

Abstract
Accumulating evidence suggests that cerebellar dysfunction early in life is associated with autism spectrum disorder (ASD), but the molecular mechanisms underlying the cerebellar deficits at the cellular level are unclear. Tuberous sclerosis complex (TSC) is a neurocutaneous disorder that often presents with ASD. Here, we developed a cerebellar Purkinje cell (PC) model of TSC with patient derived human induced pluripotent stem cells (hiPSCs) to characterize the molecular mechanism underlying cerebellar abnormalities in ASD and TSC. Our results show that hiPSCs-derived PCs from patients with pathogenic TSC2 mutations displayed mTORC1-pathway hyperactivation, defects in neuronal differentiation and RNA regulation, hypoexcitability and reduced synaptic activity when compared to those derived from controls. Our gene expression analyses revealed downregulation of several components of fragile-X mental retardation protein (FMRP) targets in TSC2-deficient hiPSC-PCs. We detected decreased expression of FMRP, glutamate receptor δ2 (GRID2) and pre- and post-synaptic markers such as synaptophysin and PSD95 in the TSC2-deficient hiPSC-PCs. The mTOR-inhibitor rapamycin rescued the deficits in differentiation, synaptic dysfunction and hypoexcitability of TSC2-mutant hiPSC-PCs in vitro. Our findings suggest that these gene expression changes and cellular abnormalities contribute to aberrant PC function during development in TSC affected individuals.
Introduction
Developmental abnormalities of the cerebellum and early cerebellar injury have both been implicated in the development of ASD. Loss of cerebellar PCs in individuals with ASD1, 2 and a higher incidence of ASD in premature infants with isolated cerebellar hemorrhage3 indicate that cerebellar dysfunction early in life contributes to the pathogenesis of ASD, but the molecular mechanisms underlying these deficits remain unclear.
TSC is an autosomal dominant monogenic disorder that leads to the development of ASD in ~50% of TSC patients. TSC is characterized by significant neurological disabilities, and it affects 1:6000 newborns yearly. Loss of function of TSC1 or TSC2 leads to dysregulation of the mechanistic target of rapamycin complex 1 (mTORC1)4, 5. TSC1 and TSC2 form a complex together with TBC1D76. TSC2 works as a GTPase-activating protein (GAP) for the Ras homolog enriched in brain (RHEB), which is an important activator of mTORC17. TSC2 catalyzes the hydrolysis of GTP that is bound on RHEB, and loss of function of either TSC1 or TSC2 destabilizes the TSC complex, leading to increased RHEB-GTP activation and subsequent mTORC1 hyperactivation 7. mTORC1 is a conserved serine/threonine kinase that is activated downstream of the PI3K/AKT pathway, and phosphorylates substrates such as S6-kinase1, S6 and 4EBP1, which are involved in mRNA translation, proliferation, and differentiation4. Inhibition of the mTOR-pathway is a target for pharmacological treatment of TSC.
Cerebellar abnormalities are also associated with a higher prevalence of ASD in individuals with TSC8, 9. The deep cerebellar nuclei are typically inhibited by the cerebellar cortex, but individuals with TSC and ASD have increased glucose metabolism in the deep cerebellar nuclei10, consistent with the hypothesis that the dysregulation of the cerebellar cortex is involved in the development of ASD in TSC patients. In addition, clinical findings suggest that cerebellar lesions negatively affect the development of the dentatothalamic-frontal circuits11. We previously created a mouse model lacking the Tsc1 gene specifically in cerebellar PCs (L7-Cre+;Tsc1f/f), which displayed autistic-like features, including deficits in social interaction and increased repetitive behaviors, as well as cellular abnormalities12. Another study also observed cellular degeneration and autistic like behavior in mice with a conditional Tsc2-mutation in mouse PCs 13. The data from individuals with TSC and Tsc1/2 conditional knock-out mice indicate that PC dysfunction may have an important role in the development of ASD, but the molecular mechanisms are unknown.
In this study, we generated hiPSC lines from individuals with TSC with or without ASD to study the abnormalities produced by specific TSC2 mutations in PCs in vitro. Since TSC mutations and dysfunctional PCs are both highly associated with the development of ASD12, 13, we differentiated the TSC2-mutant hiPSCs into cerebellar precursors and PCs using a novel differentiation protocol. Using this first human TSC-patient derived PC model, we identified several abnormalities in TSC2-deficient PCs, including differentiation deficits, increased soma size, autophagy activation, oxidative stress and cell death. In addition, we detected down regulation of FMRP expression, synaptic dysfunction and hypoexcitability of PCs derived from TSC-patients with ASD compared to control PCs. Importantly, we demonstrate that the mTOR-inhibitor rapamycin can rescue these deficits during neuronal development in vitro. Our findings suggest that these cellular abnormalities may contribute to aberrant PC function during development in TSC and related neurodevelopmental disorders.
Materials and Methods
Human iPSC line derivation from TSC patients and controls
The subjects enrolled in this study were recruited through Boston Children’s Hospital. Supplementary Table 1 describes detailed information about patients and TSC2 mutations. Protocol was approved by Boston Children’s Hospital (Boston, USA) IRB (P00008224). Informed consents were obtained from all participants and/or their parents as appropriate (See Supplementary Table S1, Figure S1 and Supplementary Materials and Methods). Briefly, hiPSC lines were derived from 3 individuals with TSC, cortical tubers, and epilepsy, (3 patients; 77, 47-01 and CRA401 cell lines, TSC2+/−), and 2 of them were diagnosed with ASD (47-01 and CRA401 patient lines), hiPSC reprogramming was done as described14. Control hiPSCs were derived either from the parents of the TSC-patients or gender matched individuals without TSC2-mutations (4 individuals, TSC2+/+)15. In addition, we studied a TALEN-engineered isogenic TSC2−/− hiPSC line (bi-allelic microdeletion in the 77-patient cell line), method described previously16. We also created isogenic control hiPSC-line by correction of the heterozygous microdeletion of TSC2 in 77-patient cell line with CRISPR-cas9 method17, 18 (Supplementary Figure S2). See Supplementary materials and methods for detailed protocols.
PC differentiation of hiPSCs
A schema of the differentiation protocol and sample collection time points is presented in Figure 1. See Supplementary materials and methods for the detailed differentiation protocol and characterization of the hiPSC-derived PCs.
Figure 1. Differentiation protocol for hiPSC-derived PCs.
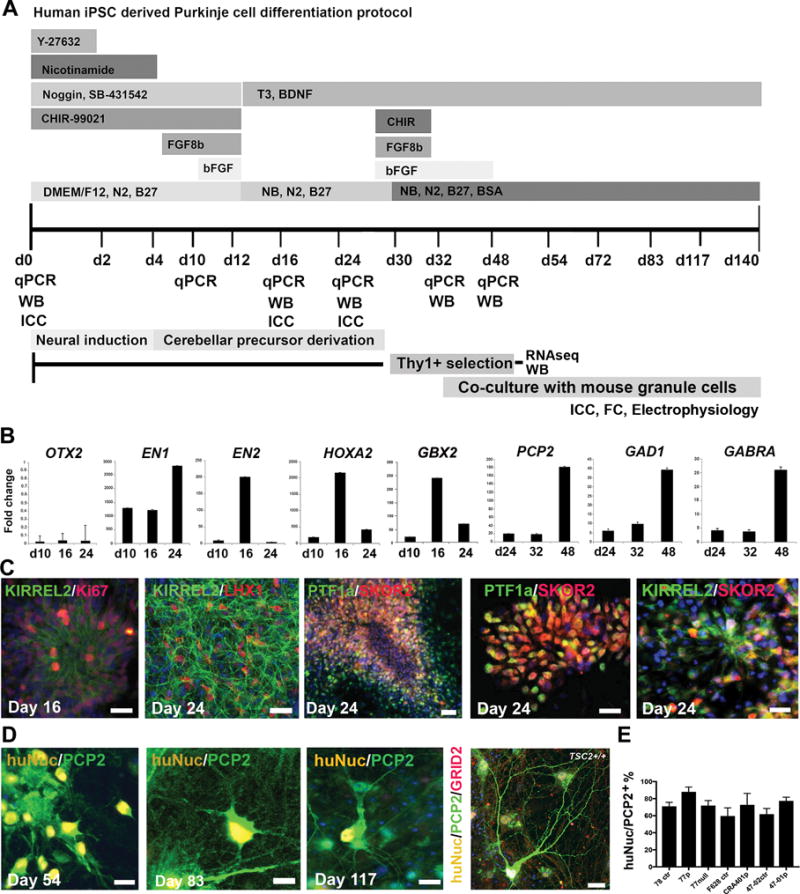
A) Schematic representation of the in vitro differentiation protocol and sample collection time points. B) Quantitative RT-PCR analyses of OTX2, EN1, EN2, HOXA2 and GBX2 during the initial cerebellar patterning of hiPSCs at days 10-24 of differentiation, and mature PC markers PCP2, GAD1 and GABRA2 at days 24-48 of differentiation, 47-02 (TSC2+/+) compared to undifferentiated hiPSCs (fold change=1). C) Representative images of hiPSC-derived PC progenitors stained with KIRREL2/Ki67 day 16 and KIRREL2/LHX1 day 24, scale bars 32 μm. PTF1a/SKOR2, scale bars 64 μm and 32 μm, and KIRREL2/SKOR2 at day 24 of differentiation scale bar 32 μm. D) Morphological development of PCP2/huNuc+ hiPSC-derived PC precursors in co-cultures with mouse granule neurons on days 54, 83, 117 and at day 140 of differentiation (cell line CRA401, TSC2+/−). Scale bars 20 μm, 20 μm, 20 μm and 50 μm, respectively. Nuclear staining (Hoechst, blue). E) Quantification of huNuc/PCP2 positive cells from the total cell population at day 125 of differentiation.
Statistical Analyses
Sample size of 15 per group (assuming independence) in imaging assays and flow cytometry provided 80% power to detect an effect size of 1.18 standard deviations (SD) while preserving the Type I error rate to its nominal value of 0.025 (after adjustment of at least two comparisons within each experiment). Similarly, sample sizes of 8 per group allowed to detect an effect size of 1.70 SD in case of western blot experiments. Statistical analysis was performed using two-sample t-test, one-way ANOVA and two-way ANOVA depending on the factors considered for each experiment described in the supplemental file. Prior to the model building, all the data were graphically plotted (histograms and box-plots) to check for normality, outliers and homogeneity in terms of variance across groups. Furthermore, we employed Anderson-Darling tests to check of normality and Levene’s test to test for equality of variances between groups. In case of two-way ANOVA model, the interaction effect was removed from the model in case it wasn’t found to be significant based on the F-test. Pre-specified post-hoc comparisons were performed to test the differences within and between the factors after multiple comparison adjustments using Sidak’s approach. Statistical analyses was performed using SAS (version 9.2 Cary, NC), and the multiple comparison adjusted p values <0.05 were considered statistically significant.
