

Abstract
Multi-drug resistant Gram-negative bacterial infections are on the rise, and there is a lack of new classes of drugs to treat these pathogens. This drug shortage is largely due to the challenge of finding antibiotics that can permeate and persist inside Gram-negative species. Efforts to understand the molecular properties that enable certain compounds to accumulate based on retrospective studies of known antibiotics have not been generally actionable in the development of Gram-negative antibiotics. A recent assessment of the ability of >180 diverse small molecules to accumulate in Escherichia coli led to predictive guidelines for compound accumulation in E. coli. These “eNTRy rules” state that compounds are most likely to accumulate if they contain a non-sterically encumbered ionizable Nitrogen (primary amines are the best), have low Three-dimensionality (Globularity ≤ 0.25), and are relatively Rigid (rotatable bonds ≤ 5). In this review, we look back through 50+ years of antibacterial research and 1000s of derivatives and assess this historical data set through the lens of these predictive guidelines. The results are consistent with the eNTRy rules, suggesting that the eNTRy rules may provide an actionable and general roadmap for the conversion of Gram-positive-only compounds into broad-spectrum antibiotics.
Keywords: Gram-negative bacteria, multi-drug resistance, antibacterials, molecular properties
The rise of drug-resistant bacteria
Bacterial resistance has been observed to every antibiotic introduced into the clinic,1 including drugs-of-last resort such as vancomycin,2-6 daptomycin,7, 8 and colistin.9-11 The increased rate of resistance, coupled with declining trends in discovery and development of new antibiotics,12-15 portends a crisis. Pathogens now exist that are resistant to all or almost all available antibiotics, resulting in an increased number of deaths from bacterial infections.16-18 In 2013, the Centers for Disease Control estimated that at least 23,000 people die per year in the United States from drug-resistant bacteria.19 Without improvements in antibacterial drug discovery and development this problem is only expected to get worse; in fact, it has been predicted that by 2050 10 million people worldwide will die per year from drug-resistant bacterial infections.20
Gram-negative bacteria as a major public health concern
Gram-negative bacteria have emerged as some of the most problematic pathogens in the last two decades.21-23 In 2008, Louis B. Rice published an editorial highlighting the pathogens that cause the most hospital-acquired infections and frequently “escape” the action of traditional therapeutics.14 The pathogens highlighted are collectively referred to as the ESKAPE pathogens: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae/Escherichia coli (Enterobacteriacaea), Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species. Four of the six ESKAPE pathogens (K. pneumoniae/E. coli, A. baumannii, P. aeruginosa, and Enterobacter species) are Gram-negative species. Increased hospital surveillance, the approval of new drugs, and improved hygiene have helped stem the rise in multidrug-resistant (MDR) Gram-positive infections,24-29 but the prevalence of MDR Gram-negative infections has continued to increase.13, 25, 30 In February of 2017, the World Health Organization issued a list of ‘Priority Pathogens’ for which new antibiotics are urgently needed.31 The list is broken down into three categories: critical, high, and medium priority. Nine out of the 12 pathogens listed are Gram-negative bacteria including all three Critical Priority Pathogens (carbapenem-resistant Enterobacteriaceae (CRE), A. baumannii, and P. aeruginosa).
No new class of antibiotics effective against Gram-negative bacteria has been introduced into the clinic since the fluoroquinolones in 1968,32 and clinicians have had to rely on later generation broad-spectrum antibiotics (that is, drugs that are effective against both Gram-positive and Gram-negative pathogens), particularly the carbapenems. Unfortunately carbapenem-resistance is also on the rise,33 and infections caused by CRE are estimated to have as much as 50% higher mortality rates than carbapenem-susceptible infections.34-36 With the increase in carbapenem-resistant infections, colistin, a drug that was previously removed from the clinic due to high toxicity, has been re-introduced to the clinic. Colistin is far from an ideal drug, as 50-60% of people taking colistin experience acute kidney injury.37-39 Additionally, colistin resistance has emerged, and alarmingly the resistance can be easily spread via horizontal gene transfer of plasmids.9-11, 40 The first plasmid associated with colistin resistance was discovered in 2015 in China,40 but has already spread throughout the world.9, 41-43 Pathogens that are resistant to both colistin and carbapenems are very challenging to treat, with mortality rates ranging from 20-70%.44-46
Gram-negative bacteria are impermeable to most small molecules
The lack of Gram-negative antibacterial discovery can be traced to the structure of the Gram-negative outer membrane. Unlike Gram-positive bacteria, Gram-negative bacteria have two cellular membranes (Fig. 1A). The outer leaflet of the outer membrane is unique to Gram-negative bacteria and is composed of lipopolysaccharides (LPS), or a similar lipid linked to a carbohydrate.47 LPS are made up of lipid A (Fig. 1B) and a long oligosaccharide.48 Lipid A molecules contain 4-7 acyl chains attached to a phosphorylated disaccharide (Fig. 1B). The oligosaccharide is attached directly to lipid A, and is usually negatively charged and differs by strain.49 The adjacent negative charges of the LPS are stabilized by divalent cations (Fig. 1B).50, 51 This stabilization, along with the fact that LPS lipid chains are predominately saturated (Fig. 1B),49 allows LPS molecules to stack together very tightly. This tight packing and the long, negatively charged oligosaccharides make passive diffusion of essentially all small molecules extremely challenging.
Figure 1.

Components of the Gram-negative cellular envelope. (A) The cellular envelope of Gram-negative bacteria (right) contains two lipid membranes, while Gram-positive bacteria (left) have only one lipid membrane. The outer leaflet of the outer-membrane of Gram-negative bacteria is composed of lipopolysaccharides (LPS). Most small molecules are unable to passively diffuse at an appreciable rate through the LPS layer. Instead, molecules enter through channel proteins called porins (yellow). Almost all compounds, however, are subject to multidrug efflux pumps. (B) The structure of lipid A. The core oligosaccharide is linked to the 6′ carbon. (C) Top view of OmpF, the prototypical porin. A negatively-charged loop extends into the center of the cavity across from a positively-charged wall, creating a narrow constriction site and preventing the passage of many compounds. The crystal structure was generated from PDB 1opf using the Molecular Operating Environment software.
As compounds cannot readily passively diffuse across the outer membrane, small molecules permeate the outer membrane through porins and/or the self-promoted uptake pathway. Porins are relatively narrow water-filled β-barrels lined interiorly with charged amino acids. OmpF is the major porin of E. coli and the prototypical porin, containing a negatively-charged loop that extends into the barrel, across from a positively-charged wall (Fig. 1C).52 This ‘constriction zone’ is approximately 7 by 11 Å,52 allowing only certain molecules to rapidly diffuse through the channel.
Certain compounds, including β-lactams, chloramphenicol, tetracyclines, and fluoroquinolones, can enter Gram-negative bacteria through porins (Fig. 2).53 Other small molecules can pass through the outer membrane via the self-promoted uptake pathway by displacing the divalent cations, and temporarily destabilizing the LPS layer;54-57 compounds that utilize the self-promoted pathway are typically polycationic,57, 58 and include the polymyxins,59, 60 the aminoglycosides,61 and azithromycin62 (Fig. 2). However, it has been difficult to discern precisely how antibiotics penetrate Gram-negative bacteria, with the belief being that some compounds likely enter through a combination of both pathways (Fig. 2). For example, it has been hypothesized that compounds with metal-binding motifs can also utilize the self-promoted pathway.63 As such, it has been proposed that fluoroquinolones, which contain a metal-binding β-ketoacid,62, 64-66 and tetracyclines, which are known to bind metals through their 1,3-keto-enol system,67-69 cross the outer membrane through both mechanisms (Fig. 2). The permeation of fluoroquinolones and tetracyclines through the self-promoted uptake pathway is debated in the literature.70, 71 Self-promoted uptake is most often determined by one of two assays. In one assay, additional Mg2+ is added to the media; if the antibiotic activity is significantly decreased, compounds are assumed to enter through the self-promoted uptake system because they can no longer compete with the level of Mg2+ present.63 In the other assay, bacteria are incubated with the antibiotic of interest and a fluorescent small molecule. Self-promoted uptake requires temporary destabilization of the LPS layer, and compounds that utilize the self-promoted pathway can cause the bacteria to uptake the fluorescent compounds.62 The activity of tetracyclines and fluoroquinolones is decreased in the presence of Mg2+, but there is no consistency in the literature whether they promote uptake of fluorescent small molecules.15, 70, 71 It is also important to note that the addition of Mg2+ also reduces the susceptibility of Gram-positive bacteria to fluoroquinolones.70 This has led to the hypothesis that the addition of magnesium simply causes increased interaction between the cell wall of both Gram-positive and Gram-negative bacteria and the fluoroquinolones, preventing diffusion into the cells.70 Clearly, better assays for self-promoted uptake and a better understanding of this pathway are needed.
Figure 2.

Antibiotic structures and spectrum of activity. Broad-spectrum antibiotics (effective against Gram-positive and Gram-negative pathogens), and Gram-positive-only antibiotics. Although information on the precise mode of entry into Gram-negative bacteria is not definitive, data suggests that antibiotics can enter Gram-negative bacteria through porins, the self-promoted uptake, or through a combination of both. *Colistin is Gram-negative only.
Compounds that can cross the outer membrane are then subject to highly promiscuous efflux pumps (Fig. 1A),72 and essentially all small molecules are believed to be efflux substrates to some extent.73 Thus, to accumulate inside Gram-negative bacteria, small molecules must cross the outer membrane faster than they are pumped out. As such, many antibiotics cannot accumulate in Gram-negative species and are inactive against these pathogens; these are termed Gram-positive-only antibiotics (Fig. 2).
Another complicating factor is periplasmic versus cytoplasmic accumulation. It has been proposed that the outer and inner membrane of Gram-negative bacteria have orthogonal sieving properties, and that compounds able to cross the inner membranes most readily are neutral and relatively hydrophobic.74 We recently conducted a study where we analyzed compound accumulation in E. coli protoplasts (cells lacking an outer membrane), and a broad range of compounds tested were able to accumulate, including acidic compounds.75 Additionally, ciprofloxacin, which contains a weak acid, is able to readily accumulate in the cytoplasm where it exerts its activity. Finally, in E. coli strains with a compromised outer membrane, fusidic acid and mupirocin, which both contain a carboxylic acid, are active. In general, investigations into the differences in the sieving properties of the outer and inner membrane are in their infancy; much more work is needed to fully address this important issue.
