

Abstract
Objective
Studies using conventional full‐field visual evoked potentials (ffVEP) have reported subtle abnormalities in patients with chronic inflammatory demyelinating polyneuropathy (CIDP). We hypothesize that these abnormalities can be detected in the majority of CIDP patients using enhanced methods.
Methods
We performed a cross‐sectional noninterventional study comparing 18 CIDP patients and 18 matched healthy controls using multifocal VEP (mfVEP) as a technique with enhanced sensitivity to detect conduction abnormalities across the spectrum of optic nerve fibers. Patients with confounding diseases (ophthalmologic, diabetes mellitus) were excluded.
Results
The mean amplitude and latency, as well as the low‐contrast visual acuity, did not differ between CIDP patients and controls. Subanalyses revealed latency differences concerning the superior sector of the visual field. Severity markers of CIDP (ODSS, motor nerve conduction velocity) were associated with mfVEP latency delay.
Interpretation
We could not adduce evidence for clinically or diagnostically relevant visual pathway involvement in CIDP. The latency differences identified were very subtle and restricted to the superior visual field which cannot be readily explained biologically, anatomically, or pathologically. In summary, we conclude that our study revealed no relevant differences in mfVEP parameters between CIDP patients and controls.
Introduction
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a subacute or chronic autoimmune neuropathy pathologically characterized by demyelination and secondary axonal degeneration of peripheral nerves.1, 2 Interestingly, previous studies suggested that central nervous system (CNS) involvement, including the visual pathway, may also occur in this disease.3, 4, 5 Of note, subclinical CNS involvement in CIDP patients, that is, pathological ffVEP measurements have been described before.3 Hawke et al.6 reported abnormal magnetic resonance imaging (MRI) characterized by high white matter signals in six out of 26 cases. Furthermore, 14 out of 28 cases were reported by Ormerod et al.7 Thomas et al.8 described a series of six cases in which a chronic demyelinating neuropathy was associated with a relapsing multifocal demyelinating CNS disorder: five of six cases showed prolonged full‐field visual evoked potentials (ffVEP) latencies in one or both eyes along with T2 signals on cranial MRI. These authors reviewing pathological reports and studies on experimental models raised the issue of shared pathogenic mechanisms explaining involvement of both the peripheral and central nervous system. In a recent nationwide study of combined central and peripheral demyelination (CCPD) in Japan, 15 out of 21 cases showed ffVEP abnormalities,9 whereas a CCPD study in China revealed pathological ffVEPs in 11 out of 22 patients.10 The pathophysiological mechanism underlying CNS involvement is not understood. It has been suggested that the CNS and PNS may share antigens to which aberrant humoral or T‐cell immune responses are directed. This has been hypothesized for circulating M‐proteins3 in monoclonal gammopathy‐associated peripheral neuropathies with CNS involvement and in combined central and peripheral demyelination in Japan where Neurofascin‐155 was reported to represent a shared antigen recognized by antibodies circulating in the blood of CIDP patients.9 In the largest conducted clinicopathological study on 100 French patients with CIDP, five patients presented with symptomatic CNS involvement.11
Of note, in the previous studies using conventional full‐field visual evoked potentials (ffVEP), CIDP patients were not compared with age‐ and sex‐matched controls and confounding ophthalmic and systemic disorders that may be relevant in these cohorts of predominantly elderly patients have not been accounted for. Therefore, we performed a cross‐sectional, noninterventional study of CIDP patients and age‐ and sex‐matched healthy controls without confounding concomitant diseases such as ophthalmic pathologies and diabetes mellitus by measuring multifocal visual evoked potentials (mfVEPs). This technique is able to detect visual pathway abnormalities with substantially enhanced sensitivity by recording simultaneously from multiple regions of the visual field.12
The primary aim of our study was to investigate the hypothesis that mfVEP, due to its sensitivity, may be suitable to detect visual system pathology even in CIDP patients without clinically overt visual dysfunction or other signs of CNS involvement.
Methods
Patients
Patients were prospectively recruited at the Department of Neurology, Heinrich‐Heine‐University in Düsseldorf, Germany. Inclusion criteria were age >18 years, probable or definite CIDP according to the EFNS/PNS CIDP Guidelines13 and response to immunomodulatory treatment. Out of 61 subjects (43 CIDP patients, 18 healthy controls), a total of 25 CIDP patients and seven eyes were excluded: nine patients and seven eyes due to ophthalmic pathologies (one glaucoma, two cataract, four bilateral drusen, one papilledema, one choroidal neovascularization, one eye with macular edema, six eyes with drusen), 13 patients because of diabetes mellitus,14 two patients due to head tremor interfering with the mfVEP assessment and one patient who was positive for anti‐MAG‐IgM. The remaining 36 subjects were 18 patients with CIDP and 18 age‐ and sex‐matched healthy controls (Fig. 1). Three CIDP patients had a monoclonal gammopathy. INCAT ODSS15 (Inflammatory Neuropathy Cause and Treatment, Overall Disability Sum Score) was assessed in all CIDP patients. All patients and controls included in the final analysis showed no abnormalities on neuroophthalmological examination including assessment of corrected visual acuity (Sloan charts), tonometry, slit lamp examination, fundoscopy, and optical coherence tomography. In addition, all patients and controls were tested for corrected low‐contrast letter recognition using 2.5% low‐contrast early treatment of diabetic retinopathy (ETDRS) charts. As nerve conduction studies of the lower limbs and sensory nerve conduction studies of the upper limbs showed a high rate of signal loss, motor nerve conduction velocity (MNCV) of the right ulnar nerve was used to investigate the association with mfVEP parameters.