Additional experimental procedures
For more detailed materials and methods, please see Supplementary Materials and Methods and Supplementary Tables 1-5.
Results
Differentiation of hiPSC-derived PCs
To model TSC patient derived PC dysfunctions in vitro, we created a specific developmental model of cerebellar tissue by differentiating hiPSCs along the cerebellar lineage into PCs (Figure 1A). Prior to differentiation, the control and TSC2-deficient hiPSCs expressed pluripotency markers, had normal karyotype and typical stem cell colony formation in vitro (Supplementary Figure S1). To guide cerebellar tissue differentiation in vitro, we utilized several growth factors and small molecules crucial in embryonic cerebellum development in vivo (Figure 1A), Wnt1 and Fgf8b19, 20. We used the Wnt-signaling activator CHIR-99021 together with FGF8b and bFGF, which induce efficient midbrain/hindbrain boundary development in vivo to differentiate hESCs and hiPSCs, as we have shown previously21. We then cultured the cells in the presence of nicotinamide, which enhances the neural commitment of stem cells22 during the neural induction with dual-Smad inhibition23. We detected upregulation of markers of midbrain/hindbrain patterning such as EN1/EN2 and caudalization such as HOXA2 by quantitative RT-PCR at day 16 of differentiation. GBX2 expression was also increased during differentiation, while rostral marker OTX2 remained low (Figure 1B). Following 24 days of differentiation, the cerebellar patterning markers EN2, HOXA2, and GBX2 were down-regulated, while markers of PCs, including the PC specific gene PCP2, and gamma-aminobutyric acid (GABA) production related GAD1 and receptor GABRA2 were up-regulated in the differentiated cerebellar cultures through days 24 to 48 (Figure 1B, Supplementary Table S5).
Using our protocol, by day 16, cerebellar precursors expressed KIRREL2/Ki67 (Figure 1C). Between days 16-24 of differentiation, the cell population started to express KIRREL2/LHX1, PTF1a/SKOR2 and KIRREL2/SKOR2 (Figure 1C). Initially, we tested KIRREL2+ sorting24 to enrich cerebellar precursors in the heterogeneous cultures (Supplementary Figure S4), but found that THY1+ selection, a method previously used to purify mouse PCs25, led to a higher purity of hiPSC-derived PCs when isolated at days 28 to 32. To enhance PC differentiation and synaptic function in hiPSC-PCs in vitro, we co-cultured purified PCs with mouse cerebellar granule neurons isolated from P4-P7 in the presence of BDNF and T325. With this protocol, hiPSC-derived PCP2+ cells first developed bipolar morphology and short branches around day 54-60, after which the extent and branching of the PC dendrites increased, and the cell soma developed an oval shape typical of PCs (day 83-90). Multi-branched maturing PCP2+ PCs could be detected on days 100-140, and these cells expressed PC specific glutamate receptor δ2 (GRID2, Figure 1D).
Previously, hESCs or hiPSC have been differentiated into PC precursors in neural suspension cultures with high concentrations of bFGF and insulin24, 26. For maturation into functional neurons, these protocols enriched for PCs by KIRREL2+ sorting and co-cultured them with rodent embryonic cerebellar cells or human cerebellar slices. However, this resulted in heterogeneous populations of neural cells with a modest cell survival rate and efficiency of PC differentiation of 10-16% PCs prior cell sorting and purity of ~60% PCP2+ cells after cell sorting and co-culturing24, 26. Our protocol described here results efficient cerebellar precursor cell and PC differentiation from hiPSCs: >22% PCP2+ cells at day 30 (Supplementary Figure S6) and purity up to 60-91% PCP2+ cells after THY1+ selection and co-culturing with mouse granule neurons in vitro (Figure 1E).
TSC2-deficient hiPSC-NPCs have increased mTOR-pathway activation and altered proliferation capacity
To study the effect of loss of function of TSC2 on PC differentiation and function, we derived hiPSC lines from three individuals with heterozygous loss of TSC2 (TSC2+/−), and clinically diagnosed to have TSC, cortical tubers, and epilepsy (3 patients) and ASD (2 patients). Since the bi-allelic loss of function TSC2 mutations are found in a subset of cells in tubers of individuals affected with TSC27, we also characterized a TALEN-engineered isogenic TSC2−/− hiPSC line. We compared these cells with healthy control cell lines derived from unaffected parent of the patient or unaffected gender-matched control lines without the TSC2-mutation (4 individuals, TSC2+/+) or CRISPR-cas9 corrected isogenic hiPSC line (from 1 individual, 3 clones), (Supplementary Table S1, Supplementary Figures S1–S2).
During the neuroectodermal stage of hiPSC differentiation, we analyzed the mTORC1 activation in hiPSC-derived neural precursor cells (NPCs) between days 24 to 38 in vitro. According to qRT-PCR analyses we detected downregulation of TSC2 in TSC2+/− and TSC2−/− hiPSC-NPCs compared to control NPCs and increased mRNA expression of mTOR-pathway activation related genes: IRS1, AKT1S1, MTOR, K-RAS, EIF4G1 in both TSC2+/− and TSC2−/− hiPSC-NPCs compared to control TSC2+/+ NPCs. In addition, we detected significantly increased expression of EIF4EBP1, PIK3CA, PRKAA1, RPS6KB1, ARAF, PTEN, CCND1, MAPK3 in TSC2−/− NPCs compared to control TSC2+/+ NPCs (Supplementary Figure S3A, Supplementary Table S5).
At the protein level, we did not detect significant increase in the phosphorylation of S6 at early stage of neural differentiation between heterozygous TSC2+/− and control TSC2+/+ cells (day 24, Supplementary Figure S3). However, we detected significant increase in pS6 levels/total S6 between TSC2+/− vs TSC2+/+ cells after further neural differentiation (Figure 2A). These data are consistent with a previous study of TSC2-deficient hESC-derived neuroectodermal rosettes that did not show a difference in pS6/S6 levels between control cells and cells with heterozygous loss of TSC2 at the early neural precursor stage, but did show increased pS6/S6 at later neuronal differentiation28.
Figure 2. mTOR-pathway hyperactivation in TSC2-deficient hiPSC-derived NPCs.
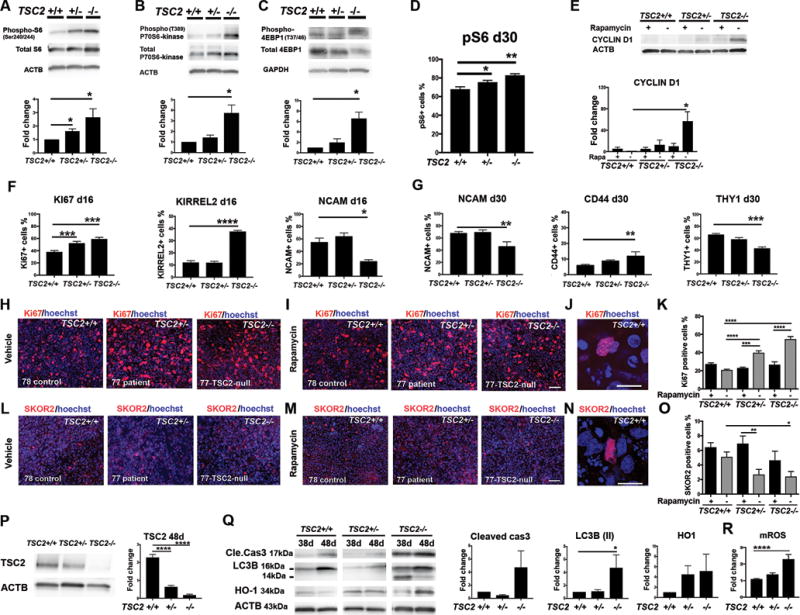
Protein expression analyses of the hiPSC-NPCs from the cell lines 78-control (TSC2+/+), 77-patient (TSC2+/−) and 77-TSC2-null (TSC2−/−). A–C) Semi-quantitative analyses of pS6/S6, p-P70S6-kinase/P70S6-kinase, and p4EBP1/4EBP1 intensities between control hiPSC-NPCs (fold change=1) and TSC-deficient NPCs. Fold changes were analyzed with one-way ANOVA and post-hoc-test between columns (*p<0.05). D) Flow cytometric analyses of pS6+ cell number at day 30, one-way ANOVA and post-hoc-test between columns (*p<0.05, **p<0.01). E) WB analyses of the CYCLIN D1 at day 24. F) Flow cytometric analyses of Ki67, KIRREL2, NCAM at day16, and G) NCAM, CD44, and THY1 at day 30 of differentiation. All data are presented as mean±SEM. One-way ANOVA, post-hoc test between groups, (****p<0.0001, ***p<0.001, **p<0.01, *p<0.05). H–I) Representative images of Ki67+ cells at day 24 of differentiation, scale bar 32 μm. J) Higher magnification of a Ki67+ cell, scale bar 10 μm. K) Quantification of Ki67+ cells. L–M) Representative images of SKOR2+ cells at day 24 of differentiation, scale bar 32 μm. N) Higher magnification of a SKOR2+ cell, scale bar 10 μm. O) Quantification of SKOR2+ cells. Nuclear staining (Hoechst, blue). Two-way ANOVA, followed by post-hoc-test for Ki67 (in K) and SKOR2 (in O) analyses. (****p<0.0001, ***p<0.001, **p<0.01, *p<0.05). Data are presented as mean±SEM. (K and O data are from cell lines; 78 control (TSC2+/+) 77 patient (TSC2+/−) and 77-TSC2-null (TSC2−/−). P) WB analyses of TSC2 expression between control (fold change=1) and TSC-deficient cerebellar cells (n=3) at day 48 of the neural differentiation of hiPSCs. Fold changes were analyzed with one-way ANOVA and post-hoc test, ****p<0.0001. Representative blots of 78 control (TSC2+/+), 77 patient (TSC2+/−) and 77-TSC2-null (TSC2−/−) cell lines are presented (P–Q). Q) Increased expression of apoptosis marker cleaved caspase 3, autophagy activation marker LC3B (II, size 14kDa) were detected in TSC2−/− cell population at day 48. Variable expression of oxidative stress marker HO-1 were detected in both TSC2+/− and TSC2−/− cells compared to control cell population (TSC2+/+). LC3B (II)/ACTB expression was significantly up-regulated in TSC2−/− cells at day 48, (one-way ANOVA, post-hoc-test, *p<0.05). R) Flow cytometric analyses of mROS in hiPSC-PCs (one-way ANOVA, post-hoc-test, ****p<0.01).
