

SUMMARY
In all organisms, replication impairments are an important source of genome rearrangements, mainly because of the formation of double-stranded DNA (dsDNA) ends at inactivated replication forks. Three reactions for the formation of dsDNA ends at replication forks were originally described for Escherichia coli and became seminal models for all organisms: the encounter of replication forks with preexisting single-stranded DNA (ssDNA) interruptions, replication fork reversal, and head-to-tail collisions of successive replication rounds. Here, we first review the experimental evidence that now allows us to know when, where, and how these three different reactions occur in E. coli. Next, we recall our recent studies showing that in wild-type E. coli, spontaneous replication fork breakage occurs in 18% of cells at each generation. We propose that it results from the replication of preexisting nicks or gaps, since it does not involve replication fork reversal or head-to-tail fork collisions. In the recB mutant, deficient for double-strand break (DSB) repair, fork breakage triggers DSBs in the chromosome terminus during cell division, a reaction that is heritable for several generations. Finally, we recapitulate several observations suggesting that restart from intact inactivated replication forks and restart from recombination intermediates require different sets of enzymatic activities. The finding that 18% of cells suffer replication fork breakage suggests that DNA remains intact at most inactivated forks. Similarly, only 18% of cells need the helicase loader for replication restart, which leads us to speculate that the replicative helicase remains on DNA at intact inactivated replication forks and is reactivated by the replication restart proteins.
KEYWORDS: recombination, replication restart, PriA, RecA, RecBC, RuvAB, RecG, replication fork reversal, chromosome terminus, double-strand break, RecBCD
INTRODUCTION
The two replication forks assembled at the replication origin of a bacterial circular chromosome progress in opposite directions until they meet in the terminus region, unless they are arrested by DNA damage or protein roadblocks. Obviously, proper chromosome replication is crucial because chromosomes can be transmitted to progeny only if they are fully replicated, but in addition, replication fork arrest has dramatic consequences for genome stability. This idea emerged in studies of bacteria from the observation that mutations affecting DNA replication exhibited a hyperrecombination phenotype and from the direct demonstration that blocked replication forks could be broken and thus become entry points for DNA degradation or recombination and, in turn, a source of DNA rearrangements (1–7). These concepts were soon extended to yeast and multicellular eukaryotes (8; for recent reviews, see references 9–12). The identification of the possible causes and consequences of accidental replication fork arrest and the description of replication restart pathways thus became the subjects of intense studies.
In this review, we first recall the molecular mechanism of homologous recombination at double-stranded DNA (dsDNA) ends in Escherichia coli and then present the three documented pathways for the formation of dsDNA ends at inactivated replication forks. After this, we discuss our recent study of the formation of spontaneous dsDNA ends in unchallenged E. coli cells and finally describe how our results suggest important differences in replication restart reactions depending on whether the DNA at an arrested replication fork is broken or remains intact.
REPAIR OF dsDNA ENDS IN E. COLI
The repair of dsDNA ends in E. coli starts by the action of RecBCD, a heterotrimeric complex with a helicase and a dsDNA exonuclease activity (reviewed in references 13–15). Its exonuclease action (Exo V) is modified by the encounter of a specific site called Chi (5′-GCTGGTGG-3′), which triggers the loading of the recombinase RecA by RecBCD onto the 3′-ended DNA strand (repair of a dsDNA end) (Fig. 1A). The RecA–single-stranded DNA (ssDNA) filament catalyzes a homology search and strand exchange, which results in an X-like structure called a Holliday junction (HJ), adjacent to a displacement loop (D-loop) (Fig. 1A). HJs are specifically recognized and bound by RuvA and RuvB, which promote their migration, extending the heteroduplex sequence until the HJ-RuvAB complex is bound by the resolvase RuvC. RuvC resolves the HJ by the cleavage of two opposite strands, and ligation produces two dsDNA recombinant molecules. Replication restart from recombination intermediates is essential for homologous recombination (16, 17). D-loops formed by strand invasion are, as replication forks, three-arm structures, with one ssDNA arm, two dsDNA arms, and a 3′ DNA end at the junction; therefore, they are targeted by PriA, the key enzyme for replication restart (18–21). This recognition of the strand invasion intermediate by PriA allows the reassembly of the replisome on the replication fork framework, formed by invasion, and triggers replication restart (Fig. 1A). The PriA protein is a 3′-to-5′ helicase, but its helicase activity is not required for restart (22). Note that double-strand breaks (DSBs) are two-ended if they occur away from replication forks, while they have a one-ended configuration when they occur at replication forks. These two types of DSBs can be differentiated experimentally (see, for example, reference 23). However, a DSB occurring close enough behind a fork is likely to be converted to a one-ended break by DNA degradation and will become indistinguishable from replication fork breakage.
FIG 1.

(A) Repair of a broken replication fork by RecBCD, RecA, Ruv, and PriA. RecBCD binds to DNA double-strand ends and degrades both strands until it encounters a Chi site at which it loads RecA onto the 3′ DNA end. The RecA-ssDNA filament invades a homologous region and promotes strand exchange, resulting in a Holliday junction (HJ), indicated by the blue and red crossing lines, and an adjacent displacement loop, also called a D-loop, schematized by the displacement of one of the red lines by the blue line end. RuvAB binding to the HJ drives branch migration. After RuvC binding, the RuvABC complex catalyzes the resolution of the HJ, resulting in a recombinant molecule. PriA restarts replication from the D-loop. (B) Formation of a “broken fork” by the encounter of a preexisting nick. A ssDNA break is drawn here on the lagging-strand template, but the same reaction occurs with a ssDNA break on the leading-strand template. DSB repair is the same as in panel A and reconstitutes a replication fork. (C) Replication fork reversal. In the first step (step a), the replication fork is arrested, and the leading- and lagging-strand ends of the newly synthesized strands anneal. The resulting structure is called a reversed fork; it has a four-arm structure akin to a HJ. Two alternative representations of this structure are shown, called open X and parallel stacked X. RecBC acts on the dsDNA end (as shown in panel A) and is essential for the resetting of the fork, either by RecA-dependent homologous recombination (steps b and c) or by DNA degradation (steps b to d). Either pathway creates a substrate for replication restart proteins (PriA and its partners), since homologous recombination leads to a D-loop, as shown in panel A, and DNA degradation restores a fork structure. In the absence of RecBCD, the resolution of the HJ causes chromosome linearization (not shown). (D) dsDNA ends formed by head-to-tail collision of replication forks. The dsDNA ends formed by rereplication are recombined as in panel A. This reaction occurs at forks blocked at an ectopic Ter site, where it requires UvrD to dislodge the Tus protein (see the text). In panel A, the blue and red continuous double lines represent two homologous DNA molecules. In panels B and C, the continuous lines represent the parental chromosome, and the dashed lines represent the newly synthesized strands. In panel D, the dashed lines represent the DNA synthesized in a second replication round. Arrowheads show DNA 3′ ends. Incised purple circles, RecBCD; small yellow circles, RecA; green circles, RuvAB.
FORMATION OF dsDNA ENDS AT REPLICATION FORKS IN E. COLI
In parallel with studies dedicated to replication restart, several investigations have aimed to understand how replication impairment can lead to the formation of dsDNA ends at forks. Although blocked forks might be inherently fragile owing to their ssDNA regions, only the seqA mutant was proposed to suffer direct breakage of ssDNA at stalled replication forks (24, 25), and it turned out that most often, arrested forks were not broken. Three main modes of dsDNA end formation at forks were reported: (i) encounter of a replication fork with a preexisting single-stranded DNA interruption in a template strand (originally called “replication fork collapse” in a seminal review by A. Kuzminov [26]) (Fig. 1B); (ii) replication fork reversal (RFR) (27), sometimes also called replication fork regression (Fig. 1C); and (iii) encounter of a replication fork with a previously arrested fork, also called head-to-tail fork collisions or fork rear-ending (28) (Fig. 1D).
Formation of a dsDNA End by the Encounter of a Replication Fork with a Single-Stranded DNA Interruption in a Template Strand
An engineered ssDNA break is converted into a dsDNA end by the arrival of a replication fork (29) (Fig. 1B). In eukaryotic organisms, site-specific or drug-induced ssDNA breaks were also shown to be converted into dsDNA breaks by the arrival of a replication fork (30, 31). The Kuzminov laboratory set out to identify mutations that increased the frequency of such chromosomal DSBs. Knowing that RecA is essential for the repair of dsDNA ends, those researchers isolated mutations that are colethal with recA inactivation (32). Two mutations that were isolated perturbed the synthesis of deoxynucleotides (tdk and rdgB) and so directly implicated DNA replication. In these mutants, noncanonical deoxynucleoside triphosphates are not removed from the DNA precursor pools and are misincorporated into DNA during chromosome replication. They are then excised by a specific endonuclease, and this can lead to the formation of dsDNA ends in two ways: (i) the formation of a two-ended DSB when two adjacent excision reactions occur, one on each DNA strand, resulting in two nearly opposite single-stranded interruptions (Fig. 2), or (ii) the formation of a single dsDNA end when a replication fork reaches a ssDNA break created by nucleotide excision in the template strand (Fig. 1B). DSBs, formed in either way, are then repaired by the successive action of RecBCD, RecA, RuvABC, and PriA (Fig. 1A). However, the former happens at an undefined position behind the replication fork, while the latter occurs at the replication fork only when it reaches the DNA interruption.
FIG 2.