Figure 1.
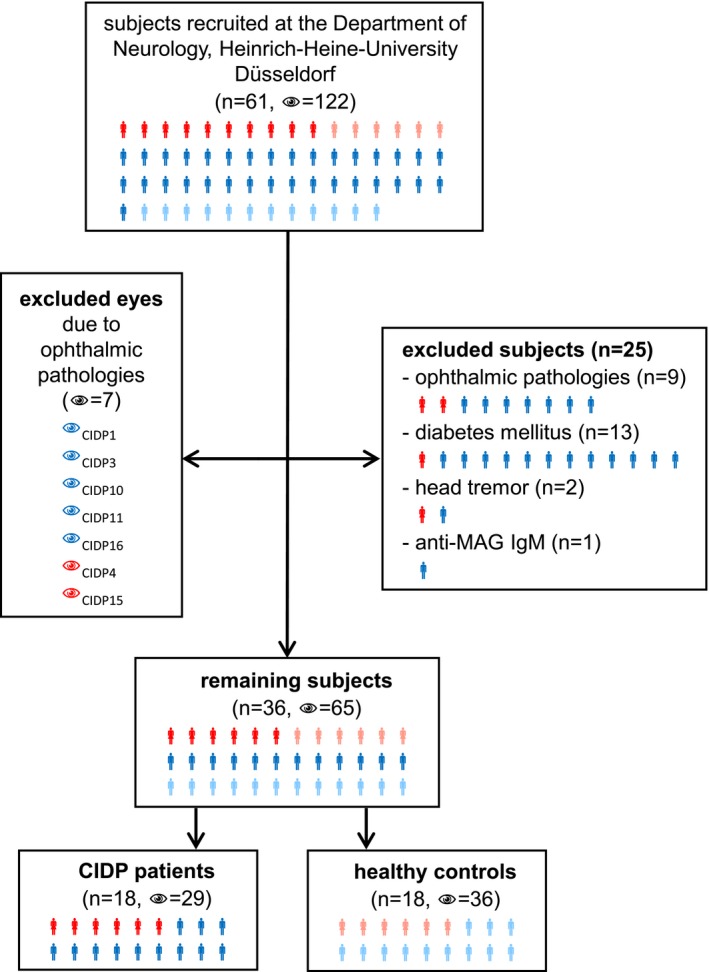
Recruitment. Flowchart of the subject recruitment at the Department of Neurology, Heinrich‐Heine‐University Düsseldorf. Twenty‐five of the 61 subjects were excluded: nine due to ophthalmic pathologies, 13 due to diabetes mellitus, two due to head tremor, and one due to positive anti‐MAG‐IgM. Seven of the 122 eyes were excluded due to ophthalmic pathologies. The remaining 36 subjects were composed of 18 healthy volunteers (36 eyes) and 18 CIDP patients (29 eyes). Red symbols represent female CIDP patients, blue symbols, male CIDP patients. Female healthy volunteers are shown in light red and male healthy volunteers in light blue.
Matched controls did not have any symptoms suspicious of polyneuropathy. An overview of the patient and healthy control cohorts is provided in Tables S1 and S2.
Multifocal visual evoked potentials
Multifocal visual evoked potentials (mfVEPs) were measured as described previously16: The Visionsearch® mfVEP device applies simultaneous multifocal stimulation of 56 segments of the visual field (24° of eccentricity) via a 68 sec pseudorandom sequence and recording a 2‐channel visual response using a custom designed occipital cross electrode holder which predetermines the four occipital electrode positions.17 Cross‐correlation of the event‐related response with the sequence itself allows for recording of evoked potentials in the nanovolt range, which originate from monocular stimulation of distinct areas of the visual field.
The 56 segments of the visual field can be assigned either to four sectors (Fig. S1A) or to five eccentricities (Fig. S1B). Regarding the sectoral division, 18 segments each belong to the superior (segments: 6–7, 16–19, 28–31, 40–43, 52–55) and inferior sector (segments: 2–3, 10–13, 22–25, 34–37, 46–49), whereas the temporal (segments: 1, 8–9, 20–21, 32–33, 44–45, 56) and nasal sector (segments: 4–5, 14–15, 26–27, 38–39, 50–51) consist of 10 segments each. Concerning the eccentricities, segments can be arranged in circles. The central circle comprises eight segments (segments: 1–8), while the peripheral circles consist of 12 segments each.