Interestingly, we detected increased phosphorylation of p70-S6-kinase and S6 in the TSC2−/− NPCs compared to control NPCs, both at the early NPCs and later time point (Figure 2A–B, Supplementary Figure S3B) and rapamycin treatment (20nM) decreased significantly the phosphorylation of these proteins (Supplementary Figure S3B). We also detected increased phospho-4EBP1/total 4EBP1 in TSC2−/− cells compared to control NPCs (Figure 2C). During further analyses, we confirmed increased pS6+ cell number with flow cytometry at day 30, which revealed significantly higher total amount of pS6+ cells in TSC2+/− (75.4±2%, *p<0.05) and TSC2−/− (82.9±1.6%, **p<0.01) cell populations compared to control TSC2+/+ (67.9±2.6%, Figure 2D).
We then assayed whether aberrant mTORC1 pathway regulation in TSC2-deficient cells lead to overactivation of cell cycle regulators and proliferation. We detected increased expression of cell cycle regulator CYCLIN D1 in TSC2−/− cells compared to controls (Figure 2E), suggesting that TSC2-deficiency leads to increased neural cell proliferation. In support of this, we detected increased expression of the cell proliferation marker Ki67 in TSC2+/− and TSC2−/− cell populations on days 16 and 24 of differentiation (Figures 2F, 2H-K, Supplementary Figures S2G, S4). In addition, we found that TSC2−/− hiPSC-derived cultures contained more KIRREL2+ progenitors at day 16 of differentiation than those from control population (TSC2+/+, Figure 2F). Rapamycin treatment for 7 or 14 days reduced the number of Ki67+ proliferative cells in the TSC2-deficient populations (Figures 2H–K, Supplementary Figures S2, S4) and decreased the expression of CYCLIN D1 in TSC2−/− cells (Figure 2E). Taken together, we found that mTORC1 over-activation in TSC2-deficient cells led to increased pS6+ cell number, increased expression of CYCLIN D1 in TSC2−/− cells, and increased proliferation of TSC2-deficient NPCs, and that these disease phenotypes could be rescued with rapamycin.
TSC2-deficient hiPSC-NPCs have decreased differentiation capacity into PCs and increased tendency to differentiate to an astroglial fate
Next, we investigated whether TSC2-deficiency affects the capacity of hiPSCs to differentiate into cerebellar neurons in a TSC2-gene dosage dependent manner. First, we analyzed mRNA expression levels of hiPSC-derived cell populations during days 0 to 48 of differentiation without THY1+ cell enrichment. During the initial cerebellar cell differentiation (days 0 to 16), we detected delayed and reduced expression of GBX2 in TSC2-deficient cell populations compared to control cells at day 16 (***p<0.001, Supplementary Figure S5A). This suggests dysfunctional hindbrain/midbrain signaling in TSC-patient derived cells that cause deficits of the cerebellar tissue development at the embryonal-neural tube formation-stage and decreased cerebellar precursor cell derivation from TSC-patient cells (TSC2+/− and TSC2−/−). During later stages of differentiation (days 24 to 48), we detected decreased PC specific marker PCP2 expression in TSC2-deficient cell populations compared to control cells at day 48 (*** *p<0.0001, Supplementary Figure S5A). This suggests that TSC2-mutations reduce the differentiation capacity of the hiPSC-derived cells towards PCs compared to control cells. Consistent with these findings, we discovered reduced expression of the non-proliferative PC precursor markers THY1 and LHX1 in TSC2−/− cells at day 24 (Figure 2G, Supplementary Figures S5B–C, S6), and the number of the PC specific SKOR2+ precursors were significantly lower in both the TSC2+/− and TSC2−/− cell populations compared to the controls at day 24 (Figures 2L–O, Supplementary Figures S2, S4). To further analyze the mTOR-pathway involvement in this process, we treated the TSC-patient derived cell populations and control cells with rapamycin for 2 weeks during differentiation. Rapamycin treatment restored the number of SKOR2+ PC precursor cells in the TSC2 deficient cell populations to similar levels with controls (Figure 2O, Supplementary Figure S2I, S4D).
Overall, we detected altered neuronal differentiation in TSC2-deficient TSC2−/− cell populations vs controls cell populations with decreased expression of NCAM (at days 16 and 30), and increased expression astroglial precursor markers CD44 and GFAP, and increased number of pS6+ cells (Figure 2F–G, Supplementary Figures S5D, S6). Taken together, these in vitro findings (Figure 2, Supplementary Figures S2-5) recapitulate several neuropathological features identified in the brains of TSC patients, by displaying hyperactivated mTORC1-pathway, deficient cerebellar patterning and decreased PC differentiation capacity, and increased number of astroglial cells in TSC2−/− cell population.
To determinate if TSC2-loss leads to hyperproliferation and decreased cerebellar precursor cell differentiation in the same genetic background, we created a CRISPR-corrected isogenic control hiPSC line from the heterozygous TSC2+/− 77-patient line, by correction of the microdeletion using specific sgRNA guide and ssODN (Supplementary Figure S2). Compared to isogenic CRISPR-corrected hiPSC-derived NPCs (TSC2+/+), we detected hyperactivated mTORC1-pathway, increased proliferation, and decreased cerebellar cell differentiation capacity of the TSC2-deficient isogenic cells (TSC2+/−, TSC2−/−, Supplementary Figure S2), and these defects were rescued with rapamycin treatment (Supplementary Figure S2). These data were consistent with the data of non-isogenic control lines vs TSC2-patient derived cells. This data confirmed that the differentiation deficits of the TSC2-mutant cells were caused due the specific loss-of TSC2 expression and mTORC1-pathway hyperactivation in hiPSC-derived isogenic cell lines, and the effect was TSC2-gene dosage dependent.
TSC2-deficient hiPSC-derived PC precursors are susceptible to oxidative stress and cell death
To characterize the effects of TSC2 loss on cellular viability and molecular function, we further characterized the hiPSC-derived PC precursor populations at days 38-48 of differentiation, prior to co-culturing the cells with mouse cerebellar granule neurons. As expected, we detected decreased expression of TSC2 protein in TSC2−/− and TSC2+/− hiPSC-derived cell populations in a gene-dosage dependent manner compared to control TSC2+/+ cells (Figure 2P). Apoptosis was increased in TSC2−/− cells as shown by increased cleaved Caspase 3 expression at days 38-48 of PC-precursor-differentiation, compared to control cells (Fold change 4.7 vs 1 of control cells, Figure 2Q). In addition, we detected increased autophagy activation and oxidative stress in TSC2−/− cells, as shown by upregulation of LC3B(II)/ACTB and increased mitochondrial reactive oxygen species (mROS) in the TSC2−/− cells compared to control cells (Figure 2Q–R). Together, these data show that the dysregulation of the TSC2 expression and mTORC1-pathway increased the vulnerability of the TSC2-deficient cerebellar cell cultures to cellular stress, autophagy and cell death, which may underlie the loss of mature PCs in individuals with TSC.
Abnormal mTORC1-pathway activation alters cellular morphology and synaptic development of the TSC2-deficient THY1+ hiPSC-derived PCs
Next, we asked whether the mTORC1 pathway hyperactivation was present in the TSC2-deficient hiPSC-PCs after long-term differentiation and co-culturing the cells with mouse granule neurons. First, we enriched the differentiated hiPSC-derived PCs and eliminated the dying cells and non-neural cell from the cell population with THY1+ selection, prior to co-culturing the human cells with mouse granule neurons (Figure 3A). We assayed the cells with human nuclear marker (huNuc) and with antibody against phosphorylated form of ribosomal protein S6 (pS6) at day 125 of differentiation (Figure 3B–C). TSC2-deficient THY1-selected huNuc+ hiPSC-PCs had significantly increased pS6 expression levels compared to control PCs (Figure 3B–C, Supplementary Figure S7), and the effect was inversely related to the number of functional TSC2 alleles (Figure 3C, Supplementary Figure S7). pS6 accumulated increasingly in the neurites of both TSC2−/− and TSC2+/− PCs (Supplementary Figure S7) but not in control cells. Accumulation of pS6 in the neurites of TSC2-mutant cells demonstrated increased mTORC1-pathway activation and abnormal regulation of protein synthesis in the neurites, which could dysregulate synapse development in TSC2-deficient PCs. No significant differences in pS6 levels were detected between different TSC-patient lines (analyzed with one-way ANOVA, Supplementary Figure 8). We confirmed pS6 co-localization specifically in PCs with co-staining of Calbindin (CALB1)/pS6 (Supplementary Figure S7G). Long-term rapamycin treatment restored the pS6 expression in the TSC2-mutant PCs to similar levels with the control PCs (Figure 3B–C, Supplementary Figure S7).
Figure 3. TSC2-deficient hiPSC-derived PCs have decreased synaptic marker expression, increased cellular stress and morphological abnormalities compared to control PCs.
