

Abstract
The promise of gene therapy for the treatment of cystic fibrosis has yet to be fully clinically realized despite years of effort toward correcting the underlying genetic defect in the cystic fibrosis transmembrane conductance regulator (CFTR). mRNA therapy via nanoparticle delivery represents a powerful technology for the transfer of genetic material to cells with large, widespread populations, such as airway epithelia. We deployed a clinically relevant lipid-based nanoparticle (LNP) for packaging and delivery of large chemically modified CFTR mRNA (cmCFTR) to patient-derived bronchial epithelial cells, resulting in an increase in membrane-localized CFTR and rescue of its primary function as a chloride channel. Furthermore, nasal application of LNP-cmCFTR restored CFTR-mediated chloride secretion to conductive airway epithelia in CFTR knockout mice for at least 14 days. On day 3 post-transfection, CFTR activity peaked, recovering up to 55% of the net chloride efflux characteristic of healthy mice. This magnitude of response is superior to liposomal CFTR DNA delivery and is comparable with outcomes observed in the currently approved drug ivacaftor. LNP-cmRNA-based systems represent a powerful platform technology for correction of cystic fibrosis and other monogenic disorders.
Keywords: cystic fibrosis, gene therapy, ion transport, nasal potential difference, mRNA therapeutics, nanoparticles
Graphical Abstract
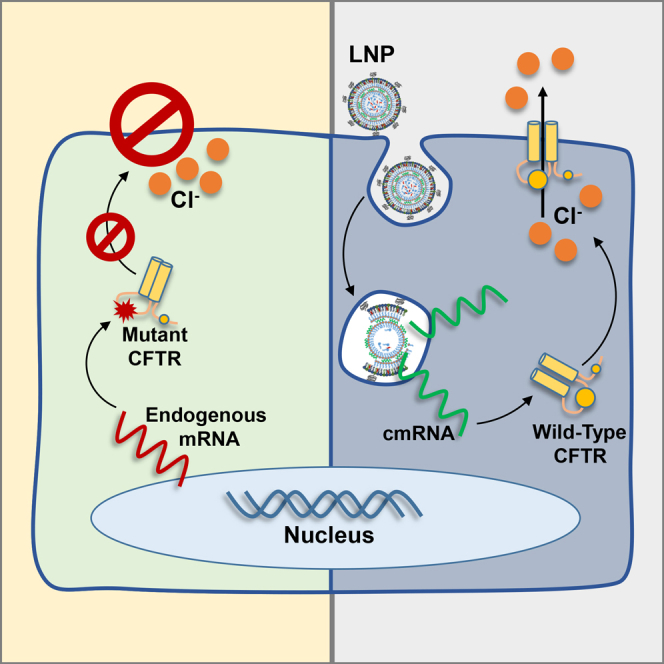
The use of lipid nanoparticles to deliver therapeutic mRNA is an increasingly attractive gene therapy modality. Here, Robinson et al. demonstrate that lipid nanoparticle transfection of mRNA can repair the central defect in cystic fibrosis, chloride transport, both in vitro and in vivo. The authors discuss why this technology is a promising treatment option for monogenic disorders such as cystic fibrosis.
Introduction
Cystic fibrosis is a monogenic lifespan-reducing disorder affecting approximately 70,000 people worldwide.1 The disease is caused by genetic variance within the coding region of the cystic fibrosis transmembrane conductance regulator (CFTR), an anion channel necessary for chloride efflux from secretory epithelial cells.2, 3, 4, 5 Over time, the resulting ion transport dysregulation induces multisystem organ failure and death. There are currently over 300 distinct disease-conferring mutations, leading to a wide variance in disease progression.6 Among other symptoms, patients experience respiratory challenges, including dehydrated pulmonary air surface liquid, impaired mucociliary clearance, permanent bacterial colonization, and lung disease.3, 7, 8, 9, 10 Restoration of normal ion homeostasis to cystic fibrosis patients represents a major goal of therapeutic agent development.