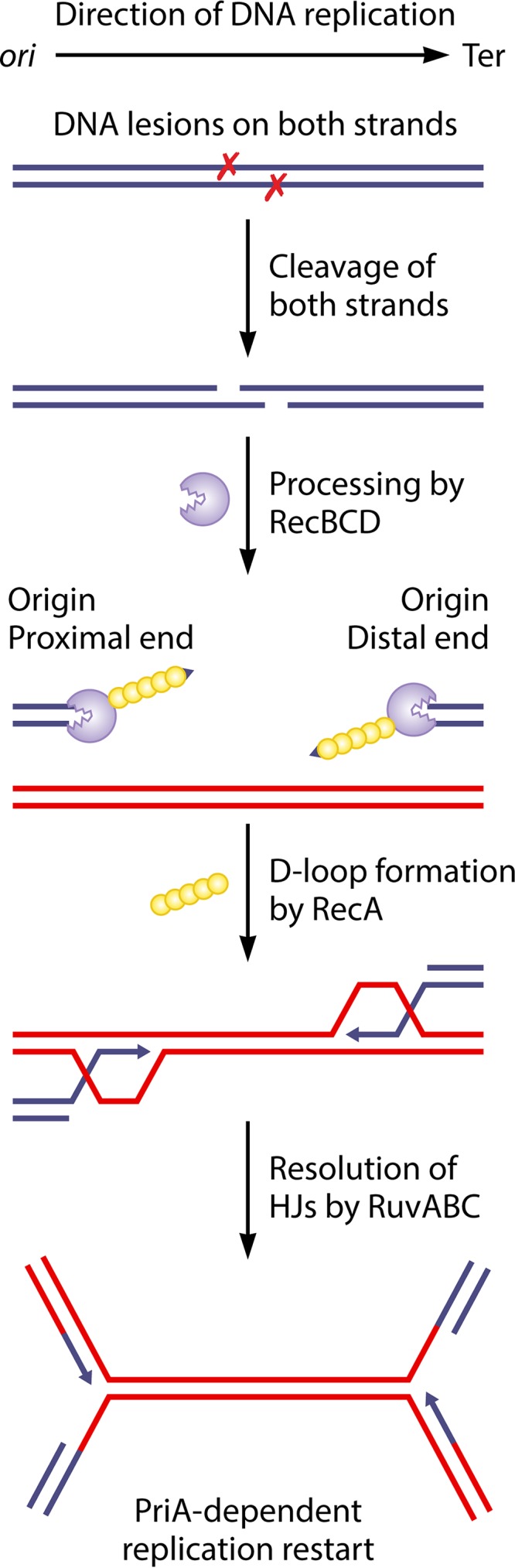
Model for two-ended break repair following the excision of two closely spaced lesions on opposite strands of the DNA. When two lesions (denoted by X) are closely spaced on opposite strands of the DNA, their nucleolytic excision can lead to a two-ended double-strand break behind the replication fork. One of the ends, closest to the origin of replication, is denoted the origin-proximal end, and the other end, closest to the terminus of replication, is denoted the origin-distal end. Each end is processed by the RecBCD enzyme, which loads the RecA protein. The RecA protein catalyzes strand invasion to form two D-loops and two HJs that are resolved by RuvABC. This converts the joint molecules to converging replication forks, which assemble new replisomes through PriA-dependent restart. The blue and red continuous double lines represent two homologous DNA molecules. Arrowheads show DNA 3′ ends.
Several studies were carried out to differentiate between DNA breakages behind the replication fork and at the replication fork. The rdgB mutant was shown to cause DSBs by triggering the incorporation of xanthine and hypoxanthine into DNA, with their subsequent excision by endo V, but the exact mode of DSB formation was not elucidated (33–35). The dut mutant, which incorporates uracil into DNA, was also colethal with recA inactivation and was shown by direct visualization of chromosomes to suffer replication-dependent DSBs (36–38). A detailed molecular analysis of chromosome breakage in a dut recB mutant suggested that a combination of persistent ssDNA interruptions in the path of replication forks (Fig. 1B) and clustered excision of misincorporated nucleotides on both strands (Fig. 2) is responsible for DSB formation (38). That study was based on the idea that in a recB mutant, replication-dependent breakage generates exclusively origin-proximal and no origin-distal dsDNA ends (the dsDNA end in Fig. 1B is linked to the origin and thus called an origin-proximal end). Origin-distal ends were observed although at a lower frequency than origin-proximal ends. Chromosome breakage in the dut recB mutant therefore results mainly from the encounter of replication forks with ssDNA interruptions and, to some extent, from clustered excision behind replication forks (38).
Ligase mutants also require repair by recombination for viability, and since it was thought that leading-strand synthesis was continuous and ligases were required to seal discontinuous synthesis of the lagging strand, ssDNA breaks were originally believed to accumulate only on the lagging strand. However, it was observed that chromosomes in a ligase mutant accumulate nicks on both strands (39, 40). This finding suggested the discontinuous synthesis of both leading and lagging strands and raised the possibility of DSBs resulting from nicking across long-lived ssDNA breaks. Although DSBs in a weak ligase mutant could result mainly from replication forks reaching unsealed ssDNA breaks, DSBs in a strongly affected ligase mutant occurred most often behind replication forks, presumably resulting from nicks in close proximity on both strands, for example, if a second nick in the opposite strand is preconditioned by, and targeted to, a first one that persisted because of the absence of ligase (41).
In conclusion, replication-dependent fork breakage events occur when forks encounter ssDNA nicks or gaps on the template DNA and were observed readily in those cells that accumulate or fail to repair such nicks or gaps. However, in certain mutants, two-ended DSBs also occur behind replication forks, in addition to the one-ended breaks that occur at replication forks.
Replication Fork Reversal
A screen for genes involved in replication fork breakage in the E. coli rep replication mutant led to the isolation of several mutations in the ruvAB operon, encoding proteins that act at Holliday junctions, and this observation gave rise to the replication fork reversal (RFR) model (27). According to this model, at certain blocked replication forks, such as in the rep mutant, the newly synthesized DNA ends anneal, forming a Holliday junction adjacent to a dsDNA end (Fig. 1C). In a cell proficient for the exonuclease V activity of RecBCD (Exo V) or homologous recombination, the dsDNA end is degraded or recombined. In cells deficient for both Exo V and homologous recombination (such as the recBC mutant), the HJ is resolved by RuvABC, and the resulting linear chromosome arm is not repaired. Importantly, the RFR model proposed for the first time the formation of recombination substrates and the action of recombination proteins at blocked forks (Fig. 1C). The hallmarks of RFR are (i) a requirement for RecBCD for viability (RecBCD can either degrade or recombine the dsDNA ends), (ii) no requirement for RecA provided that the exonuclease V action of RecBCD is active (requirement for either homologous recombination or linear DNA degradation), (iii) no measurable DNA degradation associated with replication inactivation in replication mutants that lack RecA (the degraded sequence is short), and (iv) RuvABC-dependent chromosome breakage in the absence of RecBCD.
RFR was originally observed in E. coli in two helicase mutants, the rep-null mutant that lacks an accessory replicative helicase and a dnaB(Ts) mutant where the main replicative helicase, DnaB, can be inactivated by a shift to a high temperature (27). In the rep mutant, replication was thought to be arrested by protein roadblocks, and thus, the role of Rep helicase was proposed to be to facilitate obstacle removal (27, 42). It was later shown that the primary role of Rep is to clear RNA polymerases from the path of replication forks and that Rep is present at forks by its interaction with the helicase DnaB (43–45). RFR also occurs under several other conditions of replication impairment: in replication mutants affected for different subunits of the holoenzyme polymerase III (HE Pol III) (46, 47), in the priA replication restart mutant (48), in mutants impaired for the biosynthesis of the nucleotide pool (49, 50), in UV-treated cells (51), in the presence of a topoisomerase inhibitor (52), in Pseudomonas syringae grown at a low temperature (53), and in Salmonella enterica serovar Typhimurium during nitrosative stress (54). In agreement with the original observation of RFR in an E. coli rep mutant, which lacks the main accessory helicase facilitating replication across DNA-bound proteins, such as RNA polymerases (43, 55), RFR also occurred at an engineered strong replication-transcription collision site, where replication was arrested by an oppositely oriented, highly transcribed region (56). Finally, a helicase-driven RFR reaction was reported in vivo and in vitro for phage T4 (57). Note that reversed forks were proposed to form in eukaryotic cells and to be targeted by polymerases to allow lesion bypass (58), but the molecules observed in that work were later shown to form in vitro during DNA extraction (59). The interconversion of replication and recombination intermediates in bacteria was also proposed theoretically (60).
In cells that undergo RFR, the enzyme that catalyzes fork breakage was readily identified as RuvC. Two E. coli enzymes were shown to catalyze the conversion of replication forks into HJs in vitro and in vivo, and both are homologous recombination enzymes: the recombinase RecA and the HJ branch migration enzyme RuvAB.
Replication Fork Reversal in E. coli Is Catalyzed by RecA or RuvAB
RecA was the first enzyme shown to promote replication fork reversal: RuvABC-dependent breakage was abolished in the dnaB(Ts) mutant by the inactivation of RecA. It was proposed that RecA binding to the lagging-strand template at a blocked fork could promote fork reversal by the invasion of the homologous leading strand (Fig. 3A) (61). RecA-dependent fork reversal was also observed in UV-irradiated cells (51). This intramolecular recombination reaction could be reconstituted on a DNA molecule mimicking a replication fork in vitro (62). A similar reaction was later shown to be mediated in vivo by the eukaryotic homologue of RecA, Rad51, following mild replication stress (63).
FIG 3.

(A) Model of RFR by RecA. RecA binding to the ssDNA region on the lagging-strand template of a blocked fork can promote the invasion of the homologous sequence on the leading strand. This reaction produces a reversed fork. (B) Model of RFR by RuvAB. The RuvAB complex formed on a replication fork can contain only one RuvB hexamer. Branch migration promoted by this RuvB hexamer extrudes a dsDNA end on which a second RuvB hexamer can bind, resulting in a RuvAB-HJ complex similar to the one formed during homologous recombination. RuvC resolves the HJ, which, in the case of a reversed fork, results in fork breakage. Continuous lines, parental DNA strands; dashed lines, newly synthesized DNA strands; small yellow circles, RecA; orange, trefoil RuvA tetramer; green circles, RuvB. Arrowheads show DNA 3′ ends. The small black arrows indicate the direction of strand displacement by the RuvAB complex.
