

Significance
The identification of aberrant SOD1WT species accumulating in the spinal cord during aging could reveal pathogenic species involved in sporadic (s)ALS. Using a combination of biochemical approaches, we discovered that disulfide–cross-linked SOD1WT aggregates rise before other abnormal protein species during aging and are significantly increased in sALS spinal cord tissue. We also found that endoplasmic reticulum stress stimulates accumulation of these species, with involvement of tryptophan-32 oxidation. These results establish a connection between SOD1WT aggregation and a major proteostasis network affected in ALS.
Keywords: amyotrophic lateral sclerosis, wild-type SOD1, ER stress, aging, protein aggregation
Abstract
Abnormal modifications to mutant superoxide dismutase 1 (SOD1) are linked to familial amyotrophic lateral sclerosis (fALS). Misfolding of wild-type SOD1 (SOD1WT) is also observed in postmortem tissue of a subset of sporadic ALS (sALS) cases, but cellular and molecular mechanisms generating abnormal SOD1WT species are unknown. We analyzed aberrant human SOD1WT species over the lifetime of transgenic mice and found the accumulation of disulfide–cross-linked high–molecular-weight SOD1WT aggregates during aging. Subcellular fractionation of spinal cord tissue and protein overexpression in NSC-34 motoneuron-like cells revealed that endoplasmic reticulum (ER) localization favors oxidation and disulfide-dependent aggregation of SOD1WT. We established a pharmacological paradigm of chronic ER stress in vivo, which recapitulated SOD1WTaggregation in young transgenic mice. These species were soluble in nondenaturing detergents and did not react with a SOD1 conformation-specific antibody. Interestingly, SOD1WT aggregation under ER stress correlated with astrocyte activation in the spinal cord of transgenic mice. Finally, the disulfide–cross-linked SOD1WT species were also found augmented in spinal cord tissue of sALS patients, correlating with the presence of ER stress markers. Overall, this study suggests that ER stress increases the susceptibility of SOD1WT to aggregate during aging, operating as a possible risk factor for developing ALS.
Amyotrophic lateral sclerosis (ALS) is characterized by selective degeneration of motoneurons from cerebral cortex, brainstem, and spinal cord leading to muscle weakness, atrophy, paralysis, and premature death (1, 2). Most ALS cases are considered sporadic (sALS), while 10% are familial (fALS), involving mutations in superoxide dismutase 1 (SOD1), transactive response DNA binding protein 43 (TARDBP or TDP-43), fused in sarcoma/translocated in sarcoma (FUS/TLS), and the hexanucleotide repeat expansions in C9ORF72 as the most common alterations (1–3).
More than 150 mutations in SOD1 have been linked to ALS, with varying degrees of aggressiveness and aggregation propensity (4). SOD1 folding depends on complex posttranslational modifications, including the insertion of zinc and copper ions and intramolecular disulfide-bond formation, followed by homodimerization (4). Biochemical studies have shown that fALS-linked mutations destabilize SOD1 polypeptide, preventing its posttranslational processing with resultant accumulation of aggregation-prone species (5). Mutant SOD1 toxicity may be the result of disturbed cellular homeostasis at many levels, such as axonal transport, the cytoskeleton, mitochondrial function, and the secretory pathway, in addition to cell-nonautonomous mechanisms marked by the abnormal activation of microglia and astrocytes (6, 7).
Several studies have reported the presence of abnormal SOD1WT species in postmortem sALS tissue using conformation-specific antibodies that recognize misfolded forms of the protein, possibly corresponding to small and diffusible oligomeric species that do not accumulate into high–molecular-weight (HMW) aggregates (8–10). Misfolded SOD1WT can exhibit toxic properties by impairing axonal transport, inhibiting endoplasmic reticulum (ER)-associated degradation, and self-propagating its misfolding in a prion-like fashion (9–12). At the molecular level, altered posttranslational maturation of SOD1WT can trigger its misfolding and aggregation (9, 13, 14). Interestingly, coexpression of human SOD1WT together with fALS-linked SOD1 mutants aggravates disease severity in ALS mouse models, and homozygous SOD1WT transgenic mice develop late-onset motor disease (15, 16). Despite representing an attractive hypothesis of convergent disease mechanisms of sALS and fALS, the overall relevance of SOD1WT to sALS has been questioned because of low frequency or lack of staining with conformation-specific antibodies in some studies (17, 18).