Because of steady advancements to alleviate complications, the median life expectancy of cystic fibrosis patients has quadrupled since 1962, progressing from 10 to 40 years.11 Recent breakthroughs in drug development have identified several small-molecule CFTR modulators (including ivacaftor, lumicaftor, and tezecaftor) that partially restore trafficking and chloride transport function to the endogenously expressed mutant protein.12, 13, 14, 15 Although promising, these drugs have several limitations. First, these “protein rescue” therapies only apply to specific CFTR mutations, leaving approximately 30% of patients with no protein-specific treatment options.16 Second, in cases where the disease arises from CFTR trafficking defects, these therapies have shown only modest improvements in lung function, sweat chloride, and hospitalization rates.17, 18, 19, 20 Third, a recent study suggest that 17.2% of eligible patients discontinued therapy because of side effects or perceived lack of benefit, which increased to 31.6% when subjects had less than 40% of predicted healthy lung function.20 Finally, the use of protein rescue molecules has been restricted to patients older than 6 years, in part because of toxicity concerns.16
Unlike pharmacotherapeutic interventions, gene therapy holds the potential to treat cystic fibrosis irrespective of patient genotype. mRNA has garnered significant attention in academia and industry because it acts in the cytosol, eliminating the challenges of nuclear translocation.21, 22, 23 This method offers amplified production of therapeutic proteins through rapid, repeated translation in dividing and non-dividing cells.21, 24, 25 Unfortunately, exogenous mRNA can be detected and destroyed by serum nucleases and can trigger an immune response upon cellular entry.26, 27 Current strategies utilize chemically modified mRNA (cmRNA), which contains modified nucleosides such as pseudouridine and 5-methylcytidine.28, 29, 30, 31 These chemical modifications aid in effective subversion of the innate immune response and confer enhanced stability.24, 28, 29
Because of their high negative charge, nucleic acids are unable to cross the cell membrane and, therefore, require assistance for intracellular delivery. Lipid-based nanoparticles (LNPs) are a leading delivery platform because they efficiently encapsulate, protect, and deliver nucleic acids.32, 33, 34, 35, 36, 37, 38, 39 LNPs contain an ionizable lipid that facilitates cmRNA packaging through complexation and aids in triggering endosomal escape through membrane destabilization.33, 40, 41, 42, 43, 44, 45, 46, 47 Formulations also consist of a phospholipid and cholesterol to maintain structural integrity and an outer lipid that is decorated with polyethylene glycol (PEG).40, 41, 48, 49 This coating cloaks the LNP from the host immune response, imparts serum stability, and extends in vivo circulation time.50, 51, 52 Recently, LNPs have succeeded in clinical trials to deliver small interfering RNA for treatment of metabolic disorders (patisiran) and are frontrunners in clinical development of mRNA vaccines (mRNA-1440) without noted toxic effects.53, 54, 55
We designed a formulation of LNPs containing cmRNA encoding human CFTR (cmCFTR, LNP-cmCFTR) that transfected immortalized patient-derived bronchial epithelial cells, resulting in rescue of chloride efflux. We further demonstrated that nasal application of LNP-cmCFTR to CFTR knockout (CFKO) mice restores chloride transport to the nasal airway epithelium, as determined by testing nasal potential difference (NPD). Here we present a proof of concept for the application of LNP-cmCFTR as an effective, transient, and reversible treatment for cystic fibrosis.
Results
Particle Characterization, cmRNA Uptake, and Protein Expression
LNPs were formulated through microfluidic mixing of the lipids with mRNA, which spontaneously self-assemble into small uniform nanostructures. Despite the considerable size of cmCFTR (4.5 kb), individual nanoparticle tracking analysis showed that particles of 176.6 ± 74.9 nm were formed with a concentration of 1.65 × 1011 nanoparticles/mL solution. Dynamic light scattering (DLS) showed that LNP-cmCFTR had an average hydrodynamic size of 104.2 ± 30.5 nm and was uniform, with a polydispersity index (PDI) of 0.08 (Figures 1A and 1B) and high encapsulation efficiency (>99%, data not shown). These characterizations indicate that LNPs can encapsulate large cmRNA molecules into nanoscale packages used for intracellular delivery.