The inability of most small molecules to accumulate in Gram-negative bacteria has made the discovery of new antibiotics for these pathogens extremely challenging, despite significant efforts. In 1995, the first bacterial genome was sequenced,76 leading to optimism about the discovery of new antibiotics using target-based discovery approaches (through biochemical/in vitro assays). Between 1995 and 2004, 34 different companies published high-throughput screens for novel antibiotics. In total, >125 high-throughput screens of >60 novel targets were reported, with essentially none of the screens resulting in credible candidates for new antibiotics.77 Chan and coworkers from GlaxoSmithKline note that lack of cell permeability of hits was highly problematic as no rational structure-activity-relationship (SAR) processes existed to improve compound accumulation in bacteria.77 In 2007, Payne and coworkers summarized the results of 67 high-throughput biochemical screens at GlaxoSmithKline.78 In this review, the challenge of identifying hits and progressing hits into leads was again emphasized. Only 16 of the screens resulted in hits (“hit” was defined as a chemically tractable, low-micromolar potency inhibitor of the target and, where appropriate, at least tenfold selectivity against the mammalian version of the target). Of these initial hits, only five were successfully converted into leads with whole-cell antibacterial activity, and only against Gram-positive bacteria.78 One of the major problems with identifying hits that Payne and coworkers note is a lack of molecular diversity in the screening library, as assessed by principle component analysis.79 Researchers at AstraZeneca observed that improving molecular diversity is not necessarily sufficient for improving antibacterial drug discovery efforts, as they experienced little difficulty in finding initial hits (“hit” was defined as a compound with a lack of activity in artifact assays but a reproducible dose response, typically in two separate assays with similar conditions but orthogonal detection systems).80 However, like GlaxoSmithKline, they struggled to convert hits active against purified enzymes into leads with whole-cell activity, particularly against Gram-negative bacteria.
Frustrations with target-based screens led GlaxoSmithKline, and others, to attempt whole-cell screening.81-83 Whole-cell screening has the advantage that any hits are cell-permeable (or at least have whole-cell activity), but the screening of standard compound collections for Gram-negative antibacterial leads has also proven fruitless.1, 77, 78, 80, 84 As one example, GlaxoSmithKline reported a whole-cell screen of 500,000 compounds against E. coli and S. aureus.78 Interestingly, hundreds of compounds with activity against S. aureus, but not E. coli, were identified. Most of these compounds were later ruled out as non-specific membrane-active agents.78 However, the fact that these non-specific membrane permeabilizers had no effect on E. coli demonstrates just how impenetrable the outer membranes of Gram-negative species are to small molecules. In total, three tractable hits were identified for S. aureus and no hits were identified for E. coli.78 Combined analysis of this sobering HTS data – lack of any new Gram-negative actives from the collective screening of >8 million compounds at GlaxoSmithKline, AstraZeneca, Merck and others78, 80, 84, 85 – led to the conclusion that an improved understanding of compound penetration into Gram-negative bacteria would be necessary to make meaningful progress toward rational discovery/design of broad-spectrum antibiotics,78 and this call has been echoed by others.74, 86-89
Understanding compound uptake in Gram-negative bacteria: retrospective studies
The structure of the Gram-negative cellular envelope dictates that only certain small molecules can accumulate inside these bacteria. In 2008, O'Shea and Moser quantified the molecular properties of Gram-negative antibacterials (broad-spectrum antibiotics) via a retrospective analysis of known antibacterial drugs.90 In this landmark study, physicochemical properties were calculated for 147 marketed antibiotics and several thousand non-antibacterial drugs represented by a shortened list of the Comprehensive Medicinal Chemistry Database (CMC). They determined that antibiotics with Gram-negative activity tend to be smaller than Gram-positive-only antibiotics, with a molecular weight cutoff around 600 Da, consistent with the narrow porin channel. Additionally, Gram-negative active antibiotics were found to be much more polar on average than both Gram-positive-only antibacterials and non-antibacterial drugs. The difference in polarity was most clearly observed by comparing ClogD7.4 values, which accounts for charge at physiological pH. O'Shea and Moser observed that Gram-negative antibacterials have unique physicochemical properties that generally do not follow Lipinski's rules of five. This succinct analysis showing differences in physicochemical features of approved antibiotics was a significant advance; however, there are several limitations to this study. O'Shea and Moser analyzed clinically approved antibiotics, therefore the compounds studied were from only a few drug classes. In fact, the Gram-negative-active antibiotics are predominately from only five classes (tetracyclines, sulfa drugs, Gram-negative β-lactams, fluoroquinolones, and aminoglycosides), and the properties of each class cluster together.90 In addition, this retrospective study does not explain why certain antibacterials are small and relatively polar, but are still only active against Gram-positive species. Finally, like any retrospective study relying on antibacterial data, detailed information cannot be gleaned about the types of compounds that can and cannot accumulate in Gram-negative bacteria. While information from this study has not been reliably actionable in facilitating the conversion of Gram-positive-only antibacterials into broad-spectrum agents, it did provide important insights into the properties of broad-spectrum antibiotics.
Another retrospective study was published by AstraZeneca in 2014.84 In comparison to the O'Shea and Moser study, this article focuses significantly more on the challenges of advancing initial hits into leads with respect to physicochemical properties. However, like the O'Shea and Moser study, the AstraZeneca study begins with an analysis of antibacterial properties of compounds with whole-cell antibacterial activity. AstraZeneca assessed more than 3,000 compounds generated from existing antibacterial drug programs. In the analysis, they examined each species of bacteria independently, and analyzed net charge, in addition to many of the properties O'Shea and Moser assessed. The results of this assessment are broadly similar to those of O'Shea and Moser, with antibacterials with Gram-negative activity having much lower ClogD7.4 values on average than antibacterials with Gram-positive-only activity.84 The analysis of ionic charge showed that most compounds with Gram-negative antibacterial activity are charged, and this is split between acids and bases. A limitation of this report is that the diversity of the collection cannot be determined because compound structures are not provided. While a large percentage of Gram-negative-active compounds contain carboxylic acids, the authors note that these are almost all β-lactams,84 where the acid is required for activity. The inability to differentiate properties necessary for target engagement/antibacterial activity from those necessary for accumulation is a major limitation of any study relying on antibacterial data.
Understanding compound uptake in Gram-negative bacteria: whole-cell studies
In principle, whole-cell compound accumulation studies are the ideal method to understand small-molecule accumulation in Gram-negative bacteria as they do not rely on antibacterial activity, opening up the possibility of assessing many more structural classes. However, the challenges of implementing a whole-cell compound accumulation assay on scale has, until recently, precluded systematic analyses of large numbers of structurally diverse compounds. Accumulation of small numbers of compounds has been assessed with radiolabeling or by taking advantage of certain physical or reactive properties of a single antibiotic class.91-104 For example, Nikaido and coworkers assessed the permeation of cephalosporins into E. coli using a micro-iodometric assay. The micro-iodometric assay is a colorimetric assay that relies on the deep blue color of triiodide complexed with starch.96-98 In an aqueous solution of iodine and potassium iodide, triiodide is present at equilibrium. Penicillins hydrolyzed by β-lactamases to penicilloic acid reduce iodine much more readily than intact penicillins, resulting in a decrease in triiodide and decolorization of the starch-triiodide mixture. The benefit of this assay is that the reaction can be quenched by deactivating β-lactamases, thus not requiring removal of extracellular compound before analysis. However, only β-lactams can be assessed, preventing utility of the assay in assessment of other compounds.99
In another example, Bazile and coworkers measured the accumulation of fluoroquinolones in bacteria. For this analysis, bacteria were separated from extracellular compound using silicone oil, the bacteria were lysed, and then the concentration of fluoroquinolone in the supernatant was determined by fluorescence.100 Washing bacteria by centrifuging through silicone oil is a method that was first introduced in 1965,105 and it has been validated that only a very small volume of extracellular buffer remains after centrifugation relative to the volume inside of the cell.106 A challenge is to rigorously ensure that this method removes any compound that may non-specifically interact with the outer membrane of bacteria, and Bazile and coworkers did not report controls to account for this possibility. More recently, a significant advance has been made toward a general method to analyze data from accumulation studies using liquid chromatography-tandem mass spectrometry (LC-MS/MS).107 Taking advantage of this methodology, Davis and coworkers measured the accumulation of sulfonyladenosines,108 using a similar setup as Bazile and coworkers. Instead of washing the bacteria by centrifuging through silicone oil, the bacteria were washed four times with an aqueous buffer. The number of washes was validated by determining that additional washes had no additional effect on readout.108 Additionally, in the past year, there has been an effort to understand the effect of various membrane components on net accumulation in whole cells, including studying the relationship between specific molecular changes in a substrate and permeation through an isolated porin in whole cells,109 and studying the synergy of efflux and outer membrane influx using hyperporinated strains of Gram-negative bacteria.110
We set an ultimate goal of evaluating close to 200 diverse compounds for whole-cell accumulation, thus it was critical for us to implement a rigorous accumulation assay fully validated with multiple controls. Most important was to be cognizant of the possibility of non-specific binding of compound to the membrane; that is, a compound giving a false positive signal when it is in actuality not a true accumulator. To safeguard against this we envisioned controls with well-known Gram-positive-only and broad-spectrum drugs, control experiments where compound accumulation is artificially reduced (through use of a porin knockout), and control experiments where the amount of compound accumulation is artificially increased (through use of a membrane permeabilizer such as colistin). We thus optimized an accumulation assay in E. coli using LC-MS/MS as a readout, rapidly separating the bacterial pellets from the buffer during the wash step to prevent compound loss through efflux after extracellular compound is removed. Like Bazile and coworkers, this was done by layering the bacteria on cold silicone oil in an Eppendorf tube and centrifuging; the bacteria pellets below the oil and the aqueous layers stays above the oil (Fig. 3). To validate the assay and wash procedure (depicted in Figure 3), control antibiotics were tested for accumulation. For high-accumulating controls, antibiotics were utilized with activity against E. coli, including tetracycline, ciprofloxacin, and chloramphenicol. For low-accumulating controls, antibiotics were utilized that are not active against E. coli because they cannot accumulate, including novobiocin, erythromycin, rifampicin, vancomycin, daptomycin, clindamycin, mupirocin, and fusidic acid. Ampicillin was also used as a negative-control because it is rapidly covalently modified and thus cannot be measured by LC-MS/MS. A significant increase in accumulation was observed for the high-accumulating controls over the low-accumulating controls and ampicillin (Fig. 4A) with the silicone oil wash alone; additional aqueous washes were assessed, but no improvements in signal-to-noise with the controls were observed.75 To further ensure that the assay was not just reporting on nonspecific interactions with the outer membrane, accumulation was also assessed in isogenic strains. Accumulation of the high-accumulating controls was significantly lower in a porin-deficient strain of E. coli compared to the wild-type strain (Fig. 4B).75 As a final control, the accumulation of low-accumulating controls was measured in E. coli permeabilized with colistin, and a significant increase in accumulation was observed (Fig. 4C).75 All experiments were performed in aqueous buffer (as opposed to media), with our data suggesting influx and efflux are not deleterious affected under these conditions. While, as described in the previous paragraphs, there are several variations of this accumulation assay, our experience has been that it is important to use the silicone separation step (versus multiple wash steps), and to calculate accumulation by using Colony Forming Units (CFUs) (which gives much more accurate data than use of OD600).