Altered proteostasis is a common pathogenic hallmark of both sALS and many different forms of fALS (19). One of the main nodes of the proteostasis network affected in ALS is the ER (20), the major subcellular compartment involved in protein folding and quality control mechanisms. ER stress triggers an adaptive reaction known as the unfolded protein response (UPR) to restore proteostasis by attenuating protein translation and reprogramming gene expression to up-regulate ER foldases, chaperones, and the protein degradation machinery (21). However, high levels of chronic ER stress result in apoptosis (22). In the context of mutant SOD1, ER stress is proposed as a key factor determining selective neuronal vulnerability (23). Strategies to attenuate ER stress levels have been shown to delay experimental ALS (24). SOD1 is predominantly cytosolic, and although it does not have an ER-localization signal peptide, a small fraction of the protein is released to the extracellular space through the classical secretory pathway (25, 26). Here we investigated the relationship between ER stress, SOD1WT misfolding, and aging. Our results support a pathogenic role of ER stress in sALS by inducing abnormal SOD1WT aggregation at the ER during aging.
Results
Disulfide–Cross-Linked SOD1WT Aggregates Accumulate During Aging.
The buffering capacity of the proteostasis network decreases during aging (27); we reasoned that this may increase the accumulation of misfolded SOD1 species. We performed analysis of SOD1WT aggregates with different approaches in young (4-mo), middle-aged (8-mo), and old (16-mo) heterozygous human SOD1WT transgenic mice (see schema in SI Appendix, Fig. S1A). As a positive control for the methods, we analyzed SOD1G93A transgenic mice (SI Appendix, Fig. S1 B–D). Using the filter-trap assay, we observed a progressive accumulation of HMW SOD1WT aggregates, starting at 4 mo of age, with further increase in older mice (Fig. 1A). We also assessed the possible involvement of disulfide bonds in human SOD1WT protein aggregation by treating samples with the thiol-reducing agent DTT. Disulfide-independent SOD1WT aggregates were barely detectable in younger mice, with strong accumulation at 16 mo of age only (Fig. 1A).
Fig. 1.
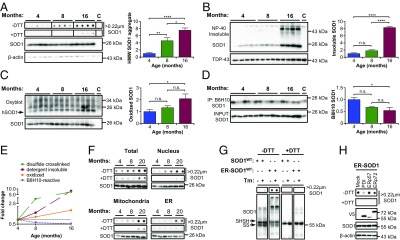
Misfolding and aggregation of SOD1WT during aging. The spinal cord of SOD1WT-Tg mice was analyzed at the indicated ages. (A) Filter-trap assay under nonreducing (−DTT) and reducing (+DTT) conditions. β-Actin was employed as loading control. (B) Western blot analysis of nonionic detergent (Nonidet P-40)-insoluble SOD1 aggregates isolated by centrifugal sedimentation. TDP-43 was used as loading control. (C) Western blot analysis of carbonylated (oxidized) proteins. SOD1 was employed as loading control. The arrow indicates oxidized SOD1WT. (D) Western blot analysis of SOD1 immunoprecipitation (IP) with B8H10 conformation-specific antibody. SOD1 in the input was used as loading control. A 16-mo-old non-Tg littermate was used in A–C as control for background (lane labeled “C”). A symptomatic SOD1G93A-Tg mouse was used in D as positive control (lane labeled C). Statistical analysis (A–D) was performed using one-way ANOVA with Tukey’s multiple comparison test. Mean ± SE is shown; P values are as follows: n.s., not significant, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001. n = 4 or 5 per group. (E) Fold-change plot for accumulation of the distinct abnormal SOD1WT species during aging. (F) Filter-trap assay of spinal cord subcellular fractions of SOD1WT-Tg mice at different ages. SOD1 was employed as loading control. (G) NSC-34 cells were transiently transfected for overexpression of SOD1WT-EGFP or ER-SOD1WT-EGFP and treated with tunicamycin (Tm; 1 μg/mL). Filter-trap and Western blot analyses were performed under nonreducing and reducing conditions after 18 h of Tm treatment. Arrows indicate reduced (SHSH) or oxidized (SS) SOD1WT monomer. (H) NSC-34 cells were transiently transfected for coexpression of SOD1WT-EGFP or ER-SOD1WT-EGFP and the indicated PDI family member. Filter-trap and Western blot analyses were performed 48 h after transfection.