Statistical evaluation
Statistical analyses were performed using SPSS Statistics 24 (IBM). Generalized estimation equation models (GEE) accounting for within‐subject intereye or intersegment correlations using an exchangeable working correlation matrix and correcting for age and sex were applied to analyze associations between mfVEP parameters and clinical data and to test for differences in the mfVEP parameters between CIDP patients and controls. Adjusted P‐values (adj. P) were calculated using Bonferroni correction in the case of multiple group analyses. Statistical power analyses were operated using G*Power 3.1.9.2. An online calculator by Psychometrica was utilized to calculate effect size, Cohen's d (dCohen) and the associated confidence intervals.
Ethics
The local ethics committee of Heinrich‐Heine‐University Düsseldorf approved this study (registry number 4849). Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Results
Primary hypothesis – difference in mean amplitude and mean latency
In patients with CIDP, mfVEP revealed a mean amplitude of 182.06 ± 48.37 nV (range = 94.55–268.98 nV) and a mean latency (first peak) of 149.65 ± 6.51 msec (range = 137.88–161.78 msec). In healthy controls, the examination revealed a mean amplitude of 183.53 ± 43.85 nV (range = 101.94–276.79 nV) and a mean latency (first peak) of 147.84 ± 5.68 msec (range = 137.65–164.11 msec). There was no single patient with pathological mean, sector or eccentricity latencies or amplitudes compared to our age‐ and sex‐matched controls. An overview is provided in Table 1. There was no statistical difference in mean amplitude (P = 0.855, mean difference = 1.47 ± 11.46 nV (95%‐CI = [−24.36–21.42]), Fig. 2A) or first peak latency (P = 0.213, mean difference = 1.81 ± 1.51 msec (95%‐CI = [−1.21–4.84]), Fig. 2B) between both groups.
Table 1.
mfVEP parameter overview of CIDP patients and healthy controls regarding the different sectors and eccentricities (STD = standard deviation, CIDP = chronic inflammatory demyelinating polyneuropathy, HC = healthy controls, Eccentr. = eccentricity, 1st per. = first peripheral, 2nd per. = second peripheral, 3rd per. = third peripheral, 4th per. = outer peripheral)
| Group | Latency (msec) | Amplitude (nV) | ||||||
|---|---|---|---|---|---|---|---|---|
| Mean | STD | Range | Mean | STD | Range | |||
| CIDP | Sector | Superior | 156.16 | 18.29 | 111.70–253.30 | 146.14 | 57.96 | 43.90–343.40 |
| Inferior | 144.45 | 15.93 | 116.70–236.70 | 214.21 | 77.49 | 57.60–500.00 | ||
| Temporal | 148.13 | 21.36 | 106.70–243.30 | 173.41 | 68.66 | 45.40–459.90 | ||
| Nasal | 148.83 | 15.57 | 120.00–231.70 | 197.50 | 74.70 | 63.50–459.40 | ||
| Eccentr. | Central | 151.36 | 20.10 | 111.70–230.00 | 175.97 | 85.27 | 45.40–459.90 | |
| 1st per. | 151.01 | 17.13 | 106.70–243.30 | 186.84 | 79.51 | 58.50–500.00 | ||
| 2nd per. | 148.43 | 15.73 | 120.00–235.00 | 192.48 | 67.81 | 61.3–396.40 | ||
| 3rd per. | 149.13 | 17.91 | 118.30–253.30 | 179.70 | 71.16 | 47.10–411.10 | ||
| 4th per. | 148.90 | 20.86 | 116.70–243.30 | 173.28 | 72.98 | 43.90–418.80 | ||
| HC | Sector | Superior | 151.29 | 19.72 | 115.00–266.70 | 147.73 | 46.72 | 54.20–334.70 |
| Inferior | 142.96 | 16.88 | 113.30–243.30 | 220.33 | 85.31 | 76.80–608.20 | ||
| Temporal | 148.12 | 19.58 | 106.70–266.70 | 171.50 | 59.93 | 47.10–394.10 | ||
| Nasal | 150.10 | 17.99 | 120.00–266.70 | 193.77 | 69.41 | 52.20–432.30 | ||
| Eccentr. | Central | 147.67 | 18.74 | 106.7–248.3 | 184.11 | 71.86 | 52.20–470.20 | |
| 1st per. | 147.57 | 16.62 | 116.70–266.70 | 192.45 | 73.41 | 50.80–493.10 | ||
| 2nd per. | 147.78 | 17.87 | 118.30–266.70 | 195.77 | 76.61 | 47.10–550.80 | ||
| 3rd. per. | 147.28 | 18.87 | 113.30–263.30 | 176.90 | 70.66 | 53.20–594.10 | ||
| 4th per. | 148.83 | 21.71 | 116.70–266.70 | 168.61 | 72.04 | 54.20–608.20 | ||
Figure 2.