Figure 1.

LNP Particle Characterization, Uptake, and Efficacy
Particle size distribution of LNP-cmCFTR was measured through (A) nanoparticle tracking analysis and (B) dynamic light scattering. (C) CF and CF-WT bronchial epithelial cells were exposed to varying concentrations of LNP-cmRNA. Luciferase expression was normalized to cell viability (relative light units [RLU]/relative fluorescent units [RFU], gray bars), and Cy5 uptake was normalized to cell count (RFU, gray line) (n = 3, mean ± SD). (D) Intranasal lung instillation of LNP-cmFLuc (0.6 mg cmRNA/kg) was conducted in BALB/c mice (n = 4). At 12 hr, organs were harvested, and luciferase intensity was measured using IVIS.
To study the in vitro uptake and efficacy of these LNPs, we employed CFBE41o− cells derived from a patient homozygous for the common disease-causing F508del mutation (CF cells). As a positive control, we utilized a CFBE41o− daughter strain stably transfected to express additional wild-type (WT) CFTR (CF-WT cells; see Materials and Methods for strain details). We tested particle efficacy in CF and CF-WT cells using a bioluminescent reporter cmRNA encoding firefly luciferase (LNP-cmFLuc; see Materials and Methods) and found that, following 24 hr exposure to LNP-cmFLuc, both cell lines exhibited a dose-dependent increase in luciferase activity (Figure 1C, bars) with negligible toxicity. As an indicator of cmRNA uptake, we performed the same experiment using LNPs encapsulating Cy5-tagged cmRNA and found that the trend of cmRNA uptake was consistent with the expression observed (Figure 1C, lines), with no difference in expression or uptake between the experimental and control cell lines.
Delivery of genetic material to the airway is complicated by the mucosal barrier of the respiratory epithelium, which facilitates ciliary clearance of particulates upon introduction to the airway.56, 57, 58, 59, 60, 61 Advances in nanoparticle engineering have shown that decorating the surface of nanoparticles with PEG prevents mucosal trapping, and a decreased size further expedites transmucosal delivery and subsequent cellular entry.57, 58, 59, 60, 61, 62 LNP-cmFLuc (0.6 mg/kg) endowed with a PEG shell was delivered to the lungs of healthy BALB/c mice through intranasal instillation. 12 hr after transfection, luciferase expression was localized to the lungs without any evidence of transfection in other organs (Figure 1D).
Exogenous cmCFTR Translates and Localizes to the Plasma Membrane In Vitro
To elucidate the mechanisms underlying CFTR malfunction, its cellular itinerary has been studied rigorously.63, 64, 65, 66, 67, 68 Nascent CFTR becomes core-glycosylated (135 kDa) in the endoplasmic reticulum and is further modified to become fully complex-glycosylated (180 kDa) as it moves through the trans-Golgi network before reaching its functional destination at the plasma membrane. The most common CFTR mutation features a complete deletion of the phenylalanine at position 508 (F508del).2, 6 The resulting misfolded protein variant is prevented from trafficking through the trans-Golgi network and is subsequently degraded before complex glycosylation, resulting in a loss of chloride secretion (Figure 2A).69, 70, 71
Figure 2.

Exposure to LNP-cmCFTR Enriches Membrane-Integrated CFTR In Vitro
(A) CFTR-WT is core-glycosylated (CG) in the endoplasmic reticulum, after which it moves through the trans-Golgi network to become complex-glycosylated (CxG) and reaches the plasma membrane, where it acts as a chloride channel. CFTR-F508del fails to achieve complex glycosylation and rapidly undergoes degradation. (B) Western blot detection of CFTR in untreated and treated CF cell lysates (lanes 3 and 4). Lanes 1 and 2 contained complex- and core-glycosylated CFTR migration standards. β-Actin was used as a loading control. (C and D) Immunocytochemistry detection of CFTR was performed on treated (CF + LNP− cmCFTR) and untreated cells (CF-WT or CF) grown either under (C) standard growth conditions or (D) at the air-liquid interface until polarization. White arrows highlight the CFTR-enriched plasma membrane. Red, CFTR; blue, nucleus; green, actin.
