

Abstract
Objective
Anti‐NMDA receptor encephalitis (ANRE) is a potentially lethal encephalitis attributed to autoantibodies against the N‐methyl‐D‐aspartate receptor (NMDAR). We sought to clone and characterize monoclonal antibodies (mAbs) from an ANRE patient.
Methods
We used a hybridoma method to clone two IgG mAbs from a female patient with ANRE without teratoma, and characterized their binding activities on NMDAR‐transfected cell lines, cultured primary rat neurons, and mouse hippocampus. We also assessed their effects on voluntary locomotor activity in mice and binding to NMDAR in vivo.
Results
The mAbs are structurally distinct and arose from distinct B‐cell lineages. They recognize different epitopes on the GluN1 amino terminal domain (ATD), yet both require amino acids important for post‐translational modification. Both mAbs bind subsets of GluN1 on cultured rat hippocampal neurons. The 5F5 mAb binds mouse brain hippocampal tissues, and the GluN1 recognized on cultured rat neurons was substantially extra‐synaptic. Antibody binding to primary hippocampal neurons induced receptor internalization. The NMDAR inhibitor MK‐801 inhibited internalization without preventing mAb binding; AP5 inhibited both mAb binding and internalization. Exposure of mice to the mAbs following permeabilization of the blood brain barrier increased voluntary wheel running activity, similar to low doses of the NMDAR inhibitor, MK‐801.
Interpretation
These mAbs recapitulate features demonstrated in previous studies of ANRE patient CSF, and exert effects on NMDAR in vitro and in vivo consistent with modulation of NMDAR activity.
Introduction
Anti‐N‐methyl‐D‐aspartate Receptor Encephalitis (ANRE) is an autoimmune syndrome resulting from autoantibodies targeting the NMDA receptor in the brain.1 Patients with ANRE exhibit heterogeneous psychiatric and neurologic symptoms including memory loss, psychosis, hallucinations, seizures, autonomic nervous system dysfunction, and catatonia.2, 3 These symptoms correlate with the presence of IgG antibodies specific for the GluN1 (NR1) subunit of NMDAR. Treatment includes immunosuppressive therapies that reduce the serum and CSF titers of anti‐NMDAR antibodies. Despite aggressive measures, a quarter of patients with anti‐NMDAR encephalitis remains severely impaired or die. For those who survive, recovery often takes years.
ANRE was originally characterized as a disease of women with ovarian teratomas, perhaps due to auto‐immune recognition of NMDAR expressed by the teratomas. It has since been appreciated that ANRE can occur without the coexistence of teratomas. ANRE has been attributed to the activity of IgG antibodies that bind the GluN1 subunit of NMDAR in the hippocampus and amygdala, and decrease NMDA clusters and synaptic currents in post‐synaptic dendrites.4 ANRE antibody binding to GluN1 on cultured neurons causes receptor internalization, which is mediated by receptor cross‐linking and results in decreased synaptic NMDAR levels, reduced synaptic NMDAR‐mediated currents, and impairment of NMDA‐dependent processes such as long‐term potentiation.4, 5 ANRE IgG binding correlates with the frequency of channel opening, and acute ANRE IgG exposure prolongs open time of the receptor, suggesting that an open channel configuration is important for antibody binding.6, 7 .
ANRE IgGs recognize the GluN1 subunit within its extracellular amino‐terminal domain (ATD), which regulates NMDAR ion channel function; including channel open probability, deactivation rate, and allosteric regulation.6, 8 The region required for GluN1 binding to ANRE IgG includes amino acids N368 and G369, which are required for post‐translational modifications.7 Monoclonal IgG antibodies isolated from ANRE‐patient CSF replicate many of the features predicted from the studies of polyclonal CSF IgGs.9 They bind NMDAR on hippocampal neurons in patterns that partly overlap with GluN1 and require the presence of N368 for binding. Furthermore, they downregulate NMDAR from the neuronal membrane and inhibit calcium influx. These studies support the hypothesis that anti‐NMDAR antibodies are pathogenic for ANRE. In addition, the observation that patients can generate multiple, independent anti‐GluN1 antibodies suggests that the varied clinical manifestations of ANRE could be explained by differences in antibody receptor binding specificity and regulatory effects.9
In this study, we cloned two mAbs from a patient with ANRE without an associated teratoma, and analyzed their antigen binding and functional features in vitro and in vivo. The two mAbs, 5F5 and 2G6, arose from independent B‐cell lineages, and they bind noncompeting epitopes on GluN1. They bind specifically to GluN1 in transfected cells and on cultured hippocampal neurons, but only a subset of the synaptic hippocampal GluN1 is recognized by the mAbs. The NMDAR inhibitor MK‐801 inhibits internalization without preventing mAb binding; AP5 inhibited both mAb binding and internalization. Exposure of mice to the mAbs following permeabilization of the blood brain barrier increased voluntary wheel running activity, similar to low doses of the NMDAR inhibitor, MK‐801. These experiments further support the role of anti‐GluN1 antibodies in the pathogenesis of ANRE and provide additional insight into the nature of the pathogenic autoantibody response.
Materials and Methods
Cell culture, hybridoma generation and antibody purification
Peripheral blood was obtained from an 18 year‐old female who presented to the Children's Hospital of Philadelphia with emotional lability, paranoia, and temporal lobe seizures, with anti‐GluN1 IgG detectable in her CSF. Peripheral blood mononuclear cells (PBMCs) were processed and cryopreserved as previously described.10 We performed a cell fusion following standard methods.10 Briefly, PBMCs expressing the CD27 antigen were isolated with anti‐CD27 magnetic beads (Miltenyi Biotec, Auburn, CA) and cultured for 8 days in the presence of human UltraCD40L (Multimeric Biotherapeutics, San Diego, CA) in advanced RPMI supplemented with 10% fetal bovine serum (FBS), cytokines and other growth factors. On day 8, cultured cells were electrofused to the B5‐6T cell line and selected with HAT. Hybridoma supernatants were screened for secretion of IgGs that bind GluN1 by whole cell ELISA (with GluN1‐GluN2a transfected HEK293T cells). Positive clones were subcloned 3 times to isolate stable hybridomas expressing GluN1‐reactive IgGs. The hybridomas were cultured in RPMI with Ultra Low IgG Fetal Bovine Serum US origin (Thermo Fisher Scientific, Waltham, MA) and IgGs were purified from the supernatants using Protein G Sepharose columns (GE Healthcare Life Sciences, Pittsburg, PA). MAb concentrations were measured with a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). HEK293T cells were cultured in DMEM 10% fetal calf serum with pen/strep/glutamine.
DNA and antibody reagents for immunocytochemistry
For HEK293T cell transfections, we used the previously described plasmids encoding GluN1a, GluN2a, AMPA receptor type 1 (GluA1), AMPA receptor type 2 (GluA2), GluN1a with the amino terminal residues 26‐382 deleted (GluN1a‐ATD) and GluN1a containing a N368Q mutation (GluN1‐N368Q).3, 7, 11 The HEK293T‐ATD cell line contains the amino terminal domain of GluN1a, fused to a MYC tag, TEV protease site, and the PDGF receptor transmembrane domain, and expressed in HEK293T cells by retroviral transduction using pBabePuro vector (manuscript submitted).7, 12
For detection of GluN1, we used the clone 54.1 mAb, which binds the extracellular loop between transmembrane regions III & IV (Millipore Cat# MAB363 RRID:AB94946), and a rabbit mAb that binds the GluN1 C‐terminal domain (Millipore Cat# AB9864 RRID:AB2112158). For detection of GluR1 or GluR2, we used rabbit anti‐GluR1 (Millipore Cat# AB1504 RRID:AB11212863) or rabbit anti‐GluR2 (Millipore Cat# 07‐598 RRID:AB11213931) polyclonal antibodies, followed by secondary anti‐rabbit IgG conjugated to Alexa 488 (Thermo Fisher Scientific Cat# A‐11034 also A11034 RRID:AB2576217). For rat neuron immunostaining, we used the anti‐MAP2 murine mAb (Thermo Fisher Scientific Cat# 13‐1500 RRID:AB2533001), mouse anti‐GluN1 (BD Biosciences Cat# 556308 RRID:AB396353) or mouse anti‐PSD95 (BD Biosciences Cat# 610496 RRID:AB397862) and rabbit anti‐GluN1 (Millipore Cat# AB9864 RRID:AB2112158). For internalization testing, mAbs were conjugated to CypHer5E NHS Ester (GE Healthcare Life Sciences) following the manufacturer's instructions. Cells were seeded on coverslips and maintained in PBS at 37°C in a humidified CO2 incubator, and visualized at the indicated time points by confocal microscopy.
