine substitution is thought to play a major role in conferring manganese transport specificity because the side chain of asparagine has a higher propensity to coordinate with manganese than zinc 26, 27. Indeed, the asparagine residue of the NXXXD motif of the bacterial protein MntE is required for manganese transport 23. Importantly, the residues of SLC30A10 that form site A are Asn-43 and Asp-47 of the second and His-244 and Asp-248 of the fifth transmembrane segments (ref. 9; Fig.1A&B). Thus, SLC30A10 also carries a canonical NXXXD motif in its second transmembrane segment (Fig.1B). This led to the hypothesis that the presence of an asparagine residue at position 43 of SLC30A10, instead of histidine, conferred manganese transport specificity to SLC30A10 9, 28.
Figure 1. Comparison of the crystal structure of YiiP (PDB: 3H90) with the predicted structure of SLC30A10.
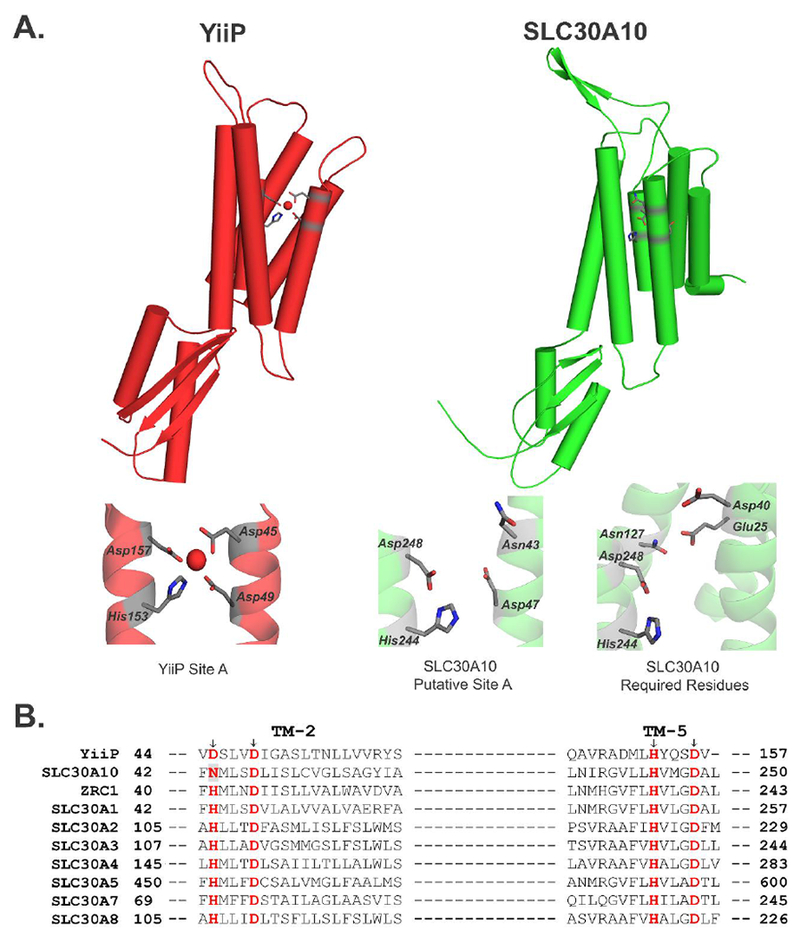
A. YiiP (Red) and SLC30A10 (Green) are viewed as a protein monomer from the membrane plane. The lower panel depicts close-up comparison of the amino acids making up Site A of YiiP, predicted Site A of SLC30A10, and those that were experimentally demonstrated to be required for the manganese transport activity of SLC30A10 in HeLa cells 9. Amino acids are shown as gray sticks with oxygen atoms depicted in red and nitrogen atoms in blue. Site A of YiiP shows a coordinated Zn2+ ion (red sphere).
B. Amino acid sequence alignment of SLC30A10 with related CDF proteins. The site A residues are highlighted in red. The Asn-43 residue of SLC30A10 is also shaded in gray. TM stands for transmembrane. Accession numbers of depicted sequences are as follows: YiiP, EIQ66948.1; SLC30A10, NP_061183.2; ZRC1, NP_013970.1; SLC30A1, NP_067017.2; SLC30A2, NP_001004434.1; SLC30A3, NP_003450.2; SLC30A4, NP_037441.2; SLC30A5, NP_075053.2; SLC30A7, NP_001138356.1; SLC30A8, NP_776250.2.
To test the above hypothesis, in 2016, we used the known crystal structure of YiiP to model a structure of SLC30A10 9. Comparison of the structural prediction of SLC30A10 with the solved structure of YiiP revealed important differences. In YiiP, side chains of Asp-45 and Asp-49 from the second transmembrane segment point toward the metal-binding pocket formed in the cavity between the second and fifth transmembrane segments (refs. 9, 21, 22; Fig. 1A). In contrast, in SLC30A10, the side chains of the corresponding Asn-43 and Asp-47 residues appear to point away from the space between the second and fifth transmembrane segments (ref.9; Fig. 1A). The structural prediction raised doubts about the requirement of Asn-43 for manganese transport. Consistent with this, experimental data revealed that Asn-43 was dispensable for manganese transport via SLC30A10 9. Indeed, among the four site A residues of SLC30A10, only one, Asp-248 was required; Asp-47 was also dispensable and His-244 was not required by itself, but played a role in association with a residue in the fourth transmembrane segment, Asn-127 (ref.9; Fig.1A). Additionally, Glu-25, from the first transmembrane segment, and Asp-40, located at the junction of the second transmembrane segment and an adjacent extracellular loop, were required (ref.9; Fig.1A). Our findings suggested that: (i) the mechanism of ion coordination within the transmembrane domain of SLC30A10 was different from that of other CDF proteins; (ii) presence of an asparagine residue in the NXXXD motif did not necessarily mean that a CDF would transport manganese; and (iii) metal specificity of CDFs may be determined by residues outside of those that constitute site A 9. For the above work, we performed experiments in human HeLa cells that transiently over-expressed SLC30A10wild-type (WT) or mutants. We validated that all SLC30A10 mutants were expressed at similar levels and localized to the cell surface. Finally, we analyzed the transport activity of each mutant by using three separate assays: comparison of levels of the intracellular manganese sensor GPP130, direct measurements of intracellular manganese using inductively-coupled plasma mass spectrometry, and assessment of cell viability in response to elevated manganese exposure 9.