Through western blot analysis, we confirmed the presence of CFTR after transfection of CF cells. As migration standards, we included complex- and core-glycosylated CFTR. Although untransfected CF cell lysates showed no CFTR-associated antibody binding, transfected CF lysates contained CFTR products throughout the range of molecular weights associated with core and complex glycosylation (Figure 2B). The presence of CFTR enrichment in lysates following nanoparticle treatment strongly indicates that exogenous cmCFTR mRNA is translated and undergoes post-translational modification.
As expected, confocal microscopy of CF-WT cells confirmed normal subcellular distribution of CFTR, including its presence in the plasma membrane (Figure 2C, left). Conversely, CF cells showed premature processing arrest of CFTR in the endoplasmic reticulum, as revealed by its perinuclear accumulation (Figure 2C, center). LNP delivery of cmCFTR to CF cells restored the plasma membrane localization of this protein (Figure 2C, right). When grown at the air-liquid interface, bronchial epithelial cells form a polarized monolayer capable of maintaining ion homeostasis mimicking that of conductive airway epithelium. Transfection of polarized CF cells with LNP-cmCFTR led to apical membrane localization of CFTR, as confirmed by z stack analysis (Figure 2D). Taken together, these data show that LNP-delivered cmCFTR undergoes successful cellular processing and that the resulting protein follows the itinerary of endogenous WT CFTR to reach its functional destination in the plasma membrane.
Transfection with LNP-cmCFTR Rescues the Chloride Efflux Phenotype In Vitro
To evaluate the chloride efflux functionality of the cmCFTR protein product, we utilized a fluorescent reporter of intracellular chloride, N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) (Figure 3A). CFTR-driven chloride efflux was stimulated by activation of cyclic AMP using forskolin and 3-isobutyl-1-methylxanthine in a zero chloride (0Cl−) solution (see Materials and Methods for details).72, 73, 74 We observed a rapid efflux in untreated CF-WT cells within 30 s of stimulation that reached steady state at 90 s (Figure 3B). A CFTR-specific inhibitor (CFTRInh-172) attenuated chloride efflux in these cells. CF cells showed a minor change in intracellular fluorescence, indicative of minimal CFTR function, which is consistent with previous studies in the same cell line.72 LNP-cmCFTR treatment induced a statistically significant increase in chloride efflux compared with untreated controls at both 30 s and 90 s (p < 0.001) (Figure 3C). Moreover, treated CF cells rescued chloride efflux to 25% that of CF-WT after 30 s, reaching comparable efflux at 90 s (Figure 3C).75 These data confirm recovery of CFTR function following transfection.
Figure 3.

In Vitro Translation of LNP-Delivered cmCFTR mRNA Yields Functional CFTR
(A) Schematic diagram of the MQAE assay, illustrating that increasing intracellular fluorescence is proportional to chloride efflux, a quantifiable byproduct of CFTR function. (B) CF-WT and CF cells were grown in chamber slides and with or without treatment with LNP-cmCFTR. The MQAE assay was performed to measure chloride efflux over 240 s (expressed as fluorescence divided by fluorescence at 0 s, Ft/F0). CF-WT cells served as a positive control for chloride efflux, and CF-WT cells treated with CFTRInh-172, a CFTR-specific inhibitor, served as a negative control. (C) Ft/F0 values obtained for each condition at 30 s and 90 s. n = 25 cells/condition; mean ± SEM; ***p < 0.005; NSD, no significant difference by unpaired t test.