Whole cell ELISA
To identify IgGs immunoreactive with GluN1, we used a whole‐cell ELISA.13 We plated 5 × 105 HEK293T cells onto 6 cm plates in 4 ml DMEM containing 10% FBS and 1% penicillin/streptomycin. One day later, we transfected 1 μg each of the GluN1a and GluN2a plasmids using X‐tremeGENE 9 DNA transfection reagent (Roche, Basel, Switzerland). Medium was supplemented with 100 μmol/L MK‐801 (SigmaAldrich, St. Louis, MO). The next day, we fed the cells with 4 mL fresh medium with MK‐801. 48 h after transfection, we trypsinized the cells, washed them with PBS, and plated them in Corning® 96 Well EIA/RIA Clear Flat Bottom Polystyrene High Bind Microplates (Corning, NY) at 1 × 105 cells/well in 100 μL PBS. The plates were spun at 350 g for 10 min, supernatants were discarded, and the plates were dried at 37°C for 20 min, then fixed with 100 μL per well of 2% paraformaldehyde in PBS for 15 min at room temp. The plates were washed three times with PBS pH 7.8 containing 0.05% Tween‐20 (PBST), then blocked with 5% bovine serum albumin (BSA) in PBST (PBST:BSA) overnight at 4°C, followed by 3 washes with PBST. Patient samples or purified mAbs were diluted in PBST:BSA and applied to the plates 1 h at 37°C (hybridoma supernatants were used without dilution). As positive and negative controls, we used an anti‐NMDAR1 rabbit mAb at 1:100 dilution (Millipore Cat# AB9864 RRID:AB2112158) and an isotype control human mAb (6A).14 After 1 h, the plates were washed 3X with PBST. As secondary antibodies, we used an HRP‐conjugated anti‐human IgG mAb (1:1500) (SouthernBiotech, Cat# 4030‐05, RRID:AB2687483) and an HRP‐conjugated, polyclonal goat anti‐rabbit IgG (1:1500) (SouthernBiotech, Cat# 9040‐05, RRID:AB2687484). OPD was used as substrate and optical density (OD) was measured at 490 nm using Synergy II plate reader (Biotek Instruments, Winooski, VT).
To determine whether 5F5 and 2G6 mAbs could simultaneously bind to GluN1, we tested binding to the HEK293T‐ATD cell line (105 cells/well) in the Whole Cell ELISA and used a luminescent detection method. We biotinylated the 5F5 and 2G6 or 5F5 using the EZ‐LinkTM Hydrazide‐Biotin kit (Thermo Fisher), then generated dilution series of 2G6 or 5F5 (50 μg/mL to 0.2 μg/mL) and added to them the plate for 1 h at 37°C, followed by three washes with PBST. We then added the biotinylated 5F5 or 2G6 mAbs at 5 μg/mL, or PBS, and incubated for 1 h at 37°C followed by three washes with PBST. The Pierce Streptavidin Poly‐HRP substrate (Thermo Fisher) was added at 1:2000 dilution and incubated for 1 h at 37°C. This was followed by SuperSignal ELISA Femto Substrate, (1:1 ratio) (Thermo Fisher) and relative luminescence values were measured using the Synergy II plate reader. Duplicate binding curves were plotted and the linear portions were used for analysis using Excel.
Immunofluorescence studies with cultured cells
HEK293T cells were cultured and transfected with plasmids expressing GluN1a, GluN2a, GluR1, GluR2, GluN1‐ATD (deleted amino acids 26‐382), and GluN1‐N368Q as described above, except that 104 cells/well were cultured on round Corning™ BioCoat™ 12 mm #1 German Glass Coverslips in 24 well plates.7 48 h after transfection, cells were stained as described.3 The cells were fixed in PBS with 4% paraformaldehyde for 10 min at room temperature, then washed with PBS, treated with 0.3% TritonX‐100 in PBS for 10 min at room temp, and washed again with PBST. The cells were blocked with 10% Goat serum + 1% BSA in PBS (PBS + G + B) for 1 h at 37°C, then washed with PBST. Cells were incubated with mAbs (5F5, 2G6, or an isotype control mAb 8E1 or 6A) at a concentration of 5 μg/mL in PBS + G + B for 1 h at room temp. ANRE patient CSF were used at 1:100 dilutions. GluN1 expression was detected with the commercial antibodies noted above. After 1 h, cells were washed twice with PBST and incubated with secondary antibodies in PBS+G+B for 1 h, 1:1000 Alexa 488 goat anti‐mouse (Thermo Fisher Scientific Cat# A‐11029 RRID:AB2534088), 1:1000 Alexa 568 goat anti‐human (A21090, Thermo Fisher) or 1:200 goat anti‐rabbit Alexa 488 (Thermo Fisher Scientific Cat# A‐11034 also A11034 RRID:AB2576217). Cells were washed once with PBS followed by dH2O and then their coverslips were mounted with ProLong Gold Antifade reagent with DAPI (Thermo Fisher) and imaged with a C2 + Nikon confocal microscope with 63x/1.3 NA oil objective; images were analyzed with ImageJ software (https://imagej.nih.gov/ij/).
High‐throughput sequencing of VH –encoding genes and phylogenetic analyses of the 5F5 and 2G6 mAb lineages
Total RNA was isolated from a subset of the CD27 + selected peripheral blood mononuclear cells that had undergone in vitro culture prior to cell fusion (see above). 500 ng RNA was used for reverse transcription according to the manufacturer's instructions using SuperScript III Enzyme (Life Technologies) and oligo‐dT primer, following standard protocols.15 After cDNA construction, VH transcripts were PCR‐amplified using FastStart Taq DNA polymerase (Sigma‐Aldrich) under the following conditions: 2 min at 95°C; 4 cycles of 92°C for 1 min, 50°C for 1 min, 72°C for 1 min; 4 cycles of 92°C for 1 min, 55°C for 1 min, 72°C for 1 min; 24 cycles of 92°C for 1 min, 63°C for 1 min, 72°C for 1 min; 72°C for 7 min; held at 4°C. The final sequencing library was sequenced using the Illumina MiSeq platform.
Raw MiSeq sequence reads were stitched using PEAR,16 and then quality filtered and annotated using MiXCR.17 Productive, full‐length VH sequences with IGHV3‐30 gene usage were grouped into 5F5‐ and 2G6‐lineages by clustering on the CDRH3 nucleotide sequences of 5F5 and 2G6 mAbs at 80% identity, respectively. Sequences in each lineage were further clustered on the full‐length nucleotide sequence at 98% identity to reduce PCR and sequencing error, and sequences with 1 read were removed. The resulting sequences were aligned by MAFFT,18 and the maximum likelihood phylogenetic trees analysis was performed using RAxML.19
Primary rat neuron and brain immunostaining
Primary rat or murine neurons were obtained from Thermo Fisher (A10841‐01) or from the Cellular Neuroscience Core Facility at the Children's Hospital of Pennsylvania.20 Cells were grown in Neurobasal® Medium supplemented with 200 mmol/L GLUTAMAX and 2% B‐27® Supplement (Thermo Fisher). A total of 105 cells/well were plated in 24‐well plate containing coverslips, and kept at 37°C for culture. After 24 h, half of the medium in each well was replaced with fresh medium and cells were grown for 14 days. The cells were then washed, fixed, and blocked as mentioned above. Cells were then incubated with 5 μg/mL of 5F5, 2G6, or CSF (1:100) in PBS + G + B. After 1 h at 37°C, cells were washed, incubated with fluorescent secondary antibodies (BD Biosciences Cat# 556308 RRID:AB396353) or mouse anti‐PSD95 (BD Biosciences Cat# 610496 RRID:AB397862) and rabbit anti‐GluN1 (Millipore Cat# AB9864 RRID:AB2112158). In the experiments with the mAbs labeled with CypHER 5E, neurons were incubated with additional 10 mmol/L glycine, 30 mmol/L glutamate, with or without either MK‐801 (50 μm) or AP5 (100 μm) for 15 min. Labeled mAbs were then added, and after 45 min the neurons were fixed and processed as noted above. Images were acquired using a Carl Zeiss LSM 510 UV META inverted confocal microscope with a Plan‐Apo 60X oil immersion lens at room temperature and processed using Zeiss AIM 4.2 SP1 software (Zeiss Microimaging, Thornwood, NY).