Contemporaneous with our work, another group also tested the role of Asn-43 of SLC30A10 and reached the opposite conclusion - that it was necessary for manganese transport 28. In these studies, WT or mutated forms of human SLC30A10 were expressed in DT40 cells, a chicken B lymphocyte-derived cell line, and manganese transport activity was analyzed using cell viability assays 28. The difference in the cell-lines utilized in the two studies may underlie the discordant results. SLC30A10WT localizes to the cell surface in numerous cell-types (e.g. HeLa, hepatic HepG2, and neuronal AF5 cells; primary mouse midbrain neurons; and body wall muscle cells and dopaminergic neurons of C. elegans) 6, 7, 9. However, in the chicken DT40 cells, a large pool of SLC30A10WT failed to traffic to the cell surface and instead remained trapped in the Golgi apparatus 28. Moreover, the Golgi calcium/manganese pump SPCA1 was deleted in the DT40 cells used 28. SPCA1 is expressed in mammalian systems, including in HeLa cells, and its depletion impacts protein glycosylation, protein trafficking, and manganese homeostasis 29, 30. Thus, among the two model systems, HeLa cells likely better represent the conditions under which SLC30A10 functions in humans.
At the organism level, however, function of SLC30A10 is important in hepatocytes, where it mediates biliary manganese excretion 7, 8, and in GABAergic neurons of the basal ganglia, which are the target cell-type injured during manganese toxicity 18, 31–33, and which robustly express SLC30A10 13, 18. Despite being of human origin, HeLa cells are not an adequate model for more complex hepatic and neuronal systems, and effects in HeLa may not be similar to those in physiologically-relevant cell-lines. As two different cell-lines, both with questionable physiologic relevance, gave divergent results about the role of Asn-43, and as lack of requirement of Asn-43 would lead to a change in thinking about the mechanisms of metal transport via CDF proteins, the goal of the current study was to rigorously test the hypothesis that Asn-43 conferred manganese transport specificity to SLC30A10 using disease-relevant hepatic and neuronal cell-lines. Added significance for performing assays in hepatic and neuronal systems comes from the fact that discovery of residues required for the manganese transport function of SLC30A10 has emerged as a useful guide to identify new disease-causing mutations. Our 2016 paper reported that Glu-25 and Asp-40 of SLC30A10 were required for manganese transport 9. In 2018, D40A, L26P, and ΔS41 mutations in SLC30A10 were reported to induce manganese toxicity in humans (Leu-26 and Ser-41 are adjacent to Glu-25 and Asp-40, respectively) 16. Thus, determining whether Asn-43 is required for manganese transport using disease-relevant models has high translational relevance as well.
Here, we generated hepatic HepG2 and neuronal AF5 cells that stably over-expressed various SLC30A10 constructs and discovered that Asn-43 was dispensable for the manganese transport activity of SLC30A10. Our current and prior results, put together, suggest that the mechanism of ion coordination within the transmembrane domain of SLC30A10 is fundamentally different from that of previously studied CDFs, and allude to as yet unappreciated complexity in the principles that guide metal selectivity in the CDF superfamily.
Results
Localization and expression of WT and mutated forms of SLC30A10 in HepG2 and AF5 cells.
As the first step in this study, we used a lentiviral system to stably over-express SLC30A10WT, SLC30A10N43A or SLC30A10E25A in HepG2 and AF5 cells. The E25A mutant was used as a negative control – we previously demonstrated that this mutation inactivated the manganese transport function of SLC30A10 in HeLa cells 9. HepG2 cells are a well-accepted cell culture model to study function of proteins in the liver. We recently used this cell-line for studies on manganese homeostasis 7. AF5 cells are a neuronal cell line routinely used to study manganese homeostasis and toxicity 6, 34, 35. Neuronal AF5 cells assume a GABAergic lineage and produce high levels of GABA when cultured in a neuronal supportive media (see Methods) 35. For studies here, we initially performed experiments in AF5 cells that had not been differentiated to become GABAergic. We then validated results in differentiated GABAergic AF5 cells. Work with undifferentiated AF5 cells provided an important proof-of-concept before more challenging GABAergic differentiation was initiated.
Immunoblot analyses using a custom antibody against the C-terminus of SLC30A10 that we recently characterized 7 revealed that, in both HepG2 and undifferentiated AF5 cells, expression of the N43A or E25A mutant was comparable to SLC30A10WT (Fig.2A). Furthermore, immunofluorescence analyses demonstrated that, similar to WT, both mutants outlined the cell surface (Fig.2B). Quantitative colocalization analyses revealed that signals for all SLC30A10 constructs were separate from those of the endoplasmic reticulum (Fig.2B). Overall, SLC30A10WT, SLC30A10N43A and SLC30A10E25A were well-expressed and localized to the cell surface in the stably-overexpressing HepG2 and undifferentiated AF5 cells generated here. The cell surface localization was consistent with our prior results 6, 7, 9.
Figure 2. Expression and localization of SLC30A10WT and mutants in different cell-lines.
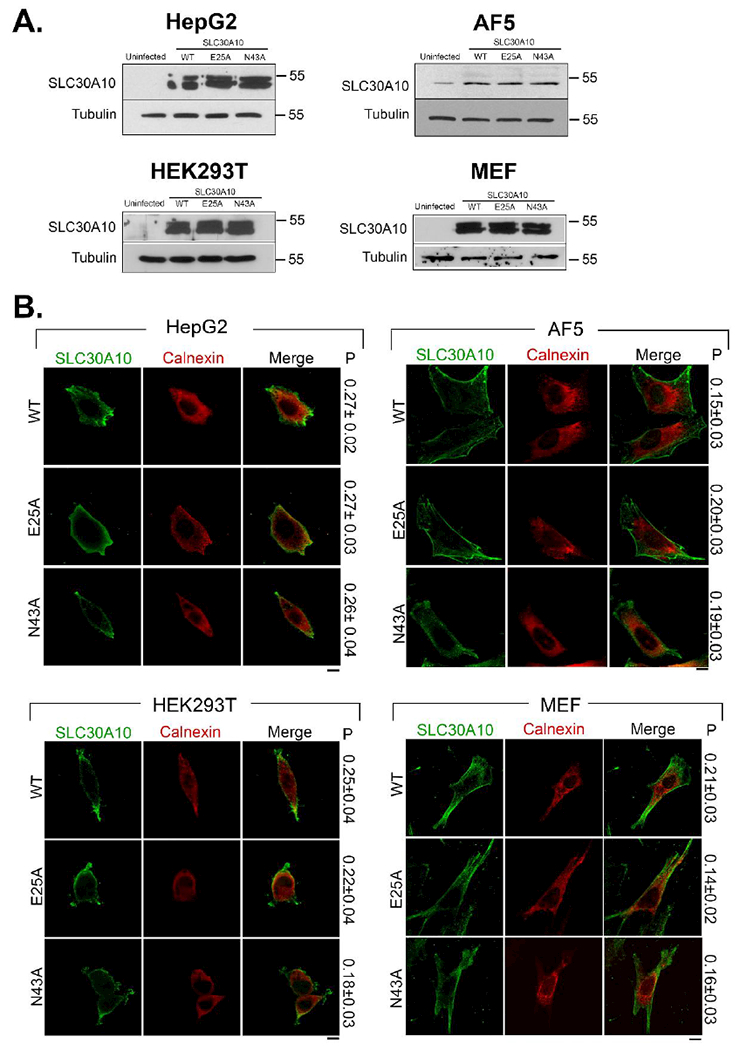
A. HepG2, undifferentiated AF5, HEK, and Slc30a10−/− MEF cells that were uninfected or that stably over-expressed indicated SLC30A10 constructs were lysed and processed for immunoblot analyses. SLC30A10 was detected using a custom polyclonal antibody against the C-terminus that we recently described 7. Tubulin was detected using a mouse monoclonal antibody.