Transfection of the Nasal Epithelium Restores CFTR-Mediated Chloride Efflux to CFKO Mice
To measure functional delivery of LNP-cmCFTR in vivo, we utilized a well-studied bi-transgenic CFKO mouse model in which the native mouse Cftr is fully knocked out and hCFTR is selectively expressed in the gastrointestinal tract to avoid intestinal complications (see Materials and Methods for strain background).76 Although CFKO mice do not spontaneously develop the lung disease or reduced air surface liquid volume characteristic of cystic fibrosis, the ion transport profile in the nasal epithelium of these mice mirrors that of human patients, making them a suitable animal model.77, 78 Because the nasal respiratory epithelium is physiologically comparable with airway epithelium, evaluation of potential differences (induced by uneven distribution of positively and negatively charged ions) across the nasal epithelium has been used to directly observe ion transport in live animals and humans.13, 77, 79, 80 NPD measurements have been in clinical use as a diagnostic tool for patients with rare disease-conferring mutations and are used as a clinical endpoint in CFTR protein rescue and gene therapy trials.78, 80
The potential difference across conductive epithelium arises from net transepithelial ion transport and is primarily driven by the activity of the epithelial sodium channel (ENaC), CFTR, and calcium-activated chloride channels (Figure 4A). Cystic fibrosis patients and CFKO mice exhibit insufficient outward transport of chloride and increased ENaC activity, causing elevated sodium absorption.67, 81, 82 Together, this results in hyperpolarization of baseline NPD and an exaggerated response to chemical inhibitors of ENaC, such as amiloride. CFTR stimulation by isoproterenol or inhibition by CFTRInh-172 evokes no response in cystic fibrosis patients and models because the protein is lacking. In all intact epithelia, ATP stimulation of calcium-activated chloride channels induces chloride efflux and a concomitant hyperpolarization and, therefore, serves as an experimental control (Figure 4A).83
Figure 4.

LNP-cmCFTR Delivery to the Nasal Epithelium of CFKO Mice Recovers Chloride Efflux
(A) Schematic diagram illustrating the correlation between NPD traces and ion transport. (B) RT-PCR analysis of CFKO mouse nostrils 24 hr after treatment with LNP-cmCFTR. Negative control, untreated mice; positive control, lysed LNP-cmCFTR; loading control, GAPDH. (C) Representative NPD traces for a single mouse preceding and following LNP-cmCFTR exposure. (D–F) NPD was recorded in a cohort of CFKO mice (pretreatment NPD) prior to exposure to LNP-cmCFTR or sham exposure. Additional NPDs were recorded on days 1, 3, 7, and 14 post-transfection. NPD response at each time point is shown in normal control mice (n = 11), sham-treated mice (days 1, 7, and 14: n = 1; day 3: n = 4), and LNP-cmCFTR CFKO mice (n = 5) for (D) baseline NPDs, (E) ENaC response, and (F) CFTR response. Mean ± SEM; ***p < 0.005, **p < 0.01, *p < 0.05; paired t test. Symbols: arrow, activation; line with bar, inhibition; dashed arrow, no effect.
After 2 days of back-to-back administration of 0.1 mg/kg/day LNP-cmCFTR, nasal brushings were taken from mouse nostrils (untreated versus treated CFKO), total RNA was extracted, and cDNA libraries were generated through RT-PCR. PCR amplification of exon 11 of hCFTR (an RT-PCR product of cmCFTR) confirmed the presence of intracellular cmCFTR in CFKO mice but was absent from untreated controls (Figure 4B). As expected, NPD studies recorded prior to LNP exposure showed characteristic hyperpolarization in baseline NPD of CFKO mice compared with normal controls (CFKO: −19.1 mV ± 1.8 mV; normal: −4.0 ± 1.0 mV), and CFKO mice exhibited an exaggerated response to blocking of ENaC (CFKO: 9.9 mV ± 0.5 mV; normal: 2.3 mV ± 0.2 mV) (Figures 4C and 4D). More importantly, when CFTR activity was stimulated, CFKO mice showed no response, whereas normal control mice exhibited a large hyperpolarization response (CFKO: 0.9 mV ± 0.7 mV; normal: −23.9 mV ± 2.4 mV) (Figure 4E). Mice received LNP-cmCFTR or sham (LNP-cmFLuc) treatment of nasal epithelia on two consecutive days (0.1 mg/kg/day), and NPDs were reevaluated on days 1, 3, 7, and 14 post-treatment (Figures 4C–4F). Although cmCFTR-treated mice exhibited no change in baseline NPD and a minor decrease in ENaC activity over the course of the study (Figures 4D and 4E), they did exhibit