With the exception of the dnaB(Ts) mutant, RFR was independent of RecA in all bacterial replication mutants tested, suggesting the existence of other pathways. Genetic studies suggested that the helicase RecG might reverse forks in UV-irradiated cells (64). This prompted the development of in vitro assays for RFR, based on the observation that in vivo HJs made by fork reversal are cleaved by the RuvABC HJ resolvase when the dsDNA end remains unprocessed (27) (Fig. 1C). Short DNA molecules that mimic replication fork structures were incubated with candidate enzymes, and the formation of a HJ was assayed by the addition of a resolvase (65, 66); later, the formation of the fourth arm of the HJ was also monitored by restriction enzyme digestion (67). These experiments showed that in vitro, RecG could catalyze the conversion of a fork structure into a resolvase substrate. However, further in vivo studies did not confirm an active role of RecG in replication restart after UV irradiation (51, 68), and, to the contrary, the inactivation of recG promoted UV-induced replication (69, 70). Furthermore, testing of RFR under different replication arrest conditions did not provide any evidence for a role of RecG in vivo (56, 71; B. Michel, unpublished results). Accordingly, the actual in vivo RecG target was shown to be joint molecules made by homologous recombination. RecG works with RuvAB to prevent the unwinding of joint molecules (presumed to be RecA-mediated D-loops) by PriA helicase activity (72). In vitro and in vivo, RecG acts at D-loops in combination with the replication restart protein PriA: RecG orients the action of PriA, and conversely, PriA binding prevents RFR by RecG (23, 73–75) (Fig. 4). Following the RecG studies, several eukaryotic helicases were also shown to catalyze fork reversal in vitro (reviewed in references 12, 75, and 76), but the lessons learned from RecG clearly warn us that these structure-specific helicases do not necessarily reverse forks in vivo.
FIG 4.

Model of the concerted action of RecG and PriA at forks. It has been shown that RecG remodels replication forks in vitro. (Ai) When RecG is alone, this remodeling causes RFR. (ii) When both RecG and PriA are present, PriA binds to the 3′ end at the fork, preventing its unwinding by RecG. This reaction is likely to also take place in vivo, since no genetic evidence for RecG-dependent RFR could be obtained. (Bi) It has been proposed that in vivo, in addition to preventing RFR, the binding of PriA in the presence of RecG leads to the correct loading of DnaB to the lagging-strand template. Because the PriA helicase domains, represented as small orange stars, bind to the lagging-strand template, replication restarts in the initial direction. (ii) In the absence of RecG, the PriA helicase domains bind to the newly synthesized lagging strand, and consequently, PriA can load DnaB incorrectly to this strand. This results in reverse restart, the assembly of a replication fork proceeding in the wrong direction. Large blue lines, template strands; red lines, newly synthesized strands; small blue lines in panel Bii, strand synthesized by reverse restart; green crescent, RecG; purple star, PriA; blue ring, DnaB replicative helicase. Arrowheads show DNA 3′ ends.
Replication fork reversal was shown to be catalyzed by RuvAB in several E. coli replication mutants (71) (Fig. 3B). Indeed, in these replication mutants, the inactivation of RuvAB prevented chromosome breakage by the alternative resolvase RusA, which indicated that RuvAB is necessary and sufficient for HJ formation at blocked forks (ruvAB inactivation abolished RFR, although all other helicases were expressed). ruvA and ruvB separation-of-function mutants were isolated, which were still fully functional for homologous recombination but unable to reverse forks (77–79). Biochemically, these RuvA mutant proteins were less efficient than wild-type RuvA for fork binding and for HJ branch migration in the presence of RuvB (77, 79). This result suggested that the conversion of a replication fork into a HJ (RFR) is a more demanding reaction than HJ branch migration. RFR is more difficult than HJ branch migration because the substrate of RFR has three DNA arms, including a ssDNA one; therefore, there are fewer RuvA tetramer contacts with the DNA, and RFR starts with one RuvB hexamer bound to the three-strand junction. In contrast, the HJ has four dsDNA arms; therefore, all RuvA monomers in the tetramers contact DNA, and two RuvB hexamers bind to the structure (Fig. 3B). Accordingly, when a RuvAB-dependent RFR reaction was reconstituted in vitro on plasmid molecules, the branch migration reaction was so efficient that the HJ intermediate could not be trapped, and the short plasmid molecule carrying a blocked fork was entirely unwound by RuvAB (80).
It was also proposed that RFR could occur independently of any enzymatic activity, promoted by an excess of positive supercoiling at blocked forks (81). This idea was tested in vivo by using a gyrase mutant [gyrB(Ts)] and a topoisomerase IV (Topo IV) mutant [parE(Ts)], in which positive supercoils created by transcription or replication are not efficiently removed at a high temperature, leading to replication fork blockage and lethality (82). The partial inactivation of gyrase or Topo IV caused replication fork arrest, as deduced from the need for the key replication restart protein PriA for viability. However, although arrested, forks blocked by positive supercoiling in vivo are not reversed, since RecBC was not essential for viability upon the partial inactivation of gyrase or Topo IV, and no increase in DSBs could be detected in the gyrB(Ts) recB mutant (83, 84). Furthermore, the in vitro experiments supporting supercoiling-driven RFR used DNA incubation with a high concentration of an intercalating agent, which is difficult to correlate with physiological conditions (81, 85, 86). Despite these reservations, the positive supercoiling-driven RFR reaction has been proposed on several occasions when the enzymes responsible for RFR could not be identified.
In conclusion, after the reconstitution of RuvAB-catalyzed RFR in vitro (80) and our recent understanding of how PriA prevents RecG-catalyzed RFR (23, 73–75), two modes of RFR reactions remain documented both in vivo and in vitro in E. coli: RecA-catalyzed strand exchange between leading- and lagging-strand ends (Fig. 3A) and the RuvAB-catalyzed unwinding of a fork, converting it into a HJ (Fig. 3B). In addition, the UvsW helicase from phage T4 was also shown to catalyze RFR both in vivo and in vitro (57).
Formation of dsDNA Ends by Head-to-Tail Fork Collision
Replication forks are naturally arrested in the chromosome terminus at specific Ter sites by the encounter of Ter-Tus complexes (87). The use of ectopic Ter sites, introduced in the chromosome to block replication progression, showed that replication forks arrested at an ectopic Ter-Tus complex remained intact for one generation (28, 88). Chromosome labeling experiments allowed us to conclude that dsDNA ends are formed when the following replication round copied the blocked fork to the end, causing a head-to-tail fork collision (28) (also called fork rear-ending [24]) (Fig. 1D). Head-to-tail collisions were also proposed to account for the observations in cells mutated for a subunit of the replicative DNA polymerase, the holoenzyme polymerase III (89); in a dnaA(Cos) mutant that suffers from hyperinitiation at oriC (90); in cells where hyperinitiation could be induced to a high level at an engineered replication origin (91); and, finally, in a seqA mutant defective for sister chromatid cohesion (25; but see also reference 24). Interestingly, repair by RecBCD-, RecA-, and RuvABC-mediated recombination of the dsDNA ends made by rereplication of forks blocked by a Ter-Tus complex was essential for viability (7, 28), suggesting that homologous recombination allowed the removal of Tus from the DNA. It turned out that replication forks restarting from recombination intermediates differed from the originally arrested forks by their accessibility to the fork-clearing helicase UvrD, which allowed restarting replication forks to progress across the Ter site by displacing the DNA-bound Tus protein (92).
THE GROWTH DEFECT OF recB MUTANT CELLS RESULTS FROM THE FORMATION OF SIGMA-REPLICATING CHROMOSOMES
In addition to DSB repair, RecA is also essential for repair by the recombination of ssDNA gaps and for the SOS response, which is the induction of more than 40 proteins by DNA damage (reviewed in references 93 and 94). Nevertheless, recB cells are less viable than recA cells, suggesting that the second function of RecBCD, dsDNA end degradation, is important for viability (95, 96). In mutants that undergo RFR, RecBCD degrades only a short tail, and DNA degradation is too limited to be detected (27, 46). In contrast, extensive RecBCD-dependent chromosome degradation is observed in the recA single mutant, suggesting that dsDNA ends form in this mutant by a reaction other than RFR (97–99).
Fork Breakage Occurs in Eighteen Percent of Unchallenged Wild-Type Cells per Generation
An important clue to the origin of the low viability of the recBC mutant came from the interesting observation of a deficit of DNA sequences in the chromosome terminus of the recB mutant (100, 101). We explored the reasons for this deficit by a combination of microscopy and marker frequency analyses, in minimal medium to prevent multifork DNA replication. We showed that terminus sequences were lost at the time of cell division, in one daughter cell only, in a division-dependent manner (102) (Fig. 5). Based on the observation that the phenomenon of terminus DNA loss was transmitted to progeny, we proposed and tested the model shown in Fig. 5 (103). In a first step, random replication fork breakage leads to the formation of a sigma-replicating chromosome, the linear and circular parts then segregate to the two cell halves, and, finally, they are separated by terminus DNA cleavage upon septum closure (Fig. 5). One of the daughter cells will never form a colony, as it contains a linear chromosome that is being degraded by nucleases, and the other one, which contains a circular chromosome with a linear tail, undergoes the same reaction again at each following generation (Fig. 5). Using fluorescence microscopy to measure the cleavage of the chromosome terminus, we showed that the frequency of initial replication fork breakage was 18% per cell per generation. As each cell harbors two replication forks, each individual replication fork has a 9% probability of being broken and not reaching the terminus. However, due to the heredity of the phenomenon, the percentage of dead cells in a recB mutant exponential-phase culture amounted to 32% (102).
FIG 5.

Model for terminus DNA loss in the recB mutant. In a first step, a replication fork broken at a random position remains unrepaired in a recB mutant, resulting in the inability to complete one chromosome. The two daughter chromosomes, one truncated and one whole, are linked by the intact replication fork and segregate to the two cell halves. In a second step, the terminus region of the truncated chromosome becomes trapped in the septum and is broken at the dif site during cell division. A nonviable cell with a linear chromosome and a viable cell with a sigma-replicating chromosome are generated. In a third step, the intact replication fork on the sigma-replicating chromosome meets a fork coming from the origin, which extends the small tail by the entire chromosome arm. This leads to the same substrate as the one generated originally by the fork breakage event, except that the end of the chromosome arm results from terminus breakage. Breakage of the new terminus DNA will occur again at the next cell division, generating a nonviable cell with a full linear chromosome and a viable cell with a sigma-replicating chromosome, and the same reaction can take place for several generations. (See reference 103 for a more detailed depiction of these events.) Light blue lines, bacteria; dark blue lines, DNA; large red arrows, DNA breaks. The positions of the replication origin oriC and of the last segregated sequence in the terminus, the dif site opposite oriC, are indicated.