B. Localization of SLC30A10 was assessed in cell-lines described in Panel A by immunofluorescence. Over-expressed SLC30A10 was detected using a monoclonal antibody against the FLAG-tag. A polyclonal antibody against calnexin was used to demarcate the endoplasmic reticulum. P represents the Pearson’s coefficient for colocalization between SLC30A10 and calnexin (mean ± S.E.; n = 10 cells per construct). Note that values for the Pearson’s coefficient for colocalization for two fluorophores can range from + 1 to −1 6, 9, 42. Values close to +1 indicate a high degree of overlap. Values close to 0 and negative values imply lack of overlap 6, 9, 42. Values obtained here indicate lack of overlap. Scale bars, 10 μm.
For added rigor, as part of these studies, we repeated all experiments in two additional cell lines: HEK cells and mouse embryonic fibroblasts (MEFs) obtained from Slc30a10−/− mice, which we recently generated and characterized 5, 7. Similar to the results with HepG2 and undifferentiated AF5 cells, in HEK cells and Slc30a10−/− MEFs, the N43A and E25A mutants were expressed at levels comparable to SLC30A10WT and all three forms of SLC30A10 trafficked to the cell surface (Fig.2A&B).
Expression of SLC30A10WT or SLC30A10N43A reduces intracellular Mn levels.
As the next step, we assayed for intracellular manganese levels. SLC30A10 is an efflux transporter 6, 8. We previously demonstrated that, in HeLa cells, expression of SLC30A10WT or the N43A mutant, but not SLC30A10E25A, reduced intracellular manganese levels 9. These results implied that the N43A mutant retained, while the E25A mutant lost, manganese efflux activity 9. For the current experiment, we first generated a manganese dose-response curve in HepG2, undifferentiated AF5, HEK, and Slc30a10−/− MEF cells that did not over-express any SLC30A10 construct. The dose-response was necessary because basal intracellular manganese levels in these cell lines are usually at or below the limit of detection of inductively coupled plasma mass spectrometry. This necessitates supplementation of the growth media with manganese. The amount of manganese that is added must increase intracellular manganese so that levels can be accurately detected, but should not induce overt toxicity. Dose-response analyses revealed that, in all cell types, the LD50 of manganese after a 16 h exposure was >1 mM (Fig.3A). Based on this, we exposed cultures to a lower concentration, 500 μM manganese for 16 h, and then measured intracellular metal levels. Note that the level of manganese used here was similar to that utilized in our previous work in HeLa cells 6, 9. In all cell types, intracellular manganese levels of cells expressing SLC30A10WT or SLC30A10N43A were comparable to each other and lower than uninfected controls (Fig.3B). Manganese levels of cells expressing SLC30A10E25A were significantly greater than those expressing SLC30A10WT or SLC30A10N43A (Fig.3B). Interestingly, compared with uninfected controls, expression of SLC30A10E25A induced a modest decrease in intracellular manganese in HEK and MEF, but not HepG2 or undifferentiated AF5 cells (Fig.3B), highlighting the fact that cell-type specific differences may exist in transporter function. Observed changes in manganese were specific because levels of zinc and iron were comparable between uninfected controls and cells expressing SLC30A10 constructs (Fig.3C&D). These results imply that in HepG2, AF5, HEK and Slc30a10−/− MEF cells, mutation of Asn-43 to alanine does not impact the manganese efflux function of SLC30A10, while that of Glu-25 to alanine does.
Figure 3. Intracellular manganese levels are comparable between cells expressing SLC30A10WT or SLC30A10N43A.
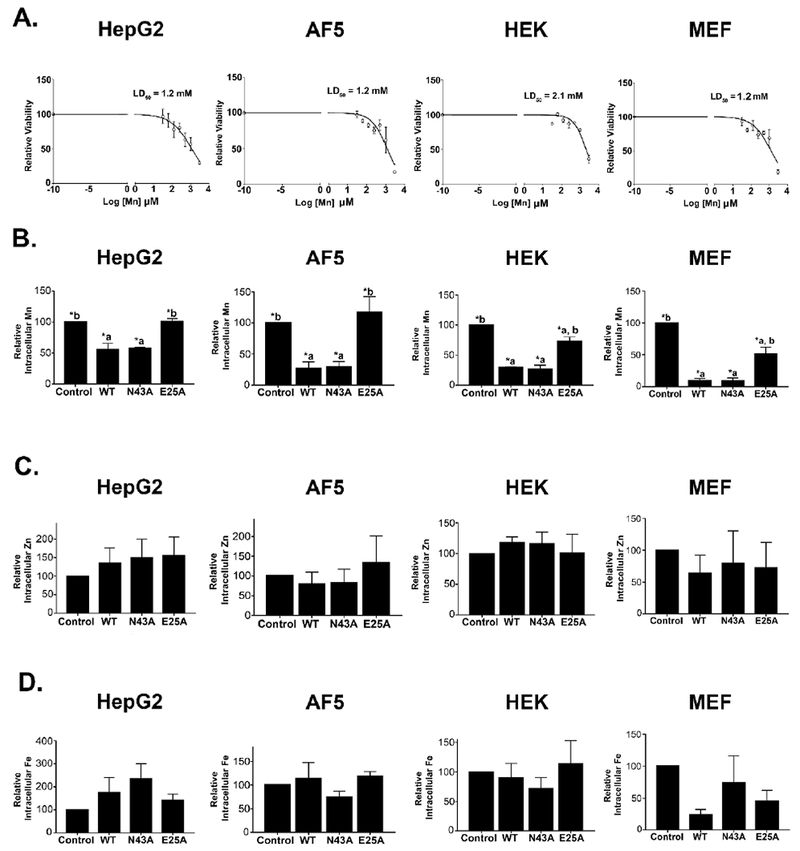
A. Viability of HepG2, undifferentiated AF5, HEK, and Slc30a10−/− MEF cells that did not express any SLC30A10 construct was assessed after exposure to indicated amounts of manganese for 16 h. Viability in the absence of manganese exposure was expressed as 100 and used for normalization (mean ± S.E.; n = 3 per manganese treatment condition).
B-D. HepG2, undifferentiated AF5, HEK, and Slc30a10−/− MEF cells that were uninfected or that stably over-expressed indicated SLC30A10 constructs were exposed to 500 μM for 16 h. After this, samples were processed for metal measurements using inductively coupled plasma mass spectrometry. For each sample, metal levels were normalized to total protein content, independently. After this, for each cell type and metal, levels in uninfected controls were expressed as 100. Levels in other infection conditions were expressed relative to uninfected controls (mean ± S.E.; n = 3 per infection condition for HepG2, AF5 and HEK cells, and 4 for MEF cells. All metals were measured in all samples. *, p < 0.05 using one-way ANOVA and Tukey–Kramer post hoc test, with a or b indicating differences in comparison with uninfected controls or SLC30A10WT-expressing cells, respectively).