We then monitored detergent-insoluble SOD1WT aggregates by centrifugal sedimentation (SI Appendix, Fig. S1C), which only showed a significant increase in aged mice (Fig. 1B). Since SOD1WT oxidation had been implicated in sALS and may contribute to its misfolding (13, 28), we measured protein carbonylation in SOD1WT transgenic mice. The identification of carbonylated SOD1WT could be easily inferred by comparison of band pattern in transgenic mice and nontransgenic littermates (Fig. 1C). Carbonylated SOD1WT was significantly accumulated in older animals, whereas the overall pattern of protein oxidation did not change across ages (Fig. 1C).
Next, we focused on detecting misfolded soluble SOD1WT species using conformation-specific antibodies that were generated using the SOD1G93A antigen but recognize modified SOD1WT species (9, 29). The monoclonal antibody B8H10 was employed since it yielded satisfactory immunoprecipitation of SOD1 (SI Appendix, Fig. S1D). We consistently detected B8H10-positive SOD1WT in transgenic mice at the three ages analyzed, with variable amounts between animals (Fig. 1D). Contrary to the trend observed for other species, B8H10-positive SOD1WT was reduced with aging (Fig. 1D). Overall, disulfide-dependent SOD1WT aggregates accumulate earlier in the aging process relative to other SOD1WT species (see summary in Fig. 1E).
Accumulation of HMW SOD1WT Aggregates in Microsomal Fractions During Aging.
To define the distribution of disulfide–cross-linked SOD1WT species during aging, we performed subcellular fractionation of spinal cord tissue of animals at different ages using differential centrifugation. Filter-trap analysis of total extracts confirmed our previous finding indicating that disulfide-dependent SOD1WT aggregates accumulate progressively during the lifetime, while disulfide-independent species are increased in aged animals (Fig. 1F). Nucleus- and mitochondria-enriched fractions contained most of the SOD1WT aggregates, with disulfide-dependent and -independent aggregates increasing with aging (Fig. 1F). On the other hand, microsomal (ER-enriched) fractions predominantly accumulated disulfide–cross-linked SOD1WT aggregates in older mice, with negligible levels of DTT-insensitive species (Fig. 1F). This result indicates that disulfide-dependent SOD1WT aggregation is favored at the ER.
We investigated whether directing expression of human SOD1WT to the ER lumen influences protein aggregation. Thus, we transiently expressed in NSC-34 cells a previously described SOD1WT fused to EGFP that is targeted to the ER lumen (ER-SOD1WT) (SI Appendix, Fig. S2A) (26). Remarkably, expression of ER-SOD1WT led to a dramatic accumulation of HMW aggregates that were sensitive to DTT treatment as detected using filter-trap and Western blot analysis (Fig. 1G). The stimulation of ER stress with tunicamycin (Tm) in vitro triggered a slight increase in these species (Fig. 1G). Disulfide bonds at the ER are catalyzed by a family of protein disulfide isomerase (PDI) (30). We tested the effects of coexpressing major PDIs with ER-SOD1WT. The overexpression of PDI or ERp72 significantly reduced the disulfide-dependent HMW aggregates (Fig. 1H and SI Appendix, Fig. S2B), indicating that altered redox folding drives SOD1WT aggregation in the ER. Taken together, these results suggest that ER localization of SOD1WT favors protein aggregation in a specific manner.
ER Stress Leads to Disulfide-Dependent Aggregation of SOD1WT.
ER stress is a salient feature of sALS, and has been proposed as a major pathological reaction in various experimental models of the disease (20). We first monitored expression levels of the ER stress-inducible genes Xbp1s, Bip, and Edem1 in the spinal cord of SOD1WT transgenic mice and littermate controls and observed a significant increase in both groups at 16 mo of age (Fig. 2A and SI Appendix, Fig. S3A). The induction of Edem1 levels with aging was even higher in SOD1WT transgenic mice, possibly reflecting disturbed ER function (Fig. 2A). We then investigated whether ER stress could trigger SOD1WT misfolding and aggregation using a pharmacological paradigm based on the injection of tunicamycin leading to activation of the UPR in the nervous system (Fig. 2 B and C and SI Appendix, Results and Fig. S3 B–F).
Fig. 2.