Figure 3.
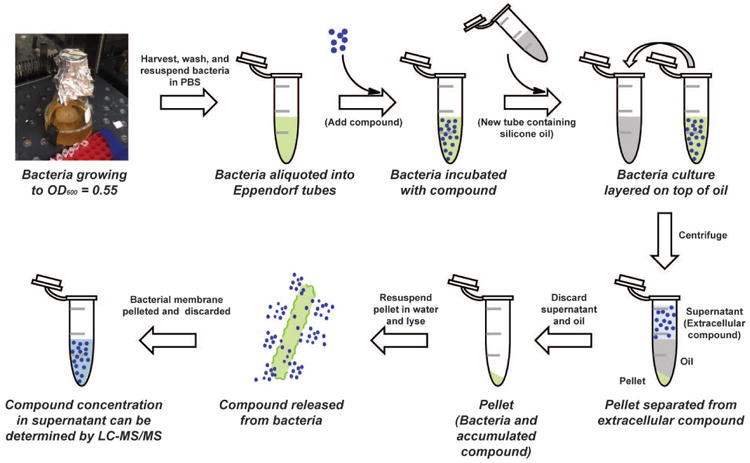
Workflow for the accumulation assay. After a culture of bacteria is grown to mid-log phase, bacteria are harvested and washed with sterile phosphate-buffered saline (PBS). The pellet is then resuspended in a much smaller volume of PBS, and aliquoted into Eppendorf tubes. The cultures are then incubated with compound of interest for 10 minutes. To separate the bacteria from extracellular compound, cultures are layered on cold silicone oil and centrifuged. The oil forms a layer between the bacterial pellet and the supernatant. After disposal of both the supernatant and the oil, the pellets are resuspended in water and the bacteria are lysed by freeze-thawing. The lysate is centrifuged to remove cellular debris, and the supernatant is analyzed by LC-MS/MS.
Figure 4.
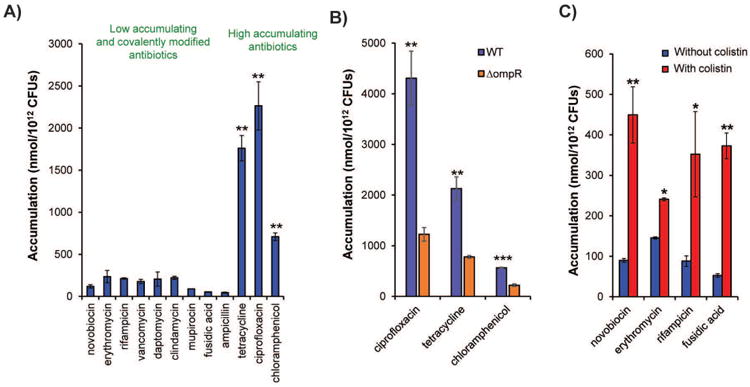
Validation of compound accumulation assay in E. coli. (A) Compounds that are active against E. coli have significantly higher accumulation than low-activity antibiotics and ampicillin. Statistical significance was determined by using a two-sample Welch's t-test (one-tailed test, assuming unequal variance) relative to the negative controls. P values relative to the average of the low-accumulating controls; (B) Accumulation comparison of ciprofloxacin, tetracycline, and chloramphenicol for E. coli ΔompR vs parental strain E. coli BW25113. (C) Co-treatment with colistin enhances the accumulation of low-accumulating antibiotics in E. coli. For B and C statistical significance was determined by using a two-sample Student's t-test (two-tailed test, assuming equal variance). *P < 0.05, *P < 0.01, *P < 0.001 All experiments were performed in biological triplicate. Error bars represent s.e.m. Data for this figure is taken from Richter et al.75 with permission.
Having validated a general assay for measuring compound accumulation in E. coli, we then assessed the ability of a diverse set of small molecules, as determined by Tanimoto similarity coefficients,111 to accumulate in E. coli, and utilized the results to develop predictive guidelines for compound accumulation in Gram-negative species, both experimentally and computationally.75 For the initial testing, we examined the accumulation of 100 compounds with diversity both in terms of structure and charge, including positively charged, negatively charged, and neutral compounds, and observed 12 accumulating compounds. All 12 accumulators were positively charged and 9 of the 12 contained a primary amine. To examine the importance of the primary amine, we performed extensive structure-activity relationship studies by systematically converting the primary amines to other functional groups, as well as more substituted amines. In all cases, we observed that the compounds with primary amines accumulated the best, with few other compounds showing any accumulation over the negative controls. However, we also observed that a primary amine was not sufficient for accumulation. To test which other molecular properties were important for accumulation, we expanded our library of primary amines, assessed accumulation, and then analyzed the data computationally. We calculated 297 molecular descriptors for each primary amine based on ensembles of conformations generated in MOE using the LowModeMD method with default settings, and these descriptors were used to train a random forest classification model that predicts amine accumulation.75 Based on these analyses the following guiding principles for compound accumulation in E. coli, called “eNTRy Rules”, were developed: compounds are most likely to accumulate if they contain a non-sterically encumbered ionizable Nitrogen (primary amines are the best in our study but retrospective studies of antibiotics suggest more substituted amines can accumulate as well), have low Three-dimensionality (Globularity ≤ 0.25), and are relatively Rigid (rotatable bonds ≤ 5) (Fig. 5A).75 Mathematically, globularity is the inverse condition number of the covariance matrix of atomic coordinates (a value of 1 indicates a perfect sphere while a value of 0 indicates a two- or one-dimensional object); more intuitively, benzene (Glob=0) and adamantane (Glob=1) represent the limits of the Glob scale. Rotatable bonds is a count of single bonds, not in a ring, bound to a non-terminal heavy atom, excluding C–N amide bonds because of their high rotational energy barrier. A final facet is that accumulation is most likely when compounds have some non-polar functionality; the computational model identified increased amphiphilic moment (vSurf_A), which is a measure of a vector pointing from the center of the hydrophobic domain to the center of the hydrophilic domain, with the vector length being proportional to the strength of the amphiphilic moment,112 as a trait favoring accumulation. Compounds lacking a strong amphiphilic moment (i.e., do not have a hydrophobic region across the molecule from the primary amine) are unlikely to accumulate. The vast majority of compounds that meet the eNTRy rules in the test set accumulate in E. coli (Fig. 5B).
Figure 5.
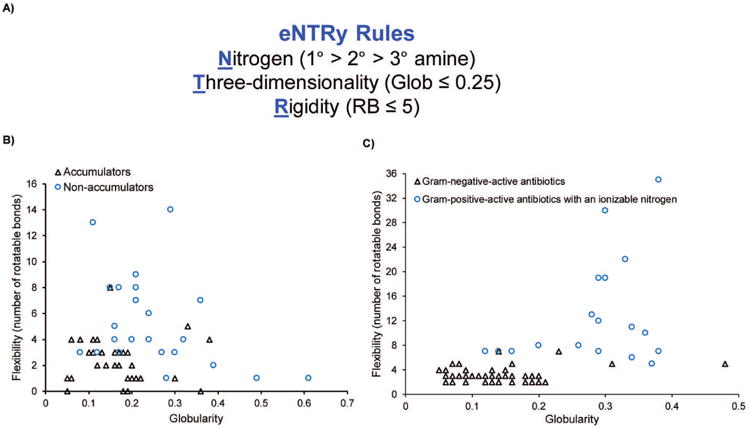
eNTRy Rules. (A) Compounds are most likely to accumulate in Gram-negative bacteria if they contain a non-sterically encumbered ionizable Nitrogen (we observed primary amines are the best), have low Three-dimensionality (Globularity ≤ 0.25), and are relatively Rigid (rotatable bonds ≤ 5). Additionally, some degree of hydrophobicity is required. (B) Flexibility (as measured by the number of rotatable bonds) plotted against globularity for the primary amines tested in Richter et al.75 with sufficient hydrophobicity. (C) Flexibility versus globularity for antibiotics. Antibiotics included are Gram-positive-only drugs containing an ionizable nitrogen (triangles), and Gram-negative actives that are believed to enter cells through porins (circles). Data for this figure is taken from Richter et al.75 with permission.
To test the validity of these guidelines, the set of antibacterials assessed by O'Shea and Moser90 was evaluated for charge, flexibility, and globularity. For the charge analysis, antibacterial drugs containing an ionizable nitrogen were accepted, and for Gram-negative antibacterials, only those predicted to enter through porins were included. For these drugs with ionizable nitrogens, compounds active against Gram-negative bacteria largely separate from those with Gram-positive-only activity based on these two physicochemical parameters, with the exception of one fluoroquinolone and two tetracyclines (Fig. 5C). No Gram-positive-only antibacterial with an ionizable nitrogen has the correct rigidity and globularity for accumulation in E. coli (Fig. 5C).
The eNTRy rules we developed differ in important ways from the conclusions from the retrospective analyses. Most obviously, the importance of an ionizable nitrogen is apparent in our study. In addition, we observe that MW is an imperfect surrogate for size and shape, as we report on head-to-head comparisons of series of compounds that have low MW (<400 Da) and a primary amine, but differ in the number of RBs, or differ in their Glob values.75 From these comparisons it can be seen that polar, low MW compounds still will not accumulate if they have high RBs or Glob. It is likely that three-dimensional measures of size/shape, such as RB and Glob, more accurately predict accumulation than MW, and the results of these studies emphasize the importance of the ability to systematically compare structurally similar compounds head-to-head in an accumulation assay.
An important lesson from the combined analysis of the retrospective studies, the failures of whole-cell screening, and whole-cell accumulation assays is the critical importance of the compound collection. Our own analysis suggests that of the ∼150,000 compounds in the Chembridge Library (a standard commercial screening collection), less than 0.1% meet the eNTRy guidelines,113 nicely explaining the failures of large-scale screens searching for Gram-negative antibiotics. The use of a set of complex and diverse compounds was absolutely essential for the development of the eNTRy rules. These compounds, produced by the complexity-to-diversity method,111, 114-116 possess structural features that are largely absent from commercial screening collections, including contiguous/overlapping ring systems and primary amines. The use of such structurally diverse compounds will be critical to future studies that seek to further refine the eNTRy rules and apply them to other microorganisms.