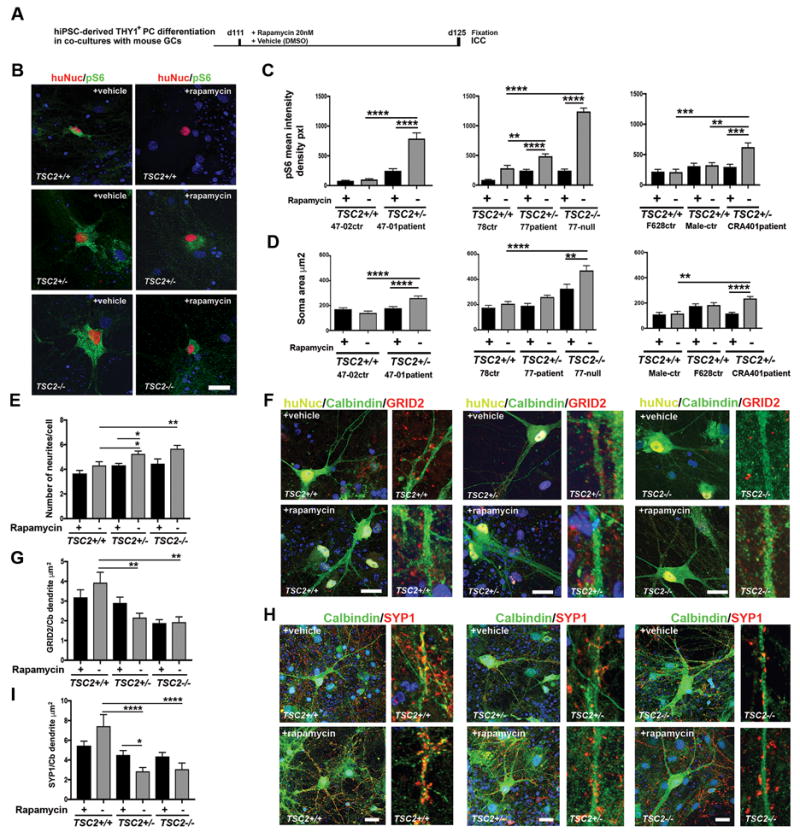
A) Schematic presentation of the rapamycin (20nM) or vehicle (DMSO) treatment at days 111-125 of THY1+ hiPSC-PC differentiation in mouse GC co-cultures in vitro. B) Representative images of pS6 expression in huNuc+ hiPSC-PCs (78-control, TSC2+/+) compared to TSC2-deficient hiPSC-PCs (77-patient TSC2+/−) and 77 TSC2-null PCs (TSC2−/−). Scale bar 40 μm. C) Quantification of pS6 mean intensity density in huNuc+ PCs. Rapamycin treatment decreased significantly the pS6 expression in TSC2-deficient patient PCs (47-01, 77, CRA401) and in 77 TSC2-null PCs during the differentiation process. Two-way ANOVA, followed by post-hoc-test, (**p<0.01, ***p<0.001, ****p<0.0001). D) Quantification of soma area of CALB1+ hiPSC-PCs. Two-way ANOVA, followed by post-hoc-test, (**p<0.01, ***p<0.001, ****p<0.0001). E) Quantification of the number of neurites growing from the soma. Two-way ANOVA, followed by post-hoc-test, (*p<0.05, **p<0.01). F). GRID2 expression (red) was detected in huNuc+ (yellow) and CALB1+ (green) control PCs (TSC2+/+). GRID2 expression was significantly lower in the TSC-patient PCs (TSC2+/−) and TSC2−/− PCs. After rapamycin treatment the TSC2+/+ and TSC2+/− PCs expressed GRID2, and low intensity of GRID2 was detected in the TSC2−/− cells. Scale bar 20 μm. Hoechst (blue, nuclear staining). G) Quantification of GRID2 puncta in CALB1-positive dendrites. Two-way ANOVA, post-hoc-test, (*p<0.05). H) SYP1 expression (red) was detected in CALB1 (green) positive control PCs (TSC2+/+). SYP1 expression was lower in the TSC-patient PCs (TSC2+/−) and TSC2−/− PCs. Rapamycin treatment increased expression of SYP1 in the TSC-patient PCs (TSC2+/−) and in the TSC2−/− PCs. Scale bar 20 μm. Hoechst (blue, nuclear staining). I) Quantification of SYP1 puncta in CALB1-positive dendrites. Two-way ANOVA, post-hoc-test, (*p<0.05). All data are presented as mean±SEM.
Further analysis of THY1-selected huNuc+/CALB1+ PCs in co-cultures with mouse granule neurons showed that the TSC2+/− and TSC2−/− PCs had significantly larger somas compared to control PCs at day 125 of differentiation (Figure 3D). There were no significant differences in soma size between different patient lines (analyzed with one-way ANOVA, Supplementary Figure 8). Long-term rapamycin treatment restored the soma size of TSC2+/− PCs to similar levels with control PCs (TSC2+/+), and partly restored the cell size of TSC2−/− cell population (Figure 3D). In addition, we detected an increased number of dendrites per cell in the TSC2+/− and TSC2−/− PCs, compared to control cells (Figure 3E, Supplementary Figure S9A). Rapamycin treatment rescued the morphological deficits of the TSC2+/− and TSC2−/− PCs (Figure 3E, Supplementary Figure S9A). In summary, our data show that in the TSC2-mutant hiPSC-PCs the absence of TSC2 leads to mTORC1 pathway hyperactivation, increased phosphorylation of S6, and abnormal morphology and cell growth. This data is consistent with the role of TSC2 as a tumor suppressor in cells, and its capacity to regulate cellular growth by mediating cellular mTORC1, which controls cell growth29.
To further characterize TSC related pathophysiology during the human cerebellar PC development in vitro, we analyzed synaptic development in TSC2-deficient hiPSC-derived PCs. First, we characterized the expression of PC specific markers during the differentiation at days 38-48 in vitro, prior to co-culturing the cells with mouse granule neurons. CALB1 expression was delayed in the TSC2−/− cell population (detected at day 48) compared to control cells (detected at days 38 and 48, Supplementary Figure S9B). The control TSC2+/+ cells expressed PC specific glutamate receptor δ2 (GRID2), post-synaptic marker PSD95 and pre-synaptic marker synaptophysin (SYP1) at day 48 of differentiation. However, the expression of GRID2 was reduced in TSC2+/− and TSC2−/− PCs at day 48, and PSD95 was reduced in TSC2−/− PCs at day 48 (Supplementary Figure S9C). Next, we assayed whether reduced synaptic marker expression that was detected at the early PC development, was also present in the hiPSC-derived PCs after long-term differentiation in co-cultures with mouse GCs (Figure 3F–I). We detected GRID2, SYP1 and PSD95 expression at day 125 of differentiation in the THY1+ selected hiPSC-derived CALB1-positive dendrites of control PCs (TSC2+/+) (Figure 3F–I, Supplementary Figure S9D–E). GRID2 and SYP1 expression was significantly decreased in the TSC2+/− and TSC2−/− PCs (Figure 3F–I), and PSD95 was decreased in the TSC2−/− PCs compared to control PCs (Supplementary Figure S9D–E). The decreased synaptic marker expression in the TSC2-deficient PCs suggests abnormal synaptic development and function of the developing human PCs. Rapamycin treatment for two weeks (20nM) partly rescued the GRID2 and SYP1 staining intensity in the TSC2-deficient TSC2+/− PCs (Figure 3F–I), suggesting mTORC1-pathway dependent regulation of synapse formation and development in the cells.
TSC2 mutant hiPSC-PCs exhibit defects in excitability and synaptic function that can be rescued by mTOR-inhibition
Next, we assayed whether the abnormal cell size, and decreased synaptic marker expression in TSC2-deficient human PCs affected the functional properties of our cell model (Figure 4). Whole cell patch clamp recordings were performed on mature, >125-day-old THY1+ human PCs (Supplementary Figure S10) in the presence of synaptic blockers. TSC2+/− human PCs exhibited a 21-33% higher membrane capacitance and a 22-24% lower input resistance compared to their respective controls (Figure 4A, Supplementary Table S4). TSC2−/− PCs also had a 2-fold increase in membrane capacitance and a 55% reduction in input resistance compared to the control (Figure 4A, Supplementary Table S4). We suggested that the alterations in passive membrane properties are likely due to the increase in cell size as shown in (Figure 3D, 4A), since there were no significant differences between genotypes after normalizing the input resistance by the capacitance in individual cells. The change in membrane capacitance and input resistance associated with TSC2 mutations were reversed by a 2 weeks treatment with rapamycin (20nM) prior to recording, while this treatment had no effect on the same parameters in control (TSC2+/+) PCs (Figure 4A, Supplementary Table S4).
Figure 4. Electrophysiological characterization of TSC2-deficient hiPSC-derived PCs.
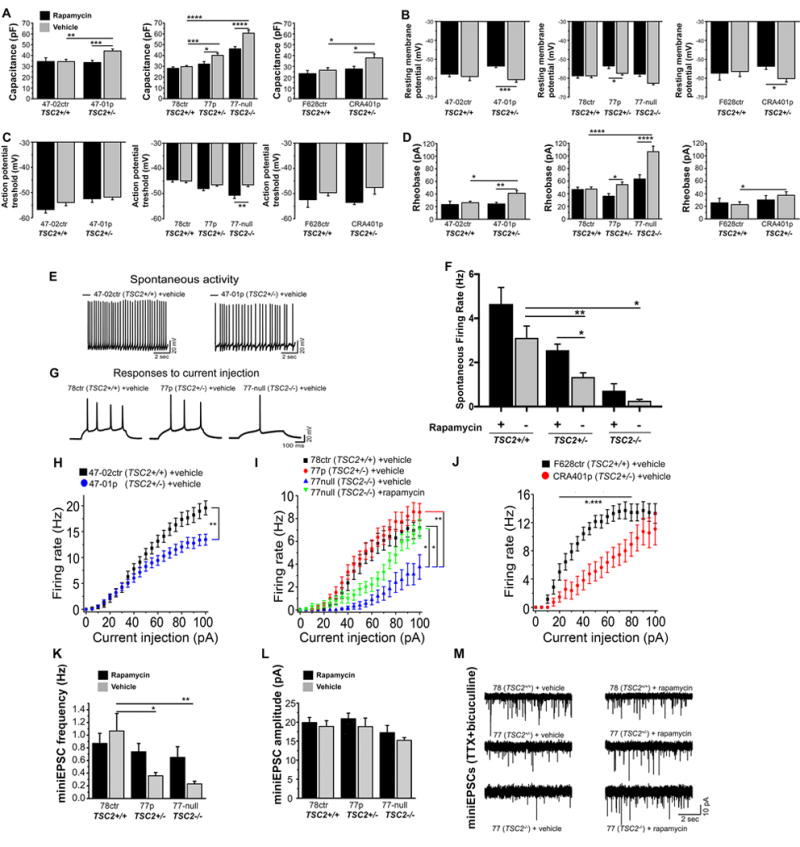
A) Capacitance of hiPSC-derived PCs. TSC2+/− and TSC2−/− PCs had a significantly larger capacitance as compared to TSC2+/+ control PCs, with the defects completely (TSC2+/− PCs) or partially (TSC2−/− PCs) rescued by rapamycin treatment. B) Resting membrane potential of hiPSC-PCs. Rapamycin treatment depolarized the RMP in TSC2+/− PCs. C) Action potential threshold of hiPSC-PCs. Rapamycin treatment lowered the action potential threshold in TSC2−/− PCs. D) Rheobase of hiPSC-PCs. Rheobase of 47-01, 77-TSC2-null and CRA401 PCs, but not 77-patient PCs, was higher compared to control PCs. Rapamycin treatment reduced the rheobase in all TSC-deficient PC lines. E) Representative traces of spontaneous activity in TSC2+/+ (47-02) control and TSC2+/− (47-01) patient PCs. 47-01-patient derived PCs display a lower spontaneous firing rate. F) Spontaneous firing rates of control and TSC2-mutant PCs, data is pooled from three control lines (47-02, 78, F628), three patient lines (47-01, 77, CRA401) and three biological replicates of TSC2-null lines. G) Representative responses of TSC2+/+ control, TSC2+/− patient, and TSC2−/− hiPSC-PCs to a 500 ms 55 pA current injection step. H–J) Excitability curves of control and TSC2-deficient hiPSC-PCs, created using 500ms 0-100pA step injections in 5pA increments. K) mEPSC frequency was lower in TSC2+/− and TSC2−/− PCs compared to control (TSC2+/+) PCs, and rapamycin treatment partly restored the frequencies. L) mEPSC amplitude was lower in TSC2−/− cells, but not TSC2+/− PCs compared to control PCs. All data are presented as mean±SEM. *p<0.05, ** p<0.01, ***p<0.001, ****p<0.0001, p-values were calculated using two-way ANOVA, followed by post-hoc-tests. M. Representative mEPSCs recorded from control and TSC-deficient PCs in the presence of 1μM TTX + 30 μM bicuculline. TSC deficient PCs displayed a lower rate of mEPSCs that was increased by rapamycin treatment.