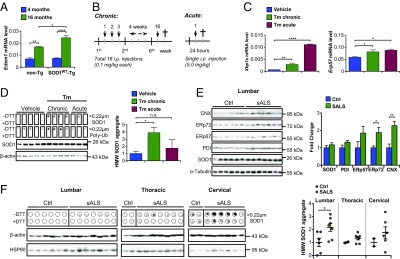
Disulfide–cross-linked SOD1WT aggregates in transgenic mice under ER stress and in sALS patients. (A) mRNA levels of the ER stress marker Edem1 were analyzed by quantitative PCR in the spinal cord of SOD1WT-Tg mice and non-Tg littermates at different ages (n = 3 to 6 per group). (B) Schematic view of the protocol for induction of chronic and acute ER stress in vivo. In the chronic paradigm, mice were treated with a total of 16 low doses of tunicamycin (Tm). In the acute protocol, mice received a single high-dose injection of Tm. (C) mRNA levels of the ER stress markers Xbp1s and Erp57 were analyzed by quantitative PCR in the spinal cord of SOD1WT-Tg mice submitted to Tm treatments. (D) Filter-trap assay of total spinal cord extracts of hSOD1WT-Tg mice described in C under nonreducing (−DTT) and reducing (+DTT) conditions for detection of HMW SOD1 and polyubiquitinated aggregates. Duplicated membranes were employed in this analysis. β-Actin was employed as loading control. Statistical analysis was performed using two-way ANOVA (A) or one-way ANOVA (C and D) with Tukey’s multiple comparison test. Mean ± SE is shown; P values are as follows: n.s., P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001. n = 3 to 6 per group. Frozen postmortem spinal cord tissue of sALS patients and controls (SI Appendix, Table S1) was processed for biochemical analysis. (E) Western blot analysis of SOD1 and ER chaperone levels in lumbar spinal cord of sALS cases and controls. α-Tubulin was employed as loading control. CNX, calnexin. (F) Filter-trap assay of total spinal cord extracts of sALS cases and controls under nonreducing (−DTT) and reducing (+DTT) conditions for detection of HMW SOD1 aggregates. β-Actin was employed as loading control. (Left) Lumbar, (Middle) thoracic, and (Right) cervical spinal cord segments. Statistical analysis was performed using Student’s t test. Mean ± SE is shown; P values are as follows: n.s., P > 0.05; *P ≤ 0.05; **P ≤ 0.01. n = 6 or 7 per group.
We compared the possible effects of chronic and acute ER stress on SOD1WT aggregation. Filter-trap analysis revealed that mice exposed to chronic ER stress presented accumulation of disulfide–cross-linked SOD1WT aggregates in spinal cord tissue at 4 mo of age (Fig. 2D). This effect was not observed in animals treated with an acute regimen of tunicamycin (Fig. 2D). Unexpectedly, neither detergent-insoluble species nor B8H10-positive SOD1WT showed significant changes under chronic or acute ER stress (SI Appendix, Fig. S3 G and H). Chronic ER stress also further increased the accumulation of disulfide-dependent SOD1WT aggregates in animals at 16 mo of age (SI Appendix, Fig. S4). Furthermore, the treatment of the NSC-34 cells with tunicamycin induced aggregation of endogenous SOD1 (SI Appendix, Fig. S5), reinforcing the significance of our findings in the transgenic model. Thus, our pharmacological paradigm for chronic ER stress in vivo was able to accelerate the appearance of a biochemical fingerprint of SOD1WT aggregation that spontaneously occurs during aging.
Increased Levels of Disulfide–Cross-Linked SOD1WT Aggregates in sALS Tissue.
We then addressed the possible association of disulfide–cross-linked SOD1WT aggregates with sALS by investigating postmortem samples. We analyzed lumbar spinal cord tissue of six control subjects and seven sALS patients, in addition to two control and seven sALS samples of thoracic and two control and six sALS samples of cervical segments as available (see the clinical and histopathological data of patients in SI Appendix, Table S1). We confirmed the occurrence of ER stress in human sALS tissue, reflected in significant up-regulation of calnexin and ERp72, using Western blot (Fig. 2E). Total SOD1 levels were not altered. We performed filter-trap assay to assess the possible accumulation of SOD1WT aggregated species. Remarkably, disulfide-dependent HMW SOD1WT aggregates were significantly augmented in the lumbar spinal cord of patients (4 positive/7 total) compared with control (0 positive/6 total) subjects (Fig. 2F). In addition, a trend to an increase of these SOD1WT species was also observed in thoracic (5 positive/7 total) and cervical (3 positive/6 total) segments of the spinal cord of sALS cases, with no relevant accumulation found in the two control samples for each region (Fig. 2F). These results suggest that HMW aggregates of SOD1WT are present in sALS spinal cord tissue and correlate with the up-regulation of ER stress-responsive chaperones.