The lack of primary amines in most commercial screening libraries is most likely due to challenges with the synthesis and purification of primary amine-containing compounds. Additionally, there can be concerns about the liabilities of primary amines (e.g., increased risk of hERG signals). To further investigate this, we recently analyzed a database of approved drugs from around the world, and observed that almost 8% of drugs in clinical use contain a primary amine, including top selling drugs given for chronic conditions, such as Lyrica and Januvia.113 As it is true that antibiotics are often taken at far higher doses than drugs for other indications, we were curious about the presence of primary amines in approved antibiotics, or antibiotics that have been shown to be safe in clinical trials. Our analysis shows that there are dozens of antibiotics that are safe for human use that contain a primary amine, including commonly employed antibiotics such as many β-lactams (ampicillin, amoxicillin, ceftolozane, cefadroxil, cephalexin, etc.), aminoglycosides (kanamycin, amikacin, sisomicin, netilmicin, etc.), vancomycin, daptomycin, colistin, and many antibiotics that progressed through various stages of clinical trials (nemonoxacin, plazomicin, surotomycin, cefilavancin, cefamulin, LTX-109, ACHN-975, epetraborole, murepavadin). This information indicates that there is no intrinsic problem with having a primary amine in an antibiotic.
Converting Gram-positive-only antibacterials into broad-spectrum agents
Most antibacterial targets are highly conserved across Gram-positive and Gram-negative species,117 and thus lack of compound entry/accumulation is the main reason antibiotics are not active against Gram-negative bacteria. This is demonstrated by the observation Gram-negative bacteria with a permeabilized outer membrane or deficient efflux pumps are generally susceptible to antibiotics that are not active against wild-type Gram-negatives.118-120 We therefore speculated that a Gram-positive-only antibiotic with the proper rigidity and three-dimensionality could be converted to a broad-spectrum antibacterial by the addition of an amine to a position that that does affect the ability of the compound to interact with its target. We applied the eNTRy rules to convert deoxynybomycin (DNM), a natural product that inhibits DNA gyrase and is only active against Gram-positive organisms,121, 122 into 6DNM-NH3, an antibiotic with activity against a diverse panel of multi-drug resistant Gram-negative pathogens (Fig. 6A).75 This conversion of DNM into broad-spectrum 6DNM-NH3 through the addition of an amine was intentional, but we were curious if other serendipitous examples existed in the literature. As described below and armed with the discovery about the importance of shape, flexibility, and the presence of a primary amine, by going back through 50+ years of antibacterial research and 1000s of derivatives, we can look for patterns in this historical data set and see if they are consistent with our predictive guidelines.
Figure 6.

Appending a sterically-unencumbered amine on relatively flat and rigid antibiotics can enhance their activity against Gram-negative bacteria. (A) Seven examples75, 128, 132-136, 151 of conversions of Gram-positive-only compounds into broad-spectrum antibiotics via addition of a primary amine, all are consistent with the eNTRy rules. (B) Two examples of conversions to broad-spectrum antibiotics that are not consistent with the eNTRy rules, both converted compounds are diamines and may enter Gram-negative bacteria through the self-promoted uptake pathway. RB = rotatable bonds; Glob = globularity; Broad-spectrum = activity against both Gram-positive and Gram-negative bacteria; PA = P. aeruginosa; MIC = minimum inhibitory concentration.
For this study, we focused on compounds with activity against Enterobacteriaceae (E. coli and Klebsiella), P. aeruginosa, and/or A. baumannii, which are all nonfastidious Gram-negative organisms. Fastidious bacteria, such as Neisseria gonorrhoeae and Helicobacter pylori, have complex nutritional requirements, and their outer membranes are significantly different than those of nonfastidious organisms.123-125 The same is true for Mycobacteria, which are often considered Gram-negative because they do not stain with Gram stain. However, unlike traditional Gram-negative species that contain a LPS layer, Mycobacteria contain a mycolic acid layer.126, 127 Due to the membrane differences between nonfastidious Gram-negative species, fastidious Gram-negative species, and Mycobacteria, the guidelines for compound accumulation are likely different. Herein, we are only considering nonfastidious Gram-negative species, and cannot conjecture about the permeability requirements of these other organisms. Throughout the rest of the review, the term “Gram-negative” will be used to describe the nonfastidious Gram-negative organisms, Enterobacteriaceae, A. baumannii, P. aeruginosa.
Successful Gram-positive to Gram-negative conversions
The most prominent example of the conversion of a Gram-positive-only antibacterial into a broad-spectrum agent through is the conversion of penicillin G to ampicillin. Penicillin G has flexibility/shape parameters that make it an outstanding candidate for conversion (RB = 4, Glob = 0.17), and addition of an amine results in ampicillin (RB = 4, Glob = 0.12), which now meets all criteria for accumulation (Fig. 6A). Penicillin G has virtually no activity against Gram-negative pathogens, whereas ampicillin kills E. coli below 4 μg/mL.128 Ampicillin was the first β-lactam with Gram-negative activity, and represented a major public health advance because other antibacterials with Gram-negative activity available in the 1960s worked poorly in vivo.129 Ampicillin is an interesting example because it is zwitterionic. As discussed in the ‘Summary and Outlook’ section, exactly how zwitterions fit into the broader context of the eNTRy rules is not yet well understood, and will require systematic studies with multiple classes of zwitterionic compounds.
The β-lactams are a prime example of why it is important to decouple antibiotic activity from accumulation when attempting to understand properties that favor accumulation. Many third and fourth generation β-lactams are relatively flexible and do not have a primary amine, but are active against Gram-negative species. However, these β-lactams have been experimentally shown to have reduced accumulation in Gram-negative bacteria compared to their positively-charged analogues that meet the flexibility/shape parameters.130 These compounds likely retain antibacterial activity due to the fact that third and fourth generation β-lactams are significantly more stable to β-lactamases than the early generation β-lactams.130
Although β-lactams are the only example of successful conversions through the addition of a primary amine that has reached the market, there are a handful of analogous conversions that can be found in the literature. One example is the DNA gyrase/topoisomerase IV inhibitors developed by RedX Pharma.131 REDX04139 (Fig. 6A) has potent activity against Gram-positive species, including fluoroquinolone-resistant S. aureus, but shows no activity (minimum inhibitory concentration (MIC) >128 μg/mL) against Gram-negative bacteria. Being relatively flat (Glob = 0.07) and rigid (RB = 2), REDX04139 is an ideal compound for conversion. REDX05931 is structurally identical to REDX04139, except for the addition of a primary amine (Fig. 6A). The amine does not affect its ability to inhibit DNA gyrase and topoisomerase IV, as demonstrated by both in vitro enzyme activity assays and activity against Gram-positive bacteria.131 REDX05931 shows significantly improved activity against E. coli with an MIC of 0.5 μg/mL. It is also moderately active against P. aeruginosa (MIC = 16 μg/mL) and Klebsiella pneumoniae (MIC = 8 μg/mL).131 An interesting aspect of the parent REDX04139 is that it contains an aniline. Anilines are the only ionizable nitrogen on certain broad-spectrum antibiotics, such as the sulfa drugs. Although most anilines are neutral at physiological pH, it could be hypothesized that in certain cases, the aniline is sufficient to aid in Gram-negative accumulation. In the case of these RedX Pharma tricyclic inhibitors, the aniline is apparently not sufficient for accumulation in Gram-negative pathogens, as evidenced by the lack of broad-spectrum activity of REDX04139 and other structurally similar compounds.131
Another class of DNA gyrase/topoisomerase inhibitors with broad-spectrum activity are a pyrimidoindole inhibitor series developed by Trius Therapeutics. The inhibitors were developed through structure guided discovery, beginning with a crystallographic fragment screen.132 The scientists at Trius Therapeutics recognized that compounds capable of Gram-negative penetration and retention would need to have low molecular weight, sufficient hydrophilic character, and functional groups with ionizable centers at physiological pH.132 They therefore chose a pyrimidoindole scaffold as a candidate for optimization because it contained a site for the introduction of charged functional groups. This scaffold is also relatively flat, rigid, and easy to derivatize; a patent on these pyrimidoindoles contains hundreds of compounds.133 Compounds lacking a primary amine are inactive against E. coli, even though they retain activity against S. aureus, such as Pyrimidoindole 812 in Figure 6A. However, compounds with a primary amine are active against Gram-negative bacteria. For example, Pyrimidoindole 799 meets the guidelines for Gram-negative accumulation (RB = 4; Glob = 0.05), and has single-digit MIC activity against Gram-negative species, including fluoroquinolone resistant E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa. Fortuitously, the amine aids in both enzyme inhibition potency and, apparently, accumulation.
LpxC is responsible for the first committed step of lipid A biosynthesis, which is necessary for the growth of most Gram-negative species, but is not present in Gram-positive species. Many LpxC inhibitors have been developed, but none have reached the market due to challenges with low solubility and in vivo toxicity.134 LpxC has a large, solvent-exposed pocket surrounded by hydrophilic amino acids. Researchers at Novartis hypothesized that they could improve existing LpxC inhibitors by linking water-solubilizing groups to extend into this pockets.134 In doing so, the number of rotatable bonds is increased and no primary amine is present. Although these flexible derivatives, such as Hydroxamic acid 1, are able to inhibit LpxC as evidenced by the retention of activity against efflux-pump knock out P. aeruginosa, they are inactive against wild-type P. aeruginosa, presumably due to limited accumulation (Fig. 6A). Hydroxamic acid 2, on the other hand, is more rigid than the other derivatives, contains a primary amine, and is active against wild-type P. aeruginosa. The different spectrum of activity between Hydroxamic acid 1 and Hydroxamic acid 2 is likely due to differences in accumulation. The molecular weight is also reduced from Hydroxamic acid 1 to Hydroxamic acid 2.
Another example that suggests addition of a primary amine to an appropriate scaffold aids in accumulation are the aminomethylbenzoxaboroles, which inhibit bacterial leucyl-tRNA synthetase (LeuRS).135 The derivative AN3016 is an ideal compound for conversion as it is relative flat and rigid (RB = 4; Glob = 0.08). The addition of amine results in AN3365 (RB = 5; Glob = 0.08) (Figure 6A).135 AN3016 and AN3365 are equally potent against a strain of E. coli lacking the outer membrane channel TolC that works in combination with efflux pumps (MIC = 2 μg/mL). However, against Gram-negative strains with efflux pumps intact, AN3365 retains activity, while AN3016 is significantly less active (Fig. 6A). The change in MIC between the derivatives is relatively modest against wild-type E. coli, with the MIC of AN3365 being 1 μg/mL, and the MIC of AN3016 being 4 μg/mL. However, against clinical isolates of Gram-negative bacteria, the difference is more pronounced, with the neutral derivative having essentially no activity.135
Like any functional group, addition of a primary amine can negatively affect target binding. The challenge of increasing accumulation and retaining target engagement is demonstrated by efforts to broaden the spectrum of oxazolidinones. The first approved oxazolidinone antibiotic, linezolid, was approved by the FDA in 2000 and is used for severe Gram-positive antibacterial infections, including methicillin-resistant S. aureus. Linezolid is a protein synthesis inhibitor that prevents tRNA from binding to the ribosome. In an effort to broaden the spectrum of oxazolididones, Takrouri and coworkers synthesized over 70 polar derivatives and tested their activity in S. aureus and E. coli.136 Cyano-analog DP-63 shows no activity in wild-type E. coli, although it is active against S. aureus (Figure 6A).136 Reduction of the cyano group of DP-63 results in a derivative with a primary amine, compound DP-326, and this derivative is the most active compound in wild-type E. coli (MIC = 51 μg/mL). However, it is more than six times less active against S. aureus than linezolid, indicating that although the amine may aid with accumulation into Gram-negative bacteria, it likely impedes target engagement (Fig. 6A).