We next measured the PC resting membrane potential and action potential threshold. TSC2−/−, but not TSC2+/− PCs exhibited a lower resting membrane potential compared to the control cells (Figure 4B, Supplementary Table S4). Rapamycin treatment had no effect on resting membrane potential in control cells, but increased the resting membrane potential of both the TSC2+/− and the TSC2−/− cells (Figure 4B). The action potential threshold was similar in TSC2+/+, TSC2+/− and TSC2−/− cells (Figure 4C, Supplementary Table S4). Rapamycin treatment decreased the threshold in TSC2−/− cells, likely increasing their excitability, but had no effect on TSC2+/+ PCs. Next, we recorded spontaneous PC activity, since it is influenced by both the resting membrane potential and the action potential threshold. We observed a decrease in spontaneous activity in TSC/ASD-patient derived PC lines (47-01 and CRA 401, TSC2+/−) compared to their respective control PC lines (47-02 and F628, TSC2+/+, Figure 4E–F, Supplementary Table S4). Rapamycin treatment increased the spontaneous activity in the CRA401 TSC2+/− line to the same level as the control F628 TSC2+/+ line, and also increased the spontaneous activity in the 77 TSC2+/− and in the 47-01 TSC2+/− line (Supplementary Table S4).
To measure PC excitability, we recorded the rheobase in hiPSC-derived PCs and found that it was significantly higher in TSC/ASD patient derived PCs (47-01 and CRA401, TSC2+/−) and TSC2−/− PCs (77 TSC2 null) than in control TSC2+/+ PCs (Figure 4D, Supplementary Table S4). A higher rheobase is generally indicative of lower excitability, and this excitability deficit was rescued by rapamycin in TSC2+/− PCs and partially rescued in TSC2−/− PCs, while rapamycin had no effect on the control neurons (Figures 4D, Table S4). In another measure of neuronal excitability, we stimulated the PCs with 500 ms current pulses of increasing amplitudes (Figure 4H–J) and recorded the number of action potentials generated with each current pulse. TSC2+/− PCs from one TSC-patient (77) responded similarly to control cells (78 ctr), while two other TSC/ASD patient derived PCs (47-01, CRA401, TSC2+/−) were significantly hypo-excitable compared to controls (Figure 4H–J, Supplementary Table S4). To generate the same frequency of AP firing in the TSC2−/− PCs (77-TSC2 null) the cells required a >3-fold larger current amplitude compared to control cells (Figure 4I, Supplementary Table S4). This excitability deficit could also be partially rescued by rapamycin treatment. Together, these data reveal a significant hypo-excitability in PCs derived from two individuals with TSC and diagnosed with ASD (47-01 and CRA401 lines, Figure 4, Supplementary Table S4), and this deficit could be rescued with rapamycin. In contrast, PCs derived from one individual with TSC without ASD did not show significant hypo-excitability (cell line 77) compared to control cells. Interestingly, after a mutation in the second allele of TSC2 allele was introduced (77 TSC2-null hiPSC-line), the cells displayed hypoexcitability, which was rescued with rapamycin treatment (Figure 4I). Taken together, our data show that specific TSC2-mutations affects functionality of PCs, suggesting that TSC2 expression is important for development of normal electrophysiological properties of human PCs.
We further characterized synaptic activity of the PCs, by first recording spontaneous excitatory postsynaptic currents (sEPSCs) and inhibitory post-synaptic currents (sIPSCs) from the hiPSC-derived PCs (Supplementary Figure S10A). PCs received inhibitory GABAergic synaptic input that was blocked by bicuculline, a selective GABAA receptor antagonist, and excitatory glutamatergic input that was blocked by CNQX, a selective AMPA receptor antagonist (Supplemental Figure S10A). Together, these data show that GABAA and AMPA receptors mediate inhibitory and excitatory synaptic inputs onto hiPSC-derived PCs, a finding supported by previous studies of hESC-derived PCs24.
To evaluate the impact of TSC2 deficiency on glutamatergic synapse formation and excitatory synaptic transmission, we recorded miniature EPSCs from control and TSC2-deficient hiPSC-derived PCs in the presence of 1 μM TTX and the GABAA-receptor antagonist bicuculline (30μM). The frequency of mEPSCs was significantly reduced in gene-dosage-dependent manner in TSC2+/− and TSC2−/− PCs relative to control lines, and could be partially rescued with rapamycin treatment (Figure 4K, Supplementary Table S4). Compared to control cells, there was no change in mEPSC amplitude in TSC2+/− PCs, but there was a small decrease in mEPSC amplitude in the TSC2−/− cells (Figure 4L, Supplementary Table S4). These data demonstrate that there are decreased number of functional glutamatergic synapses in the TSC2-deficient PCs compared to controls, which is in line with decreased expression of GRID2 and synaptophysin in TSC2+/− and TSC2−/− PCs (Figure 3). There was no change in EPSC decay kinetics (Supplementary Table S4), implying that glutamate receptor properties were similar at the synapses of both control and TSC2-deficient neurons. Thus, changes in neuronal excitability and excitatory synaptic function are evident in TSC2-deficient hiPSC-derived PCs. Our data show that these defects can be corrected by inhibition of mTORC1 at a time when human PCs and granule neurons have established a synaptic network.
To analyze also the effects of specific antagonists of both mTORC1 and mTORC2 in hiPSC-derived PCs, we treated the cells with Torin1 25nM for 1 week after long-term differentiation with mouse GCs. Torin1 is a selective ATP-competitive inhibitor that can block phosphorylation of both mTORC1 and mTORC2. As expected, the Torin1 reduced significantly the overactivation of mTORC1-pathway in TSC2-deficient patient cells by decreasing the pS6 levels to similar level with control cells (Supplementary Figure S11). In addition, Torin1 increased the expression of synaptic markers GRID2 and SYP1 in the TSC2-deficient CALB1+ cells (Supplementary Figure S11). We detected increased cell size and capacitance of TSC2+/− patient cells compared to control cells (Figure 4A), which was rescued with Torin1 treatment to similar level with control cells (Supplementary Figure S11). In conclusion, our data show that the effect of Torin1 in TSC2-deficient PCs were consistent with the rescue effects of rapamycin in TSC2-deficient PCs.
Gene expression analyses of TSC2-deficient hiPSC-PCs
To identify the molecular mechanisms underlying the morphological and physiological changes, we performed RNA sequencing in five different hiPSC-line derived THY1+ PCs: two control lines (TSC2+/+), two TSC–patient lines (TSC2+/−): one TSC-patient without ASD and one TSC/ASD patient (TSC2+/−), and one TSC2-null line, (n=3 for each cell-line) (Supplementary Table 1, Supplementary Figure 12). We compared gene expression between each patient line (TSC2+/−) and the related controls (TSC2+/+), as well as the TSC2−/− line to its related control. First, we removed the genes expressed differently between genders, by using previously obtained microarray data from control hiPSC-neurons derived from male and female individuals, described in 30. After removal of genes differentially expressed due to gender differences, we identified 117 and 30 genes differentially expressed between patient and related control cells and 585 differentially expressed genes between the TSC2−/− line and control cells (FDR < 0.01; Figure 5A, Supplementary Table S6). We compared the differentially expressed genes identified in this analysis to a previous study of hESC-neuronal cultures (mixture of glutamatergic and GABAergic neural cells) from control TSC2+/+ and engineered TSC2+/− and TSC2−/− cells31. We identified a significant overlap with the differentially expressed genes between the TSC2−/− line and control (p=2.56e-07) demonstrating a consistent cellular response to the complete loss of TSC2 in neurons (Supplementary Figure S13). Since the TSC2−/− cells displayed more severe phenotype than the TSC2+/− cells, we decided to perform weighted gene co-expression network analysis to identify gene expression patterns that were related to TSC2 allele dosage. We identified two modules of co-expressed genes that had the greatest positive (M67) and negative (M99) correlation with TSC2-genotype in hiPSC-PCs (Figure 5B–C, Supplementary Tables S7-8).
Figure 5. Gene expression profiling of TSC2 deficient hiPSC-derived PCs.