Selective Activation of Astrocytes in SOD1WT Transgenic Mice Under ER Stress.
To explore the possible occurrence of pathological changes induced by chronic ER stress in the context of human SOD1WT overexpression, we investigated lumbar spinal cord alterations using histological methods. We performed immunohistochemical analysis with choline acetyltransferase (ChAT) antibody to quantify motoneuron number. This analysis did not reveal any evident neuronal loss in mice submitted to ER stress (Fig. 3A). We then evaluated glial responses by monitoring the activation of microglia and astrocytes using immunofluorescence. Remarkably, we observed selective and marked activation of astrocytes in the spinal cord ventral horn of SOD1WT transgenic mice under ER stress (Fig. 3B). Nontransgenic animals did not show any activation of astrocytes under the same conditions (Fig. 3B). In contrast, ER stress did not induce the differential activation of microglia in any experimental group (Fig. 3C). These results suggest a correlation between SOD1WT aggregation and abnormal astrocyte activation under ER stress.
Fig. 3.

Histopathological alterations in SOD1WT transgenic mice exposed to ER stress. SOD1WT-Tg mice and non-Tg littermates were submitted to chronic tunicamycin or vehicle treatment and histopathological analysis was performed in lumbar spinal cord. (A) Representative images of ChAT staining for visualization of motoneurons in the spinal cord ventral horn. Graphs show quantification of motoneuron number in 200-μm-spaced serial sections spanning from L2 to L5 lumbar spinal cord. (B) Representative images of GFAP staining for detection of astrocytes. Graphs show the relative mean of gray matter area covered by astrocytes as quantified in four 800-μm-spaced serial sections spanning from L2 to L5 lumbar spinal cord. (C) Representative images of Iba1 staining for detection of microglia. Graphs show the relative mean of gray matter area covered by microglia as quantified in four 800-μm-spaced serial sections spanning from L2 to L5 lumbar spinal cord. (Scale bars, 200 μm.) Statistical analysis was performed using two-way ANOVA with Tukey’s multiple comparison test. Mean ± SE is shown; P values are as follows: n.s., P > 0.05; ***P ≤ 0.001; ****P ≤ 0.0001. n = 6 to 11 per group.
Tryptophan-32 Oxidation Enhances SOD1WT Aggregation at the ER.
To gain insights about the molecular alterations that could contribute to SOD1WT aggregation during ER stress, we first measured protein carbonylation of SOD1WT since it can trigger its misfolding and aggregation in vitro (9, 13). Nonetheless, both chronic and acute ER stress caused an increment of carbonylated SOD1WT (Fig. 4A). Total levels of carbonylation did not change with overexpression of human SOD1WT (Fig. 4A).
Fig. 4.

Posttranslational modification and aggregation of SOD1WT in the ER. (A) Western blot analysis of carbonylated (oxidized) proteins in total extracts of SOD1WT-Tg mice and non-Tg littermates submitted to tunicamycin treatments as indicated. The arrow points to oxidized SOD1WT. Ponceau S staining was employed as loading control. Statistical analysis was performed using two-way ANOVA (Upper) or one-way ANOVA (Lower) with Tukey’s multiple comparison test. Mean ± SE is shown; P values are as follows: n.s., P > 0.05; *P ≤ 0.05. n = 3 to 5 per group. (B and C) Mass spectrometry analysis of posttranslational modifications of SOD1WT isolated from spinal cord subcellular fractions of SOD1WT-Tg mice submitted to chronic Tm treatment. Seven animals were pooled in each sample. (B) Liquid chromatograms of a double-charged precursor ion with m/z = 353.20. Arrows indicate the peptide with oxidized tryptophan 32 [VW(O)GSIK]. Chromatograms were aligned to facilitate visualization. (C) Quantification of the percentage of SOD1 containing oxidized W32. (D) NSC-34 cells were transiently transfected for overexpression of SOD1WT-EGFP, SOD1W32F-EGFP, ER-SOD1WT-EGFP, or ER-SOD1W32F-EGFP. Filter-trap and Western blot analyses under nonreducing (−DTT) and reducing (+DTT) conditions were performed 48 h after transfection. Arrows indicate reduced (SHSH) or oxidized (SS) SOD1WT monomer. The graph shows the ratio of SOD1W32F to SOD1WT total protein levels. Statistical analysis was performed using Student’s t test. Mean ± SE is shown; P values are as follows: n.s., P > 0.05; **P ≤ 0.01. n = 3. (E) Schematic model for SOD1WT aggregation in the ER. Dashed arrows indicate unidentified pathways.