While compounds in the examples cited above meet the eNTRy rules (primary amine, RB ≤ 5, Glob ≤ 0.25) and thus would be predicted to accumulate in Gram-negative bacteria, there are also examples where the flexibility and/or globularity parameters do not fit these parameters. One example is a series of pleuromutilin derivatives developed at Nabriva Therapeutics. By inverting the stereocenter at carbon 12 of pleuromutilin (Fig. 6B, numbered in pink), and appending two amines off position 12, pleuromutilin can be converted into a broad-spectrum agent.137 For example, Pleuromutilin derivative 1 is active only against Gram-positive bacteria. Pleuromutilin 66, on the other hand contains a long tail with a secondary amine and a primary amine, and has potent broad-spectrum activity (Fig. 6B). Pleuromutilin 66 would not be predicted to accumulate based on flexibility (RB = 13) and globularity (Glob = 0.41). A similar pattern is observed with the tetrahydropyran-based bacterial topoisomerase inhibitors.138 Tetrahydropyran 4 and Tetrahydropyran 21 are identical except the alcohol on Tetrahydropyran 4 is replaced with a primary amine on Tetrahydropyran 21 (Fig. 6B). Both derivatives would not be predicted to accumulate based on flexibility (RB = 7), but have moderate activity against Gram-negative species (Fig. 6B). With the addition of the primary amine, however, Tetrahydropyran 21 is 4-8× more potent against Gram-negative species than Tetrahydropyran 4 (Fig. 6B).
A key difference between the pleuromutilin and tetrahydropyran derivatives and the examples in Figure 6A is that the pleuromutilin and tetrahydropyran derivatives are both polycationic. Polycationic compounds are prone to enter via the self-promoted uptake pathway. While it remains to be investigated, it is quite possible that these pleuromutilin and tetrahydropyran derivatives do not follow the same guidelines as they may have a non-porin mediated mode of uptake.
Examples when the addition of an amine does not result in broad-spectrum activity
A primary amine on an antibacterial is not always sufficient to aid in Gram-negative accumulation as our data suggests that the primary amine needs to be embedded on a compound imbued with proper flexibility and shape parameters, unless the compound is polycationic and can accumulate via the self-promoted uptake pathway. As a result, there are many examples of Gram-positive-only antibacterials containing primary amines, including vancomycin (RB = 13; Glob = 0.28), daptomycin (RB = 35; Glob = 0.38), and the DNA gyrase inhibitor cyclothialidine139-141 (RB = 10; Glob = 0.23) (Fig. 7).
Figure 7.

A sterically-unencumbered amine on large and flexible Gram-positive-only antibiotics does not enhance their activity against Gram-negative bacteria. (A) Existing Gram-positive-only antibiotics that have a primary amine but do not meet the shape requirements for broad-spectrum activity. (B) Addition of a primary amine to compounds that do not meet the shape requirement does not broaden the spectrum of activity.140-143, 148, 149 RB = rotatable bonds; Glob = globularity.
The need for proper flexibility and shape parameters nicely explains unsuccessful attempts at broadening the spectrum of the macrolides and pleuromutilins. As one example from the macrolide class, there are no differences in the spectrum of activity observed for erythromycin versus 9(S)-erythromycylamine;142, 143 these compounds do not possess the appropriate RB and/or Glob parameters for accumulation (Fig. 7). It is worth noting that the macrolide azithromycin (Fig 2, RB = 7; Glob = 0.24) is potently active against E. coli and is one of the recommended antibacterials for E. coli infections,144-147 but azithromycin enters Gram-negative bacteria through the self-promoted pathway.62 Pleuromutilin derivatives containing a single primary amine (and no other cationic groups) also lack Gram-negative antibacterial activity, including valnemulin, which is approved for veterinary use.148 A direct comparison of a pleuromutilin derivative with a cyano-group on the side-chain versus an amino-group on the side-chain shows no change in antibiotic-spectrum (pleuromutilin-CN vs pleuromutilin-amine, Figure 7).149 These studies suggest the importance of considering flexibility and shape parameters when attempting to broaden the spectrum of Gram-positive only antibiotics.
Summary and outlook
Broad-spectrum antibacterial discovery has been hindered by a lack of understanding of the types of compounds that can accumulate in Gram-negative species. A recent unbiased assessment of the ability of diverse compounds to accumulate in E. coli led to the development of predictive guidelines for compound accumulation. These results suggest that compounds are most likely to accumulate if they contain a non-sterically encumbered ionizable Nitrogen (primary amines are the best), have low Three-dimensionality (Globularity ≤ 0.25), and are relatively Rigid (rotatable bonds ≤ 5).108 With these experimentally determined eNTRy rules, it is now possible to to provide further insights into past screening as a to complement analyses of these failures in previous reviews.13, 78, 80, 89, 90 A key aspect of the eNTRy rules is the presence of an ionizable nitrogen, particularly a primary amine. Primary amines are essentially absent in large screening libraries, with about 0.1% of compounds in a standard commercial screening collection containing a primary amine.113 It should now be possible to synthesize collections of compounds that meet the eNTRy rules (and are hence strongly biased for accumulation in E. coli and likely other Gram-negatives), and such compounds could be then used in whole-cell screens for Gram-negative active antibiotics. Furthermore, use of the eNTRy rules should help guide efficient optimization of hits from target-based screens and Gram-positive-only leads into compounds with broad-spectrum activity. Finally, the eNTRy rules explain why the spectrum of certain antibiotics can be broadened with the addition of a primary amine, while others cannot.
One of the biggest questions moving forward is how well the eNTRy rules apply to diverse species of Gram-negative bacteria. The eNTRy rules were developed with E. coli, and based on the eNTRy rules, we designed and synthesized 6DNM-NH3, and observed that it was active against a broad range of clinical isolates, including strains of E. coli, Klebsiella pneumoniae, and A. baumannii; this result suggests that there is at least some overlap between compounds that can accumulate in the different Gram-negative species.75 However, the extent of this overlap is not yet defined, and a full understanding will require similar accumulation studies performed for other classes of bacteria. Choice of an appropriate compound collection will be most critical to any study of accumulation, as large amounts of data indicate that compounds in standard commercial screening collections contain vanishingly few accumulators in Gram-negative bacteria.75
An additional avenue that needs to be explored is what form of positive charge is accepted. For the development of the eNTRy rules, primary amines were thoroughly assessed, but ionizable nitrogens can take other forms. Further exploration of the other forms the ionizable nitrogen can take, including anilines, heterocycles, N-oxides, secondary amines, etc, will be critical. Exploration of zwitterions is especially intriguing as some broad-spectrum antibiotics are zwitterionic (e.g. β-lactams and fluoroquinolones), and most porin channels have juxtaposed positively-charged and negatively-charged regions, suggesting zwitterions may interact very well within the porin cavity.150 It will be interesting to see how the spatial relationship of the amine and the carboxylic acid affects accumulation, and if this relationship is variable from species-to-species with distinctly different porin cavities. Information on such issues will be best gleaned from detailed studies on series of compounds where these functional groups are systematically placed a range of distances away from each other. These studies and others will aid in the understanding of how zwitterions fit in the broader context of the eNTRy rules.
Finally, a major remaining question is the role of self-promoted uptake in compound accumulation, and its relationship with porin-mediated uptake. It does seem that there is some overlap in the physiochemical features of compounds that go through the self-promoted and porin-mediated pathways, most notably the presence of an amine (or multiple amines). Further definition of the molecular properties required for self-promoted uptake could lead to the systematic exploitation of this mechanism for the creation of broad-spectrum antibiotics. However, this will first require a reliable and rigorously validated assay that definitively reports on self-promoted uptake into Gram-negative bacteria.
Unbiased accumulation studies, where accumulation is decoupled from antibacterial activity, have been extremely useful in helping to understanding compound accumulation in Gram-negative bacteria. The guidelines developed help to explain many of the past successes and failures in antibiotic drug development. Although large-scale accumulation studies on multiple different Gram-negative pathogens will take time and require a significant resource investment, such experiments will lead to a fuller picture of the molecular properties required for compound accumulation and speed antibiotic development.
Acknowledgments
We thank the University of Illinois, Urbana-Champaign for funding our work, Bryon Drown for performing computational calculations and for providing insight, and Dr. Elizabeth Parkinson for generating the OmpF structure in the Molecular Operating Environment software. We are grateful to the NIH (R01GM118575) and the University of Illinois for funding this work.