polarization in response to CFTR stimulation (Figure 4F). This effect was fully reversible by addition of CFTRInh-172, confirming that the observed response was CFTR-dependent (Figure 4C). Chloride efflux was significant at each time point (p < 0.05) and peaked on day 3, when 2 of 5 mice surpassed 50% restoration of chloride efflux (cohort average: 32%) (Figure 4F). A modest CFTR response persisted through day 15 (Figure 4F). CFKO mice treated with sham LNP-cmFLuc showed no significant difference in response to CFTR stimulation between pre- and post-treatment evaluations, eliminating the possibility that exposure to LNPs or control cmRNA facilitated chloride efflux through an alternate route (Figure 4F). In all cases, ATP stimulation resulted in an abrupt hyperpolarization event, followed by refractory depolarization, confirming the integrity of the nasal epithelium (Figure 4C). The small but statistically significant decrease in ENaC activity observed on days 7 and 14 of this study suggests that ENaC regulation does not occur immediately upon restoration of CFTR function. These findings are consistent with the results of clinical trials using small-molecule potentiators, where patient chloride secretion improved but sodium absorption remained dysregulated.84, 85, 86
Discussion
Since the initial identification and cloning of the CFTR gene in 1989, which was rapidly followed by its in vitro delivery in 1990, gene therapy was forecasted be a promising technology for correction of the basic defect in cystic fibrosis.2, 87, 88 Until recently, development focused primarily on delivery of DNA. This method had modest success because of the large size of the CFTR gene, inefficient viral and non-viral delivery vectors, the inability of DNA to transfect non-dividing airway epithelia, and the potential for insertional mutagenesis and genotoxicity.23, 86, 89, 90, 91, 92, 93 Although adeno-associated viral delivery of CFTR cDNA has been well tolerated and shown to partially restore CFTR function, the resulting expression of CFTR cDNA is weak, with diminishing efficacy on repeat exposure.94, 95, 96, 97 One recent phase 2b trial delivered CFTR DNA using a liposomal delivery system (GL67A/pGM169) demonstrated that, after repeat nebulization monthly for 1 year, treatment groups of cystic fibrosis patients exhibited stabilized lung function, whereas the placebo group experienced a decline.98 Notably, only 30% of patients showed a response in NPD following CFTR stimulation, with a maximum of −7.0 mV. Our initial data presented here demonstrate that all treated mice exhibited a chloride response for 2 weeks post-exposure to LNP-cmCFTR, with a maximum response of −12.6 mV on day 3 (55% of the normal control response). We attribute this improvement to avoiding the pitfalls of DNA-based gene therapy by using LNP-delivered cmRNA, which is immediately translated in the cytosol independent of the cell cycle.21
LNPs represent a versatile platform that has been successfully tuned for cmRNA delivery by an array of classical drug administration routes.29, 37, 39 Further, microfluidic technology has facilitated ease of production, allowing optimization of uniform nanoparticles with seamless scale-up.99, 100, 101, 102 Nanoparticles introduced intranasally must travel through the changing airway topology and traverse highly crosslinked mucus. To this end, they can be tailored to a reduced size with enhanced mucosal penetration through dense coating with PEG.50, 57, 58, 59, 60, 61 Alternatively, LNPs can be introduced through systemic intravenous administration and actively transported across pulmonary barriers by modulation of charge density or through surface decoration using antibodies, peptides, or small-molecule ligands.103, 104, 105 A remaining challenge with LNP delivery of cmRNA is that, following uptake, the cmRNA must escape the endocytic system to reach its cytosolic target. This process represents an important barrier to effective nucleic acid delivery because, typically, only 1%–2% of nucleic acid cargo reaches the cytosol, whereas the remainder undergoes lysosomal degradation.34, 106, 107, 108, 109 Previous and ongoing work has successfully yielded particles specifically engineered for endosomal escape, resulting in increased cmRNA payload and boosted protein expression.42, 110 Exploitation of additional particle dissociation mechanisms and endosome-perturbing pathways can be employed to enhance this effect.