Next, we focused on identifying specific SOD1WT posttranslational modifications that could potentially impart aggregation-prone motifs to the protein. We purified SOD1WT using immunoprecipitation of subcellular fractions of spinal cord tissue under basal and chronic ER stress conditions, followed by enzymatic digestion and mass spectrometry analysis. Screening for modifications in key residues previously known to be involved in SOD1WT misfolding and aggregation (31) revealed higher levels of oxidized tryptophan 32 (W32) in SOD1WT purified from microsomal fractions (Fig. 4 B and C and SI Appendix, Fig. S6). Upon ER stress, increased W32 oxidation was also detected in cytosolic and mitochondrial pools of the protein (Fig. 4 B and C).
To understand the impact of W32 oxidation on SOD1WT aggregation, we performed site-directed mutagenesis to substitute W32 for phenylalanine (W32F), an amino acid with aromatic properties but resistant to oxidative modifications. Using overexpression of SOD1WT and ER-targeted SOD1WT in NSC-34 cells, we determined that W32F substitution significantly diminishes total protein levels and the formation of HMW disulfide-dependent SOD1WT aggregates in the ER (Fig. 4D). These results suggest that the ER environment significantly contributes to abnormal posttranslational modification of SOD1WT and its aggregation.
Discussion
A pathogenic role of SOD1WT has been inferred from the detection of misfolded forms of the protein in postmortem tissue of sALS patients (12). The current evidence supporting SOD1WT misfolding in sALS is mainly based on immunohistochemical staining of paraffin-embedded formalin-fixed spinal cord tissue of patients using conformation-specific antibodies (8–10). This approach is inherently difficult since misfolded SOD1 reactive toward conformation-specific antibodies is mainly constituted by soluble species (9, 32), yielding a diffuse staining pattern in sALS tissue (8, 9). Some studies have challenged the participation of SOD1WT in sALS pathogenesis based on the lack of staining with conformationally sensitive antibodies (18, 32, 33). These reports showed that antigen-retrieval treatments can enhance staining of SOD1WT and mutant SOD1 with diverse conformation-specific antibodies and detect protein inclusions in SOD1-positive fALS, but not sALS, cases (18, 32, 33). However, the staining pattern of mutant SOD1 in patient samples may actually reflect the high protein concentration in SOD1-positive inclusions and should not be used as a main argument to rule out SOD1WT involvement in sALS. As an alternative approach, immunoprecipitation with conformationally sensitive antibodies has been used to detect misfolded SOD1WT in frozen sALS tissue, resulting in divergent conclusions (10, 18). Although misfolded and oxidized SOD1WT species have been shown to have adverse consequences to motoneurons (9, 10, 28), their detection in sALS is not ubiquitous and may depend on the antibody employed and patient cohort (8–10, 17, 18). Moreover, there is scarce knowledge about the mechanisms contributing to SOD1WT misfolding and aggregation in vivo.
To tackle this question, we employed transgenic mice overexpressing human SOD1WT to investigate whether normal aging and ER stress could contribute to SOD1WT aggregation and the appearance of disease features. Remarkably, we discovered that disulfide–cross-linked SOD1WT aggregates accumulate in middle-aged mice whereas other aberrant SOD1WT forms are detected only late, in aged animals. Subcellular fractionation of spinal cord showed that microsomes exclusively accumulate disulfide-dependent SOD1WT aggregates with aging. Moreover, the ER-targeted overexpression of SOD1WT greatly favored protein aggregation through disulfide cross-links, which is significantly inhibited by enforcing PDI expression. Together, these data indicate that ER localization favors SOD1WT aggregation due to altered redox folding. ER stress is an early and transversal pathogenic mechanism in ALS that may explain the differential neuronal vulnerability (23). In this context, we investigated the relationship between ER stress and SOD1WT misfolding and aggregation in vivo. Here we identified the selective accumulation of HMW disulfide–cross-linked SOD1WT aggregates as the molecular signature of chronic ER stress in vivo. Thus, we reason that normal aging and chronic ER stress possibly share molecular mechanisms causing SOD1WT aggregation in the ER (Fig. 4E). Similarly, we recently reported the presence of HMW disulfide–cross-linked aggregates of mutant TDP-43 in transgenic mice (34).