Footnotes
Competing interests: The authors declare no competing interests
References
- 1.Silver LL. Challenges of Antibacterial Discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uttley AH, Collins CH, Naidoo J, George RC. Vancomycin-resistant enterococci. Lancet. 1988;1:57–8. doi: 10.1016/s0140-6736(88)91037-9. [DOI] [PubMed] [Google Scholar]
- 3.Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med. 1988;319:157–61. doi: 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 4.Frieden TR, Munsiff SS, Low DE, Willey BM, Williams G, et al. Emergence of vancomycin-resistant enterococci in New York City. Lancet. 1993;342:76–9. doi: 10.1016/0140-6736(93)91285-t. [DOI] [PubMed] [Google Scholar]
- 5.Boyce JM. Vancomycin-resistant enterococci: pervasive and persistent pathogens. Infect Control Hosp Epidemiol. 1995;16:676–9. doi: 10.1086/647040. [DOI] [PubMed] [Google Scholar]
- 6.Centers for Disease C, Prevention. Nosocomial enterococci resistant to vancomycin--United States, 1989-1993. Morb Mortal Wkly Rep. 1993;42:597–9. [PubMed] [Google Scholar]
- 7.Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–78. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humphries RM, Kelesidis T, Tewhey R, Rose WE, Schork N, et al. Genotypic and phenotypic evaluation of the evolution of high-level daptomycin nonsusceptibility in vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother. 2012;56:6051–3. doi: 10.1128/AAC.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGann P, Snesrud E, Maybank R, Corey B, Ong AC, et al. Escherichia coli Harboring mcr-1 and blaCTX-M on a Novel IncF Plasmid: First Report of mcr-1 in the United States. Antimicrob Agents Chemother. 2016;60:4420–1. doi: 10.1128/AAC.01103-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pragasam AK, Shankar C, Veeraraghavan B, Biswas I, Nabarro LE, et al. Molecular Mechanisms of Colistin Resistance in Klebsiella pneumoniae Causing Bacteremia from India-A First Report. Front Microbiol. 2016;7:2135. doi: 10.3389/fmicb.2016.02135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16:161–8. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 12.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, et al. The Epidemic of Antibiotic-Resistant Infections: A Call to Action for the Medical Community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155–64. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 13.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, et al. Bad bugs, no drugs: no ESKAPE! An update from the infectious diseases society of america. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 14.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis. 2008;197:1079–81. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 15.Laxminarayan R, Duse A, Wattal C, Zaidi AKM, Wertheim HFL, et al. Antibiotic resistance—the need for global solutions. Lancet Infect Dis. 2013;13:1057–98. doi: 10.1016/S1473-3099(13)70318-9. [DOI] [PubMed] [Google Scholar]
- 16.Boucher HW, Corey GR. Epidemiology of Methicillin-Resistant Staphylococcus aureus. Clin Infect Dis. 2008;46:S344–S9. doi: 10.1086/533590. [DOI] [PubMed] [Google Scholar]
- 17.Falagas ME, Bliziotis IA. Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era? Int J Antimicrob Agents. 2007;29:630–6. doi: 10.1016/j.ijantimicag.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Falagas ME, Tansarli GS, Karageorgopoulos DE, Vardakas KZ. Deaths attributable to carbapenem-resistant Enterobacteriaceae infections. Emerg Infect Dis. 2014;20:1170–5. doi: 10.3201/eid2007.121004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.CDC. Antibiotic resistance threats in the United State, 2013 2013 [Google Scholar]
- 20.O'Neill J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. London: Review on Antimicrobial Resistance, 2014; 2014. [Google Scholar]
- 21.Vasoo S, Barreto JN, Tosh PK. Emerging issues in gram-negative bacterial resistance: an update for the practicing clinician. Mayo Clin Proc. 2015;90:395–403. doi: 10.1016/j.mayocp.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Peleg AY, Hooper DC. Hospital-acquired infections due to gram-negative bacteria. N Engl J Med. 2010;362:1804–13. doi: 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zowawi HM, Harris PNA, Roberts MJ, Tambyah PA, Schembri MA, et al. The emerging threat of multidrug-resistant Gram-negative bacteria in urology. Nat Rev Urol. 2015;12:570–84. doi: 10.1038/nrurol.2015.199. [DOI] [PubMed] [Google Scholar]
- 24.Hacek DM, Paule SM, Thomson RB, Jr, Robicsek A, Peterson LR. Implementation of a universal admission surveillance and decolonization program for methicillin-resistant staphylococcus aureus (MRSA) reduces the number of MRSA and total number of S. aureus isolates reported by the clinical laboratory. J Clin Microbiol. 2009;47:3749–52. doi: 10.1128/JCM.01223-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson AP. Surveillance of antibiotic resistance. Philos Trans R Soc B. 2015;370:20140080. doi: 10.1098/rstb.2014.0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson AP, Pearson A, Duckworth G. Surveillance and epidemiology of MRSA bacteraemia in the UK. J Antimicrob Chemother. 2005;56:455–62. doi: 10.1093/jac/dki266. [DOI] [PubMed] [Google Scholar]
- 27.Kavanagh KT, Calderon LE, Saman DM, Abusalem SK. The use of surveillance and preventative measures for methicillin-resistant staphylococcus aureus infections in surgical patients. Antimicrob Resist Infect Control. 2014;3:18. doi: 10.1186/2047-2994-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, Fortin E, Tremblay C, Ngenda-Muadi M, Garenc C, et al. Hospital-Acquired Methicillin-Resistant Staphylococcus aureus Bloodstream Infections in Quebec: Impact of Guidelines. Infect Control Hosp Epidemiol. 2017:1–8. doi: 10.1017/ice.2017.81. [DOI] [PubMed] [Google Scholar]
- 29.Pearson A, Chronias A, Murray M. Voluntary and mandatory surveillance for methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S. aureus (MSSA) bacteraemia in England. J Antimicrob Chemother. 2009;64:11–7. doi: 10.1093/jac/dkp260. [DOI] [PubMed] [Google Scholar]
- 30.Agata X, EM C. Rapidly rising prevalence of nosocomial Multidrug-Resistant, Gram-Negative Bacilli: A 9-Year Surveillance Study. Infect Control Hosp Epidemiol. 2004;25:842–6. doi: 10.1086/502306. [DOI] [PubMed] [Google Scholar]
- 31.Willyard C. The drug-resistant bacteria that pose the greatest health threats. Nature. 2017;543:15. doi: 10.1038/nature.2017.21550. [DOI] [PubMed] [Google Scholar]
- 32.Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12:371–87. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 33.Vital signs: carbapenem-resistant Enterobacteriaceae. Morb Mortal Wkly Rep. 2013;62:165–70. [PMC free article] [PubMed] [Google Scholar]
- 34.Borer A, Saidel-Odes L, Riesenberg K, Eskira S, Peled N, et al. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol. 2009;30:972–6. doi: 10.1086/605922. [DOI] [PubMed] [Google Scholar]
- 35.Patel G, Huprikar S, Factor SH, Jenkins SG, Calfee DP. Outcomes of carbapenem-resistant Klebsiella pneumoniae infection and the impact of antimicrobial and adjunctive therapies. Infect Control Hosp Epidemiol. 2008;29:1099–106. doi: 10.1086/592412. [DOI] [PubMed] [Google Scholar]
- 36.Schwaber MJ, Klarfeld-Lidji S, Navon-Venezia S, Schwartz D, Leavitt A, Carmeli Y. Predictors of Carbapenem-Resistant Klebsiella pneumoniae Acquisition among Hospitalized Adults and Effect of Acquisition on Mortality. Antimicrob Agents Chemother. 2008;52:1028–33. doi: 10.1128/AAC.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zavascki AP, Nation RL. Nephrotoxicity of Polymyxins: Is There Any Difference between Colistimethate and Polymyxin B? Antimicrob Agents Chemother. 2017;61 doi: 10.1128/AAC.02319-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Nation RL, Milne RW, Turnidge JD, Coulthard K. Evaluation of colistin as an agent against multi-resistant Gram-negative bacteria. Int J Antimicrob Agents. 2005;25:11–25. doi: 10.1016/j.ijantimicag.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Carlson-Banning KM, Chou A, Liu Z, Hamill RJ, Song Y, Zechiedrich L. Toward repurposing ciclopirox as an antibiotic against drug-resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae. PLoS One. 2013;8:e69646. doi: 10.1371/journal.pone.0069646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasman H, Hammerum AM, Hansen F, Hendriksen RS, Olesen B, et al. Detection of mcr-1 encoding plasmid-mediated colistin-resistant Escherichia coli isolates from human bloodstream infection and imported chicken meat, Denmark 2015. Euro Surveill. 2015;20 doi: 10.2807/1560-7917.ES.2015.20.49.30085. [DOI] [PubMed] [Google Scholar]
- 42.Falgenhauer L, Waezsada SE, Yao Y, Imirzalioglu C, Käsbohrer A, et al. Colistin resistance gene mcr in extended-spectrum b-lactamase-producing and carbapenemase-producing Gram-negative bacteria in Germany. Lancet Infect Dis. 2016;16:282–3. doi: 10.1016/S1473-3099(16)00009-8. [DOI] [PubMed] [Google Scholar]
- 43.Malhotra-Kumar S, Xavier BB, Das AJ, Lammens C, Hoang HTT, et al. Colistin-resistant Escherichia coli harbouring mcr-1 isolated from food animals in Hanoi, Vietnam. Lancet Infect Dis. 2016;16:286–7. doi: 10.1016/S1473-3099(16)00014-1. [DOI] [PubMed] [Google Scholar]
- 44.Tumbarello M, Viale P, Viscoli C, Trecarichi EM, Tumietto F, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis. 2012;55:943–50. doi: 10.1093/cid/cis588. [DOI] [PubMed] [Google Scholar]
- 45.Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev. 2012;25:682–707. doi: 10.1128/CMR.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daikos GL, Tsaousi S, Tzouvelekis LS, Anyfantis I, Psichogiou M, et al. Carbapenemase-producing Klebsiella pneumoniae bloodstream infections: lowering mortality by antibiotic combination schemes and the role of carbapenems. Antimicrob Agents Chemother. 2014;58:2322–8. doi: 10.1128/AAC.02166-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamio Y, Nikaido H. Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase C and cyanogen bromide activated dextran in the external medium. Biochemistry. 1976;15:2561–70. doi: 10.1021/bi00657a012. [DOI] [PubMed] [Google Scholar]
- 48.Wilkinson SG. Bacterial lipopolysaccharides-themes and variations. Prog Lipid Res. 1996;35:283–343. doi: 10.1016/s0163-7827(96)00004-5. [DOI] [PubMed] [Google Scholar]
- 49.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotra LP, Golemi D, Amro NA, Liu GY, Mobashery S. Dynamics of the Lipopolysaccharide Assembly on the Surface of Escherichia coli. J Am Chem Soc. 1999;121:8707–11. [Google Scholar]