Adult mouse brains were fixed for 24 h in 4% paraformaldehyde, then stored in PBS. Brains were embedded in a 4% agarose block, sectioned using a vibratome (20 μm sections), and collected in anti‐freeze (30% ethylene glycol, 30% glycerol, 30% MilliQ water, 10% 10X PBS) as floating sections, washed 5X in PBST, and blocked with 5% normal goat serum. Before staining, mouse sections were incubated with Vector M.O.M. diluent prepared in M.O.M mouse IgG Blocking Reagent (Vector Laboratories cat# BMK‐2202), then incubated overnight with patient CSF (1:5) or mAb (100 μg/mL) and mouse‐anti GluN1 (BD Biosciences cat# 556308) prepared in Blocking Reagent. Sections were washed five times with Blocking Reagent and incubated with secondary antibodies (goat anti‐human IgG Alexa Fluor 488 and goat anti‐mouse 568) prepared in Blocking Reagent. Sections were washed four times in PBST, once with PBS, and then mounted on slides for visualization on a Leica DMi8 confocal microscope.
Effect of NMDAR antibodies on mouse wheel running activity
Female 6–8 week‐old Swiss Webster mice (Taconic Biosciences; Germantown, NY) were housed at the AAALAC‐certified animal facility at the Lankenau Institute for Medical Research. Experiments were approved by the Main Line Health Institutional Animal Care and Use Committee (IACUC). Mice were housed in pairs, in cages fitted with running wheels connected to a microchip and a magnetic wheel revolution counter (Mini‐Mitter Co. Inc., Bend, OR). They were acclimated to the cages 10‐14 days, during which time their baseline daily wheel revolutions were recorded. The mice then received (i.p) injections of 1.5 mg/kg Lipopolysaccharide (Sigma‐Aldrich). Three hours later, pairs of mice each received i.p injections of one or two human IgG, either 500 μg 6A, 500 μg 5F5, 500 μg 2G6, or 250 μg 5F5 with 250 μg 2G6. Twenty mice were tested in each group (ten cages), except for 10 mice tested in the 6A group (5 cages). The wheel running activity was counted daily following the injection for up to 25 days. The LPS significantly reduced mouse activity for ~3 days. Therefore, we compared the average daily revolutions from the 4 days prior to the LPS injection with the new steady state level following recovery from the LPS (also averaged over a 4 day period). Statistical significance was estimated by one‐way ANOVA (GraphPad). Groups of four mice (two per cage) were also tested for voluntary wheel running activity following i.p. administration of low doses of MK‐801 (100 μg/kg or 50 μg/kg),21 comparing the 4 day period prior to injection with the 4 day period afterwards.
Assessment of mAb binding to murine hippocampus following intravenous injection
To assess binding of the 5F5 and 2G6 mAbs to hippocampal tissues in vivo, Swiss Webster mice age 6–8 weeks were injected i.p with LPS (1.5 mg/kg) and 15 min later, i.v. with 250 μg 5F5 and 250 μg 2G6 combined, or 500 μg 6A. One hour later, mice were euthanized with CO2. The hippocampal and cerebellar tissues were dissected, embedded in freezing media (Tissue Tek O.C.T, Sakura Finetek, Torrance, CA), and frozen in liquid N2. Seven μm sections were cut with Microm HM505E microtome. Slides were fixed with cold Acetone (Fisher Scientific, cat #A18P‐4) for 10 min at 4°C and then stored at −20°C. For staining, slides were washed with PBS then blocked with 1% PBS‐BSA, 5% goat serum for 2 h at room temperature. Alexa Fluor 555 Goat anti‐human IgG (Invitrogen, Eugene, OR, cat # A21433) at 10 μg/mL was added, incubated for 1 h at room temperature. Slides were washed and mounted with ProLong Gold Antifade reagent with DAPI (Thermo Fisher) and imaged with a C2 + Nikon confocal microscope with 63x 1.3NA lenses. The confocal microscope setting was optimized according to the signal intensity of the negative control mAb (6A). The images were analyzed using Image J software (http://imagej.nih.gov/ij).
Methods to prevent bias
Cell binding experiments were repeated multiple times using duplicate and triplicate samples. Mice were randomly selected to receive antibody and drug treatments. DNA sequence analysis was performed in two laboratories. Neuron staining and internalization studies were performed in two laboratories with neurons from different sources.
Results
Isolation of ANRE patient‐derived mAbs that are immunoreactive with GluN1
We obtained peripheral mononuclear cells from an 18 year‐old female who presented with emotional lability, paranoia, and temporal lobe seizures, without an ovarian teratoma, and was found to have anti‐NMDAR IgG antibodies in her CSF. We used standard hybridoma methods to obtain two IgG mAbs reactive with the NMDAR‐expressing 293T cells.7, 14 Following purification, we tested 5F5 and 2G6 for binding to N1a/N2b‐transfected HEK293T by ELISA, confirming that binding depended on the expression of N1a/N2b (Fig. 1). We further assessed 5F5 and 2G6 binding to N1a/N2b‐transfected HEK293T cells by immunofluorescence microscopy, in comparison to ANRE patient CSF, a murine anti‐GluN1 mAb, and a control human IgG (8E1) (Fig. 2). 5F5 and 2G6 both showed diffuse, bright, punctate staining in the N1a/N2b‐transfected cells only, whereas 8E1 did not stain. The ANRE patient CSF stained the cells in a similar, but more diffuse pattern. We costained these cells with the commercial anti‐GluN1 mAb and either 5F5, 2G6, or ANRE patient CSF (Fig. 3). The two mAbs and the patient CSF (red) both colocalized substantially with the commercial mAb (green).
Figure 1.
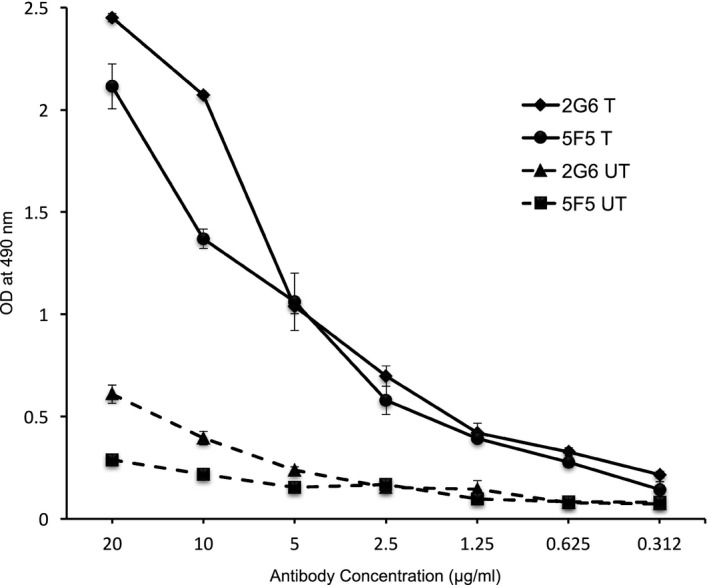
Monoclonal IgG antibodies from ANRE patients bind HEK 293T cells expressing GluN1/2a. Triplicate serial dilutions of the 5F5 and 2G6 mAbs were assessed for binding to HEK293T cells transfected with GluN1a and GluN2a expression plasmids (T) or untransfected (UT) in a whole cell lysate ELISA. Both 5F5 and 2G6 bind preferentially to GluN1/GluN2a expressing cells. OD, optical density.
Figure 2.