The transmission of sigma-replicating chromosomes to progeny explains why most DSBs in a recB mutant occur in the terminus region, although the original DSBs occur at replication forks (two-ended chromosome DSBs, occurring elsewhere than at forks, would not lead to heritable terminus breakage [103]). As one event of replication fork breakage triggered several rounds of terminus breakage, the model also explained why a high level of replication fork impairment was not observed in the recBC mutant (102, 104). However, while we now know that replication forks break at a frequency of 18% per cell per generation (9% per replication fork), we still do not know the molecular mechanism of fork breakage.
Spontaneous Fork Breakage May Result from the Encounter of a Replication Fork with a Single-Stranded DNA Interruption in a Template Strand
A putative role of RFR in the 18% spontaneous replication fork breakage was tested by microscopy, using the loss of labeled terminus DNA as an indication of chromosome terminus breakage, and by comparing recB with recB ruvAB mutant cells and recA recB with recA recB ruvAB mutant cells. The levels of spontaneous replication fork breakage were identical in ruv mutants and Ruv+ strains (21%), which strongly argues against replication fork reversal (103). Intriguingly, when linear DNA formation was measured by pulsed-field gel electrophoresis (PFGE), the inactivation of ruvAB in a recA recB mutant decreased linear DNA formation 2-fold, suggesting that fork breakage in this mutant occurred in part following RFR (47, 48). However, in our study of terminus DNA loss, ruvAB inactivation only reduced the transmission of chromosome terminus breakage to the subsequent generations (from 84% in recA recB cells to 60% in the recA recB ruvAB mutant), which remains unexplained. It did not affect spontaneous replication fork breakage (103), suggesting that RFR is not involved in fork breakage in wild-type E. coli growing in minimal medium; this presumably results from a high efficiency of the Rep helicase for the removal of protein roadblocks from the path of replication forks.
On the other hand, the fork rear-ending model implies dsDNA end formation by the rereplication of blocked forks, thus one generation after replication fork blockage. Therefore, it is expected to cause a delay in cell division, while no cell division delay or cell elongation was observed prior to terminus DNA loss (103). Thus, the most probable hypothesis remaining is that dsDNA end formation by the replication of preexisting ssDNA interruptions is the principal source of spontaneous double-strand breaks detected in a recB mutant. These events occur in 18% of cells per generation; therefore, on the 4.6-Mb E. coli chromosome, ssDNA interruptions would be present with a frequency of 3.8 × 10−8 per base pair (1 per 26 Mb). Whether these ssDNA interruptions result from the repair of spontaneous DNA damage is presently unknown, but recB mutants are particularly sensitive to oxidative damage, and ssDNA breaks are putative intermediates in oxidative lesion repair (105, 106; reviewed in reference 107).
The repair of broken replication forks is predicted to lead to dimer chromosome formation when fork breakage occurs on the lagging strand but not when it occurs on the leading strand (108). Dimers are resolved to monomers by dif-dif recombination catalyzed by XerCD, and thus, the frequency of the formation of dimers was deduced from the frequency of exchanges between dif sequences (109, 110). Results from these two studies were slightly different but allow an estimation of 2% to 7.6% RecB-dependent dimer formation at each generation (by subtracting RecF-dependent dimers from the 10 to 16% total dimers measured in wild-type cells). These results suggest that fork breakage occurs on both strands, with leading-strand breakage (caused by the encounter of a ssDNA break on the previous lagging strand) occurring more often than lagging-strand breakage.
REPLICATION RESTART PATHWAYS
In E. coli, replication initiation at positions other than the replication origin can take place at inactivated intact replication forks (17), at recombination intermediates (16, 17) (Fig. 1A), and at R-loops in certain specific mutants, a phenomenon called constitutive stable DNA replication (cSDR) (111). An R-loop is a three-arm structure that results from the stable pairing of a ssRNA molecule with one of the two dsDNA strands and displacement of the homologous DNA strand. R-loops are recognized by PriA, as a 3′ RNA end is present at the junction and, as at replication forks and D-loops, one arm is single stranded and two arms are double stranded. oriC-independent replication is affected by mutations in the priA, priB, priC, dnaT, and dnaC genes, alone or in combination (reviewed in references 18–21). DnaC is also required for replication initiation from the replication origin oriC, and accordingly, it catalyzes the loading of DnaB at the chromosome origin in vitro (112, 113). In contrast, PriA, PriB, PriC, and DnaT do not act at oriC but are specific for replication restart. PriA-dependent replication initiation was reconstituted in vitro on D-loops formed by RecA-mediated strand invasion (PriA substrates schematized at the bottom of Fig. 1A) and on naked DNA structures that mimic replication forks (intact replication forks schematized at the top of Fig. 1C). In both situations, this reaction required the sequential action of PriA, PriB, DnaT, and DnaC: PriA targets D-loops or fork structures and promotes the binding of PriB and DnaT, which recruit the DnaC-DnaB complex for the loading of the replicative helicase DnaB on ssDNA (114–117). The similarity of the requirements for replication initiation from D-loops and forked structures in vitro led to the idea that in vivo, restart from both inactivated intact forks and recombination intermediates requires the same set of proteins. As explained below, this may not be the case.
Replication Restarts Mainly from Inactivated Intact Forks in Wild-Type Untreated Cells
A priA mutant does not propagate in rich medium, and it grows slowly on minimum medium, while a priB priC double mutant is dead (17, 118). In contrast, all recombination mutants are viable on rich and on minimal media. The reduced plating efficiency of recA and recB mutants is not as severe as that of a priA mutant grown on minimal medium (or the complete loss of viability of the priB priC double mutant). Furthermore, the reduced plating efficiency of these recombination mutants results in large part from their own defect in homologous recombination, which triggers terminus DSBs (Fig. 5) (103). Thus, the low viability of replication restart mutants cannot be explained by a lack of replication initiation from recombination intermediates and suggests that they fail to restart blocked forks that have not recombined. Here, we call such replication forks “inactivated intact forks,” whose DNA remains intact although replication elongation is arrested. The low viability of replication restart mutants compared to recombination mutants thus suggests that these inactivated intact forks are the main substrate for replication restart proteins in untreated wild-type cells.
Epistatic interactions of mutations that inactivate the priA, priB, or priC gene were used to define replication restart pathways in otherwise wild-type cells (originally proposed in reference 119; reviewed in references 18 and 20) (Table 1). The main replication restart pathway requires PriA, DnaT, DnaC, and either PriB or PriC (pathway 1A) (called PriA-PriB and PriA-PriC below) (Table 1). The alternative PriC-DnaC pathway is poorly active, since it supports the viability of the priA mutant only poorly; other pathways are activated by dnaC mutations (Table 1). The PriA-PriC pathway (not reconstituted in vitro) is as efficient as the PriA-PriB pathway in wild-type cells, since the single priB mutant has no deleterious phenotype. However, the PriA-PriC pathway requires the helicase activity of PriA, specifically inactivated in a priA300 mutant (22, 120). Indeed, the individual priB, priC, and priA300 mutants are fully viable, while the combination of priB inactivation with priC or priA300 strongly affects viability, thereby suggesting that replication restart from inactivated intact forks is affected in these double mutants (118, 120).
TABLE 1.
Model of replication restart pathwaysa
| Pathway subcategory | Description |
|
|---|---|---|
| Restart from inactivated intact replication forks (essential for viability) (pathway 1) | Restart from D-loops, reversed replication forks, and R-loops (pathway 2) | |
| A | Wild-type pathways, PriA-PriB or PriC-DnaT-(DnaC) | Wild-type pathway, PriA-PriB-DnaT-DnaC |
| B | Minor pathway, PriC-DnaC | Inactive |
| C | Suppressor pathway C, PriB or PriC-DnaC809 | Inactive |
| D | Mutant dnaC1331 pathway, PriA-PriC-DnaC1331 | Inactive |
| E | Suppressor pathways E, (PriA)-DnaC824 | Suppressor pathway E, (PriA)-DnaC824 |
| F | Suppressor pathway F, DnaC809,820 | Suppressor pathway F, (PriB)-DnaC809,820 |
Replication restart pathways were originally defined previously (119; later modified in reference 18). We propose here that different pathways operate depending on whether replication restarts from inactivated intact forks (pathway 1) or from D-loops, reversed forks, or R-loops (pathway 2). For pathway A, in wild-type cells, replication can restart from inactivated intact forks via either of two pathways: PriA-PriB-DnaT or PriA-PriC-DnaT. DnaC is not needed for replication restarts from inactivated intact forks in a dnaC2(Ts) mutant and a dnaC28(Ts) mutant at a restrictive temperature. In contrast, both PriB and DnaC are essential for restart from D-loops, reversed forks, and R-loops (together with the helicase function of PriA [not shown here]). For pathway B, in the absence of PriA, a minor PriC-DnaC pathway allows restart from inactivated intact forks but not from D-loops, reversed forks, and R-loops. For pathway C, in the dnaC809 mutant, this pathway is strongly activated; it can use either PriB or PriC but does not need PriA and DnaT. For pathway D, the dnaC1331 mutation blocks the PriA-PriB replication restart pathway, leaving only the PriA-PriC pathway at inactivated intact forks (DnaT was not tested). For pathway E, the dnaC824 mutation bypasses the need for PriB and PriC and only partially bypasses PriA at inactivated intact replication forks (DnaT was not tested). It bypasses PriB and PriC at D-loops and reversed forks (PriA and R-loops were not tested). For pathway F, the dnaC809 dnaC820 (dnaC809,820) mutation bypasses all other replication restart proteins at inactivated intact forks as at D-loops and reversed forks but needs PriB for replication initiation at R-loops. The inactivation of proteins shown in parentheses had nearly no effect (DnaC in pathway 1A) or had a partial effect (PriA in pathway 1E and 2E and PriB in pathway 2F).