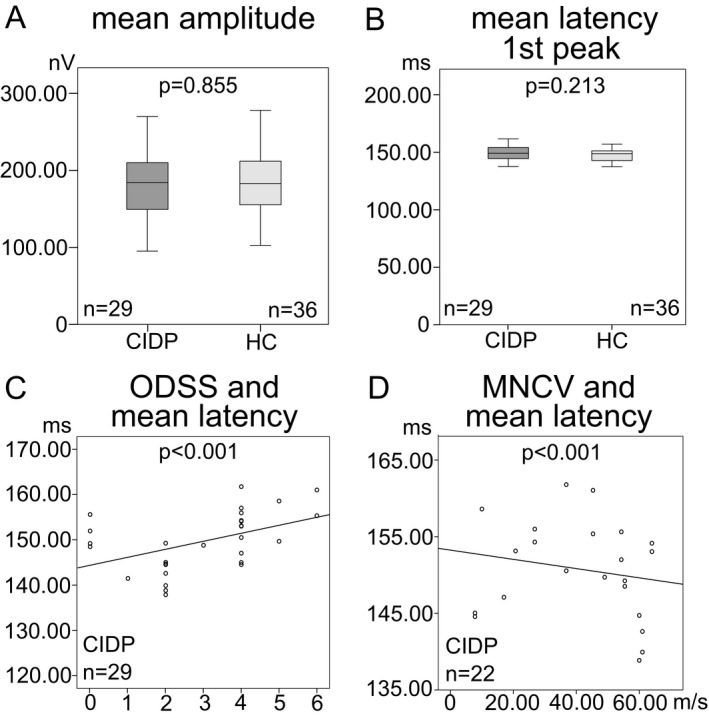
mfVEP parameters of CIDP patients and healthy controls (HC). Box‐and‐whisker plots of the mean amplitude in nV (A) and the mean latency of the first peak in msec (B). The black line within the box indicates the mean, the box marks the interquartile range and the whiskers represent the minimum and maximum. Scatter plots of the association between the ODSS (overall disability sum score) and the mean latency (C), and the MNCV (motor nerve conduction velocity) of the right ulnar nerve in m/sec and the mean latency (D). Each eye is shown as a dot. Regression lines, numbers of eyes, and P ‐values (GEE analysis) are provided for the different mfVEP parameters.
Due to the rather small sample size we cannot exclude subtle differences between CIDP patients and controls. Therefore, we performed a power analysis to determine the sample size needed to detect possible significant differences based on our data. Our results for the amplitude difference reached an effect size of 0.022 (power = 0.05, d Cohen = 0.032 (95%‐CI = [−0.46–0.52]) and for the latency difference an effect size of 0.36 (power = 0.30, d Cohen = 0.30 (95%‐CI: [−0.19–0.79]), This suggests that 121 and 32,598 patients would be needed to reach a power (1−β) of 0.8 for latencies and amplitudes, respectively.
Secondary outcomes – subanalyses
First, we created heatmap‐graphics for amplitude (Fig. 3A) and latency (Fig. 3B) showing the differences in each segment of the visual field between CIDP patients and healthy controls. These were calculated using the following formula:
Figure 3.
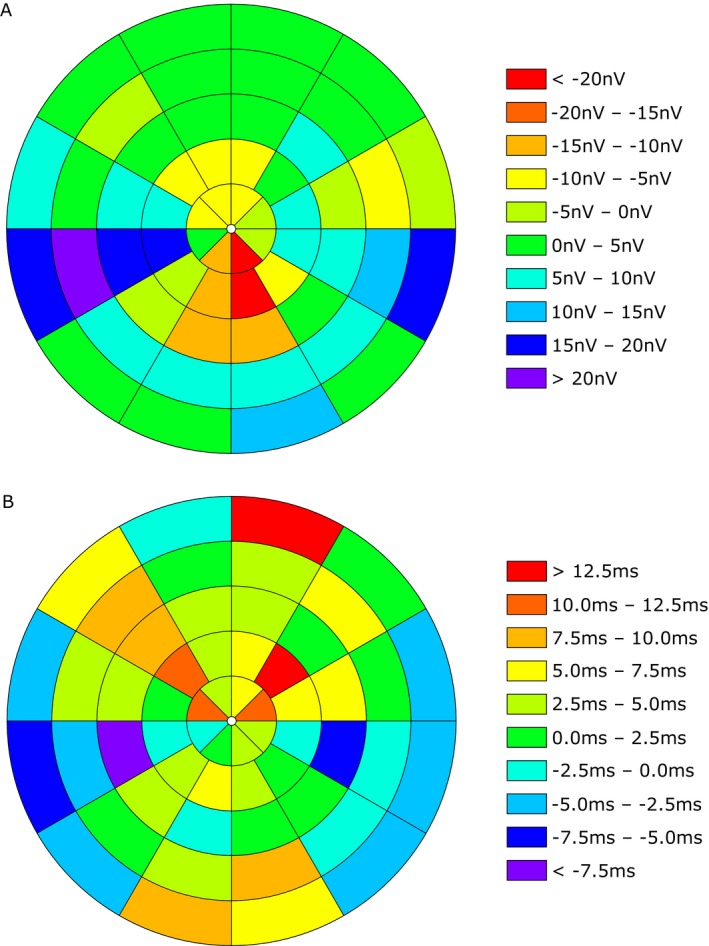
Differences in each segment of the visual field between CIDP patients and healthy controls (HC). Heat maps show the amplitude in nV (A) and the latency in msec (B). Different colors represent the positive or negative value of the differences (CIDP – HC).