- 51.Coughlin RT, Tonsager S, McGroarty EJ. Quantitation of metal cations bound to membranes and extracted lipopolysaccharide of Escherichia coli. Biochemistry. 1983;22:2002–7. doi: 10.1021/bi00277a041. [DOI] [PubMed] [Google Scholar]
- 52.Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, et al. Crystal structures explain functional properties of two E. coli porins. Nature. 1992;358:727–33. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 53.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hancock RE, Falla T, Brown M. Cationic bactericidal peptides. Adv Microb Physiol. 1995;37:135–75. doi: 10.1016/s0065-2911(08)60145-9. [DOI] [PubMed] [Google Scholar]
- 55.Hancock RE. Peptide antibiotics. Lancet. 1997;349:418–22. doi: 10.1016/S0140-6736(97)80051-7. [DOI] [PubMed] [Google Scholar]
- 56.Vaara M. Agents that increase the permeability of the outer-membrane. Microbiol Rev. 1992;56:395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaara M, Vaara T. Polycations as Outer Membrane-Disorganizing Agents. Antimicrob Agents Chemother. 1983;24:114–22. doi: 10.1128/aac.24.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hancock RE. Alterations in outer membrane permeability. Annu Rev Microbiol. 1984;38:237–64. doi: 10.1146/annurev.mi.38.100184.001321. [DOI] [PubMed] [Google Scholar]
- 59.Newton BA. The properties and mode of action of the polymyxins. Bacteriol Rev. 1956;20:14–27. doi: 10.1128/br.20.1.14-27.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, Dhillon P, Yan H, Farmer S, Hancock REW. Interactions of Bacterial Cationic Peptide Antibiotics with Outer and Cytoplasmic Membranes of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000;44:3317–21. doi: 10.1128/aac.44.12.3317-3321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hancock RE, Farmer SW, Li ZS, Poole K. Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of Escherichia coli. Antimicrob Agents Chemother. 1991;35:1309–14. doi: 10.1128/aac.35.7.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farmer S, Li ZS, Hancock RE. Influence of outer membrane mutations on susceptibility of Escherichia coli to the dibasic macrolide azithromycin. J Antimicrob Chemother. 1992;29:27–33. doi: 10.1093/jac/29.1.27. [DOI] [PubMed] [Google Scholar]
- 63.Chapman JS, Georgopapadakou NH. Routes of quinolone permeation in Escherichia coli. Antimicrob Agents Chemother. 1988;32:438–42. doi: 10.1128/aac.32.4.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sánchez BM, Cabarga MM, Navarro AS, Hurlé ADG. A physico-chemical study of the interaction of ciprofloxacin and ofloxacin with polivaient cations. Int J Pharm. 1994;106:229–35. [Google Scholar]
- 65.Teixeira MH, Vilas-Boas LF, Gil VM, Teixeira F. Complexes of ciprofloxacin with metal ions contained in antacid drugs. J Chemother. 1995;7:126–32. doi: 10.1179/joc.1995.7.2.126. [DOI] [PubMed] [Google Scholar]
- 66.Turel I. The interactions of metal ions with quinolone antibacterial agents. Coord Chem Rev. 2002;232:27–47. [Google Scholar]
- 67.Lambs L, Decock-Le Reverend B, Kozlowski H, Berthon G. Metal ion-tetracycline interactions in biological fluids. 9. Circular dichroism spectra of calcium and magnesium complexes with tetracycline, oxytetracycline, doxycycline, and chlortetracycline and discussion of their binding modes. Inorg Chem. 1988;27:3001–12. [Google Scholar]
- 68.Kohn KW. Determination of Tetracyclines by Extraction of Fluorescent Complexes. Application to Biological Materials. Anal Chem. 1961;33:862–6. [Google Scholar]
- 69.White JP, Cantor CR. Role of magnesium in the binding of tetracycline to Escherichia coli ribosomes. J Mol Biol. 1971;58:397–400. doi: 10.1016/0022-2836(71)90255-5. [DOI] [PubMed] [Google Scholar]
- 70.Marshall AJ, Piddock LJ. Interaction of divalent cations, quinolones and bacteria. J Antimicrob Chemother. 1994;34:465–83. doi: 10.1093/jac/34.4.465. [DOI] [PubMed] [Google Scholar]
- 71.Lindner B, Wiese A, Brandenburg K, Seydel U, Dalhoff A. Lack of Interaction of Fluoroquinolones with Lipopolysaccharides. Antimicrob Agents Chemother. 2002;46:1568–70. doi: 10.1128/AAC.46.5.1568-1570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun J, Deng Z, Yan A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun. 2014;453:254–67. doi: 10.1016/j.bbrc.2014.05.090. [DOI] [PubMed] [Google Scholar]
- 73.Elkins CA, Nikaido H. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominantly by two large periplasmic loops. J Bacteriol. 2002;184:6490–8. doi: 10.1128/JB.184.23.6490-6498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Silver LL. A Gestalt approach to Gram-negative entry. Bioorg Med Chem. 2016;24:6379–89. doi: 10.1016/j.bmc.2016.06.044. [DOI] [PubMed] [Google Scholar]
- 75.Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature. 2017;545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 77.Chan PF, Holmes DJ, Payne DJ. Finding the gems using genomic discovery: antibacterial drug discovery strategies – the successes and the challenges. Drug Discov Today Ther Strateg. 2004;1:519–27. [Google Scholar]
- 78.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 79.Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inf Comput Sci. 2003;43:218–27. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 80.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov. 2015;14:529–42. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 81.Alksne LE, Burgio P, Hu W, Feld B, Singh MP, et al. Identification and analysis of bacterial protein secretion inhibitors utilizing a SecA-LacZ reporter fusion system. Antimicrob Agents Chemother. 2000;44:1418–27. doi: 10.1128/aac.44.6.1418-1427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeVito JA, Mills JA, Liu VG, Agarwal A, Sizemore CF, et al. An array of target-specific screening strains for antibacterial discovery. Nat Biotechnol. 2002;20:478–83. doi: 10.1038/nbt0502-478. [DOI] [PubMed] [Google Scholar]
- 83.Fischer HP, Brunner NA, Wieland B, Paquette J, Macko L, et al. Identification of antibiotic stress-inducible promoters: a systematic approach to novel pathway-specific reporter assays for antibacterial drug discovery. Genome Res. 2004;14:90–8. doi: 10.1101/gr.1275704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brown DG, May-Dracka TL, Gagnon MM, Tommasi R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J Med Chem. 2014;57:10144–61. doi: 10.1021/jm501552x. [DOI] [PubMed] [Google Scholar]
- 85.Chan PF, Holmes DJ, Payne DJ. Finding the gems using genomic discovery: antibacterial drug discovery strategies – the successes and the challenges. Drug Discov Today Ther Strateg. 2004;1:519–27. [Google Scholar]
- 86.Silver L. Topics in Medicinal Chemistry. Springer; Berlin, Heidelberg: 2017. The Antibiotic Future. [Google Scholar]
- 87.Singh SB, Young K, Silver LL. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem Pharmacol. 2017;133:63–73. doi: 10.1016/j.bcp.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 88.Macielag MJ. Chemical Properties of Antimicrobials and Their Uniqueness. In: Dougherty TJ, Pucci MJ, editors. Antibiotic Discovery and Development. Boston, MA: Springer US; 2012. pp. 793–820. [Google Scholar]
- 89.Dougherty TJ, Pucci MJ. Antibiotic discovery and development. Springer Science & Business Media; 2011. [Google Scholar]
- 90.O'Shea R, Moser HE. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem. 2008;51:2871–8. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 91.Celesk RA, Robillard NJ. Factors influencing the accumulation of ciprofloxacin in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1989;33:1921–6. doi: 10.1128/aac.33.11.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piddock LJV, Johnson MM. Accumulation of 10 Fluoroquinolones by Wild-Type or Efflux Mutant Streptococcus pneumoniae. Antimicrob Agents Chemother. 2002;46:813–20. doi: 10.1128/AAC.46.3.813-820.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mortimer PG, Piddock LJ. A comparison of methods used for measuring the accumulation of quinolones by Enterobacteriaceae, Pseudomonas aeruginosa and Staphylococcus aureus. J Antimicrob Chemother. 1991;28:639–53. doi: 10.1093/jac/28.5.639. [DOI] [PubMed] [Google Scholar]
- 94.Li XZ, Nikaido H, Poole K. Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1995;39:1948–53. doi: 10.1128/aac.39.9.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Piddock LJ, Williams KJ, Ricci V. Accumulation of rifampicin by Mycobacterium aurum, Mycobacterium smegmatis and Mycobacterium tuberculosis. J Antimicrob Chemother. 2000;45:159–65. doi: 10.1093/jac/45.2.159. [DOI] [PubMed] [Google Scholar]
- 96.Novick RP. Micro-iodometric assay for penicillinase. Biochem J. 1962;83:236–40. doi: 10.1042/bj0830236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sneath PH, Collins JF. A method for the chromatographic detection of penicillins and related compounds and of penicillinase. Biochem J. 1961;79:512–4. doi: 10.1042/bj0790512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zimmermann W, Rosselet A. Function of the outer membrane of Escherichia coli as a permeability barrier to beta-lactam antibiotics. Antimicrob Agents Chemother. 1977;12:368–72. doi: 10.1128/aac.12.3.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nikaido H, Rosenberg EY, Foulds J. Porin channels in Escherichia coli: studies with beta-lactams in intact cells. J Bacteriol. 1983;153:232–40. doi: 10.1128/jb.153.1.232-240.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bazile S, Moreau N, Bouzard D, Essiz M. Relationships among antibacterial activity, inhibition of DNA gyrase, and intracellular accumulation of 11 fluoroquinolones. Antimicrob Agents Chemother. 1992;36:2622–7. doi: 10.1128/aac.36.12.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Piddock LJ, Ricci V. Accumulation of five fluoroquinolones by Mycobacterium tuberculosis H37Rv. J Antimicrob Chemother. 2001;48:787–91. doi: 10.1093/jac/48.6.787. [DOI] [PubMed] [Google Scholar]
- 102.Piddock LJV, Jin YF, Ricci V, Asuquo AE. Quinolone accumulation by Pseudomonas aeruginosa, Staphylococcus aureus and Escherichia coli. J Antimicrob Chemother. 1999;43:61–70. doi: 10.1093/jac/43.1.61. [DOI] [PubMed] [Google Scholar]
- 103.Ricci V, Piddock L. Accumulation of garenoxacin by Bacteroides fragilis compared with that of five fluoroquinolones. J Antimicrob Chemother. 2003;52:605–9. doi: 10.1093/jac/dkg418. [DOI] [PubMed] [Google Scholar]