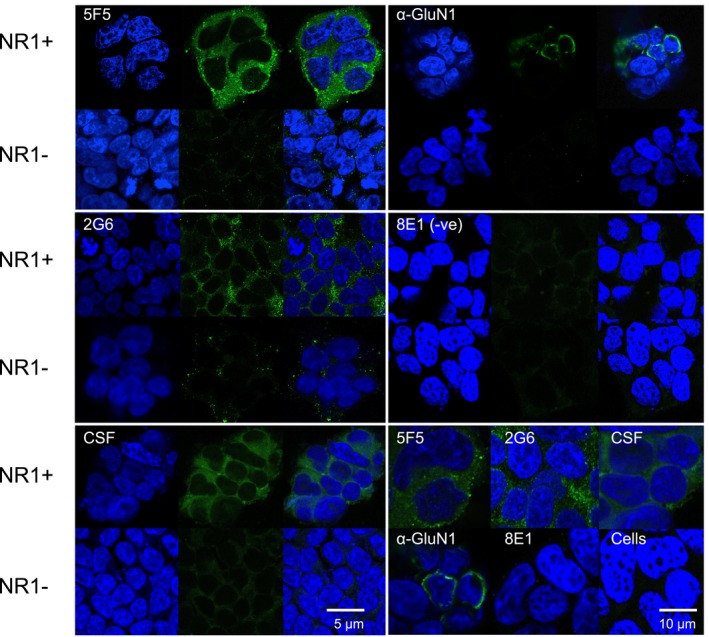
Fluorescent immunostaining by the 5F5 and 2G6 mAbs of HEK293T cells expressing GluN1/2a. HEK293T cells with (NR1 + ) or without (NR1‐) transient expression of GluN1 and GluN2a were immunostained with 5F5 (top left panel), 2G6 (middle left panel), ANRE patient CSF (bottom left panel), murine anti‐GluN1 (top right panel), or 8E1 non‐specific control IgG (middle right panel), followed by the corresponding anti‐human or anti‐mouse Alexa 488 secondary antibody (green) and nuclear DAPI stain (blue), and visualized by confocal microscopy. For each antibody are shown, from left to right: DAPI, mAb‐only, and merged images. Scale bar = 5 μm. The bottom right panel shows higher magnification merged images of 5F5, 2G6, ANRE CSF, anti‐GluN1, 8E1, as well as a control sample not exposed to human antibody (Cells). 5F5, 2G6, and patient CSF bind preferentially to GluN1/GluN2a expressing cells. Scale bar = 10 μm.
Figure 3.
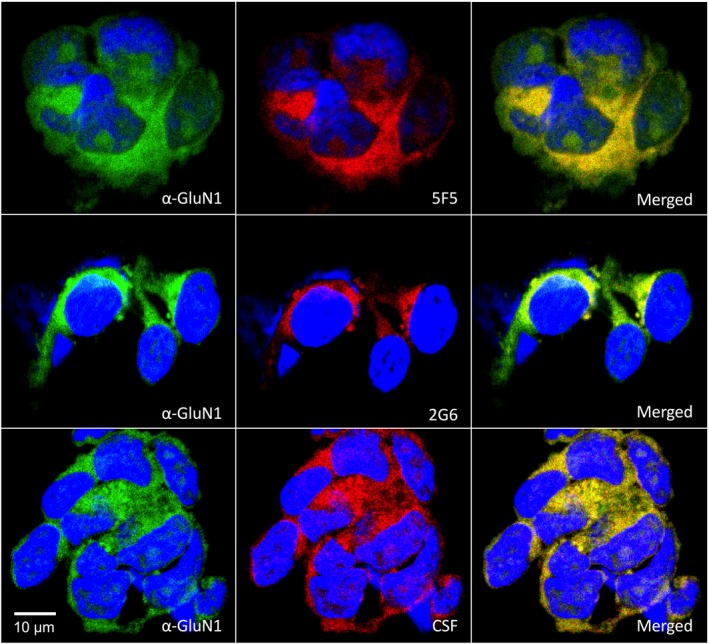
Colocalization of the 5F5 and 2G6 human mAbs with a murine anti‐GluN1 mAb on HEK293T cells expressing GluN1/GluN2a. HEK 293T cells expressing GluN1 and GluN2a were co‐immunostained with either 5F5, 2G6 or CSF (red) and the murine anti‐GluN1 antibody (green). In each row, from left to right are shown cells stained with the anti‐GluN1 mAb, human mAb or CSF, and merged images. Nuclei were visualized by DAPI staining. Colocalization of the GluN1 antigens recognized by the mAbs is demonstrated by the yellow fluorescence in the merged images. Scale bar = 10 μm.
ANRE patient IgGs bind a conformational epitope on the GluN1 ATD.1, 7 We assessed mAb binding to two GluN1 mutants known to affect pathogenic IgG binding: an almost complete ATD deletion (aa 1‐382) and the GluN1‐N368Q mutant, and tested binding of 5F5, 2G6, and patient CSF (Fig. 4). We saw no binding to either of the altered GluN1 proteins, whereas a C‐terminal‐specific antibody did bind. We next tested binding to an ATD fusion protein, which contains the 541 amino acid ATD fused to the transmembrane domain of the PDGF receptor in HEK293T cells (Fig. 5) (manuscript submitted). Both the 5F5 and 2G6 mAbs bound the ATD fusion, whereas the 8E1 did not. Furthermore, none of the mAbs bound to the AMPA class of ionotropic NMDA receptors, GluA1 or GluA222 (data not shown).
Figure 4.
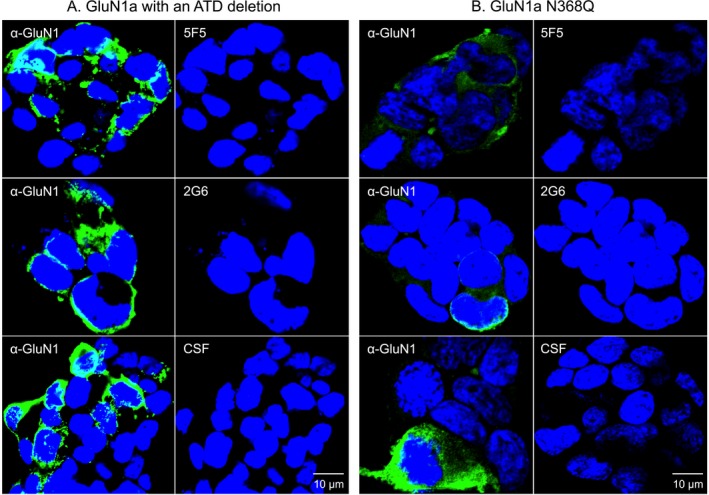
GluN1 structural changes known to impair antigen binding by ANRE patient CSF IgGs also inhibit 5F5 and 2G6 binding. HEK293T cells expressing mutant GluN1 proteins were stained with a commercial anti‐GluN1 antibody (green), followed by 5F5, 2G6, or CSF (red). Nuclei were stained with DAPI. (A) The GluN1 amino terminal deletion mutant protein (ATD). (B) GluN1 with the N368Q mutation. Neither mutant GluN1 protein was recognized by 5F5, 2C6, or CSF. Scale bars = 10 μm.
Figure 5.
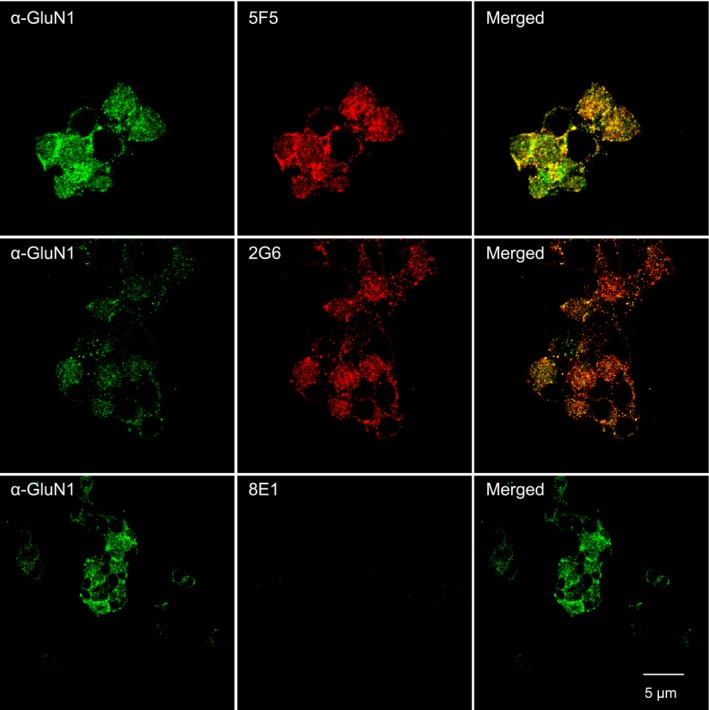
Binding of the 5F5 and 2G6 mAbs to the GluN1 Amino Terminal Domain (ATD). The GluN1‐ATD, fused to the PDGF receptor transmembrane domain, was stably expressed on the surface of HEK 293T cells. Cells were immunostained with a commercial anti‐GluN1 antibody (green), followed by 5F5, 2G6, or the 8E1 negative control mAb (red). Both 5F5, 2C6 mAbs bind to the GluN1 ATD, whereas the 8E1 does not. Scale bar = 5 μm.