Expression of SLC30A10WT or SLC30A10N43A protects cells against manganese toxicity.
As an independent test of manganese efflux activity, we assayed for the viability of cells after exposure to elevated manganese. Our previous studies showed that expression of either the WT form of SLC30A10 or the N43A mutant protected HeLa cells against manganese-induced cell death, but expression of SLC30A10E25A did not 9. In current studies, as expected, in all four cell types, expression of SLC30A10WT robustly protected against manganese-induced cell death (Fig.4). A similar protection was evident in cells expressing SLC30A10N43A (Fig.4). Importantly, the E25A mutant failed to protect (Fig.4). Thus, results of the cell viability assay are consistent with those of the metal measurement experiments. Put together, results in Figs.2-4 indicate that the side-chain of Asn-43 is not required for the manganese efflux activity of SLC30A10 in multiple different cell types, while that of Glu-25 is required.
Figure 4. Expression of SLC30A10WT or SLC30A10N43A protects against manganese-induced cell death.
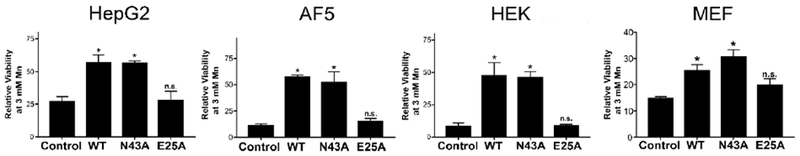
HepG2, undifferentiated AF5, HEK, and Slc30a10−/− MEF cells that were uninfected or that stably over-expressed indicated SLC30A10 constructs were treated with or without 3 mM manganese for 16 h. Viability was then assessed. For each cell type and infection condition, viability after manganese exposure was expressed relative to that in the absence of manganese treatment normalized to 100 (mean ± S.E.; n = 3 for each infection condition; *, p < 0.05 using one-way ANOVA and Tukey–Kramer post hoc test for the comparison between uninfected control and other infection conditions. N.S. denotes that there was no difference between the uninfected control and SLC30A10E25A groups for any cell-type. There were no differences between WT and N43A groups for any cell-type).
Validation of results in differentiated AF5 cells.
After this, we validated our results in differentiated AF5 cells. Intracellular levels of manganese in cells expressing SLC30A10WT or SLC30A10N43A were comparable to each other and significantly lower than uninfected controls (Fig.5A). There were no changes in intracellular zinc or iron levels (Fig.5B&C). Expression of SLC30A10WT or SLC30A10N43A also protected against manganese-induced cell death (Fig.5D). The decrease in intracellular manganese and protection against manganese toxicity was not evident in cells expressing SLC30A10E25A (Fig.5A&D). Thus, Asn-43 is not required for the manganese efflux activity of SLC30A10 in differentiated GABAergic AF5 cells.
Figure 5. Validation of results in differentiated AF5 cells.
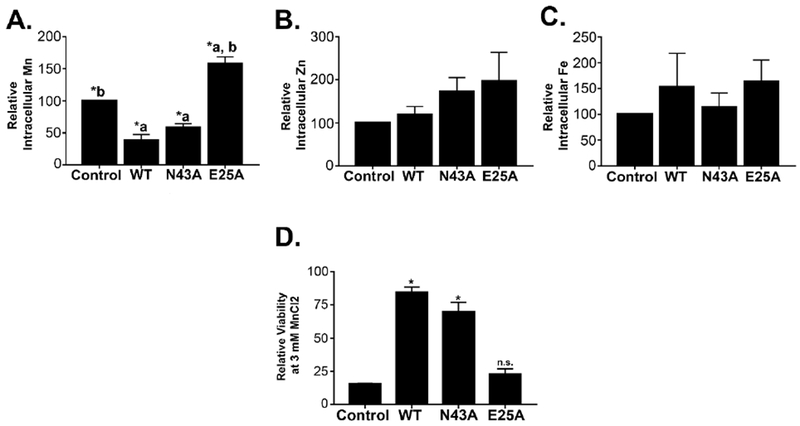
A-C. AF5 cells infected with various SLC30A10 constructs or left uninfected were differentiated as described in Methods. Samples were then processed for measurement of intracellular metals exactly as described in Fig.3. (mean ± S.E.; n = 3 per infection condition; *, p < 0.05 using one-way ANOVA and Tukey–Kramer post hoc test, with a or b indicating differences in comparison with uninfected controls or SLC30A10WT-expressing cells).
D. Differentiated AF5 cells were exposed to 0 or 3 mM manganese for 16 h. Viability was then assessed. Relative viability after manganese exposure was expressed as described in Fig.4 (mean ± S.E.; n = 3 for each infection condition; *, p < 0.05 using one-way ANOVA and Tukey–Kramer post hoc test for the comparison between uninfected control and other infection conditions. N.S. denotes that there was no difference between the uninfected control and SLC30A10E25A groups. There were no differences between cells expressing SLC30A10WT or SLC30A10N43A).
His-43 of SLC30A1/ZnT1 is required for zinc transport.
The lack of requirement of Asn-43 of SLC30A10 for manganese transport led us to wonder whether residues that correspond to Asn-43 in other SLC30 proteins, which mediate zinc efflux, were necessary for zinc transport. In other SLC30 proteins, a histidine residue corresponds to Asn-43 of SLC30A10 and contributes to site A (Fig.1B). Review of the literature revealed that while the role of site A residues had been tested for some zinc-transporting SLC30 proteins 24, 25, requirement of the histidine residue, by itself, was unclear. To address this issue, we focused on SLC30A1/ZnT1, which is closely related to SLC30A10 by phylogeny, but is a zinc-transporter 1, 4, 25. Using the algorithms of the PHYRE 2.0 server 36, we obtained a predicted structure of SLC30A1/ZnT1. Site A of SLC30A1/ZnT1 is predicted to be formed by residues His-43 and Asp-47 of the second and His-251 and Asp-255 of the fifth transmembrane segments (Fig.6A). Comparison of this structure with the predicted structure of SLC30A10 and the solved structure of YiiP immediately revealed that, unlike SLC30A10, the side chain of His-43 of SLC30A1/ZnT1 points towards the putative transmembrane metal binding cavity (Fig.6A). This increased the likelihood that His-43 of SLC30A1/ZnT1 may be required for metal transport. To obtain experimental data, we used a transient-transfection system and over-expressed SLC30A1/ZnT1WT or SLC30A1/ZnT1H43A in HeLa cells, and subsequently, indirectly measured zinc efflux by assaying for protection against zinc toxicity. We could not use intracellular zinc measurements for assessment of transporter function because cellular zinc levels are far higher than manganese, and exposure of cells to low levels of zinc induces cell death before changes in intracellular zinc can be detected. Indeed, in our hands, treatment of cells with 200 μM zinc for 16 h induced robust cell death, but failed to produce a detectable increase in intracellular zinc (Fig.6B&C). Also note that these experiments were performed in HeLa cells because we previously demonstrated that transient over-expression of SLC30A1/ZnT1WT in HeLa cells protected against zinc toxicity, but did not reduce cellular manganese levels, implying that the zinc-specific transport activity of SLC30A1/ZnT1WT was recapitulated in this cell-line 9. Both SLC30A1/ZnT1WT and the H43A mutant were expressed at comparable levels and trafficked to the cell surface (Fig.6D). Cell surface localization of SLC30A1/ZnT1WT was consistent with prior results 9, 25. Importantly, while expression of the WT protein robustly protected against zinc-induced cell death, expression of the H43A mutant did not (Fig.6E). Therefore, the side chain of His-43 is necessary for zinc transport activity of SLC30A1/ZnT1.