- 104.Ricci V, Piddock LJV. Accumulation of Norfloxacin by Bacteroides fragilis. Antimicrob Agents Chemother. 2000;44:2361–6. doi: 10.1128/aac.44.9.2361-2366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hurwitz C, Braun CB, Peabody RA. Washing Bacteria by Centrifugation Through a Water-Immiscible Layer of Silicones. J Bacteriol. 1965;90:1692–5. doi: 10.1128/jb.90.6.1692-1695.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pozueta-Romero J, Frehner M, Akazawa T. Filtering centrifugation through two layers of silicone oil: a method for the kinetic analysis of rapid metabolite transport in organelles. Cell Struct Funct. 1991;16:357–63. doi: 10.1247/csf.16.357. [DOI] [PubMed] [Google Scholar]
- 107.Cai H, Rose K, Liang LH, Dunham S, Stover C. Development of a liquid chromatography/mass spectrometry-based drug accumulation assay in Pseudomonas aeruginosa. Anal Biochem. 2009;385:321–5. doi: 10.1016/j.ab.2008.10.041. [DOI] [PubMed] [Google Scholar]
- 108.Davis TD, Gerry CJ, Tan DS. General platform for systematic quantitative evaluation of small-molecule permeability in bacteria. ACS Chem Biol. 2014;9:2535–44. doi: 10.1021/cb5003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iyer R, Sylvester MA, Velez-Vega C, Tommasi R, Durand-Reville TF, Miller AA. Whole-Cell-Based Assay To Evaluate Structure Permeation Relationships for Carbapenem Passage through the Pseudomonas aeruginosa Porin OprD. ACS Infect Dis. 2017;3:310–9. doi: 10.1021/acsinfecdis.6b00197. [DOI] [PubMed] [Google Scholar]
- 110.Krishnamoorthy G, Leus IV, Weeks JW, Wolloscheck D, Rybenkov VV, Zgurskaya HI. Synergy between Active Efflux and Outer Membrane Diffusion Defines Rules of Antibiotic Permeation into Gram-Negative Bacteria. mBio. 2017;8 doi: 10.1128/mBio.01172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huigens RW, Morrison KC, Hicklin RW, Flood TA, Richter MF, Hergenrother PJ. A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat Chem. 2013;5:195–202. doi: 10.1038/nchem.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mannhold R, Kubinyi H, Folkers G, Cruciani G. Molecular interaction fields: applications in drug discovery and ADME prediction. John Wiley & Sons; 2006. [Google Scholar]
- 113.Richter MF, Hergenrother PJ. Reaction: Broad-Spectrum Antibiotics, a Call for Chemists. Chem. 2017;3:10–3. [Google Scholar]
- 114.Garcia A, Drown BS, Hergenrother PJ. Access to a Structurally Complex Compound Collection via Ring Distortion of the Alkaloid Sinomenine. Org Lett. 2016;18:4852–5. doi: 10.1021/acs.orglett.6b02333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hicklin RW, Lopez Silva TL, Hergenrother PJ. Synthesis of bridged oxafenestranes from pleuromutilin. Angew Chem Int Ed. 2014;53:9880–3. doi: 10.1002/anie.201404765. [DOI] [PubMed] [Google Scholar]
- 116.Rafferty RJ, Hicklin RW, Maloof KA, Hergenrother PJ. Synthesis of complex and diverse compounds through ring distortion of abietic acid. Angew Chem Int Ed. 2014;53:220–4. doi: 10.1002/anie.201308743. [DOI] [PubMed] [Google Scholar]
- 117.Zgurskaya HI, Löpez CA, Gnanakaran S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect Dis. 2015;1:512–22. doi: 10.1021/acsinfecdis.5b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, et al. Antibiotic Susceptibility Profiles of Escherichia coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob Agents Chemother. 2001;45:1126–36. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pérez A, Poza M, Fernández A, del Carmen Fernández M, Mallo S, et al. Involvement of the AcrAB-TolC Efflux Pump in the Resistance, Fitness, and Virulence of Enterobacter cloacae. Antimicrob Agents Chemother. 2012;56:2084–90. doi: 10.1128/AAC.05509-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tamae C, Liu A, Kim K, Sitz D, Hong J, et al. Determination of Antibiotic Hypersensitivity among 4,000 Single-Gene-Knockout Mutants of Escherichia coli. J Bacteriol. 2008;190:5981–8. doi: 10.1128/JB.01982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Parkinson EI, Bair JS, Nakamura BA, Lee HY, Kuttab HI, et al. Deoxynybomycins inhibit mutant DNA gyrase and rescue mice infected with fluoroquinolone-resistant bacteria. Nat Commun. 2015;6:6947. doi: 10.1038/ncomms7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hiramatsu K, Igarashi M, Morimoto Y, Baba T, Umekita M, Akamatsu Y. Curing bacteria of antibiotic resistance: reverse antibiotics, a novel class of antibiotics in nature. Int J Antimicrob Agents. 2012;39:478–85. doi: 10.1016/j.ijantimicag.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 123.Burch CL, Danaher RJ, Stein DC. Antigenic variation in Neisseria gonorrhoeae: production of multiple lipooligosaccharides. J Bacteriol. 1997;179:982–6. doi: 10.1128/jb.179.3.982-986.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hill SA, Masters TL, Wachter J. Gonorrhea - an evolving disease of the new millennium. Microbial Cell. 2016;3:371–89. doi: 10.15698/mic2016.09.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Britigan BE, Cohen MS, Sparling PF. Gonococcal infection: a model of molecular pathogenesis. N Engl J Med. 1985;312:1683–94. doi: 10.1056/NEJM198506273122606. [DOI] [PubMed] [Google Scholar]
- 126.Marrakchi H, Laneelle MA, Daffe M. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol. 2014;21:67–85. doi: 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 127.Daffe M, Draper P. The envelope layers of mycobacteria with reference to their pathogenicity. Adv Microb Physiol. 1998;39:131–203. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- 128.Acred P, Brown DM, Turner DH, Wilson MJ. Pharmacology and chemotherapy of ampicillin--a new broad-spectrum penicillin. Br J Pharmacol Chemother. 1962;18:356–69. doi: 10.1111/j.1476-5381.1962.tb01416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hanson DJ. Local toxic effects of broad-spectrum antibiotics following injection. Antibiot Chemother (Northfield Ill) 1961;11:390–404. [PubMed] [Google Scholar]
- 130.Yoshimura F, Nikaido H. Diffusion of beta-lactam antibiotics through the porin channels of Escherichia coli K-12. Antimicrob Agents Chemother. 1985;27:84–92. doi: 10.1128/aac.27.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Savage VJ, Charrier C, Salisbury AM, Moyo E, Forward H, et al. Biological profiling of novel tricyclic inhibitors of bacterial DNA gyrase and topoisomerase IV. J Antimicrob Chemother. 2016;71:1905–13. doi: 10.1093/jac/dkw061. [DOI] [PubMed] [Google Scholar]
- 132.Tari LW, Li X, Trzoss M, Bensen DC, Chen Z, et al. Tricyclic GyrB/ParE (TriBE) Inhibitors: A New Class of Broad-Spectrum Dual-Targeting Antibacterial Agents. PLOS ONE. 2013;8:e84409. doi: 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bensen D. U.S. Patent WO2014043272. 2014 Mar 20;
- 134.Piizzi G, Parker DT, Peng Y, Dobler M, Patnaik A, et al. Design, Synthesis, and Properties of a Potent Inhibitor of Pseudomonas aeruginosa Deacetylase LpxC. J Med Chem. 2017;60:5002–14. doi: 10.1021/acs.jmedchem.7b00377. [DOI] [PubMed] [Google Scholar]
- 135.Hernandez V, Crepin T, Palencia A, Cusack S, Akama T, et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother. 2013;57:1394–403. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Takrouri K, Cooper HD, Spaulding A, Zucchi P, Koleva B, et al. Progress against Escherichia coli with the Oxazolidinone Class of Antibacterials: Test Case for a General Approach To Improving Whole-Cell Gram-Negative Activity. ACS Infect Dis. 2016;2:405–26. doi: 10.1021/acsinfecdis.6b00003. [DOI] [PubMed] [Google Scholar]
- 137.Thirring K, Heilmayer W, Riedl R, Kollmann H, Ivezic-Schoenfeld Z, et al. U.S. Patent WO2015110481A1. 2015 Jul 30;
- 138.Surivet JP, Zumbrunn C, Bruyère T, Bur D, Kohl C, et al. Synthesis and Characterization of Tetrahydropyran-Based Bacterial Topoisomerase Inhibitors with Antibacterial Activity against Gram-Negative Bacteria. J Med Chem. 2017;60:3776–94. doi: 10.1021/acs.jmedchem.6b01831. [DOI] [PubMed] [Google Scholar]
- 139.Kamiyama T, Shimma N, Ohtsuka T, Nakayama N, Itezono Y, et al. Cyclothialidine, a novel DNA gyrase inhibitor. II. Isolation, characterization and structure elucidation. J Antibiot (Tokyo) 1994;47:37–45. doi: 10.7164/antibiotics.47.37. [DOI] [PubMed] [Google Scholar]
- 140.Nakada N, Shimada H, Hirata T, Aoki Y, Kamiyama T, et al. Biological characterization of cyclothialidine, a new DNA gyrase inhibitor. Antimicrob Agents Chemother. 1993;37:2656–61. doi: 10.1128/aac.37.12.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lewis RJ, Singh OM, Smith CV, Maxwell A, Skarzynski T, et al. Crystallization of inhibitor complexes of an N-terminal 24 kDa fragment of the DNA gyrase B protein. J Mol Biol. 1994;241:128–30. doi: 10.1006/jmbi.1994.1480. [DOI] [PubMed] [Google Scholar]
- 142.Counter FT, Ensminger PW, Preston DA, Wu CY, Greene JM, et al. Synthesis and antimicrobial evaluation of dirithromycin (AS-E 136; LY237216), a new macrolide antibiotic derived from erythromycin. Antimicrob Agents Chemother. 1991;35:1116–26. doi: 10.1128/aac.35.6.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Massey EH, Kitchell BS, Martin LD, Gerzon K. Antibacterial activity of 9(S)-erythromycylamine-aldehyde condensation products. J Med Chem. 1974;17:105–7. doi: 10.1021/jm00247a018. [DOI] [PubMed] [Google Scholar]
- 144.DuPont HL. Azithromycin for the Self-Treatment of Traveler's Diarrhea. Clin Infect Dis. 2007;44:347–9. doi: 10.1086/510594. [DOI] [PubMed] [Google Scholar]
- 145.Rakita RM, Jacques-Palaz K, Murray BE. Intracellular activity of azithromycin against bacterial enteric pathogens. Antimicrob Agents Chemother. 1994;38:1915–21. doi: 10.1128/aac.38.9.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de la Cabada Bauche J, DuPont HL. New Developments in Traveler's Diarrhea. Gastroenterol Hepatol (NY) 2011;7:88–95. [PMC free article] [PubMed] [Google Scholar]
- 147.Hill DR, Beeching NJ. Travelers' diarrhea. Curr Opin Infect Dis. 2010;23:481–7. doi: 10.1097/QCO.0b013e32833dfca5. [DOI] [PubMed] [Google Scholar]
- 148.Stipkovits L, Ripley PH, Tenk M, Glavits R, Molnar T, Fodor L. The efficacy of valnemulin (Econor) in the control of disease caused by experimental infection of calves with Mycoplasma bovis. Res Vet Sci. 2005;78:207–15. doi: 10.1016/j.rvsc.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 149.Egger H, Reinshagen H. New pleuromutilin derivatives with enhanced antimicrobial activity. II. Structure-activity correlations. J Antibiot (Tokyo) 1976;29:923–7. doi: 10.7164/antibiotics.29.923. [DOI] [PubMed] [Google Scholar]
- 150.Nestorovich EM, Danelon C, Winterhalter M, Bezrukov SM. Designed to penetrate: Time-resolved interaction of single antibiotic molecules with bacterial pores. Proc Natl Acad Sci USA. 2002;99:9789–94. doi: 10.1073/pnas.152206799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Azam MA, Thathan J, Jubie S. Dual targeting DNA gyrase B (GyrB) and topoisomerse IV (ParE) inhibitors: A review. Bioorg Chem. 2015;62:41–63. doi: 10.1016/j.bioorg.2015.07.004. [DOI] [PubMed] [Google Scholar]