Second, we performed systematical analyses regarding the different sectors and eccentricities. The statistical analysis of the sectors showed significant difference in first peak latency concerning the superior sector (P = 0.005, adj. P = 0.015, Fig. S2A) between both groups. There was no statistical difference regarding the remaining parameters. An overview of the results is provided in Table 2.
Table 2.
Overview of secondary outcome subanalyses. Table showing association analyses of the mean mfVEP parameters with the ODSS and MNCV, as well as analyses regarding the sectors and eccentricities (GEE analysis)
| Primary measure | Secondary measure | Tertiary measure | Latency | Amplitude | ||||
|---|---|---|---|---|---|---|---|---|
| P | adj. P | B | P | adj. P | B | |||
| Mean | ODSS ass. | <0.001 | 1.318 | 0.296 | 6.129 | |||
| MNCV ass. | <0.001 | −0.081 | 0.738 | −0.170 | ||||
| Sector | Superior | 0.005 | 0.015 | 5.205 | 0.684 | n.s. | −4.266 | |
| Inferior | 0.226 | n.s. | 1.712 | 0.782 | n.s. | −5.271 | ||
| Temporal | 0.896 | n.s. | 0.313 | 0.940 | n.s. | 1.112 | ||
| Nasal | 0.531 | n.s. | −1.483 | 0.781 | n.s. | 4.630 | ||
| Superior | MNCV ass. | 0.418 | n.s. | −0.039 | 0.188 | n.s. | −0.567 | |
| Inferior | MNCV ass. | 0.092 | n.s. | −0.073 | 0.995 | n.s. | 0.004 | |
| Temporal | MNCV ass. | 0.006 | 0.018 | −0.160 | 0.830 | n.s. | 0.123 | |
| Nasal | MNCV ass. | 0.084 | n.s. | −0.096 | 0.849 | n.s. | −0.129 | |
| Eccentricity | Central | 0.173 | n.s. | 3.622 | 0.723 | n.s. | −6.896 | |
| First peripheral | 0.104 | n.s. | 3.626 | 0.787 | n.s. | −4.960 | ||
| Second peripheral | 0.679 | n.s. | 0.811 | 0.791 | n.s. | −4.212 | ||
| Third peripheral | 0.224 | n.s. | 2.081 | 0.916 | n.s. | 1.369 | ||
| Outer peripheral | 0.917 | n.s. | 0.168 | 0.754 | n.s. | 3.867 | ||
| Central | MNCV ass. | 0.659 | n.s. | −0.027 | 0.589 | n.s. | −0.419 | |
| First peripheral | MNCV ass. | 0.003 | 0.012 | −0.119 | 0.807 | n.s. | −0.166 | |
| Second peripheral | MNCV ass. | 0.085 | n.s. | −0.079 | 0.951 | n.s. | −0.031 | |
| Third peripheral | MNCV ass. | 0.011 | 0.044 | −0.100 | 0.796 | n.s. | −0.136 | |
| Outer peripheral | MNCV ass. | 0.293 | n.s. | −0.049 | 0.608 | n.s. | −0.272 | |
Bonferroni Correction was used for adjusted P‐values. Significant results are highlighted in bold and light gray. (P = P‐value, adj. P = adjusted P ‐value, n.s. = not significant, ass. = association, MNCV = motor nerve conduction velocity, ODSS = overall disability sum score).
To examine if the severity of CIDP relates to mfVEP parameters, we performed analyses concerning ODSS and the motor nerve conduction velocity (MNCV, Table 2).
The ODSS in CIDP patients ranged from 0 to 6 (median = 4, mean = 3.11 ± 1.69). There was no significant association of amplitude with ODSS (P = 0.296, B = 6.129), but a significant positive association between first peak latency and ODSS (P < 0.001, B = 1.318, Fig. 2C).