The identification of SOD1WT aggregates in mice undergoing ER stress may be of relevance for sALS, since we were able to detect a significant increase in the levels of these SOD1WT species in a subset of sALS patients (4 positive/7 total) accompanied by altered levels of ER chaperones. Moreover, disulfide–cross-linked SOD1 aggregation is a salient feature of mutant SOD1 ALS mouse models that sharply increases with disease progression and severity (35–38). It remains to be determined if the occurrence of SOD1WT misfolding triggered by ER stress enhances its spreading through the nervous system. Although the induction of ER stress in human SOD1WT transgenic mice was not sufficient to trigger motoneuron loss in the time frame of the study, it caused a selective activation of astrocytes in these animals. Since astrocyte activation is a relevant factor contributing to ALS pathogenesis (1), our observations suggest that the interplay between ER stress and SOD1WT aggregation may influence cell-nonautonomous pathogenic mechanisms.
The analysis of posttranslational modifications of SOD1WT identified microsomal SOD1WT containing oxidized W32, indicating that the ER environment contributes to generation of aggregation-prone forms of the protein. The pattern of W32-oxidized SOD1WT distribution under ER stress reveals a complex scenario where SOD1WT may leak from the ER into mitochondria and cytosol, likely propagating protein aggregation in the intracellular milieu. This hypothesis remains to be explored. However, ER permeabilization under ER stress has been described to mediate the release of intraluminal components into the cytosol (39). Our results suggest that SOD1 trafficking through the ER is susceptible to W32 oxidation, facilitating disulfide-dependent protein aggregation (Fig. 4E). Importantly, W32 oxidation has also been associated with mutant SOD1 toxicity (40). Overall, this study uncovered an unexpected link between ER stress and SOD1WT aggregation with implications for understanding sALS pathogenesis.
Materials and Methods
Human Samples.
Frozen postmortem spinal cord tissue from sALS patients and control subjects was obtained from the Alzheimer Disease Research Center at Massachusetts General Hospital and the R.H.B. laboratory at the Department of Neurology of University of Massachusetts Medical School under an approved institutional review board protocol (FWA 00004009). Written informed consent was obtained from all donors before tissue collection. The analysis of the human samples was also authorized by the Ethics Committee of the Faculty of Medicine of the University of Chile.
Animals.
The experimental procedures using animals were approved by the Institutional Review Board for Animal Care of the Faculty of Medicine of the University of Chile (protocol 0503). The human SOD1WT-Tg (transgenic) and SOD1G93A-Tg mouse lines were purchased from The Jackson Laboratory and maintained on a C57BL/6 background. Details are given in SI Appendix, Materials and Methods.
Supplementary Material
Acknowledgments
We thank Isabel Constantino and Diane McKenna-Yasek for their procurement of human tissue samples. We also thank John Leszyk and Scott Shaffer of the Proteomics and Mass Spectrometry Facility, University of Massachusetts Medical School. This work was funded by FONDAP (Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias) program 15150012, Millennium Institute P09-015-F, FONDECYT (Fondo Nacional de Desarrollo Científico y Tecnológico) 1180186, ALS Therapy Alliance 2014-F-059, Muscular Dystrophy Association 382453, Department of Defense ALS Research Program Award 81XWH-16-1-0112 (to C.H.), FONDECYT 3130351 and 11150579 (to D.B.M.), FONDECYT 3110067 and 1150743 (to U.W.), and National Institutes of Health Grant R01NS067206 (to D.A.B.). R.H.B. receives support from the National Institute of Neurological Disorders and Stroke (NINDS), the ALS Association, ALS Finding a Cure, ALS ONE, the Angel Fund for ALS Research, the Celluci Endowment for ALS Research, and Project ALS.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Raw data available at www.hetzlab.cl/data/.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1801109115/-/DCSupplemental.