Replication Restart from Inactivated Intact Forks and Broken Forks Has Different Protein Requirements
The operation of these replication restart pathways can be deduced from the analysis of replication mutants in which PriA-mediated restart is essential for viability: (i) restart from inactivated intact replication forks is essential in a gyrB(Ts) mutant, which does not require homologous recombination for growth (83); (ii) restart from recombination intermediates is essential in a dam mutant, which requires homologous recombination for viability (121); (iii) restart after RFR is essential in the rep and holD mutants, which require only RecBC for viability (119, 122); and (iv) restart from R-loops is essential for the viability of a dnaA(Ts) rnh mutant, which uses R-loops for replication initiation (111, 123, 124). PriB is essential for the growth of dam, rep, holD, and dnaA(Ts) rnh cells, while gyrB(Ts) priB cells are viable and affected for growth only in rich medium (83, 119, 122–124). Therefore, the PriA-PriB pathway is essential for replication restart from D-loops, reversed forks, and R-loops but not for replication restart from inactivated intact forks. Similarly, the priA300 mutation that inactivates PriA helicase activity barely affected the viability of gyrB(Ts) cells, whereas rep priA300 and holD priA300 mutants showed a strongly reduced colony size, and dnaA rnh priA300 cells were dead (83, 119, 122, 123). These observations suggest that replication restart from D-loops, reversed forks, and R-loops requires PriB and the helicase function of PriA (pathway 2A) (Table 1), while restart from inactivated intact forks does not (pathway 1A) (Table 1). Nevertheless, the requirement for PriB and PriA helicase activity for homologous recombination is not absolute, since priB and priA300 mutants individually are not deficient for P1 transduction and are not sensitive to low UV doses, in contrast to the priB priA300 double mutant (120). There is either a qualitative difference between recombination after P1 transduction and in mutant strains that require recombination for viability (the PriA target is for some reason not exactly the same) or a quantitative difference (priB and priA300 mutants can manage a few recombination events, such as during P1 transduction or at low UV doses, but cannot manage several recombination events per cell cycle as in dam, rep, or holD mutants).
Studies of dnaC mutants further support the idea that restart from D-loops, reversed forks, and R-loops does not require the same functions as restart from inactivated intact replication forks. Two dnaC(Ts) mutations inactivated replication initiation from oriC at a high temperature while allowing most ongoing replication rounds to finish, which led to their use in replication synchronization experiments [dnaC2(Ts) and dnaC28(Ts)] (125). This suggests that either a mutated DnaC protein with residual replication restart activity was synthesized in these dnaC(Ts) mutants at a high temperature or DnaC was not essential for replication restart from inactivated intact forks. To determine the proportion of cells that need intact DnaC protein for replication restart, these dnaC(Ts) mutations were used to synchronize replication in a cell population, and the proportion of chromosomes that were unable to complete a single round of replication was measured by flow cytometry (126). Eighteen percent of chromosomes remained partially replicated in this mutant, suggesting that replication was interrupted and did not restart in 18% of cells at each generation in these dnaC(Ts) mutants. This is similar to the percentage of cells that suffer fork breakage (103) (Fig. 5), which suggests that DnaC may be required in wild-type cells only for replication initiation at the origin and for replication restart from recombination intermediates (pathway 2A) (Table 1) (126) [note that the dnaC2(Ts) mutant also carries a dnaT mutation, but similar results were obtained with a dnaC28(Ts) mutant, excluding a role for the dnaT mutation]. Accordingly, in the rep mutant, where blocked forks are reversed, almost the whole cell population was unable to complete a single round of replication in a dnaC2(Ts) mutant, indicating that reversed replication forks required wild-type DnaC for replication restart (126). In two other studies, replication restart occurred in the dnaC2(Ts) mutant after induced replication arrest. First, when replication fork arrest was increased by a gyrase ATPase inhibitor (which blocks replication without causing fork breakage or reversal [83]), no chromosomes were fully replicated in a priA mutant, but 60% of chromosomes were fully replicated in a dnaC2(Ts) mutant (127). Second, in a study where replication forks were blocked by the encounter of a series of repressor-operator complexes, the removal of these obstacles allowed 61% of replication forks to restart in a dnaC2(Ts) mutant, versus 81% in the wild type and only 17% in a dnaB(Ts) mutant (128). Note that direct replication restart was quantified after a short time of replication arrest, while prolonged replication inhibition by these protein roadblocks led to RFR (128), as previously observed (56).
The behavior of the dnaC2(Ts) mutation points to a pivotal role of DnaC function in differentiating between inactivated intact forks and D-loops, reversed forks, or R-loops. This idea is supported by the properties of dnaC point mutants that activate PriA-independent pathways of replication restart or affect PriA-dependent replication restart (Table 1). dnaC809 is a gain-of-function mutation that fully restores the viability of priA and gyrB(Ts) priA mutants, therefore allowing replication restart from inactivated intact forks in the absence of PriA (17, 83). In contrast, DnaC809 does not bypass the need for PriA in cells that undergo RFR or initiate replication from R-loops (119, 122, 123), although it restores P1 transduction in the priA mutant (17) (possibly because of a qualitative or a quantitative difference in the needs for replication restart between mutants that require it for viability and during P1 transduction [see above]). Therefore, the dnaC809 mutation allows replication restart in the absence of PriA from inactivated intact forks but not from D-loops, reversed forks, or R-loops, which supports the idea that the role of DnaC is not the same in these different situations (pathway 1C) (Table 1).
The dnaC1331 mutation was isolated as affecting the replication of plasmids that initiate replication from R-loops (Table 1) (pathway 1D) (129). It does not affect the viability of otherwise wild-type cells, which indicates that it does not affect restart from inactivated intact forks. However, it is colethal with rep and dam mutations, indicating that, in addition to restart from R-loops, it also affects restart from reversed forks and D-loops (124). This phenotype is similar to that of the priB mutant described above, and accordingly, dnaC1331 is also strongly deleterious in combination with a priA300 mutation (124). In conclusion, dnaC2(Ts), dnaC28(Ts), dnaC1331, and dnaC809 are all dissociation-of-function mutations. dnaC2(Ts) and dnaC28(Ts) mutations inactivate replication initiation from the origin oriC and replication restart from D-loops at a high temperature while allowing replication restart from inactivated intact forks. dnaC1331 inactivates replication restart from D-loops, reversed forks, and R-loops while allowing replication initiation at the origin and replication restart from inactivated intact forks. dnaC809 is a gain-of-function mutation that allows replication restart in the absence of PriA from inactivated intact forks but not from D-loops, reversed forks, or R-loops. It should be noted that screening for suppressor mutations in a priB priC or rep priB mutant yielded, as expected, dnaC alleles that did not show a dissociation-of-function phenotype but similarly bypassed replication restart proteins at inactivated intact forks and at D-loops, reversed forks, or R-loops (dnaC824 and dnaC809 dnaC820 [dnaC809,820]) (pathways 1E and 1F) (Table 1) (reviewed in references 18 and 20).
We can speculate that the PriA PriC pathway is active only at intact inactivated forks (Table 1) (pathway 1A) and may not require an intact DnaC protein because it reactivates a DnaB helicase left on DNA after replication arrest. DnaB forms a hexameric complex that encircles the lagging-strand template, and to date, no mechanism of removal of the DnaB helicase from the DNA has been described. Furthermore, in vitro, DnaB progresses very slowly on DNA in the absence of the holoenzyme polymerase III (HE Pol III) (130), and a slight progression of DnaB at inactivated intact forks would render both DNA template strands single stranded, creating the preferred substrate for PriC, or might allow the progression of the lagging-strand end beyond the leading-strand end, accounting for the need for PriA helicase activity for replication restart by the PriA-PriC pathway (116, 131). If DnaB remains bound to inactivated intact forks, it may allow PriA-dependent replication restart in a dnaC2(Ts) mutant (Table 1) (pathway 1A) and PriA- or PriB-independent restart in a dnaC809 or dnaC1331 mutant, respectively (pathways 1C and 1D) (Table 1). In vitro, a preloaded DnaB helicase is sufficient for replication initiation in the presence of primase and HE Pol III (132, 133). We hypothesize that in vivo, this reaction would need some replication restart proteins.
Three Other Observations Support the Idea that Different Pathways May Restart Replication at Inactivated Intact Forks and at D-Loops, Reversed Forks, or R-Loops
First, in a microscopy study of the priA mutant, two types of cells could be observed: 84% of cells looked like wild-type cells, while 16% were elongated, with a poorly partitioned chromosome (134, 135). This phenotype was eliminated by mutation of either recA or recB, strongly suggesting that the cells with poorly partitioned chromosomes might have suffered replication fork breakage (in a priA mutant, homologous recombination will be blocked after the formation of a D-loop, owing to the lack of replication restart [Fig. 1A]). The observation of 84% normal cells was surprising considering the low viability of the priA mutant. This experiment suggests that blocking replication restart from RecA-made D-loops has severe consequences for chromosome partitioning, in contrast to blocking replication restart from inactivated intact forks.
Second, in contrast to the major fork-clearing helicase Rep, which interacts with DnaB and presumably acts directly at intact inactivated forks (45), two observations suggest that the alternative fork-clearing helicase UvrD can access blocked replication forks only after recombination or reversal. UvrD is present and active in the rep mutant, yet at the same time, fork reversal occurs in the rep mutant, showing that UvrD can replace Rep only after RFR has taken place (27, 43). UvrD is also essential for replication restart at forks blocked by an ectopic replication terminator, but again, it does not remove the Tus protein directly from blocked forks, since homologous recombination is required for its action (92). Further studies showed that yet another accessory helicase, called DinG, acts with UvrD at restarting replication forks blocked by RNA polymerases in a rep mutant (43). This raised the proposal that only replication forks reassembled after reversal or homologous recombination may be specifically accessible to certain fork-clearing proteins.
Third, yet another helicase, RecG, acts at replication forks; moreover, suppressors of the recG mutant defects mapped to priA (136). However, the nature of the interplay between RecG and PriA remained unclear for a long time (137). Recently, marker frequency analysis by genomic sequencing and analysis of RecA binding by chromatin immunoprecipitation sequencing (ChIP-seq) were carried out with a recG mutant and revealed DNA synthesis proceeding in the direction opposite that predicted for the repair of a DSB (Fig. 4). This reaction, called “reverse restart,” was observed specifically in the absence of RecG, at a DSB generated at the site of a long DNA palindrome cleaved by SbcCD, and between dsDNA ends located at the termination sites TerA and TerB in the terminus of the chromosome (23). Based on the biochemical demonstration of the correct loading of PriA at a replication fork substrate in the presence of RecG and the prevention of RecG-mediated replication fork reversal by PriA in vitro (73, 74), a specific role for RecG in reverse restart was proposed (Fig. 4) (23, 75). In the absence of RecG, PriA could be loaded incorrectly at a replication fork or a D-loop generated by recombination at the site of a DSB and at similar structures generated at Ter sites in the chromosome terminus (57). This reverse-restart reaction explains the overreplication previously observed in the absence of RecG following UV irradiation and in the chromosome terminus (70, 100, 101, 138). In agreement with the reverse-restart model and with the idea that PriB and the helicase function of PriA are required for de novo DnaB loading (see above) (Table 1), these two functions were required for overreplication in the chromosome terminus of the recG mutant (100).