Figure 6. Glu-25, His-43, and Asn-127 of SLC30A1/ZnT1 are required for zinc efflux activity.
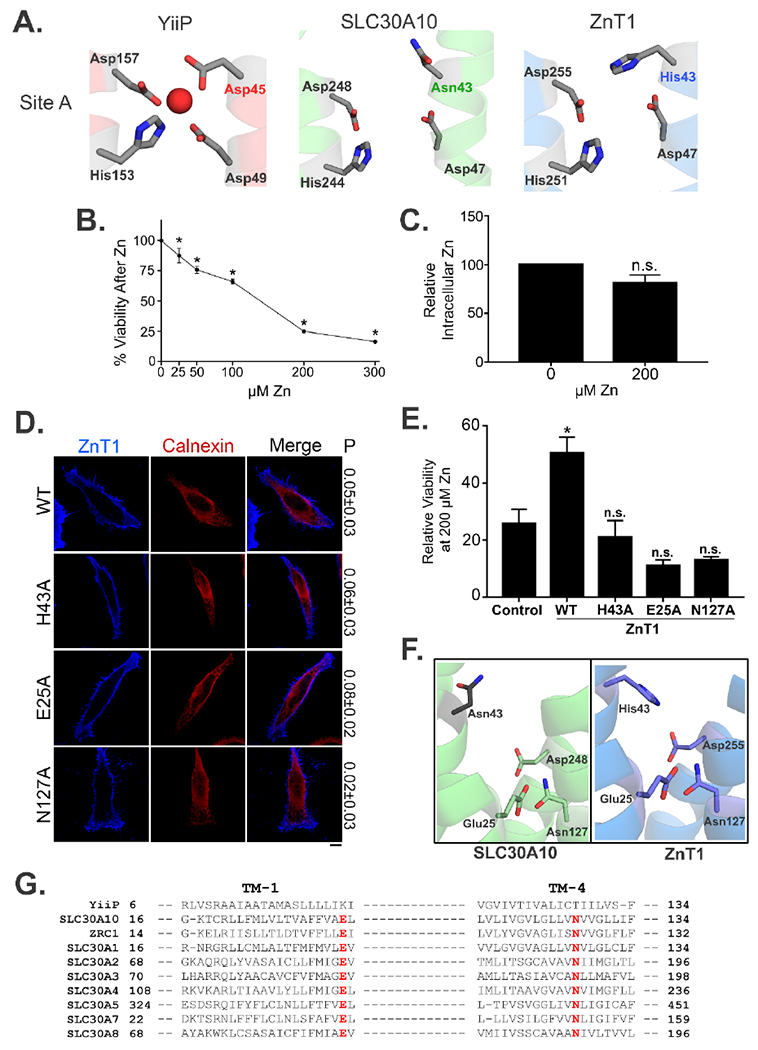
A. Structural comparison of the amino acids making up Site A in YiiP (Red) with the putative Site A in the predicted structures of SLC30A10 (Green) and ZnT1 (Blue). Amino acid residues are shown as gray sticks with oxygen atoms colored in red and nitrogen atoms in blue.
B. HeLa cells were treated with indicated amounts of zinc for 16 h and viability was then assessed. Viability in the absence of zinc treatment was normalized to 100 (mean ± S.E.; n = 3; *, p < 0.05 for the difference between viability at 0 μM zinc and other zinc concentrations by one-way ANOVA and Dunnett’s post hoc test).
C. Intracellular zinc levels were measured in HeLa cells exposed to 0 or 200 μM zinc for 16 h. In each sample, zinc levels were normalized to total protein content. Levels in cells that did not receive zinc treatment were expressed as 100 (mean ± S.E.; n = 3; n.s., not significant by t-test).
D. HeLa cells were transfected with indicated SLC30A1/ZnT1 constructs. Twenty four hours after transfection, cultures were fixed and processed for immunofluorescence. SLC30A1/ZnT1 was detected using a monoclonal antibody against the FLAG epitope. A polyclonal antibody against calnexin was used to demarcate the endoplasmic reticulum. P represents the Pearson’s coefficient for colocalization between SLC30A1/ZnT1 and calnexin (mean ± S.E.; n = 10 cells per construct). Scale bar, 10 μm.
E. HeLa cells were transfected with a control plasmid or various SLC30A1/ZnT1 constructs. One day after transfection, cultures were treated 0 or 200 μM zinc for 16 h. Viability was then assessed. For each transfection condition, viability in the absence of zinc treatment was normalized to 100 and used to express viability after zinc exposure (mean ± S.E.; n = 3 for each construct; *, p < 0.05 for the difference between control and all other transfection conditions by one-way ANOVA and Dunnett’s post hoc test; n.s., not significant).
F. Comparison of the predicted structures of SLC30A10 (Green) and ZnT1 (Blue) showing similar orientations of Glu-25 and Asn-127 residues.
G. Alignment of the primary amino acid sequence corresponding to the first and fourth transmembrane (TM) segments of CDF proteins depicted in Fig.1. Residues corresponding to Glu-25 and Asn-127 of SLC30A10 are highlighted in red. Accession numbers are identical to that in Fig.1.
Side chains of Asn-127 and Glu-25 of SLC30A1/ZnT1 are also required.
We previously demonstrated that Glu-25 and Asn-127 of SLC30A10 were required for manganese transport 9. Side chains of these residues are in proximity to the required site A residue Asp-248 (ref.9; Fig.6F). Therefore, one possibility was that the side chains of Glu-25 and Asn-127, along with that of Asp-248, created a novel manganese binding site within the transmembrane domain of SLC30A10. However, the asparagine and glutamate residues are conserved in other SLC30 proteins (Fig.6G). Furthermore, prior work showed that Glu-31 of the bacterial zinc-transporter CzcD and Asn-135 of another bacterial zinc transporter ZitB, which correspond to Glu-25 and Asn-127 of SLC30A10 respectively, were required for transport activity 3, 37. Thus, an alternate possibility was that Glu-25 and Asn-127 of SLC30A10 did not confer manganese specificity, but instead, played a more general role in facilitating metal transport via CDF proteins. To test this idea, we examined whether Asn-127 and Glu-25 of SLC30A1/ZnT1, which correspond to Asn-127 and Glu-25 of SLC30A10 and have a similar orientation in the predicted structure (Fig.6F&G), were required for zinc transport. Similar to the H43A mutant, both the N127A and E25A mutants were also expressed at levels comparable to SLC30A1/ZnT1WT and localized to the cell surface (Fig.6D). Importantly, unlike SLC30A1/ZnT1WT, expression of the N127A or E25A mutant failed to protect against zinc-induced cell death (Fig.6E). Thus, side chains of Asn-127 and Glu-25 are necessary for zinc transport via SLC30A1/ZnT1, reducing the likelihood that the corresponding Asn-127 and Glu-25 residues provide manganese transport specificity to SLC30A10.