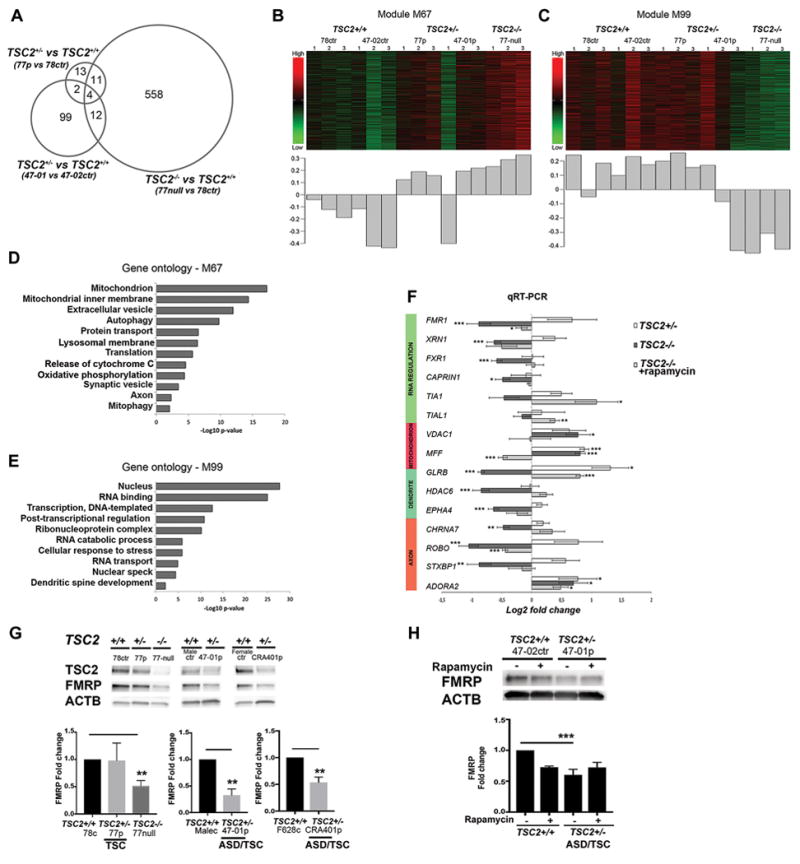
RNA sequencing analysis of hiPSC-PCs from controls (TSC2+/+) (47-02, 78), patients (TSC2+/−) (47-01 and 77) and TSC2-null cell line (TSC2−/−). A. Venn diagram demonstrating the number of differentially expressed genes for each comparison (FDR < 0.01), and their relationship to each other. B–C). Heatmaps and first principal components of the co-expressed groups of transcripts with the strongest positive (M67) and negative correlation with genotype (M99). The heatmap depicts scaled expression of all transcripts within the co-expression group, where green is low relative expression and red is high relative expression. The bar graph shows the first principal component of the expression of the transcripts within the co-expression module. D–E) Gene ontology analysis for genes belonging to the co-expression modules with strongest positive and negative correlation with genotype. Gene ontology terms were selected for specificity and non-redundancy. The x-axis represents –log10 p-value for enrichment of the GO term calculated by DAVID. F) qRT-PCR analyses from pooled data of two patients (47-01 and 77), two controls (47-02 and 78) and three replicates of TSC2-null lines, and rapamycin treated TSC2-null cells. G) FMRP expression in THY1+ hiPSC-PCs from three individual control lines (78 control, and sex-matched male-control and female-control line), vs three patient lines (77 patient, 47-01 patient, CRA401patient) and TSC2-null cell line (n=3). ASD/TSC2-patient derived PCs (47-01 and CRA401) and TSC2-null cells had significantly reduced FMRP expression compared to control cells. One-way ANOVA followed by post-hoc analyses and t-test between two groups, **p<0.01. H) FMRP expression in vehicle or rapamycin treated hiPSC-derived PCs at day 48 of differentiation. Fold changes were analyzed with two-way ANOVA followed by post-hoc-test, (***p<0.001).
Consistent with the differential expression analysis, we found a significant overlap for both groups with differentially expressed genes identified in TSC2−/− hESC-neural cells (M67 p = 8.60e-17, M99 p = 1.65e-4)31 (Supplementary Figure S13). To gain functional insight into these groups of genes, we performed gene ontology analysis. In the positively correlated group, there were several significantly enriched categories, including mitochondria, protein transport, and autophagy (Figure 5D), whereas the negatively correlated group showed enrichment of genes related to mRNA processing and transport (Figure 5E). Given the over-representation of genes associated with RNA processing and transport, we compared this group to known targets of FMRP32, 33. We found a significant over-representation of FMRP target genes within this group (p= 2.79E-78 compared to dataset 32 and p=0.015 compared to dataset 33), as well as targets of FXR1 (p=2.34E-30 compared to dataset 32), which is present within this group of genes.
To confirm these findings, we performed qRT-PCR using RNA collected from THY1+-sorted hiPSC-PCs (Figure 5F). We selected genes with high connectivity in either co-expression module or in the enriched functional categories that were identified with gene ontology analysis. Among the up-regulated genes in the TSC2-deficient cells were VDAC1 and MFF, which affect mitochondrial membrane potential and function. In the down-regulated group of genes in TSC2−/− cells were RNA binding proteins that are components of RNA granules within dendrites, including FMR1, FXR1, TIA1, TIA1L, XRN1, (Figure 5F, Supplementary Table S5). Rapamycin treatment restored the expression levels of these mRNAs in TSC2−/− cells. RNA processing dysregulation implied dysfunctional protein synthesis and synaptic development in TSC2-deficient PCs. We confirmed FMRP down regulation at protein level in TSC2-deficient ASD-patient derived PCs (patients 47-01 and CRA401) and TSC2-77-null PCs (Figure 5G). Rapamycin treatment partially rescued downregulation of the FMRP-levels in 47-01 cells, but didn’t completely restore it at day 48 (Figure 5H). Taken together, these findings indicate that loss of TSC2 function in TSC-patient derived PCs results in downregulation of RNA regulators, which affect synaptic development and function of the developing human PCs in TSC and ASD patients.
Discussion
Cerebellar dysfunction, degeneration and PC atrophy have been reported to be associated with ASD1-3, and cerebellar dysfunction has been implicated in abnormal social behavior in TSC 10. Therefore, gaining insights into the role of loss of function of TSC in human cerebellar PC development is a crucial step for identifying novel therapeutic targets for TSC and ASD. To study the cellular deficits underlying this disorder, we established here the first developmental model of cerebellar PC differentiation from individuals diagnosed with TSC and ASD. We identified mTORC1 pathway hyperactivation in TSC-patient derived PCs, which affected the neuronal differentiation, morphology, synaptic development, and electrophysiological properties of the TSC2-deficient PCs in co-culture with their cerebellar target neurons.
During embryonic brain development, mTOR pathway plays different roles depending on the cellular identity, differentiation stage and cellular functionality. mTOR has been shown to be necessary for neural stem cell proliferation in rodents 34 and the TSC complex is known to be a crucial regulator of mTOR in neural stem cells. We detected mTOR pathway overactivation and hyperproliferation of TSC2-deficient NPCs compared to controls. Cerebellum development and PC differentiation is regulated by Cbx2 expression, previously described in mouse model 35. Here we detected delayed cerebellar induction and deficient hiPSC-derived NPC and PC development with decreased GBX2 and PCP2 expression, and decreased number of SKOR2-, LHX1-precursor cells, and THY1-positive cells among TSC2-deficient cell populations. Our data in human patient derived NPCs and neural cells is consistent with previous discoveries of the Tsc-deficient rodent NPC development, where mTORC1 hyperactivation has been shown to lead to increased proliferation and reduced neuronal differentiation 36-38. Here we also detected an upregulation of astroglial precursor markers CD44 and GFAP in the TSC2−/− NPC population, which indicated an increase in the differentiation of astroglial cells from TSC2 deficient human stem cells. Our data is consistent with previous clinical studies that show astrogliosis in cortical39 and cerebellar tubers40 in TSC, and increased astroglial precursor differentiation of TSC2-mutant hESC derived cells in vitro 31. In Tsc-animal models, the mTOR-pathway hyperactivation have been linked to increased astroglial differentiation and reduced oligodendrocyte differentiation of the precursor cells 41, 42. In addition, these differentiation abnormalities are associated with hypomyelination in Tsc-deficient animal models 43, 44, and also non-cell autonomous mechanisms have been detected in Tsc-deficient neurons that affect dysfunction of the surrounding oligodendrocytes causing deficits in myelination 45, 46. Moreover, patients with TSC have abnormalities in white matter tracts on neuroimaging 47-49. In summary, these previous studies indicate that mTOR-pathway overactivation in TSC affects astroglial, oligodendroglial, and neuronal cell fate during development, and disturbs myelination of the axons in CNS. Differentiation of human oligodendrocytes from pluripotent stem cells requires specific growth factors such as PDGF-AA, SHH, RA, IGF-1, and takes over 75-120 days in vitro 50-54. Thus, we don’t anticipate spontaneous differentiation of oligodendrocytes in our cerebellar PC differentiation protocol with different growth factors, especially at the early cerebellar precursor phase on days 24-38 that is too early for oligodendrocyte differentiation, or at the later time point after enrichment of PCs with THY1-antibody that doesn’t bind to oligodendrocytes. However, we detected spontaneous astrogliosis in TSC2 deficient hiPSC-derived cell populations during cerebellar NPC differentiation in vitro. These data from TSC2-deficient cells are consistent with post-mortem studies of ASD patients, which have shown increased astroglial cells in the cerebellar white matter and reduced numbers of PCs in the cerebellum 55-57. Taken together, our human stem cell based model recapitulates effectively the deficits of the early embryonal stage cerebellar tissue development and increased astroglial differentiation in TSC and ASD.
We detected increased cleaved Caspase 3 expression and oxidative stress in TSC2−/− hiPSC-derived post-mitotic PC population, which may cause increased apoptosis of differentiating PCs, a finding that is consistent with decreased volume of the cerebellar cortex of TSC patients 58. Moreover, increased inflammation has been previously associated with TSC2 mutation in hESC-derived neurons31. Increased astroglial differentiation observed in our TSC patient derived NPCs in vitro and astrogliosis in patient brain in vivo could also be caused by reactive cells to the to the oxidative stress, inflammation, and cell death that is associated with loss of function of TSC. Here we showed that treatment of the TSC2 deficient hiPSC-derived neural cells with rapamycin in vitro restored their normal proliferation and differentiation capacity at the early stages of differentiation.
All of the TSC-patient derived hiPSC lines studied here were derived from individuals with cortical tubers (cell lines TSC2+/− 47-01, 77, CRA401), which are a hallmark of TSC. Cerebellar tubers demonstrate similar pathology to cortical tubers, including ectopic neurons with abnormal morphology, gliosis and balloon cells40 with increased phosphorylation of S659. Here we detected mTORC1-pathway overactivation and significantly enlarged cell size and number of neurites in TSC2+/− and TSC2−/− PCs compared to control cells after long-term culture in vitro, which is consistent with the presence of enlarged dysplastic neuronal cells and giant cells in human tubers60, 61 and enlarged neural cells and abnormal dendrites in Tsc1/2 mice models and mTOR-deficient rat cells 12, 38, 62, 46, 63, 64, 65. We also detected a significant increase in proliferation of TSC2+/− and TSC2−/− Ki67+ neural precursor cells, and we observed the overexpression of the cell proliferation regulator CYCLIND1 in TSC2−/− neural cells compared to controls. Previous studies have shown that mutations in CYCLIND1/D2 cause the disorder megalencephaly-polymicrogyria-polydactyly-hydrocephalus66, which demonstrates similar neuropathological findings to TSC, such as enlarged balloon cells and neuronal tissue overgrowth67. Therefore, our results suggest that the PI3K-AKT-mTOR-pathway and CYCLIND1 activity may converge, leading to common pathological abnormalities in these disorders.