Several observations suggest that by driving the progression of replication forks from the terminus to the origin, reverse restart should cause a growth defect (56, 139). However, in the recG mutant, the proportion of cells with a growth defect is close to the proportion of cells that suffer fork breakage, around 15% (140, 141). This is in agreement with the idea that RecG acts during DSB repair (23) but not at inactivated intact forks. We propose that RecG is not needed for the proper binding of PriA at intact inactivated replication forks because DnaB is already present and in the correct position for restart.
CONCLUSIONS AND PERSPECTIVES
We demonstrated recently that replication fork breakage is, as suspected, the major source of spontaneous DSBs in E. coli and that preventing broken fork repair triggers heritable cell division-dependent DSBs in the chromosome terminus (103). The molecular mechanism of DSB formation in the terminus during cell division remains to be identified. Our results lead us to propose that replication fork breakage results mainly from the encounter of nicks or gaps in the template strands. These might form during the repair of oxidative DNA damage, but this hypothesis needs further investigation. The observation that similar proportions of E. coli cells undergo replication fork breakage (103) and require helicase reloading to complete replication (126) leads us to speculate that the loading of a new helicase is needed only after replication fork breakage. Consequently, we propose that when the DNA at inactivated replication forks is intact, the replication restart proteins may reactivate the helicase left on DNA after replication arrest, by promoting the binding of a new replisome to the abandoned helicase. This new proposal will of course need to be explored in the future to be validated.
ACKNOWLEDGMENTS
Work in the laboratory of B.M. is supported by Agence Nationale de la Recherche (ANR) grant 11 BSVS5 006 01. Work in the laboratory of D.R.F.L. is supported by grant MR/M019160/1 from the Medical Research Council (UK). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Bierne H, Ehrlich SD, Michel B. 1991. The replication termination signal terB of the Escherichia coli chromosome is a deletion hot spot. EMBO J 10:2699–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bierne H, Ehrlich SD, Michel B. 1997. Deletions at stalled replication forks occur by two different pathways. EMBO J 16:3332–3340. doi: 10.1093/emboj/16.11.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vilette D, Uzest M, Ehrlich SD, Michel B. 1992. DNA transcription and repressor binding affect deletion formation in Escherichia coli plasmids. EMBO J 11:3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vilette D, Ehrlich SD, Michel B. 1995. Transcription-induced deletions in Escherichia coli plasmids. Mol Microbiol 17:493–504. doi: 10.1111/j.1365-2958.1995.mmi_17030493.x. [DOI] [PubMed] [Google Scholar]
- 5.Michel B, Ehrlich SD, Uzest M. 1997. DNA double-strand breaks caused by replication arrest. EMBO J 16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horiuchi T, Fujimura Y, Nishitani H, Kobayashi T, Hidaka M. 1994. DNA replication fork blocked at the Ter site may be an entrance for the RecBCD enzyme into duplex DNA. J Bacteriol 176:4656–4663. doi: 10.1128/jb.176.15.4656-4663.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horiuchi T, Fujimura Y. 1995. Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J Bacteriol 177:783–791. doi: 10.1128/jb.177.3.783-791.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flores-Rozas H, Kolodner RD. 2000. Links between replication, recombination and genome instability in eukaryotes. Trends Biochem Sci 25:196–200. doi: 10.1016/S0968-0004(00)01568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolinjivadi AM, Sannino V, de Antoni A, Techer H, Baldi G, Costanzo V. 2017. Moonlighting at replication forks—a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett 591:1083–1100. doi: 10.1002/1873-3468.12556. [DOI] [PubMed] [Google Scholar]
- 10.Branzei D, Szakal B. 2017. Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 52:381–394. doi: 10.1080/10409238.2017.1304355. [DOI] [PubMed] [Google Scholar]
- 11.Quinet A, Lemacon D, Vindigni A. 2017. Replication fork reversal: players and guardians. Mol Cell 68:830–833. doi: 10.1016/j.molcel.2017.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neelsen KJ, Lopes M. 2015. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- 13.Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michel B, Leach D. 11 September 2012, posting date. Homologous recombination—enzymes and pathways. EcoSal Plus 2012 doi: 10.1128/ecosalplus.7.2.7. [DOI] [PubMed] [Google Scholar]
- 15.Smith GR. 2012. How RecBCD enzyme and Chi promote DNA break repair and recombination: a molecular biologist's view. Microbiol Mol Biol Rev 76:217–228. doi: 10.1128/MMBR.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kogoma T, Cadwell GW, Barnard KG, Asai T. 1996. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J Bacteriol 178:1258–1264. doi: 10.1128/jb.178.5.1258-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandler SJ, Samra HS, Clark AJ. 1996. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics 143:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michel B, Sandler SJ. 2017. Replication restart in bacteria. J Bacteriol 199:e00102-17. doi: 10.1128/JB.00102-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabbai CB, Marians KJ. 2010. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Windgassen TA, Wessel SR, Bhattacharyya B, Keck JL. 2018. Mechanisms of bacterial DNA replication restart. Nucleic Acids Res 46:504–519. doi: 10.1093/nar/gkx1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marians KJ. 3 January 2018. Lesion bypass and the reactivation of stalled replication forks. Annu Rev Biochem doi: 10.1146/annurev-biochem-062917-011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zavitz KH, Marians KJ. 1992. ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J Biol Chem 267:6933–6940. [PubMed] [Google Scholar]
- 23.Azeroglu B, Mawer JS, Cockram CA, White MA, Hasan AM, Filatenkova M, Leach DR. 2016. RecG directs DNA synthesis during double-strand break repair. PLoS Genet 12:e1005799. doi: 10.1371/journal.pgen.1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rotman E, Khan SR, Kouzminova E, Kuzminov A. 2014. Replication fork inhibition in seqA mutants of Escherichia coli triggers replication fork breakage. Mol Microbiol 93:50–64. doi: 10.1111/mmi.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen IB, Helgesen E, Flatten I, Fossum-Raunehaug S, Skarstad K. 2017. SeqA structures behind Escherichia coli replication forks affect replication elongation and restart mechanisms. Nucleic Acids Res 45:6471–6485. doi: 10.1093/nar/gkx263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzminov A. 1995. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol 16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 27.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. 1998. RuvAB acts at arrested replication forks. Cell 95:419–430. doi: 10.1016/S0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 28.Bidnenko V, Ehrlich SD, Michel B. 2002. Replication fork collapse at replication terminator sequences. EMBO J 21:3898–3907. doi: 10.1093/emboj/cdf369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuzminov A. 2001. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A 98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arcangioli B. 1998. A site- and strand-specific DNA break confers asymmetric switching potential in fission yeast. EMBO J 17:4503–4510. doi: 10.1093/emboj/17.15.4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. 2000. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 20:3977–3987. doi: 10.1128/MCB.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kouzminova EA, Rotman E, Macomber L, Zhang J, Kuzminov A. 2004. RecA-dependent mutants in Escherichia coli reveal strategies to avoid chromosomal fragmentation. Proc Natl Acad Sci U S A 101:16262–16267. doi: 10.1073/pnas.0405943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradshaw JS, Kuzminov A. 2003. RdgB acts to avoid chromosome fragmentation in Escherichia coli. Mol Microbiol 48:1711–1725. doi: 10.1046/j.1365-2958.2003.03540.x. [DOI] [PubMed] [Google Scholar]
- 34.Lukas L, Kuzminov A. 2006. Chromosomal fragmentation is the major consequence of the rdgB defect in Escherichia coli. Genetics 172:1359–1362. doi: 10.1534/genetics.105.051144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budke B, Kuzminov A. 2010. Production of clastogenic DNA precursors by the nucleotide metabolism in Escherichia coli. Mol Microbiol 75:230–245. doi: 10.1111/j.1365-2958.2009.06994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kouzminova EA, Kuzminov A. 2004. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol Microbiol 51:1279–1295. doi: 10.1111/j.1365-2958.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- 37.Kouzminova EA, Kuzminov A. 2006. Fragmentation of replicating chromosomes triggered by uracil in DNA. J Mol Biol 355:20–33. doi: 10.1016/j.jmb.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 38.Kouzminova EA, Kuzminov A. 2008. Patterns of chromosomal fragmentation due to uracil-DNA incorporation reveal a novel mechanism of replication-dependent double-stranded breaks. Mol Microbiol 68:202–215. doi: 10.1111/j.1365-2958.2008.06149.x. [DOI] [PubMed] [Google Scholar]
- 39.Amado L, Kuzminov A. 2006. The replication intermediates in Escherichia coli are not the product of DNA processing or uracil excision. J Biol Chem 281:22635–22646. doi: 10.1074/jbc.M602320200. [DOI] [PubMed] [Google Scholar]
- 40.Amado L, Kuzminov A. 2013. Low-molecular-weight DNA replication intermediates in Escherichia coli: mechanism of formation and strand specificity. J Mol Biol 425:4177–4191. doi: 10.1016/j.jmb.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kouzminova EA, Kuzminov A. 2012. Chromosome demise in the wake of ligase-deficient replication. Mol Microbiol 84:1079–1096. doi: 10.1111/j.1365-2958.2012.08076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bidnenko V, Seigneur M, Penel-Colin M, Bouton MF, Ehrlich SD, Michel B. 1999. sbcS sbcC null mutations allow RecF-mediated repair of arrested replication forks in rep recBC mutants. Mol Microbiol 33:846–857. doi: 10.1046/j.1365-2958.1999.01532.x. [DOI] [PubMed] [Google Scholar]