DNA sequence and competitive binding analysis of the 5F5 and 2G6 mAbs
We next assessed the relatedness of the 5F5 and 2G6 mAbs. We tested whether they bind overlapping epitopes on GluN1, using a competitive binding assay with HEK293T‐ATD cells. We measured binding of one mAb, biotinylated, in the presence of increasing concentrations of the other (Fig. 6).23 In this assay, each mAb competed with itself for binding, but neither mAb interfered with the binding of the other, even at fivefold excess, indicating that the mAbs bind to different sites on the GluN1 ATD.
Figure 6.
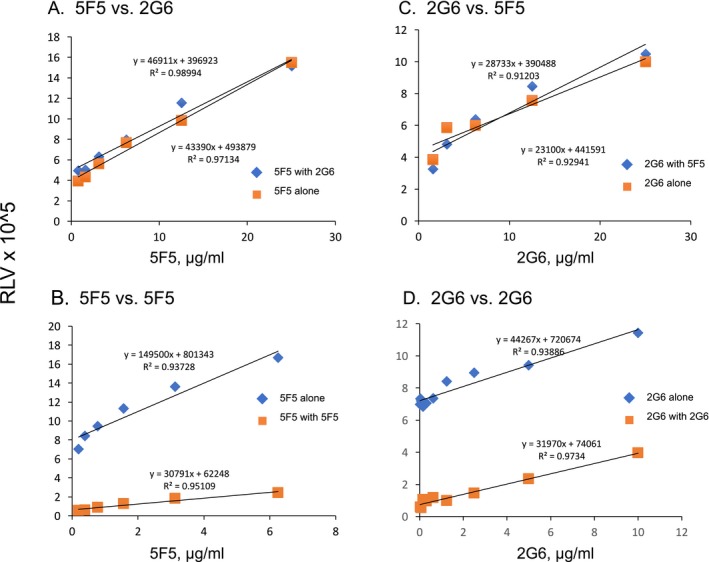
Assessment of potential antigen binding competition between 5F5 and 2G6 on the HEK293T‐ATD cell line. 5F5 and 2G6 mAbs were each biotinylated and tested for binding to the HEK293T‐ATD cell line in the presence of increasing concentrations of the other mAb, and relative luminescence values were measured (RLV). In each experiment, potential competition was measured with a value of 100% competition defined as reduction in binding seen with the homologous mAb. (A) 5F5‐biotin binding vs. increasing 2G6. (B) 5F5 biotin vs. increasing 5F5. (C) 2G6‐biotin vs. increasing 5F5. (D) 2G6‐biotin vs. increasing 2G6.
We sequenced the Ig heavy chain (HC) and light chain (LC) variable regions of the 5F5 and 2G6 mAbs and analyzed them using the IMGT program24 (Table 1). Both are IgG lambda mAbs, derived from the IGHV3‐30 HC gene family, but with different LC genes. They do not share any substantial similarity to each other in their variable domains, or to any of the previously published anti‐NMDAR mAbs.9 Both mAbs are heavily mutated; 5F5 and 2G6 showed somatic hypermutation rates of 9.0% and 8.7% in their HC variable regions, and 6.3% and 2.5% in their LC variable regions, respectively. Both have relatively long CDR‐H3 (complementarity determining region 3): 21 amino acids in 5F5 and 24 amino acids in 2G6.
Table 1.
Assignment of the 5F5 and 2G6 variable domain DNA sequences to their closest germline counterparts
| Heavy chain | Light chain | |||||||
|---|---|---|---|---|---|---|---|---|
| Antibody | CDR3 length | VH gene | % identity | J‐gene | D‐gene | VH gene | % identity | J‐gene |
| 5F5 | 21 | IGHV3‐30*03, IGHV3‐30*18, IGHV3‐30‐3*01 or IGHV3‐30‐5*01 | 90.97 | IGHJ4*02 | IGHD6‐19*01 | IGLV1‐47*01 | 93.68 | IGLJ2*01 or IGLJ3*01 |
| 2G6 | 24 | IGHV3‐30*03, IGHV3‐30*18, IGHV3‐30‐3*01 or IGHV3‐30‐5*01 | 91.32 | IGHJ6*02 | IGHD3‐10*01 | IGLV3‐10*01 | 97.49 | IGLJ2*01 or IGLJ3*01 |
We performed a lineage analysis of the 5F5 and 2G6 mAbs by sequencing the Ig heavy chain repertoire of CD27 + B cells assessed at the time of cell fusion (i.e. following in vitro expansion) (Fig. 7). We defined potential common mAb lineage members as those that used the IGHV3‐30 gene and had >80% nucleotide sequence identity in their CDR‐H3 regions. We excluded incomplete IgG sequence reads, as well as those with only one sequence read, leaving 271,896 reads, of which 3300 were related to 5F5 and 8934 to 2G6. Sequence alignment analysis reveals that the 5F5 and 2G6 mAbs arose from independent lineages, and that they are single members of two diverse families, including 9 (5F5) and 19 (2G6) relatives that have exactly the same CDR‐H3 sequences, which is the predominant determinant of IgG binding specificity.25 These data indicate that the 5F5 and 2G6 mAbs arose from independent B‐cell clones that have undergone multiple rounds of antigen‐driven somatic hypermutation.
Figure 7.
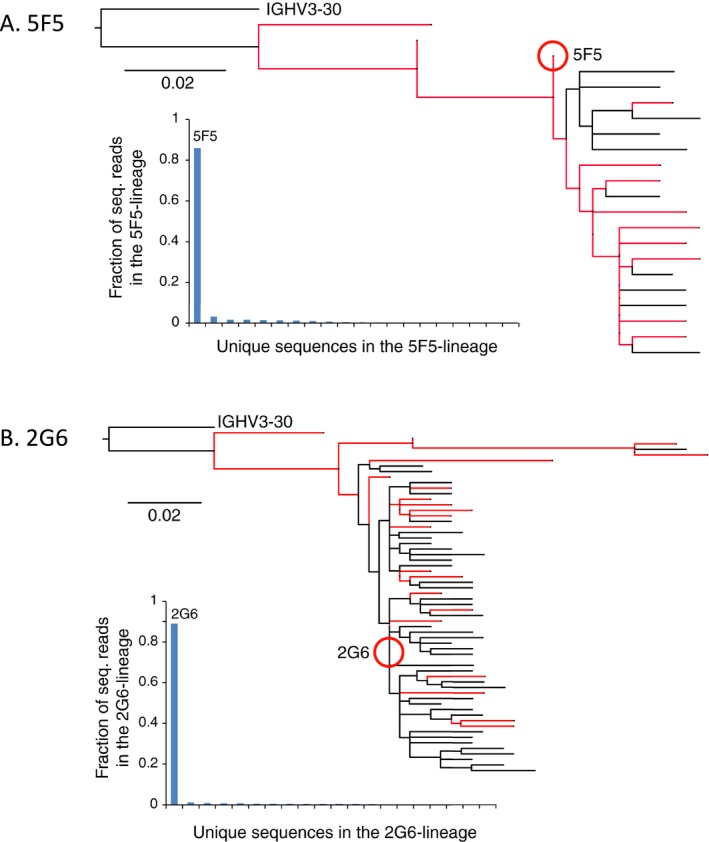
Phylogenetic analyses of the 5F5 and 2G6 mAb lineages. The patient's peripheral blood B‐cell population was sampled, after in vitro proliferation and prior to cell fusion, and analyzed by Ig heavy chain sequencing. Lineages were defined to include sequences with >80% nucleotide sequence homology in CDRH3 domain, and were analyzed by Clustal sequence analysis. Sequences with identical CDRH3 domains are shown in red. Below each dendrogram is plotted the fraction of total sequencing reads for each lineage member. (A) 5F5. (B) 2G6.