We identified several molecular abnormalities in the TSC2-deficient developing human PCs. Consistent with our previous studies with rodent and human TSC2-deficient cortical neurons68, 69,we identified upregulation of genes associated with mitochondria and autophagy activation in the TSC2−/− human PCs. Interestingly, we also detected decreased expression of genes associated with mRNA processing and transport, which were enriched in FMRP targets. FMRP has been associated with regulation of GABA receptor function70, 71, including decreased inhibitory synaptic transmission in Fragile X Syndrome72. Thus, FMRP downregulation in TSC2-deficient PCs may contribute to the aberrant reduced synaptic development and hypoexcitability of the TSC2 mutant PCs described in this study. FMRP may be regulated post-translationally by the mTOR pathway73. Therefore, dysregulation of dendritic mRNA transport and translation may represent a common pathway between TSC and Fragile X syndrome in the development of ASD. Our study suggests that modulators of FMRP signaling could be targets for development of pharmacological therapies for patients with TSC and ASD.
Our study also showed that the developmental and functional deficits of the TSC2-mutant hiPSC-derived PCs could be rescued with specific mTORC1 inhibitor rapamycin in vitro. We treated the hiPSC-derived developing PCs with rapamycin for several days, which may also have affected other molecular pathways in addition to mTORC1, and compensatory mechanisms due to the prolonged nature of the treatment may have occurred. In addition, our results of the Torin1-treatment in the TSC2-deficient hiPSC-PCs resembled the effects of rapamycin in these cells, suggesting that long term rapamycin treatment may result in inhibition of both mTORC1 and mTORC2 pathway, as described in different cell models74. Rapamycin and other rapalogs are currently being tested in clinical trials for treating neurological symptoms of children with TSC. The primary outcome measures of these studies are focused on improvements in neurocognition and behavioral changes (reviewed in75). Given that we observe TSC2-deficient PCs death in vitro, we predict that rapamycin treatment may also rescue the loss of cerebellum volume in patients with TSC2-mutations58. Our cellular data suggest that mTOR-inhibitors may be beneficial for the rescuing of the developmental deficits of the cerebellum, especially of PCs, and could improve the formation of functional signaling network in the brain.
While we specifically focused on the function of PCs from individuals with TSC, our discovery of the intrinsic hypo-excitability and excitatory synaptic dysfunction in PCs from individuals with ASD may have implications for the broader field of autism. In terms of brain circuitry, PCs are the only inhibitory output neuron from the cerebellum and connect to the thalamus and frontal cortex via the dentatothalamic tract. Decreased inhibitory signaling of PCs can cause an imbalance between inhibitory and excitatory signaling during TSC patient brain development, which can dysregulate neural circuitry formation and accelerate pathophysiology of TSC and ASD. Our results support the hypothesis that PCs are dysfunctional in patients with TSC and ASD. In addition, our results suggest that the phenotype of the patient derived cell culture model established here could be studied in the future in large cohort of TSC-patient derived hiPSC-lines to determinate if FMRP-downregulation and hypo-excitability of PCs in TSC could indicate a risk of developing ASD. Although additional studies are needed in the future, our hiPSC-derived PC culture model could also be suitable for screening of novel compounds that can rescue the PC dysfunction and for development of personalized therapies for these patients.
Supplementary Material
SM Fig 1. Pluripotency marker expression in hiPSCs.
SM Fig 2. CRISPR-mediated gene correction of TSC2-mutation in hiPSC-line and cerebellar cell differentiation capacity of isogenic cells with TSC2-deficiency.
SM Fig 3. Characterization of mTOR-pathway activation in TSC2-deficient hiPSC-derived NPCs.
SM Fig 4. Characterization of TSC2-deficient hiPSC-derived NPCs.
SM Fig 5. Differentiation capacity of TSC2-deficient hiPSC-derived NPCs into PCs.
SM Fig 6. Flow cytometric analyses of TSC2-deficient hiPSC-derived cerebellar cell populations.
SM Fig 7. mTORC1-pathway activation in THY1+ hiPSC-PCs.
SM Fig 8. Comparison of mTORC1-pathway activation and soma size between different TSC-patient hiPSC-derived PCs.
SM Fig 9. TSC2-deficient THY1+ hiPSC-PCs have abnormal morphology and synaptic marker expression compared to control cells.
SM Fig 10. Electrophysiological characterization of TSC2-deficient hiPSC-PCs.
SM Fig 11. Characterization of Torin1 treated TSC2-deficient hiPSC-derived PCs.
SM Fig 12. Identification of TSC2-mutations in hiPSC-derived THY1+ PCs.
SM Fig 13. Transcriptional profiling of TSC2-deficient THY1+ hiPSC-PCs compared to hESC-derived neural cell populations.
Table S1. Clinical information about the patient and control subjects enrolled for the hiPSC-derivation.
Table S2. Primary and secondary antibodies used for immunocytochemical staining and flow cytometry.
Table S3. Antibodies used for western blot analyses.
Table S4. Electrophysiological characterization of hiPSC-PCs.
Table S5. Human specific primers for qRT-PCR.
Table S6. A-C. Transcriptional gene-expression analyses of THY1+ hiPSC-PCs.
Table S7. Module 67 gene-expression analyses of THY1+ hiPSC-PCs.
Table S8. Module 99 gene-expression analyses of THY1+ hiPSC-PCs.
Acknowledgments
We thank the individuals affected by TSC who took part in this study. We are grateful to Dr. Kleiman, Dr. Chen and Dr. Buttermore for helpful discussion, and Kerem Muslu for assistance in qRT-PCR analyses. We would like to thank research associate Christopher Stoddard (UConn Health, Human Genome Editing Core, US) for assistance with CRISPR-gene modification of hiPSCs. The hiPSC-line F628 was provided by Dr. George Daley (Boston Children’s Hospital). We thank BCH IDDRC Cellular Imaging Core for help with confocal imaging (U54 HD090255). This study was funded by U.S. Army Medical Research Tuberous Sclerosis Complex Research Program (W81XWH-15-1-0189), Nancy Lurie Marks Family Foundation, Harvard Stem Cell Institute, and the Children’s Hospital Boston Translational Research Program (to M.S.). K.W. is supported by R25 NS07068207S1. The project was co-sponsored by the Iris and Jumming Le Foundation and the Rockefeller University Center for Clinical and Translational Science #UL1 TR000043 (NCATS, NIH), CTSA and NIH R21 NS093540-01 (to M.E.H.).
Footnotes
Conflict of interest: authors declare no conflict of interest.
Author contributions: M.S.: PC differentiation protocol development for hiPSC, experimental design, PC differentiation, phenotyping in vitro, FC, transcriptional profiling, data analyses, writing of manuscript. I.T.: electrophysiology, data analyses, writing of manuscript. D.B.: PC differentiation protocol development for hESC. K.W.: transcriptional expression analyses, writing of manuscript. V.K.: confocal microscopy. K.K.: statistical analyses. D.C.: cell analyses. D.T.: technical assistance. M.J.H.: hiPSC-line derivation, C.J.W. and M.E.H.: discussion of data. M.S., MEH: experimental design, writing of manuscript.
Supplementary information is available at Molecular Psychiatry’s website.