MNCV of the right ulnar nerve was used for analysis. CIDP patients showed a mean MNCV of 39.08 ± 19.96 m/sec (range: 7.80–64.00 m/sec). There was no significant association between MNCV and amplitude (P = 0.738, B = −0.170), but a significant negative association between MNCV and mfVEP latency (P < 0.001, B = −0.081, Fig. 2D). Therefore, we performed subanalyses regarding sectors and eccentricities. Temporal sector (P = 0.006, adj. P = 0.018, B = −0.160, Fig. S2B), first peripheral eccentricity (P = 0.003, adj. P = 0.012, B = −0.119, Fig. S2C), and third peripheral eccentricity (P = 0.011, adj. P = 0.044, B = −0.100, Fig. S2D) showed significant negative associations, but none of the other parameters (Table 2).
Furthermore, we performed an analysis of the 2.5% low‐contrast visual acuity (LCV) between both groups and an association analysis of the LCV versus the mfVEP parameters.
In CIDP patients, there was a mean LCV of 0.33 ± 0.15 (range: 0.05–0.60) and in healthy controls 0.36 ± 0.11 (range: 0.12–0.63), the analysis revealed no significant difference between both groups (P = 0.500). CIDP patients showed a significant association of LCV with amplitude (P = 0.040, B = 108.549, Fig. S2E), but not between LCV and latency (P = 0.111, B = −8.219). In healthy controls, neither amplitude (P = 0.607, B = −20.136, Fig. S2E), nor latency (P = 0.102, B = −10.132) revealed significant associations with LCV.
Discussion
Certain inflammatory neuropathies like Miller Fisher syndrome and Bickerstaff brainstem encephalitis are associated with involvement of cranial nerves and the central nervous system, respectively.18, 19 In classic CIDP, clinical symptoms are usually restricted to the peripheral nervous system. Identifying CNS involvement in CIDP, which might be detectable by sensitive methods, would be of interest, for example, to evaluate progression in severe cases where peripheral nerve conduction studies are sometimes challenging due to a lack of action potentials. In contrast to previous studies3, 4, 5 that used traditional ffVEP, we found no difference of mean mfVEP amplitudes or latencies in our cohort of patients without clinical signs of CNS involvement compared to matched controls. Therefore, our primary hypothesis of altered mean mfVEP latencies or amplitudes has to be refuted.
In an exploratory subanalysis, we found a significant delay in latencies in the superior sector of the visual field and associations of mfVEP measures with the clinical ODSS and the mean MNCV of our CIDP patients. However, since there is no simple biological, anatomical, or pathological explanation for these results; these exploratory findings need to be interpreted with great caution and we cannot rule out a false‐positive result despite the Bonferroni correction for multiple testing. Of note, if the Bonferroni correction is performed for the total number of tests performed, instead of only correcting for the number of subanalyses, the difference in latency of the superior sector loses significance. The fact that we observed a significant association of mean mfVEP latency with ODSS and MNCV is surprising considering that mean latency did not differ between patients and controls. Possible explanations are CIDP‐independent effects, for example, metabolic factors influencing both mfVEP latency and MNCV/CIDP severity, and/or a subtle involvement of the visual system only in severe cases of CIDP with ODSS above 4. Larger studies on severely affected CIDP patients including MNCV assessments also in healthy controls would be needed to address these points. However, we have to acknowledge, that the association remains weak and therefore a clinical relevance cannot be postulated.
Therefore, considering all these caveats, the significant differences observed are unlikely to be meaningful in a clinical setting.
We conclude that despite carefully selecting the patient cohort and using both enhanced mfVEP techniques, adequate inclusion and exclusion criteria and suitable statistical methods for visual outcome measures to account for intereye within‐patient dependencies20 a clinically relevant visual pathway involvement in CIDP cannot be elicited. Reasons for the positive results of the previous studies using full‐field VEPs3, 4, 5 remain subject to discussion. Possible explanations include the lack of age‐ and sex‐matched controls and differences in the composition the cohorts studied, for example, including also patients with signs of CNS involvement, or patients with other peripheral neuropathies, for example, hereditary neuropathy with pressure palsies (HNPP) and Charcot–Marie–Tooth disease type 1A (CMT1A).1 Further well‐controlled longitudinal studies involving larger cohorts of different disease entities and higher numbers of severely affected patients are warranted to refute or corroborate these results and investigate their relevance. All in all, considering the data presented here, mfVEPs do not seem promising as a monitoring parameter for CNS involvement in CIDP in the clinical routine.
Author Contributions
Jonas Graf contributed to the study concept/design, acquisition/analysis/interpretation of data, and drafting of the manuscript. Lea Kristina Jansen contributed to the study concept/design, acquisition/analysis/interpretation of data. Jens Ingwersen contributed to the acquisition/analysis/interpretation of data, critical revision of the manuscript. Marius Ringelstein contributed to the analysis/interpretation of data, critical revision of the manuscript. Alexander Klistorner contributed to the analysis/interpretation of data. Jens Harmel, Jana Rybak, Laura Rhöse, and Lena Gemerzki contributed to the acquisition of data. John‐Ih Lee contributed to the acquisition of data and critical revision of the manuscript. Robert Kolbe and Rainer Guthoff contributed to the analysis/interpretation of data. Hans‐Peter Hartung contributed to the study concept, input, and critical revision of the manuscript. Orhan Aktas contributed to the study concept/design, critical revision of the manuscript. Philipp Albrecht contributed to the study concept/design, acquisition/analysis/interpretation of data, drafting, and revision of the manuscript.