- 43.Boubakri H, de Septenville AL, Viguera E, Michel B. 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J 29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baharoglu Z, Lestini R, Duigou S, Michel B. 2010. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol Microbiol 77:324–336. doi: 10.1111/j.1365-2958.2010.07208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkinson J, Gupta MK, Rudolph CJ, Bell H, Lloyd RG, McGlynn P. 2011. Localization of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Res 39:949–957. doi: 10.1093/nar/gkq889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flores MJ, Bierne H, Ehrlich SD, Michel B. 2001. Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. EMBO J 20:619–629. doi: 10.1093/emboj/20.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grompone G, Seigneur M, Ehrlich SD, Michel B. 2002. Replication fork reversal in DNA polymerase III mutants of Escherichia coli: a role for the beta clamp. Mol Microbiol 44:1331–1339. doi: 10.1046/j.1365-2958.2002.02962.x. [DOI] [PubMed] [Google Scholar]
- 48.Grompone G, Ehrlich D, Michel B. 2004. Cells defective for replication restart undergo replication fork reversal. EMBO Rep 5:607–612. doi: 10.1038/sj.embor.7400167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guarino E, Jimenez-Sanchez A, Guzman EC. 2007. Defective ribonucleoside diphosphate reductase impairs replication fork progression in Escherichia coli. J Bacteriol 189:3496–3501. doi: 10.1128/JB.01632-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guarino E, Salguero I, Jimenez-Sanchez A, Guzman EC. 2007. Double-strand break generation under deoxyribonucleotide starvation in Escherichia coli. J Bacteriol 189:5782–5786. doi: 10.1128/JB.00411-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan SR, Kuzminov A. 2012. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J Biol Chem 287:6250–6265. doi: 10.1074/jbc.M111.322990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sutherland JH, Tse-Dinh YC. 2010. Analysis of RuvABC and RecG involvement in the Escherichia coli response to the covalent topoisomerase-DNA complex. J Bacteriol 192:4445–4451. doi: 10.1128/JB.00350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinha AK, Pavankumar TL, Kamisetty S, Mittal P, Ray MK. 2013. Replication arrest is a major threat to growth at low temperature in Antarctic Pseudomonas syringae Lz4W. Mol Microbiol 89:792–810. doi: 10.1111/mmi.12315. [DOI] [PubMed] [Google Scholar]
- 54.Schapiro JM, Libby SJ, Fang FC. 2003. Inhibition of bacterial DNA replication by zinc mobilization during nitrosative stress. Proc Natl Acad Sci U S A 100:8496–8501. doi: 10.1073/pnas.1033133100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, Moon PB, van Knippenberg IC, Cadman CJ, Dillingham MS, Lloyd RG, McGlynn P. 2009. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell 36:654–666. doi: 10.1016/j.molcel.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Septenville AL, Duigou S, Boubakri H, Michel B. 2012. Replication fork reversal after replication-transcription collision. PLoS Genet 8:e1002622. doi: 10.1371/journal.pgen.1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long DT, Kreuzer KN. 2009. Fork regression is an active helicase-driven pathway in bacteriophage T4. EMBO Rep 10:394–399. doi: 10.1038/embor.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Higgins NP, Kato K, Strauss B. 1976. A model for replication repair in mammalian cells. J Mol Biol 101:417–425. doi: 10.1016/0022-2836(76)90156-X. [DOI] [PubMed] [Google Scholar]
- 59.Tatsumi K, Strauss B. 1978. Production of DNA bifilarly substituted with bromodeoxyuridine in the first round of synthesis: branch migration during isolation of cellular DNA. Nucleic Acids Res 5:331–347. doi: 10.1093/nar/5.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morgan AR, Severini A. 1990. Interconversion of replication and recombination structures: implications for terminal repeats and concatemers. J Theor Biol 144:195–202. doi: 10.1016/S0022-5193(05)80318-2. [DOI] [PubMed] [Google Scholar]
- 61.Seigneur M, Ehrlich SD, Michel B. 2000. RuvABC-dependent double-strand breaks in dnaBts mutants require RecA. Mol Microbiol 38:565–574. doi: 10.1046/j.1365-2958.2000.02152.x. [DOI] [PubMed] [Google Scholar]
- 62.Robu ME, Inman RB, Cox MM. 2001. RecA protein promotes the regression of stalled replication forks in vitro. Proc Natl Acad Sci U S A 98:8211–8218. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGlynn P, Lloyd RG. 2000. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 65.McGlynn P, Lloyd RG. 2001. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci U S A 98:8227–8234. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McGlynn P, Lloyd RG, Marians KJ. 2001. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc Natl Acad Sci U S A 98:8235–8240. doi: 10.1073/pnas.121007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ralf C, Hickson ID, Wu L. 2006. The Bloom's syndrome helicase can promote the regression of a model replication fork. J Biol Chem 281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- 68.Donaldson JR, Courcelle CT, Courcelle J. 2004. RuvAB and RecG are not essential for the recovery of DNA synthesis following UV-induced DNA damage in Escherichia coli. Genetics 166:1631–1640. doi: 10.1534/genetics.166.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asai T, Sommer S, Bailone A, Kogoma T. 1993. Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. EMBO J 12:3287–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rudolph CJ, Upton AL, Harris L, Lloyd RG. 2009. Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73:352–366. doi: 10.1111/j.1365-2958.2009.06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baharoglu Z, Petranovic M, Flores MJ, Michel B. 2006. RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J 25:596–604. doi: 10.1038/sj.emboj.7600941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mawer JS, Leach DR. 2014. Branch migration prevents DNA loss during double-strand break repair. PLoS Genet 10:e1004485. doi: 10.1371/journal.pgen.1004485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka T, Masai H. 2006. Stabilization of a stalled replication fork by concerted actions of two helicases. J Biol Chem 281:3484–3493. doi: 10.1074/jbc.M510979200. [DOI] [PubMed] [Google Scholar]
- 74.Tanaka T, Mizukoshi T, Sasaki K, Kohda D, Masai H. 2007. Escherichia coli PriA protein, two modes of DNA binding and activation of ATP hydrolysis. J Biol Chem 282:19917–19927. doi: 10.1074/jbc.M701848200. [DOI] [PubMed] [Google Scholar]
- 75.Azeroglu B, Leach DRF. 2017. RecG controls DNA amplification at double-strand breaks and arrested replication forks. FEBS Lett 591:1101–1113. doi: 10.1002/1873-3468.12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Atkinson J, McGlynn P. 2009. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res 37:3475–3492. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baharoglu Z, Bradley AS, Le Masson M, Tsaneva I, Michel B. 2008. ruvA mutants that resolve Holliday junctions but do not reverse replication forks. PLoS Genet 4:e1000012. doi: 10.1371/journal.pgen.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Masson M, Baharoglu Z, Michel B. 2008. ruvA and ruvB mutants specifically impaired for replication fork reversal. Mol Microbiol 70:537–548. doi: 10.1111/j.1365-2958.2008.06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bradley AS, Baharoglu Z, Niewiarowski A, Michel B, Tsaneva IR. 2011. Formation of a stable RuvA protein double tetramer is required for efficient branch migration in vitro and for replication fork reversal in vivo. J Biol Chem 286:22372–22383. doi: 10.1074/jbc.M111.233908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gupta S, Yeeles JT, Marians KJ. 2014. Regression of replication forks stalled by leading-strand template damage. I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the Holliday junctions formed by RecG preferentially. J Biol Chem 289:28376–28387. doi: 10.1074/jbc.M114.587881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Postow L, Ullsperger C, Keller RW, Bustamante C, Vologodskii AV, Cozzarelli NR. 2001. Positive torsional strain causes the formation of a four-way junction at replication forks. J Biol Chem 276:2790–2796. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- 82.Khodursky AB, Peter BJ, Schmidt MB, DeRisi J, Botstein D, Brown PO, Cozzarelli NR. 2000. Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc Natl Acad Sci U S A 97:9419–9424. doi: 10.1073/pnas.97.17.9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grompone G, Ehrlich SD, Michel B. 2003. Replication restart in gyrB Escherichia coli mutants. Mol Microbiol 48:845–854. doi: 10.1046/j.1365-2958.2003.03480.x. [DOI] [PubMed] [Google Scholar]
- 84.Grompone G, Bidnenko V, Ehrlich SD, Michel B. 2004. PriA is essential for viability of the Escherichia coli topoisomerase IV parE10(Ts) mutant. J Bacteriol 186:1197–1199. doi: 10.1128/JB.186.4.1197-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fierro-Fernandez M, Hernandez P, Krimer DB, Schvartzman JB. 2007. Replication fork reversal occurs spontaneously after digestion but is constrained in supercoiled domains. J Biol Chem 282:18190–18196. doi: 10.1074/jbc.M701559200. [DOI] [PubMed] [Google Scholar]
- 86.Fierro-Fernandez M, Hernandez P, Krimer DB, Stasiak A, Schvartzman JB. 2007. Topological locking restrains replication fork reversal. Proc Natl Acad Sci U S A 104:1500–1505. doi: 10.1073/pnas.0609204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neylon C, Kralicek AV, Hill TM, Dixon NE. 2005. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiol Mol Biol Rev 69:501–526. doi: 10.1128/MMBR.69.3.501-526.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sharma B, Hill TM. 1995. Insertion of inverted Ter sites into the terminus region of the Escherichia coli chromosome delays completion of DNA replication and disrupts the cell cycle. Mol Microbiol 18:45–61. doi: 10.1111/j.1365-2958.1995.mmi_18010045.x. [DOI] [PubMed] [Google Scholar]
- 89.Nordman J, Skovgaard O, Wright A. 2007. A novel class of mutations that affect DNA replication in E. coli. Mol Microbiol 64:125–138. doi: 10.1111/j.1365-2958.2007.05651.x. [DOI] [PubMed] [Google Scholar]
- 90.Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM. 2004. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol Microbiol 51:349–358. doi: 10.1046/j.1365-2958.2003.03842.x. [DOI] [PubMed] [Google Scholar]
- 91.Khan SR, Mahaseth T, Kouzminova EA, Cronan GE, Kuzminov A. 2016. Static and dynamic factors limit chromosomal replication complexity in Escherichia coli, avoiding dangers of runaway overreplication. Genetics 202:945–960. doi: 10.1534/genetics.115.184697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bidnenko V, Lestini R, Michel B. 2006. The Escherichia coli UvrD helicase is essential for Tus removal during recombination-dependent replication restart from Ter sites. Mol Microbiol 62:382–396. doi: 10.1111/j.1365-2958.2006.05382.x. [DOI] [PubMed] [Google Scholar]