Binding of the 5F5 and 2G6 mAbs to primary hippocampal neurons and murine brain
We next explored interaction of the mAbs with native NMDAR on primary tissues and in vivo. We tested binding of the 5F5 and 2G6 mAbs to cultured rat hippocampal neurons. Commercially obtained hippocampal neurons were cultured for 14 days and confirmed to express MAP2 and GluN1 (data not shown). We fixed the neurons and stained them with 5F5, 2G6, 8E1, or ANRE patient CSF (Fig. 8A). The CSF and the 5F5 and 2G6 mAbs bound to the neurons, whereas 8E1 did not. Co‐immunostaining of the neurons indicated substantial overlap between the signals from the 5F5 and 2G6 mAbs and the murine anti‐GluN1 mAb (Fig. 8B).
Figure 8.
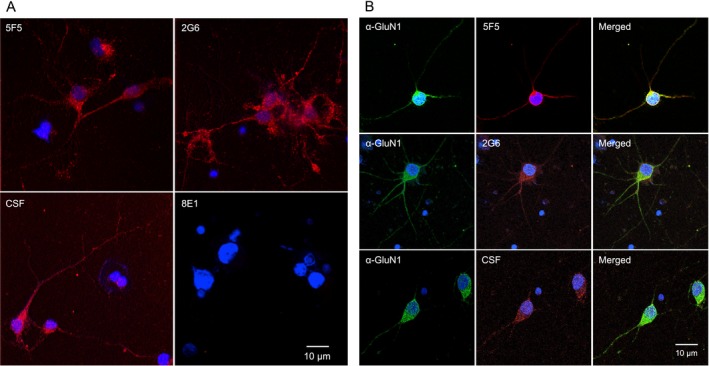
The 5F5 and 2G6 mAbs bind GluN1 on rat hippocampal neurons. (A) Neurons were cultured for 14 days and stained with ANRE CSF or mAbs (red). Top left, 5F5. Top right, 2G6. Bottom left, CSF. Bottom right, 8E1. Nuclei were stained with DAPI. Scale bar = 10 μm. (B) Neurons were stained with ANRE CSF or mAbs (red), and costained with murine anti‐GluN1 antibody (green). Rows: Top, 5F5. Middle, 2G6. Bottom, CSF. Columns: Left, GluN1. Middle, CSF or mAbs. Right, Merged images. Nuclei were stained with DAPI. Scale bar = 10 μm.
We next assessed the sites of 5F5 binding on murine primary hippocampal neurons that had not undergone fixation. Double‐labelling images showed overlapping patterns with a rabbit GluN1 antibody, though to a lesser degree than IgG from ANRE patient CSF. In contrast to patient CSF, 5F5 immunoreactivity did not consistently colocalize with PSD‐95, suggesting that it labels a subgroup of NMDAR that are primarily presynaptic or extrasynaptic (Fig. 9). We next stained floating sections of murine cortex and hippocampus with the 5F5 mAb, CSF (Fig. 10). 5F5 mAb colocalized with GluN1, particularly in the lower levels of the cortex. Less colocalization was noted in the hippocampus. No staining was noted for 8E1, but high levels of colocalization were found between patient IgG and GluN1. Taken together, the binding activities of the 5F5 and 2G6 mAbs are consistent with recognition of a subset of GluN1 in cultured neurons and in murine brain.
Figure 9.
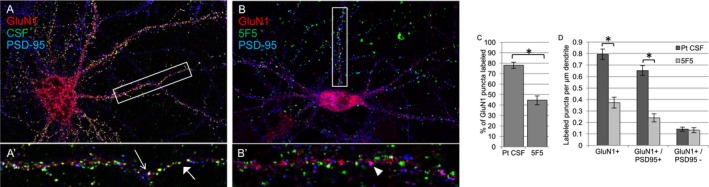
mAb 5F5 recognizes a subset of GluN1+ puncta on neurons. Live rat hippocampal neurons at 14 population doublings were stained with either (A, A’) ANRE CSF or (B, B’) 5F5 (green), and then with the commercial anti‐GluN1 mAb (red) and an anti‐PSD‐95 antibody (blue), to label synapses. ANRE CSF labels almost 80% of GluN1 puncta (A, A’, C). Most puncta are colocalized with PSD‐95 (blue); white in overlay (open arrow). Some ANRE CSF+/GluN1+ puncta are not colocalized with PSD‐95; yellow in overlay (closed arrow). (B, B’, C) 5F5 labels less than half of the GluN1 puncta. (D) Mean labeled puncta per μm dendrite, ±SEM *P < 0.0001, Student's t‐test with Bonferroni correction. N = 4 neurons, 10 dendrites, per condition. Less frequent 5F5 binding to neurons, relative to ANRE CSF, reflects different staining frequencies at synaptic sites (GluN1+/PSD‐95+), rather than extrasynaptic sites (PSD‐95‐/GluN1+). (A, B) 200X. (A’, B’) 400X.
Figure 10.
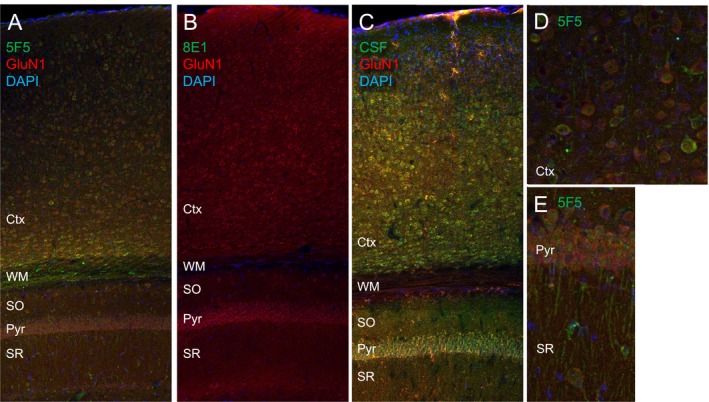
Staining of murine brain with the 5F5 mAb. Murine hippocampal sections were immunostained with the 5F5 or 8E1 mAbs, or ANRE CSF (green), in combination with the commercial anti‐GluN1 mAb (red), and DAPI. (A) 5F5, 200X. (B) 8E1, 200X. (C) ANRE patient CSF, 200X. (D) 5F5 on cortex, 400X. (E) 5F5 on the pyramidal cell layer, 400X. Ctx, cortex; WM, white matter; SO, stratum oriens; Pyr, pyramidal cell layer; SR, stratum radiatum.
ANRE patient CSF reduces the surface density of NMDAR on cultured neurons.3, 4 We conjugated the 5F5, 2G6, and 6A mAbs with CypHer5E, a pH‐sensitive dye that fluoresces upon internalization into acidic endosomes,26 and incubated the mAbs with cultured neurons (Fig. 11A). Cells were first exposed to supplemental glycine and glutamine, with or without the NMDAR inhibitors MK‐801 or AP5, for 15 min, and then exposed to the mAbs for 45 min. Both of the ANRE mAbs were internalized, whereas the control 6A mAb was not. Internalization was inhibited by treatment with the NMDAR inhibitors MK‐801 and AP5 (Fig. 11A). Notably, MK‐801 did not inhibit binding of the mAbs to the neurons, whereas AP5 did (Fig. 11B). This suggests that 5F5 and 2G6 binding alone is not sufficient for internalization, in the absence of receptor activation. Furthermore, it indicates that the closed configuration induced by AP5 masks the 5F5 and 2G6 binding epitopes.
Figure 11.