Conflicts of Interest
Jonas Graf, Lea Kristina Jansen, Jens Ingwersen, Jens Harmel, Jana Rybak, John‐Ih Lee, Laura Rhöse, Lena Gemerzki, and Robert Kolbe report no conflicts of interest. Marius Ringelstein has received consulting and speaker honoraria as well as travel reimbursements from Bayer Healthcare, Biogen, Genzyme, Teva, Merz, and Novartis. Alexander Klistorner reports grants from Biogen, and Novartis. Rainer Guthoff received speaker honoraria and travel/accommodation/meeting expenses from Novartis, Roche, and Bayer Schering. Hans‐Peter Hartung received, with approval of the Rector of Heinrich‐Heine‐University and the CEO of University of Düsseldorf Hospital, honoraria for consulting, serving on steering committees and speaking from Biogen, Geneuro, Genzyme, Medimmune, Merck, Novartis, Opexa, Receptos/Celgene, Roche, Sanofi, and Teva. Orhan Aktas received, with approval of the Rector of Heinrich‐Heine‐University, grants from the German Research Foundation (DFG), the German Ministry for Education and Research (BMBF) as part of the German Competence Network Multiple Sclerosis (KKNMS; for NEMOS Nation NMO‐PAT FKZ 01GI1602B), the Eugène Devic European Network (EU‐FP7), honoraria and travel/accommodation/meeting expenses from Almirall, Bayer, Biogen, Medimmune, Merck Serono, Novartis, Roche, Sanofi‐Genzyme, and Teva. Philipp Albrecht received grants, personal fees, and nonfinancial support from Allergan, Biogen, Ipsen, Merz Pharmaceuticals, Novartis, and Roche; personal fees and nonfinancial support from Bayer Healthcare, Merck, and Sanofi‐Aventis/Genzyme, outside the submitted work.
Supporting information
Figure S1. Visual fields, segments, sectors, and eccentricities of each eye. Graphics represent the classification into sectors (A) and eccentricities (B). Each segment is labeled by a number (1–56). The different colors of the segments mark the related sector in (A) (superior = green, nasal = pink, inferior = dark blue, temporal = purple) and the eccentricity in (B) (central = yellow, first peripheral = orange, second peripheral = red, third peripheral = light blue, fourth peripheral = gray).
Figure S2. mfVEP parameters of CIDP patients and healthy controls (HC). Box‐and‐whisker plot of the latency of the superior sector in msec (A). The black line within the box marks the mean, the box shows the interquartile range and the whiskers indicate the minimum and maximum. Scatter plots of the association between the MNCV (motor nerve conduction velocity) of the right ulnar nerve in m/sec and the latency of the temporal sector (B), the first (C) and third peripheral eccentricity (D), as well as the LCV (low‐contrast visual acuity 2,5% provided in decimals) versus the mean amplitude (E). Each eye is shown as a dot. Regression lines, numbers of eyes (B–E) or visual fields (A), P‐values (GEE analysis), and adjusted P‐values (Bonferroni Correction) are provided for the different mfVEP parameters. LCV data were available for CIDP patients and controls. Therefore, the associations between mean amplitude and LCV are provided for both groups presenting CIDP patients in green and healthy controls in black (E).
Table S1. Detailed overview of multifocal visual evoked potential and visual acuity data in CIDP patients in Düsseldorf.
Table S2. Detailed overview of multifocal visual evoked potential and visual acuity data in healthy control subjects in Düsseldorf.
Funding Information
No funding information is provided.