Discussion
Results in this manuscript confirm that the side chain of Asn-43 is dispensable for the manganese efflux activity of SLC30A10. These results imply that CDF proteins with an asparagine residue in Site A, as part of the NXXXD motif, can no longer automatically be considered to be manganese transporters. A related implication is that while bioinformatic and sequence analyses may be a useful starting point to study transporter function, they are not a substitute for well-designed and carefully controlled mechanistic assays.
Thus far, Asp-248 is the only site A residue of SLC30A10 known to be required for manganese transport 9. Therefore, another important ramification is that the mechanism by which manganese is coordinated within the transmembrane domain of SLC30A10 is fundamentally different from that of CDF proteins studied previously, and that residues outside of site A may be involved in metal coordination. Two transmembrane non-site A residues, Glu-25 and Asn-127, are required for the manganese transport activity of SLC30A10 (ref.9; Figs.3&4). Proximity of Glu-25 and Asn-127 to the required site A residue Asp-248 raised the possibility that these residues may be involved in coordinating manganese. However, these residues are conserved in other CDFs, and here we discovered that corresponding asparagine and glutamate residues of SLC30A1/ZnT1 are also required for zinc transport. Thus, it is unlikely that Glu-25 and Asn-127 play a direct role in coordinating manganese or conferring manganese transport specificity to SLC30A10. A more likely possibility is that these residues play important roles in allowing CDF proteins to transport metals in general. How then could SLC30A10 gain manganese transport capability? One possibility is that residues unique to the transmembrane domain of SLC30A10 (i.e. that are not conserved in other SLC30 proteins) may create a manganese binding site that excludes other metals. Alternatively, and perhaps counter-intuitively, the manganese binding site may be formed by residues that are conserved in other SLC30 proteins. Work on ZRC1 revealed that single amino acid changes near a putative transmembrane metal binding site may affect substrate specificity 38. Comparison of primary sequence information shows that some amino acids near Asp-248, Glu-25, and Asn-127 of SLC30A10, which are required for manganese transport, are different from those in other SLC30 proteins (Fig.1B&6G). Due to these differences, residues that coordinate manganese within the transmembrane domain of SLC30A10 may be oriented in a manner that favors manganese binding while simultaneously disfavoring binding of other metals, such as zinc.
Results about the lack of requirement of Asn-43 in the current study validate our prior work in HeLa cells 9, but differ from results obtained in DT40 cells 28. Proteins that regulate/influence transporter function are likely differentially expressed in different cell types. Indeed, even in our studies here, expression of SLC30A10E25A led to a modest reduction in intracellular manganese levels in HEK and MEF cells (Fig.3B). These findings highlight the importance of using disease relevant cell lines for addressing translationally-important questions related to transporter function.
Notably, our studies do not imply that residues corresponding to Asn-43 of SLC30A10 will be dispensable for activity of other manganese-transporting CDFs. As described earlier, in the bacterial manganese transporter MntE, the asparagine residue that corresponds to Asn-43 of SLC30A10 is required 23. Crystallization of SLC30A10 will probably provide clear understanding of the mechanisms of metal coordination and transport employed by this unique protein.
There is now a wealth of evidence to show that SLC30A10 is a specific manganese efflux transporter and that its function plays a pivotal role in regulating manganese homeostasis at the cellular and organism level 5–9. In multiple cell lines, SLC30A10WT localizes to the cell surface, reduces intracellular manganese levels, and protects against manganese-induced cell death, but does not impact zinc homeostasis 6, 7, 9. In primary mouse midbrain neurons, expression of SLC30A10WT protects against manganese-induced neurotoxicity 6. In neuronal AF5 cells, depletion of SLC30A10 increases intracellular manganese and enhances manganese toxicity 6. In C. elegans, expression of SLC30A10WT, but not a disease-causing mutant, protects against manganese toxicity 6. Finally, Slc30a10 knockout mice have elevated tissue manganese levels while levels of other metals (zinc, iron and copper) are essentially normal 5, 7. These knockouts develop severe manganese-induced hypothyroidism; this can be rescued either by feeding the animals a reduced manganese diet or by depleting the manganese importer Slc39a14, which reduces thyroid manganese levels of Slc30a10−/− mice 5, 7. The importance of SLC30A10 in regulating manganese homeostasis and detoxification raises the possibility that targeting this protein may be therapeutically useful for the management of manganese-induced disease in humans. Studies on the mechanism of ion transport via SLC30A10 may aid in the development of small molecules that enhance the manganese transport activity of SLC30A10. In this discussion it is important to note that while SLC30A10 lacks zinc transport activity in cells and organisms, liposome-based transport assays are essential to determine whether the transporter has minimal zinc transport activity that is masked by the existence of other high affinity zinc transporters.
In conclusion, unlike other CDF proteins, a critical site A asparagine residue is not required for the transport activity of SLC30A10, suggesting that the mechanisms by which proteins of this superfamily transport metals may be more complex than previously appreciated.
Methods
Growth of HepG2, HEK, AF5 and HeLa cells.
Cell culture was performed as described by us previously 6, 7, 9, 39, 40. Briefly, HeLa, HEK293T and HepG2 cells were maintained in minimum essential media (Corning, Corning NY) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 100 IU/ml penicillin-G, and 100 μg/ml streptomycin (both from Corning). AF5 cells were grown in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (Life Technologies) with 10% fetal bovine serum, 2 mm L-glutamine, 100 IU/ml penicillin-G, and 100 μg/ml streptomycin (maintenance media). GABAergic differentiation of AF5 cells was performed by transferring cultures to Neurobasal media supplemented with serum-free human B27 (Life Technologies) and 2mM L-glutamine for 48–72 h, as described by us previously 6.
Production and growth of MEFs.
Embryos were obtained from Slc30a10−/− mice that had been euthanized for other experimental purposes not related to this project. All work with animals was approved by our Institutional Animal Use and Care Facility. Each isolated embryo was rinsed in phosphate buffered saline followed by removal of the head, heart and liver. Rest of the tissue was finely dissected using a razor blade, transferred to a sterile 10 ml tube containing 1 ml trypsin, and incubated in a 37°C water bath for 10 min, with shaking. Subsequently, the tissue was further homogenized by passing through a P-1000 pipette. Then, 3 ml of Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 IU/ml penicillin-G, and 100 μg/ml streptomycin was added. The tube was allowed to remain undisturbed for 5 min at 37°C. The supernatant, consisting of single cells and cell clusters, was then transferred to a 10 cm tissue culture dish and cultured using the growth media described above. Each embryo was genotyped using tissue not used for MEF generation; primers and PCR conditions were as described in our recent publications 5, 7.