Supplementary Materials
References
- 1.Casanova MF. The neuropathology of autism. Brain Pathol. 2007;17(4):422–433. doi: 10.1111/j.1750-3639.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008;7(3):406–416. doi: 10.1007/s12311-008-0043-y. [DOI] [PubMed] [Google Scholar]
- 3.Limperopoulos C, Bassan H, Gauvreau K, Robertson RL, Jr, Sullivan NR, Benson CB, et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics. 2007;120(3):584–593. doi: 10.1542/peds.2007-1041. [DOI] [PubMed] [Google Scholar]
- 4.Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84(2):275–291. doi: 10.1016/j.neuron.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 6.Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47(4):535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eluvathingal TJ, Behen ME, Chugani HT, Janisse J, Bernardi B, Chakraborty P, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol. 2006;21(10):846–851. doi: 10.1177/08830738060210100301. [DOI] [PubMed] [Google Scholar]
- 9.Weber AM, Egelhoff JC, McKellop JM, Franz DN. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord. 2000;30(6):511–517. doi: 10.1023/a:1005679108529. [DOI] [PubMed] [Google Scholar]
- 10.Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R, et al. Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology. 2001;57(7):1269–1277. doi: 10.1212/wnl.57.7.1269. [DOI] [PubMed] [Google Scholar]
- 11.Ertan G, Arulrajah S, Tekes A, Jordan L, Huisman TA. Cerebellar abnormality in children and young adults with tuberous sclerosis complex: MR and diffusion weighted imaging findings. J Neuroradiol. 2010;37(4):231–238. doi: 10.1016/j.neurad.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488(7413):647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2013;51:93–103. doi: 10.1016/j.nbd.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A, et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33(1):58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39(21):9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horii T, Tamura D, Morita S, Kimura M, Hatada I. Generation of an ICF syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. Int J Mol Sci. 2013;14(10):19774–19781. doi: 10.3390/ijms141019774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110(39):15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Q, Li K, Sunmonu NA, Li JY. Fgf8b-containing spliceforms, but not Fgf8a, are essential for Fgf8 function during development of the midbrain and cerebellum. Dev Biol. 2010;338(2):183–192. doi: 10.1016/j.ydbio.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selvadurai HJ, Mason JO. Wnt/beta-catenin signalling is active in a highly dynamic pattern during development of the mouse cerebellum. PLoS One. 2011;6(8):e23012. doi: 10.1371/journal.pone.0023012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundberg M, Bogetofte H, Lawson T, Jansson J, Smith G, Astradsson A, et al. Improved cell therapy protocols for Parkinson’s disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells. 2013;31(8):1548–1562. doi: 10.1002/stem.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchholz DE, Pennington BO, Croze RH, Hinman CR, Coffey PJ, Clegg DO. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl Med. 2013;2(5):384–393. doi: 10.5966/sctm.2012-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10(4):537–550. doi: 10.1016/j.celrep.2014.12.051. [DOI] [PubMed] [Google Scholar]
- 25.Heuer H, Mason CA. Thyroid hormone induces cerebellar Purkinje cell dendritic development via the thyroid hormone receptor alpha1. J Neurosci. 2003;23(33):10604–10612. doi: 10.1523/JNEUROSCI.23-33-10604.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S, Wang B, Pan N, Fu L, Wang C, Song G, et al. Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci Rep. 2015;5:9232. doi: 10.1038/srep09232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crino PB, Aronica E, Baltuch G, Nathanson KL. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology. 2010;74(21):1716–1723. doi: 10.1212/WNL.0b013e3181e04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa V, Aigner S, Vukcevic M, Sauter E, Behr K, Ebeling M, et al. mTORC1 Inhibition Corrects Neurodevelopmental and Synaptic Alterations in a Human Stem Cell Model of Tuberous Sclerosis. Cell Rep. 2016;15(1):86–95. doi: 10.1016/j.celrep.2016.02.090. [DOI] [PubMed] [Google Scholar]
- 29.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grabole N, Zhang JD, Aigner S, Ruderisch N, Costa V, Weber FC, et al. Genomic analysis of the molecular neuropathology of tuberous sclerosis using a human stem cell model. Genome Med. 2016;8(1):94. doi: 10.1186/s13073-016-0347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492(7429):382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cloetta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, et al. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci. 2013;33(18):7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagan N, Guarente J, Ellisor D, Zervas M. The Temporal Contribution of the Gbx2 Lineage to Cerebellar Neurons. Front Neuroanat. 2017;11:50. doi: 10.3389/fnana.2017.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, et al. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014;7(5):1626–1639. doi: 10.1016/j.celrep.2014.04.048. [DOI] [PubMed] [Google Scholar]
- 37.Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. 2011;9(5):447–462. doi: 10.1016/j.stem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Normand EA, Crandall SR, Thorn CA, Murphy EM, Voelcker B, Browning C, et al. Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron. 2013;78(5):895–909. doi: 10.1016/j.neuron.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grajkowska W, Kotulska K, Jurkiewicz E, Matyja E. Brain lesions in tuberous sclerosis complex. Review. Folia Neuropathol. 2010;48(3):139–149. [PubMed] [Google Scholar]
- 40.Jay V, Edwards V, Musharbash A, Rutka JT. Cerebellar pathology in tuberous sclerosis. Ultrastruct Pathol. 1998;22(4):331–339. doi: 10.3109/01913129809103354. [DOI] [PubMed] [Google Scholar]
- 41.Way SW, McKenna J, 3rd, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. 2009;18(7):1252–1265. doi: 10.1093/hmg/ddp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mietzsch U, McKenna J, 3rd, Reith RM, Way SW, Gambello MJ. Comparative analysis of Tsc1 and Tsc2 single and double radial glial cell mutants. J Comp Neurol. 2013;521(16):3817–3831. doi: 10.1002/cne.23380. [DOI] [PubMed] [Google Scholar]
- 43.Carson RP, Kelm ND, West KL, Does MD, Fu C, Weaver G, et al. Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann Clin Transl Neurol. 2015;2(12):1041–1054. doi: 10.1002/acn3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lebrun-Julien F, Bachmann L, Norrmen C, Trotzmuller M, Kofeler H, Ruegg MA, et al. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci. 2014;34(25):8432–8448. doi: 10.1523/JNEUROSCI.1105-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ercan E, Han JM, Di Nardo A, Winden K, Han MJ, Hoyo L, et al. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J Exp Med. 2017;214(3):681–697. doi: 10.1084/jem.20160446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27(21):5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Makki MI, Chugani DC, Janisse J, Chugani HT. Characteristics of abnormal diffusivity in normal-appearing white matter investigated with diffusion tensor MR imaging in tuberous sclerosis complex. AJNR Am J Neuroradiol. 2007;28(9):1662–1667. doi: 10.3174/ajnr.A0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arulrajah S, Ertan G, Jordan L, Tekes A, Khaykin E, Izbudak I, et al. Magnetic resonance imaging and diffusion-weighted imaging of normal-appearing white matter in children and young adults with tuberous sclerosis complex. Neuroradiology. 2009;51(11):781–786. doi: 10.1007/s00234-009-0563-2. [DOI] [PubMed] [Google Scholar]
- 49.Peters JM, Sahin M, Vogel-Farley VK, Jeste SS, Nelson CA, 3rd, Gregas MC, et al. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol. 2012;19(1):17–25. doi: 10.1016/j.acra.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Douvaras P, Wang J, Zimmer M, Hanchuk S, O’Bara MA, Sadiq S, et al. Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Reports. 2014;3(2):250–259. doi: 10.1016/j.stemcr.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. 2013;12(2):252–264. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stacpoole SR, Spitzer S, Bilican B, Compston A, Karadottir R, Chandran S, et al. High yields of oligodendrocyte lineage cells from human embryonic stem cells at physiological oxygen tensions for evaluation of translational biology. Stem Cell Reports. 2013;1(5):437–450. doi: 10.1016/j.stemcr.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sundberg M, Hyysalo A, Skottman H, Shin S, Vemuri M, Suuronen R, et al. A xeno-free culturing protocol for pluripotent stem cell-derived oligodendrocyte precursor cell production. Regen Med. 2011;6(4):449–460. doi: 10.2217/rme.11.36. [DOI] [PubMed] [Google Scholar]
- 54.Nistor GI, Totoiu MO, Haque N, Carpenter MK, Keirstead HS. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia. 2005;49(3):385–396. doi: 10.1002/glia.20127. [DOI] [PubMed] [Google Scholar]
- 55.Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23(2–3):183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 56.Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain. 1998;121(Pt 5):889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- 57.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 58.Weisenfeld NI, Peters JM, Tsai PT, Prabhu SP, Dies KA, Sahin M, et al. A magnetic resonance imaging study of cerebellar volume in tuberous sclerosis complex. Pediatr Neurol. 2013;48(2):105–110. doi: 10.1016/j.pediatrneurol.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boer K, Troost D, Jansen F, Nellist M, van den Ouweland AM, Geurts JJ, et al. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology. 2008;28(6):577–590. doi: 10.1111/j.1440-1789.2008.00920.x. [DOI] [PubMed] [Google Scholar]
- 60.Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19(9):716–725. doi: 10.1177/08830738040190091301. [DOI] [PubMed] [Google Scholar]
- 61.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56(4):510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- 62.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8(12):1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 63.Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52(3):285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 64.Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25(49):11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25(49):11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mirzaa GM, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet. 2014;46(5):510–515. doi: 10.1038/ng.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mirzaa G, Dodge NN, Glass I, Day C, Gripp K, Nicholson L, et al. Megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus: a rare brain malformation syndrome associated with mental retardation and seizures. Neuropediatrics. 2004;35(6):353–359. doi: 10.1055/s-2004-830497. [DOI] [PubMed] [Google Scholar]
- 68.Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet. 2014;23(14):3865–3874. doi: 10.1093/hmg/ddu101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ebrahimi-Fakhari D, Saffari A, Wahlster L, DiNardo A, Turner D, Lewis TL, Jr, et al. Impaired Mitochondrial Dynamics And Mitophagy In Neuronal Models Of Tuberous Sclerosis Complex. Cell Rep. 2016;17(8):2162. doi: 10.1016/j.celrep.2016.10.051. [DOI] [PubMed] [Google Scholar]
- 70.Adusei DC, Pacey LK, Chen D, Hampson DR. Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology. 2010;59(3):167–171. doi: 10.1016/j.neuropharm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 71.Curia G, Papouin T, Seguela P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex. 2009;19(7):1515–1520. doi: 10.1093/cercor/bhn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sabanov V, Braat S, D’Andrea L, Willemsen R, Zeidler S, Rooms L, et al. Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice. Neuropharmacology. 2017;116:71–81. doi: 10.1016/j.neuropharm.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 73.Narayanan U, Nalavadi V, Nakamoto M, Thomas G, Ceman S, Bassell GJ, et al. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem. 2008;283(27):18478–18482. doi: 10.1074/jbc.C800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 75.Sundberg M, Sahin M. Cerebellar Development and Autism Spectrum Disorder in Tuberous Sclerosis Complex. J Child Neurol. 2015;30(14):1954–1962. doi: 10.1177/0883073815600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SM Fig 1. Pluripotency marker expression in hiPSCs.
SM Fig 2. CRISPR-mediated gene correction of TSC2-mutation in hiPSC-line and cerebellar cell differentiation capacity of isogenic cells with TSC2-deficiency.
SM Fig 3. Characterization of mTOR-pathway activation in TSC2-deficient hiPSC-derived NPCs.
SM Fig 4. Characterization of TSC2-deficient hiPSC-derived NPCs.
SM Fig 5. Differentiation capacity of TSC2-deficient hiPSC-derived NPCs into PCs.
SM Fig 6. Flow cytometric analyses of TSC2-deficient hiPSC-derived cerebellar cell populations.
SM Fig 7. mTORC1-pathway activation in THY1+ hiPSC-PCs.
SM Fig 8. Comparison of mTORC1-pathway activation and soma size between different TSC-patient hiPSC-derived PCs.
SM Fig 9. TSC2-deficient THY1+ hiPSC-PCs have abnormal morphology and synaptic marker expression compared to control cells.
SM Fig 10. Electrophysiological characterization of TSC2-deficient hiPSC-PCs.
SM Fig 11. Characterization of Torin1 treated TSC2-deficient hiPSC-derived PCs.
SM Fig 12. Identification of TSC2-mutations in hiPSC-derived THY1+ PCs.
SM Fig 13. Transcriptional profiling of TSC2-deficient THY1+ hiPSC-PCs compared to hESC-derived neural cell populations.
Table S1. Clinical information about the patient and control subjects enrolled for the hiPSC-derivation.
Table S2. Primary and secondary antibodies used for immunocytochemical staining and flow cytometry.
Table S3. Antibodies used for western blot analyses.
Table S4. Electrophysiological characterization of hiPSC-PCs.
Table S5. Human specific primers for qRT-PCR.
Table S6. A-C. Transcriptional gene-expression analyses of THY1+ hiPSC-PCs.
Table S7. Module 67 gene-expression analyses of THY1+ hiPSC-PCs.
Table S8. Module 99 gene-expression analyses of THY1+ hiPSC-PCs.