References
- 1. Köller H, Kieseier BC, Jander S, Hartung H‐P. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med 2005;352:1343–1356. [DOI] [PubMed] [Google Scholar]
- 2. Mathey EK, Park SB, Hughes RAC, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatr 2015;86:973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stojkovic T, de Seze J, Hurtevent JF, et al. Visual evoked potentials study in chronic idiopathic inflammatory demyelinating polyneuropathy. Clin Neurophysiol 2000;111:2285–2291. [DOI] [PubMed] [Google Scholar]
- 4. Knopp M, Leese RJ, Martin‐Lamb D, Rajabally YA. Optic and auditory pathway dysfunction in demyelinating neuropathies. Acta Neurol Scand 2014;130:53–57. [DOI] [PubMed] [Google Scholar]
- 5. Lehmann HC, Hoffmann FR, Meyer Zu Hörste G, et al. Central nervous system involvement in patients with monoclonal gammopathy and polyneuropathy. Eur J Neurol 2010;17:1075–1081. [DOI] [PubMed] [Google Scholar]
- 6. Hawke SH, Hallinan JM, McLeod JG. Cranial magnetic resonance imaging in chronic demyelinating polyneuropathy. J Neurol Neurosurg Psychiatr 1990;53:794–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ormerod IE, Waddy HM, Kermode AG, et al. Involvement of the central nervous system in chronic inflammatory demyelinating polyneuropathy: a clinical, electrophysiological and magnetic resonance imaging study. J Neurol Neurosurg Psychiatr 1990;53:789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas PK, Walker RW, Rudge P, et al. Chronic demyelinating peripheral neuropathy associated with multifocal central nervous system demyelination. Brain 1987;110(Pt 1):53–76. [DOI] [PubMed] [Google Scholar]
- 9. Ogata H, Matsuse D, Yamasaki R, et al. A nationwide survey of combined central and peripheral demyelination in Japan. J Neurol Neurosurg Psychiatr 2016;87:29–36. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y‐Q, Chen H, Zhuang W‐P, Li H‐L. The clinical features of combined central and peripheral demyelination in Chinese patients. J Neuroimmunol 2018;317:32–36. [DOI] [PubMed] [Google Scholar]
- 11. Bouchard C, Lacroix C, Planté V, et al. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology 1999;52:498–503. [DOI] [PubMed] [Google Scholar]
- 12. Alshowaeir D, Yiannikas C, Klistorner A. Multifocal visual evoked potential (mfVEP) and pattern‐reversal visual evoked potential changes in patients with visual pathway disorders: a case series. Neuroophthalmology 2015;39:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Joint Task Force of the EFNS and the PNS . European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society–first revision. J Peripher Nerv Syst 2010;15:295–301. [DOI] [PubMed] [Google Scholar]
- 14. Heravian J, Ehyaei A, Shoeibi N, et al. Pattern visual evoked potentials in patients with type II diabetes mellitus. J Ophthalmic Vis Res 2012;7:225–230. [PMC free article] [PubMed] [Google Scholar]
- 15. Merkies ISJ, Schmitz PIM, van der Meché FGA, et al. Clinimetric evaluation of a new overall disability scale in immune mediated polyneuropathies. J Neurol Neurosurg Psychiatr 2002;72:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartmann CJ, Klistorner AI, Brandt AU, et al. Axonal damage in papilledema linked to idiopathic intracranial hypertension as revealed by multifocal visual evoked potentials. Clin Neurophysiol 2015;126:2040–2041. [DOI] [PubMed] [Google Scholar]
- 17. Klistorner AI, Graham SL. Electroencephalogram‐based scaling of multifocal visual evoked potentials: Effect on intersubject amplitude variability. Invest Ophthalmol Vis Sci 2001;42:2145–2152. [PubMed] [Google Scholar]
- 18. Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti‐GQ1b antibody syndrome. J Neurol Neurosurg Psychiatr 2013;84:576–583. [DOI] [PubMed] [Google Scholar]
- 19. Wakerley BR, Uncini A, Yuki N. Guillain‐Barré and Miller Fisher syndromes–new diagnostic classification. Nat Rev Neurol 2014;10:537–544. [DOI] [PubMed] [Google Scholar]
- 20. Cruz‐Herranz A, Balk LJ, Oberwahrenbrock T, et al. The APOSTEL recommendations for reporting quantitative optical coherence tomography studies. Neurology 2016;86:2303–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Visual fields, segments, sectors, and eccentricities of each eye. Graphics represent the classification into sectors (A) and eccentricities (B). Each segment is labeled by a number (1–56). The different colors of the segments mark the related sector in (A) (superior = green, nasal = pink, inferior = dark blue, temporal = purple) and the eccentricity in (B) (central = yellow, first peripheral = orange, second peripheral = red, third peripheral = light blue, fourth peripheral = gray).
Figure S2. mfVEP parameters of CIDP patients and healthy controls (HC). Box‐and‐whisker plot of the latency of the superior sector in msec (A). The black line within the box marks the mean, the box shows the interquartile range and the whiskers indicate the minimum and maximum. Scatter plots of the association between the MNCV (motor nerve conduction velocity) of the right ulnar nerve in m/sec and the latency of the temporal sector (B), the first (C) and third peripheral eccentricity (D), as well as the LCV (low‐contrast visual acuity 2,5% provided in decimals) versus the mean amplitude (E). Each eye is shown as a dot. Regression lines, numbers of eyes (B–E) or visual fields (A), P‐values (GEE analysis), and adjusted P‐values (Bonferroni Correction) are provided for the different mfVEP parameters. LCV data were available for CIDP patients and controls. Therefore, the associations between mean amplitude and LCV are provided for both groups presenting CIDP patients in green and healthy controls in black (E).
Table S1. Detailed overview of multifocal visual evoked potential and visual acuity data in CIDP patients in Düsseldorf.
Table S2. Detailed overview of multifocal visual evoked potential and visual acuity data in healthy control subjects in Düsseldorf.