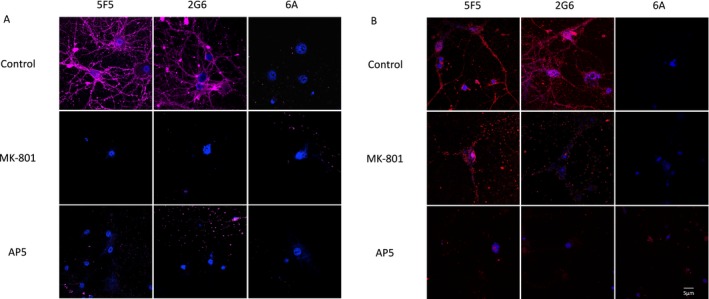
Internalization of the 5F5 and 2G6 mAbs by hippocampal neurons and the effects of MK‐801 and AP5. (A) Rat hippocampal neurons were incubated with 5F5, 2G6, or 6A mAbs conjugated to the pH‐sensitive fluorescent dye, CypHer5E, which is activated by the low pH in endosomes, alone and in the presence of MK‐801 or AP5. (B) Neurons treated with MK‐801 or AP5 were assessed for binding of the 5F5, 2G6, or 6A mAbs. Scale bar = 5 μm.
In vivo effects of the 5F5 and 2G6 mAbs
We assessed the effects mAbs on mouse voluntary locomotor activity by measuring the distance traveled by mice on a running wheel.27 Groups of 10 mice, housed in pairs, received i.p. injections of lipopolysaccharide (LPS), which induces blood brain barrier permeability.28 Three hours later, they received i.p. injections of 6A, 5F5, 2G6, or 5F5 and 2G6 combined. Because the LPS causes approximately 3 days of hypo‐activity, we compared pre‐ and post‐injection steady state activity levels, and plotted the average change in the number of daily revolutions for the mice (Fig. 12A). The post‐injection increase in wheel running activity for three of the mAb intervention groups were substantially higher than for the LPS alone group. Baseline activity did not differ among the groups, at approximately 14,000 revolutions per day (approximately 5 km). Daily wheel revolutions increased by 313 revolutions for the 6A control group (P = 0.6), 1490 for 5F5 (P = 0.026), 1448 for 2G6 (P = 0.033), and 2051 for 5F5 + 2G6 (P = 0.0005). To compare against the effects of specific NMDAR inhibition, we treated additional mice with low doses of MK‐801 (Fig. 11B). Similar to the ANRE mAbs, MK‐801 increased voluntary by over 2000 revolutions per day at both 2.5 μg and 1.25 μg/kg doses, compared to untreated mice (P < 0.0001).
Figure 12.
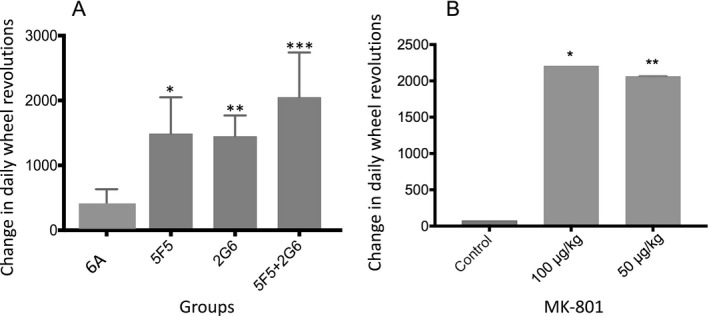
Alterations in voluntary running activity induced by 5F5 and 2G6 mAbs. (A) Voluntary running activity was measured in mice before and after receiving 5F5, 2G6, or both mAbs. Prior to mAb administration, the mice received a dose of LPS to open the blood brain barrier. Baseline levels were recorded for 4 days prior to LPS/mAb administration, and compared to the 4 day steady state period following recovery from LPS toxicity. The differences in the average number of daily wheel revolutions are shown. One‐way ANOVA *P = 0.026, **P = 0.033, ***P = 0.0005. (B) Voluntary running activity was measured in mice before and after receiving MK‐801 (100 μg/kg or 50 μg/kg). Baseline levels were recorded for 4 days prior to MK‐801 injection, and compared to the 4 days following the injection. The differences in the average numbers of daily wheel revolutions are shown. One‐way ANOVA *P = 0.0001, **P = 0.0001. Error bars indicate S.E.M.
We next assessed whether these biological effects correlated with the ability of the mAbs to bind hippocampal tissues following an intravenous injection. Groups of 6 mice received an LPS injection, followed 15 min later by 6A or 5F5 with 2G6. One hour later, they were euthanized and frozen sections of the dissected hippocampi were stained for human IgG. Representative images are shown in Figure 13. No human IgG was detected in the 6A‐injected mice, whereas widespread human IgG staining was seen in the mice that received 5F5 + 2G6.
Figure 13.
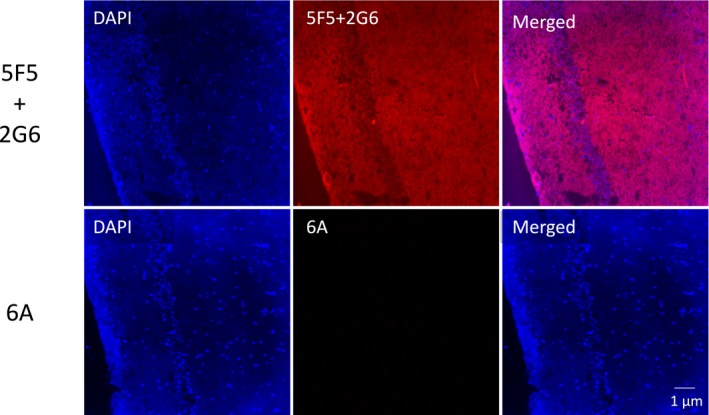
Interaction of the 5F5 and 2G6 mAbs with murine hippocampus following intravenous injection. Mice received a dose of LPS, followed 15 min later by either the 6A mAb or a combination of 5F5 and 2G6. One hour later, hippocampal frozen sections were prepared and stained for human IgG (red). Top row, 5F5 and 2G6. Bottom row, 6A. Scale bar = 1 μm.
Discussion
We isolated and characterized two IgG monoclonal antibodies from a patient with ANRE not associated with ovarian teratoma. The 5F5 and 2G6 mAbs bind GluN1 expressed by cultured hippocampal neurons, and replicate many of the activities previously described for IgGs in the CSF of ANRE patient. They bind GluN1 expressed in HEK293T cells, as well as an isolated NMDAR ATD, and they require the GluN1 N368, a site of post‐translational modification required for ANRE patient IgG binding. The mAbs bind to cultured hippocampal neurons and internalize. Extended study of the 5F5 mAb showed binding to murine hippocampus. Lastly, the mAbs induced a sustained increase in voluntary locomotor activity, similar to the effect seen with MK‐801. These data suggest that the 5F5 and 2G6 mAbs may have contributed to the development of ANRE in this patient.
The 5F5 and 2G6 mAbs arose from distinct B‐cell clones that bind non‐overlapping epitopes on NMDAR. Kreye et al.9 observed multiple NMDAR‐reactive IgG lineages in some of their patients.9 As murine experiments with a single ANRE‐patient‐derived IgG showed that a single mAb is capable of inducing ANRE‐like symptoms,29 further studies are needed to elucidate whether different ANRE antibodies differ substantially in their pathogenic effects. But the fact that 5F5 and 2G6 can simultaneously bind NMDAR suggests that some cases of ANRE may integrate the effects of multiple antibodies. Neither mAb completely colocalized with the GluN1 detected by patient IgGs or non‐human, commercial antibodies in any of the tissues or cell lines we examined. Furthermore, 5F5 preferentially bound extrasynaptic NMDAR, while ANRE patient IgGs as a whole bind more to synaptic receptors.4 Although synaptic NMDAR signaling is crucial for synaptic plasticity, learning, and memory; extrasynaptic NMDAR signaling links to excitotoxicity and cell death.8 This supports a model in which the symptoms of ANRE reflect the integration of excitatory/inhibitory imbalances of neuronal circuit function and the balance between synaptic and extrasynaptic NMDAR. These effects would depend on the types and titers of different antibodies expressed in each patient.