- 93.Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Michel B. 2005. After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol 3:e255. doi: 10.1371/journal.pbio.0030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Capaldo FN, Ramsey G, Barbour SD. 1974. Analysis of the growth of recombination-deficient strains of Escherichia coli K-12. J Bacteriol 118:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miranda A, Kuzminov A. 2003. Chromosomal lesion suppression and removal in Escherichia coli via linear DNA degradation. Genetics 163:1255–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Willetts NS, Clark AJ. 1969. Characteristics of some multiply recombination-deficient strains of Escherichia coli. J Bacteriol 100:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Skarstad K, Boye E. 1993. Degradation of individual chromosomes in RecA mutants of Escherichia coli. J Bacteriol 175:5505–5509. doi: 10.1128/jb.175.17.5505-5509.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kuzminov A, Stahl FW. 1997. Stability of linear DNA in recA mutant Escherichia coli cells reflects ongoing chromosomal DNA degradation. J Bacteriol 179:880–888. doi: 10.1128/jb.179.3.880-888.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rudolph CJ, Upton AL, Stockum A, Nieduszynski CA, Lloyd RG. 2013. Avoiding chromosome pathology when replication forks collide. Nature 500:608–611. doi: 10.1038/nature12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wendel BM, Courcelle CT, Courcelle J. 2014. Completion of DNA replication in Escherichia coli. Proc Natl Acad Sci U S A 111:16454–16459. doi: 10.1073/pnas.1415025111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sinha AK, Durand A, Desfontaines JM, Iurchenko I, Auger H, Leach DRF, Barre FX, Michel B. 2017. Division-induced DNA double strand breaks in the chromosome terminus region of Escherichia coli lacking RecBCD DNA repair enzyme. PLoS Genet 13:e1006895. doi: 10.1371/journal.pgen.1006895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sinha AK, Possoz C, Durand A, Desfontaines JM, Barre FX, Leach DRF, Michel B. 2018. Broken replication forks trigger heritable DNA breaks in the terminus of a circular chromosome. PLoS Genet 14:e1007256. doi: 10.1371/journal.pgen.1007256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Syeda AH, Atkinson J, Lloyd RG, McGlynn P. 2016. The balance between recombination enzymes and accessory replicative helicases in facilitating genome duplication. Genes (Basel) 7:E42. doi: 10.3390/genes7080042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mackey BM, Seymour DA. 1987. The effect of catalase on recovery of heat-injured DNA-repair mutants of Escherichia coli. J Gen Microbiol 133:1601–1610. [DOI] [PubMed] [Google Scholar]
- 106.Morimyo M. 1982. Anaerobic incubation enhances the colony formation of a polA recB strain of Escherichia coli K-12. J Bacteriol 152:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cromie GA, Leach DRF. 2000. Control of crossing over. Mol Cell 6:815–826. doi: 10.1016/S1097-2765(05)00095-X. [DOI] [PubMed] [Google Scholar]
- 109.Steiner WW, Kuempel PL. 1998. Sister chromatid exchange frequencies in Escherichia coli analyzed by recombination at the dif resolvase site. J Bacteriol 180:6269–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perals K, Capiaux H, Vincourt JB, Louarn JM, Sherratt DJ, Cornet F. 2001. Interplay between recombination, cell division and chromosome structure during chromosome dimer resolution in Escherichia coli. Mol Microbiol 39:904–913. doi: 10.1046/j.1365-2958.2001.02277.x. [DOI] [PubMed] [Google Scholar]
- 111.Masai H, Asai T, Kubota Y, Arai K, Kogoma T. 1994. Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO J 13:5338–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wahle E, Lasken RS, Kornberg A. 1989. The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J Biol Chem 264:2469–2475. [PubMed] [Google Scholar]
- 113.Wechsler JA, Gross JD. 1971. Escherichia coli mutants temperature-sensitive for DNA synthesis. Mol Gen Genet 113:273–284. doi: 10.1007/BF00339547. [DOI] [PubMed] [Google Scholar]
- 114.Liu JI, Xu LW, Sandler SJ, Marians KJ. 1999. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc Natl Acad Sci U S A 96:3552–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu L, Marians KJ. 2003. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell 11:817–826. doi: 10.1016/S1097-2765(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 116.Heller RC, Marians KJ. 2005. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol Cell 17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 117.Lopper M, Boonsombat R, Sandler SJ, Keck JL. 2007. A hand-off mechanism for primosome assembly in replication restart. Mol Cell 26:781–793. doi: 10.1016/j.molcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sandler SJ, Marians KJ, Zavitz KH, Coutu J, Parent MA, Clark AJ. 1999. dnaC mutations suppress defects in DNA replication- and recombination-associated functions in priB and priC double mutants in Escherichia coli K-12. Mol Microbiol 34:91–101. doi: 10.1046/j.1365-2958.1999.01576.x. [DOI] [PubMed] [Google Scholar]
- 119.Sandler SJ. 2000. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics 155:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sandler SJ, McCool JD, Do TT, Johansen RU. 2001. PriA mutations that affect PriA-PriC function during replication restart. Mol Microbiol 41:697–704. doi: 10.1046/j.1365-2958.2001.02547.x. [DOI] [PubMed] [Google Scholar]
- 121.Marinus MG. 2000. Recombination is essential for viability of an Escherichia coli dam (DNA adenine methyltransferase) mutant. J Bacteriol 182:463–468. doi: 10.1128/JB.182.2.463-468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Flores MJ, Ehrlich SD, Michel B. 2002. Primosome assembly requirement for replication restart in the Escherichia coli holDG10 replication mutant. Mol Microbiol 44:783–792. doi: 10.1046/j.1365-2958.2002.02913.x. [DOI] [PubMed] [Google Scholar]
- 123.Sandler SJ. 2005. Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics 169:1799–1806. doi: 10.1534/genetics.104.036962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boonsombat R, Yeh SP, Milne A, Sandler SJ. 2006. A novel dnaC mutation that suppresses priB rep mutant phenotypes in Escherichia coli K-12. Mol Microbiol 60:973–983. doi: 10.1111/j.1365-2958.2006.05147.x. [DOI] [PubMed] [Google Scholar]
- 125.Withers HL, Bernander R. 1998. Characterization of dnaC2 and dnaC28 mutants by flow cytometry. J Bacteriol 180:1624–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Maisnier-Patin S, Nordstrom K, Dasgupta S. 2001. Replication arrests during a single round of replication of the Escherichia coli chromosome in the absence of DnaC activity. Mol Microbiol 42:1371–1382. doi: 10.1046/j.1365-2958.2001.02718.x. [DOI] [PubMed] [Google Scholar]
- 127.Saifi B, Ferat JL. 2012. Replication fork reactivation in a dnaC2 mutant at non-permissive temperature in Escherichia coli. PLoS One 7:e33613. doi: 10.1371/journal.pone.0033613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mettrick KA, Grainge I. 2016. Stability of blocked replication forks in vivo. Nucleic Acids Res 44:657–668. doi: 10.1093/nar/gkv1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Harinarayanan R, Gowrishankar J. 2004. A dnaC mutation in Escherichia coli that affects copy number of ColE1-like plasmids and the PriA-PriB (but not Rep-PriC) pathway of chromosomal replication restart. Genetics 166:1165–1176. doi: 10.1534/genetics.166.3.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim S, Dallmann HG, McHenry CS, Marians KJ. 1996. Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell 84:643–650. doi: 10.1016/S0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 131.Heller RC, Marians KJ. 2005. Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J Biol Chem 280:34143–34151. doi: 10.1074/jbc.M507224200. [DOI] [PubMed] [Google Scholar]
- 132.Tanner NA, Hamdan SM, Jergic S, Schaeffer PM, Dixon NE, van Oijen AM. 2008. Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat Struct Mol Biol 15:170–176. doi: 10.1038/nsmb.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Graham JE, Marians KJ, Kowalczykowski SC. 2017. Independent and stochastic action of DNA polymerases in the replisome. Cell 169:1201.e17–1213.e17. doi: 10.1016/j.cell.2017.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McCool JD, Sandler SJ. 2001. Effects of mutations involving cell division, recombination, and chromosome dimer resolution on a priA2:: kan mutant. Proc Natl Acad Sci U S A 98:8203–8210. doi: 10.1073/pnas.121007698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McCool JD, Ford CC, Sandler SJ. 2004. A dnaT mutant with phenotypes similar to those of a priA2::kan mutant in Escherichia coli K-12. Genetics 167:569–578. doi: 10.1534/genetics.103.025296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Al-Deib AA, Mahdi AA, Lloyd RG. 1996. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J Bacteriol 178:6782–6789. doi: 10.1128/jb.178.23.6782-6789.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lloyd RG, Rudolph CJ. 2016. 25 years on and no end in sight: a perspective on the role of RecG protein. Curr Genet 62:827–840. doi: 10.1007/s00294-016-0589-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rudolph CJ, Upton AL, Briggs GS, Lloyd RG. 2010. Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 9:210–223. doi: 10.1016/j.dnarep.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 139.Ivanova D, Taylor T, Smith SL, Dimude JU, Upton AL, Mehrjouy MM, Skovgaard O, Sherratt DJ, Retkute R, Rudolph CJ. 2015. Shaping the landscape of the Escherichia coli chromosome: replication-transcription encounters in cells with an ectopic replication origin. Nucleic Acids Res 43:7865–7877. doi: 10.1093/nar/gkv704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lloyd RG, Buckman C. 1991. Genetic analysis of the recG locus of Escherichia coli K-12 and of its role in recombination and DNA repair. J Bacteriol 173:1004–1011. doi: 10.1128/jb.173.3.1004-1011.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ishioka K, Iwasaki H, Shinagawa H. 1997. Roles of the recG gene product of Escherichia coli in recombination repair: effects of the delta-recG mutation on cell division and chromosome partition. Genes Genet Syst 72:91–99. doi: 10.1266/ggs.72.91. [DOI] [PubMed] [Google Scholar]