Plasmids, lentivirus production, and transient transfections.
For generating lentivirus, we sub-cloned SLC30A10WT from the FLAG-SLC30A10 plasmid that we used for transient transfections in our previous publications 6, 9 to a third generation lentivirus transfer plasmid (Addgene Plasmid #34611) 41. Briefly, full length SLC30A10 was amplified using 5’ GAT ATC GCT AGC ATG GGC CGC TAC TCT G 3’ (forward) and 5’ CTG TCT GAA TTC TTA AAA ATG CGT TCT GTT GAC 3’ (reverse) primers, digested with 5’ Nhe1 and 3’ EcoR1 enzymes, and ligated into the transfer plasmid. The FLAG tag was lost during sub-cloning; therefore, a single N-terminal FLAG tag was looped into the transfer plasmid using 5’ GAT CCG CTA GCA TGG ATT ACA AGG ATG ACG ACG ATA AGG GCC GCT ACT CTG GC 3’ (forward) and 5’ GCC AGA GTA GCG GCC CTT ATC GTC GTC ATC CTT GTA ATC CAT G 3’ (reverse) primers, and the loop-in modification of the QuikChange protocol (Agilent Technologies, Santa Clara, CA). Mutations were introduced into the transfer plasmid coding for SLC30A10WT using QuikChange. Further generation of lentivirus and infection of cells was exactly as described by us recently 39. We have previously described the SLC30A1/ZnT1WT plasmid used here 9. Point mutations were introduced into this plasmid using QuikChange.
Antibodies.
We recently described the custom rabbit polyclonal antibody against the C-terminal domain of SLC30A10 7. In immunoblots, this antibody recognizes SLC30A10 as a ~50 kD band in cell-lines that express SLC30A10, but not in those that do not. Antibodies against the FLAG epitope (mouse), calnexin (rabbit), and tubulin (mouse) were also described by us recently 6, 9.
Transient transfections, metal treatments, viability assays, immunoblots, and metal measurements.
Transient transfections were performed using JetPEI reagent (VWR) as described by us previously 6, 9, 39, 40. Manganese and zinc treatments were also as described previously 6, 9. Briefly, freshly prepared MnCl2 or ZnSO4 in ultrapure water was added to the culture media to achieve desired final concentrations. Viability assessment using the methylthiazolyldiphenyltetrazolium bromide (MTT) reagent, immunoblot analyses, and measurement of intracellular levels of metals using inductively coupled plasma mass spectrometry were as described by us previously 6, 9, 39.
Immunofluorescence and microscopy.
Immunofluorescence imaging was performed as described by us previously 6, 9, 39, 40. All images were captured using a Nikon swept-field confocal and a 100× oil immersion objective with a numerical aperture of 1.45 (both from Nikon Inc., Melville, NY). An iXon3 X3 DU897 EM-CCD camera (Andor Technology, Belfast, UK) was used for image capture. Images were captured as Z-stacks with 0.5 μm spacing between individual frames. Depicted images are maximum intensity projections from the stacks. Quantification was using the NIS Elements software (Nikon), essentially as described by us previously 6, 9. Within each experiment, all images were captured using identical settings. For calculating Pearson’s correlation coefficient, one frame from the Z-stack was used. In each case, the selected frame had the largest expanse of the cell being analyzed. For quantification, an outline was drawn around the cell, and the Pearson’s coefficient was calculated using the colocalization function in NIS elements software.
Statistical analyses.
The Prism 6 software (GraphPad, La Jolla, CA) was used. All experiments were repeated at least three times, independently. Comparisons between multiple groups were performed using one-way ANOVA and appropriate post hoc tests. Comparisons between two groups were performed using Student’s t-test. Nonlinear regression was used to calculate LD50 of manganese. P < 0.05 was considered to be significant. Asterisks in graphs, where present, denote statistically significant differences.
Significance to Metallomics.
Cation diffusion facilitators (CDF) are metal transporters present in all kingdoms of life. Prior studies suggested that metal specificity was conferred by amino acids that formed a transmembrane metal binding site (Site A). In particular, presence of an asparagine residue in Site A was believed to be the hallmark of manganese transporters. We show that the critical asparagine residue in Site A of SLC30A10, a human manganese transporter that is mutated in familial manganese-induced parkinsonism, is dispensable for transport activity. Our results suggest that factors beyond Site A may confer metal specificity to CDFs.
Acknowledgements
We thank Dr. Nathaniel Miller (Jackson School of Geosciences, UT Austin) for performing all inductively-coupled plasma mass spectrometry assays reported here; Dr. Chunyi Liu for assistance with generation of mouse embryonic fibroblasts; and Dr. Steven Hutchens for help with genotyping. Supported by NIH/NIEHS grants R00-ES020844 and R01-ES024812 (S.M.) and NIH/NIEHS predoctoral fellowship 1F31-ES027317 (C.E.Z.).