Our previous studies of GluN1 binding in ANRE showed that the antibodies preferentially bind to the NMDAR in its open state.4, 7 Consistent with these results, 5F5 and 2G6 both bind in the presence of MK‐801, which stabilizes NMDAR in the open state, but not AP5, which prevents NMDAR opening.30 Our observation that MK‐801 inhibits internalization, but not binding, suggests that receptor activation by 5F5 or 2G6 is required for internalization. This contrasts with the previous observation that AP5 did not impede receptor down‐modulation induced by ANRE CSF.31 However, important methodlogical differences exist, in that Moscato et al. measured NMDAR internalization induced by CSF over 12 h, whereas we measured mAb internalization at 45 min. In ANRE, chronic anti‐NMDAR IgG treatment of neurons is proposed to lead to internalization and destruction of NMDARs, resulting in reduced synaptic NMDAR currents and impairment of NMDAR‐dependent processes such as long‐term potentiation (LTP).4, 5, 32
The central hypothesis is that ANRE patient antibodies directly cause their symptoms through NMDAR hypofunction, resulting in the amnesia and psychosis seen in anti‐NMDAR encephalitis. The two mAbs described here suggest that these antibodies can result from NMDAR antigen‐dependent somatic hypermutation, even in the absence of a teratoma. NMDAR antagonists induce psychosis in humans33 and NMDAR hypofunction has been associated with schizophrenia in mouse models.33, 34, 35, 36 However, other features of ANRE, such as seizures and dyskinesias, are not readily explained by global NMDAR hypofunction, as these can also reflect overactivation of NMDAR.37 If NMDAR internalization by IgGs requires receptor activation, it follows that the initial presentation of ANRE may reflect overactivation prior to hypofunction. In the case of the 5F5 mAb, this may reflect overactivation primarily at extrasynaptic sites. The availability of cloned mAbs from ANRE patients offers the opportunity to explore the mechanisms that underlie the protean manifestations of ANRE.
We measured the effects of the mAbs on mouse voluntary locomotor activity using the mouse wheel‐running test, a relatively nonspecific study. We used it to assess whether global effects on behavior would be altered in a manner consistent with NMDAR hypofunction. In this crude assessment, following an injection of LPS, both mAbs increased the total daily running activity, which was sustained for several days and reflected the increase in activity experienced by mice receiving low‐dose MK‐801. This suggests that the antibodies identified here may down‐regulated NMDAR activity, consistent with the primary state observed in ANRE.
Conflict of Interest
The authors declare no competing financial interests.
Acknowledgments
The work was supported by NIH grant R21 NS088148 (DRL and SKD); the Lankenau Institute for Medical Research; and CAPES, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (LFF). Reagents, monoclonal antibodies, and DNA sequence information will be shared with interested investigators upon request.
Funding Statement
This work was funded by NIH grant R21 NS088148; Lankenau Institute for Medical Research grant ; CAPES grant ; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil grant .
Contributor Information
David R. Lynch, Email: lynchd@mail.med.upenn.edu
Scott K. Dessain, Email: dessain@limr.org.
References
- 1. Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti‐N‐methyl‐D‐aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armangue T, Titulaer MJ, Malaga I, et al. Pediatric anti‐N‐methyl‐D‐aspartate receptor encephalitis‐clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;162:850–6e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti‐NMDA receptor encephalitis. J Neurosci 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Q, Tanaka K, Sun P, et al. Suppression of synaptic plasticity by cerebrospinal fluid from anti‐NMDA receptor encephalitis patients. Neurobiol Dis 2012;45:610–615. [DOI] [PubMed] [Google Scholar]
- 6. Regan MC, Romero‐Hernandez A, Furukawa H. A structural biology perspective on NMDA receptor pharmacology and function. Curr Opin Struct Biol 2015;33:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gleichman AJ, Spruce LA, Dalmau J, et al. Anti‐NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci 2012;32:11082–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev 2013;14:383–400. [DOI] [PubMed] [Google Scholar]
- 9. Kreye J, Wenke NK, Chayka M, et al. Human cerebrospinal fluid monoclonal N‐methyl‐D‐aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016;139(Pt 10):2641–2652. [DOI] [PubMed] [Google Scholar]
- 10. Puligedda RD, Kouiavskaia D, Adekar SP, et al. Human monoclonal antibodies that neutralize vaccine and wild‐type poliovirus strains. Antiviral Res 2014;108:36–43. [DOI] [PubMed] [Google Scholar]
- 11. Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ho M, Pastan I. Display and selection of scFv antibodies on HEK‐293T cells. Methods Mol Biol 2009;562:99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Erdile LF, Smith D, Berd D. Whole cell ELISA for detection of tumor antigen expression in tumor samples. J Immunol Methods 2001;258(1–2):47–53. [DOI] [PubMed] [Google Scholar]
- 14. Adekar SP, Jones RM, Elias MD, et al. Hybridoma populations enriched for affinity‐matured human IgGs yield high‐affinity antibodies specific for botulinum neurotoxins. J Immunol Methods 2008;333(1–2):156–166. [DOI] [PubMed] [Google Scholar]
- 15. Lim TS, Mollova S, Rubelt F, et al. V‐gene amplification revisited ‐ An optimised procedure for amplification of rearranged human antibody genes of different isotypes. New Biotechnol 2010;27:108–117. [DOI] [PubMed] [Google Scholar]
- 16. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina paired‐end reAd mergeR. Bioinformatics 2014;30:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 2015;12:380–381. [DOI] [PubMed] [Google Scholar]
- 18. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002;30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 2014;30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buchhalter JR, Dichter MA. Electrophysiological comparison of pyramidal and stellate nonpyramidal neurons in dissociated cell culture of rat hippocampus. Brain Res Bull 1991;26:333–338. [DOI] [PubMed] [Google Scholar]
- 21. van der Staay FJ, Rutten K, Erb C, Blokland A. Effects of the cognition impairer MK‐801 on learning and memory in mice and rats. Behav Brain Res 2011;220:215–229. [DOI] [PubMed] [Google Scholar]
- 22. Karakas E, Regan MC, Furukawa H. Emerging structural insights into the function of ionotropic glutamate receptors. Trends Biochem Sci 2015;40:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Al‐Saleem FH, Sharma R, Puligedda RD, et al. RBC adherence of immune complexes containing botulinum toxin improves neutralization and macrophage uptake. Toxins 2017;9:e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lefranc MP. IMGT, the International ImMunoGeneTics Information System. Cold Spring Harb Protoc 2011;2011:595–603. [DOI] [PubMed] [Google Scholar]
- 25. Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 2000;13:37–45. [DOI] [PubMed] [Google Scholar]
- 26. Adie EJ, Kalinka S, Smith L, et al. A pH‐sensitive fluor, CypHer 5, used to monitor agonist‐induced G protein‐coupled receptor internalization in live cells. Biotechniques 2002;33:1152–4, 1156–7. [DOI] [PubMed] [Google Scholar]
- 27. Joshi SG, Elias M, Singh A, et al. Modulation of botulinum toxin‐induced changes in neuromuscular function with antibodies directed against recombinant polypeptides or fragments. Neuroscience 2011;179:208–222. [DOI] [PubMed] [Google Scholar]
- 28. Jangula A, Murphy EJ. Lipopolysaccharide‐induced blood brain barrier permeability is enhanced by alpha‐synuclein expression. Neurosci Lett 2013;551:23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malviya M, Barman S, Golombeck KS, et al. NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann Clin Transl Neurol. 2017;4:768–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lynch DR, Guttmann RP. Excitotoxicity: perspectives based on N‐methyl‐D‐aspartate receptor subtypes. J Pharmacol Exp Ther 2002;300:717–723. [DOI] [PubMed] [Google Scholar]
- 31. Moscato EH, Peng X, Jain A, et al. Acute mechanisms underlying antibody effects in anti‐N‐methyl‐D‐aspartate receptor encephalitis. Ann Neurol 2014;76:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross‐talk between NMDA and Ephrin‐B2 receptors in anti‐NMDA encephalitis. Brain 2012;135(Pt 5):1606–1621. [DOI] [PubMed] [Google Scholar]
- 33. Javitt DC. Glutamate and schizophrenia: phencyclidine, N‐methyl‐D‐aspartate receptors, and dopamine‐glutamate interactions. Int Rev Neurobiol 2007;78:69–108. [DOI] [PubMed] [Google Scholar]
- 34. Belforte JE, Zsiros V, Sklar ER, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia‐like phenotypes. Nat Neurosci 2010;13:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Masdeu JC, Dalmau J, Berman KF. NMDA receptor internalization by autoantibodies: a reversible mechanism underlying psychosis? Trends Neurosci 2016;39:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999;98:427–436. [DOI] [PubMed] [Google Scholar]
- 37. Barker‐Haliski M, White HS. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med 2015;5:a022863. [DOI] [PMC free article] [PubMed] [Google Scholar]