References
- 1.Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: From genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner MR, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Chalabi A, Hardiman O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 4.Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 5.Sheng Y, Chattopadhyay M, Whitelegge J, Valentine JS. SOD1 aggregation and ALS: Role of metallation states and disulfide status. Curr Top Med Chem. 2012;12:2560–2572. doi: 10.2174/1568026611212220010. [DOI] [PubMed] [Google Scholar]
- 6.Peters OM, Ghasemi M, Brown RH., Jr Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125:2548. doi: 10.1172/JCI82693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- 8.Forsberg K, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5:e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grad LI, et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 2014;111:3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujisawa T, et al. A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann Neurol. 2012;72:739–749. doi: 10.1002/ana.23668. [DOI] [PubMed] [Google Scholar]
- 12.Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. doi: 10.3389/fncel.2013.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakhit R, et al. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 14.Medinas DB, Gozzo FC, Santos LFA, Iglesias AH, Augusto O. A ditryptophan cross-link is responsible for the covalent dimerization of human superoxide dismutase 1 during its bicarbonate-dependent peroxidase activity. Free Radic Biol Med. 2010;49:1046–1053. doi: 10.1016/j.freeradbiomed.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Jaarsma D, et al. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- 16.Graffmo KS, et al. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:51–60. doi: 10.1093/hmg/dds399. [DOI] [PubMed] [Google Scholar]
- 17.Liu H-N, et al. Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2009;66:75–80. doi: 10.1002/ana.21704. [DOI] [PubMed] [Google Scholar]
- 18.Da Cruz S, et al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 2017;134:97–111. doi: 10.1007/s00401-017-1688-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medinas DB, Valenzuela V, Hetz C. Proteostasis disturbance in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26:R91–R104. doi: 10.1093/hmg/ddx274. [DOI] [PubMed] [Google Scholar]
- 20.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13:477–491. doi: 10.1038/nrneurol.2017.99. [DOI] [PubMed] [Google Scholar]
- 21.Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 22.Hetz C, Papa F. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–181. doi: 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Rozas P, Bargsted L, Martínez F, Hetz C, Medinas DB. The ER proteostasis network in ALS: Determining the differential motoneuron vulnerability. Neurosci Lett. 2017;636:9–15. doi: 10.1016/j.neulet.2016.04.066. [DOI] [PubMed] [Google Scholar]
- 24.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi H, et al. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci USA. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner BJ, et al. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J Neurosci. 2005;25:108–117. doi: 10.1523/JNEUROSCI.4253-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015;21:1406–1415. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 28.Guareschi S, et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci USA. 2012;109:5074–5079. doi: 10.1073/pnas.1115402109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urushitani M, Ezzi SA, Julien J-P. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2007;104:2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatahet F, Ruddock LW. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850, and erratum (2010) 12:322. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 31.Schmitt ND, Agar JN. Parsing disease-relevant protein modifications from epiphenomena: Perspective on the structural basis of SOD1-mediated ALS. J Mass Spectrom. 2017;52:480–491. doi: 10.1002/jms.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brotherton TE, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci USA. 2012;109:5505–5510. doi: 10.1073/pnas.1115009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayers JI, et al. Conformational specificity of the C4F6 SOD1 antibody; low frequency of reactivity in sporadic ALS cases. Acta Neuropathol Commun. 2014;2:55. doi: 10.1186/2051-5960-2-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bargsted L, et al. Disulfide cross-linked multimers of TDP-43 and spinal motoneuron loss in a TDP-43A315T ALS/FTD mouse model. Sci Rep. 2017;7:14266. doi: 10.1038/s41598-017-14399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furukawa Y, Fu R, Deng H-X, Siddique T, O’Halloran TV. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci USA. 2006;103:7148–7153. doi: 10.1073/pnas.0602048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jonsson PA, et al. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain. 2006;129:451–464. doi: 10.1093/brain/awh704. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Xu G, Borchelt DR. Mapping superoxide dismutase 1 domains of non-native interaction: Roles of intra- and intermolecular disulfide bonding in aggregation. J Neurochem. 2006;96:1277–1288. doi: 10.1111/j.1471-4159.2005.03642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng H-X, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pihán P, Carreras-Sureda A, Hetz C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24:1478–1487. doi: 10.1038/cdd.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor DM, et al. Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282:16329–16335. doi: 10.1074/jbc.M610119200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.