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of the article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Kambe T, Tsuji T, Hashimoto A and Itsumura N, The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism, Physiol Rev, 2015, 95, 749–784. [DOI] [PubMed] [Google Scholar]
- 2.Kolaj-Robin O, Russell D, Hayes KA, Pembroke JT and Soulimane T, Cation Diffusion Facilitator family: Structure and function, FEBS Lett, 2015, 589, 1283–1295. [DOI] [PubMed] [Google Scholar]
- 3.Montanini B, Blaudez D, Jeandroz S, Sanders D and Chalot M, Phylogenetic and functional analysis of the Cation Diffusion Facilitator (CDF) family: improved signature and prediction of substrate specificity, BMC Genomics, 2007, 8, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang L and Tepaamorndech S, The SLC30 family of zinc transporters - a review of current understanding of their biological and pathophysiological roles, Mol Aspects Med, 2013, 34, 548–560. [DOI] [PubMed] [Google Scholar]
- 5.Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR and Mukhopadhyay S, Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice, J Biol Chem, 2017, 292, 9760–9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M and Mukhopadhyay S, SLC30A10 Is a Cell Surface-Localized Manganese Efflux Transporter, and Parkinsonism-Causing Mutations Block Its Intracellular Trafficking and Efflux Activity, J Neurosci, 2014, 34, 14079–14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR and Mukhopadhyay S, Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production, J Biol Chem, 2017, 292, 16605–16615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukhopadhyay S, Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14, Neurotoxicology, 2018, 64, 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zogzas CE, Aschner M and Mukhopadhyay S, Structural Elements in the Transmembrane and Cytoplasmic Domains of the Metal Transporter SLC30A10 Are Required for Its Manganese Efflux Activity, J Biol Chem, 2016, 291, 15940–15957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen P, Bowman AB, Mukhopadhyay S and Aschner M, SLC30A10: A novel manganese transporter, Worm, 2015, 4, e1042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aschner M, Erikson KM, Herrero Hernandez E and Tjalkens R, Manganese and its role in Parkinson’s disease: from transport to neuropathology, Neuromolecular Med, 2009, 11, 252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulab S, Kayyali HR and Al-Said Y, Atypical Neurologic Phenotype and Novel SLC30A10 Mutation in Two Brothers with Hereditary Hypermanganesemia, Neuropediatrics, 2018, 49, 72–75. [DOI] [PubMed] [Google Scholar]
- 13.Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP and Bonifati V, Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease, Am J Hum Genet, 2012, 90, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuschl K, Clayton PT, Gospe SM Jr., Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK and Mills PB, Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man, Am J Hum Genet, 2012, 90, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M and Clayton PT, Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia--a new metabolic disorder, J Inherit Metab Dis, 2008, 31, 151–163. [DOI] [PubMed] [Google Scholar]
- 16.Zaki MS, Issa MY, Elbendary HM, El-Karaksy H, Hosny H, Ghobrial C, El Safty A, El-Hennawy A, Oraby A, Selim L and Abdel-Hamid MS, Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: Six novel SLC30A10 mutations and further phenotype delineation, Clin Genet, 2018, 93, 905–912. [DOI] [PubMed] [Google Scholar]
- 17.Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, Kori I, Hattiholi V, Jain P, Aneja S, Kumar A, Gulati P, Goel M, Talukdar B and Bonifati V, Manganese transport disorder: novel SLC30A10 mutations and early phenotypes, Mov Disord, 2015, 30, 996–1001. [DOI] [PubMed] [Google Scholar]
- 18.Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW and Gospe SM Jr., Pathology of inherited manganese transporter deficiency, Ann Neurol, 2014, 75, 608–612. [DOI] [PubMed] [Google Scholar]
- 19.Hoch E, Lin W, Chai J, Hershfinkel M, Fu D and Sekler I, Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity, Proc Natl Acad Sci U S A, 2012, 109, 7202–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei Y and Fu D, Selective metal binding to a membrane-embedded aspartate in the Escherichia coli metal transporter YiiP (FieF), J Biol Chem, 2005, 280, 33716–33724. [DOI] [PubMed] [Google Scholar]
- 21.Lu M, Chai J and Fu D, Structural basis for autoregulation of the zinc transporter YiiP, Nat Struct Mol Biol, 2009, 16, 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu M and Fu D, Structure of the zinc transporter YiiP, Science, 2007, 317, 1746–1748. [DOI] [PubMed] [Google Scholar]
- 23.Martin JE and Giedroc DP, Functional Determinants of Metal Ion Transport and Selectivity in Paralogous Cation Diffusion Facilitator Transporters CzcD and MntE in Streptococcus pneumoniae, J Bacteriol, 2016, 198, 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohana E, Hoch E, Keasar C, Kambe T, Yifrach O, Hershfinkel M and Sekler I, Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter, J Biol Chem, 2009, 284, 17677–17686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shusterman E, Beharier O, Shiri L, Zarivach R, Etzion Y, Campbell CR, Lee IH, Okabayashi K, Dinudom A, Cook DI, Katz A and Moran A, ZnT-1 extrudes zinc from mammalian cells functioning as a Zn(2+)/H(+) exchanger, Metallomics, 2014, 6, 1656–1663. [DOI] [PubMed] [Google Scholar]
- 26.Barber-Zucker S, Shaanan B and Zarivach R, Transition metal binding selectivity in proteins and its correlation with the phylogenomic classification of the cation diffusion facilitator protein family, Sci Rep, 2017, 7, 16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dokmanic I, Sikic M and Tomic S, Metals in proteins: correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination, Acta Crystallogr D Biol Crystallogr, 2008, 64, 257–263. [DOI] [PubMed] [Google Scholar]
- 28.Nishito Y, Tsuji N, Fujishiro H, Takeda TA, Yamazaki T, Teranishi F, Okazaki F, Matsunaga A, Tuschl K, Rao R, Kono S, Miyajima H, Narita H, Himeno S and Kambe T, Direct Comparison of Manganese Detoxification/Efflux Proteins and Molecular Characterization of ZnT10 Protein as a Manganese Transporter, J Biol Chem, 2016, 291, 14773–14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay S and Linstedt AD, Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity, Proc Natl Acad Sci U S A, 2011, 108, 858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos-Castaneda J, Park YN, Liu M, Hauser K, Rudolph H, Shull GE, Jonkman MF, Mori K, Ikeda S, Ogawa H and Arvan P, Deficiency of ATP2C1, a Golgi ion pump, induces secretory pathway defects in endoplasmic reticulum (ER)-associated degradation and sensitivity to ER stress, J Biol Chem, 2005, 280, 9467–9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerfen CR and Surmeier DJ, Modulation of striatal projection systems by dopamine, Annu Rev Neurosci, 2011, 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olanow CW, Manganese-induced parkinsonism and Parkinson’s disease, Ann N Y Acad Sci, 2004, 1012, 209–223. [DOI] [PubMed] [Google Scholar]
- 33.Perl DP and Olanow CW, The neuropathology of manganese-induced Parkinsonism, J Neuropathol Exp Neurol, 2007, 66, 675–682. [DOI] [PubMed] [Google Scholar]
- 34.Crooks DR, Welch N and Smith DR, Low-level manganese exposure alters glutamate metabolism in GABAergic AF5 cells, Neurotoxicology, 2007, 28, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez JF, Crooks DR, Lee CT, Schoen CJ, Amable R, Zeng X, Florival-Victor T, Morales N, Truckenmiller ME, Smith DR and Freed WJ, GABAergic lineage differentiation of AF5 neural progenitor cells in vitro, Cell Tissue Res, 2006, 324, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelley LA and Sternberg MJ, Protein structure prediction on the Web: a case study using the Phyre server, Nat Protoc, 2009, 4, 363–371. [DOI] [PubMed] [Google Scholar]
- 37.Anton A, Weltrowski A, Haney CJ, Franke S, Grass G, Rensing C and Nies DH, Characteristics of zinc transport by two bacterial cation diffusion facilitators from Ralstonia metallidurans CH34 and Escherichia coli, J Bacteriol, 2004, 186, 7499–7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin H, Kumanovics A, Nelson JM, Warner DE, Ward DM and Kaplan J, A single amino acid change in the yeast vacuolar metal transporters ZRC1 and COT1 alters their substrate specificity, J Biol Chem, 2008, 283, 33865–33873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selyunin AS, Iles LR, Bartholomeusz G and Mukhopadhyay S, Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking, J Cell Biol, 2017, 216, 3249–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selyunin AS and Mukhopadhyay S, A Conserved Structural Motif Mediates Retrograde Trafficking of Shiga Toxin Types 1 and 2, Traffic, 2015, 16, 1270–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y and Sabatini DM, mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase, Science, 2011, 334, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunn KW, Kamocka MM and McDonald JH, A practical guide to evaluating colocalization in biological microscopy, Am J Physiol Cell Physiol, 2011, 300, C723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]