When free in the endosome, foreign RNA can be detected by several Toll-like receptors, resulting in initiation of the innate immune response.26, 27 This outcome can be avoided using an arsenal of chemically modified nucleosides.28 Such substitutions make therapeutic RNA delivery possible and can be adjusted to prolong nucleic acid degradation and afford precise regulation of rates of translation.21, 29, 111, 112 Here we demonstrate that cmCFTR delivered via LNPs is translated to a protein product that successfully traffics to the plasma membrane in bronchial epithelial cells. Although the expression of complex glycosylated protein appears relatively sparse, it is sufficient to restore its function in vitro, resulting in a net chloride efflux comparable with CF-WT cells. We further showed in vivo functional correction of CFTR in the nasal epithelium of an animal model. We evaluated changes in electrophysiology using the NPD assay, a well-documented method used in human trials to reflect CFTR function. The magnitude of the response we measured in our experimental group was comparable with the response measured in an ivacaftor clinical trial, where patients exhibited a corresponding improvement in lung function.113
It has been estimated that restoring 10% of CFTR mRNA or 10%–35% of protein function would be sufficient to ameliorate disease.4, 114 The preliminary results presented here suggest that exposure to LNP-cmCFTR on two consecutive days meets this benchmark, restoring approximately one third of normal chloride efflux with effects for at least 2 weeks. Although our results show promise for this platform, there are many possibilities for enhancement of the observed effects. We posit that cmCFTR-based gene transfer could be supplemented with dietary flavonoids that improve the half-life of CFTR or with CFTR potentiators known to improve its conductivity.115, 116 Additionally, the cmRNA composition remains to be optimized. Although the cmRNA utilized in our studies contains full replacement of uridine with pseudouridine and cytidine with 5-methylcytidine, the ratio and positioning of these modifications can be optimized to retain immune system subversion but extend the half-life of the molecule. A recent preprint communication suggests that intratracheal administration of optimized CFTR cmRNA packaged inside biodegradable nanoparticles can improve force expiratory volume in mouse models of cystic fibrosis, indicating recovery of lung function.112
Although dense PEGylation can avert trapping of nanoparticles in the thick cystic fibrosis mucus, their ability to cross these barriers in a patient remains a major concern for the success of gene therapy applications in this disease.58, 59 Such surface modifications add to the complexity of effective delivery because they can alter particle uptake and intracellular dissociation, leading to decreased transfection efficacy. The inherent failing of animal models to emulate thick mucous membrane and airway differentiation limits the applicability of these systems. If ex vivo studies can screen for nanoparticles that traverse patient sputum and deliver functional CFTR mRNA, and those particles can be further modified to withstand inhalation delivery systems, the most efficient means for further therapeutic development may be through patient studies rather than through animal models.35, 84, 85, 98 With continued development, cmRNA-based gene therapy will become a viable treatment by halting disease progression, offering benefits to the entire cystic fibrosis patient population, including small children and fetal cases for which no corrective therapies currently exist.16, 117, 118
Materials and Methods
Formulation
All particles contained cmCFTR mRNA (NCBI: NM_000492.3, custom-made by Trilink Biotechnologies), cmFLuc mRNA (CleanCap FLuc mRNA, TriLink Biotechnologies), or Cy5-cmEGFP mRNA (CleanCap Cyanine 5 EGFP mRNA, TriLink Biotechnologies), each with fully substituted uridine by pseudouridine and cytidine by 5-methylcytidine. LNPs were prepared by combining an aqueous phase (cmRNA diluted in 50 mM citrate buffer, pH 4) and organic phase (Table 1) using a microfluidic mixer (Precision Nanosystems).33, 99 Formulations were washed, buffer-exchanged into PBS (pH 7.2), and concentrated to 250 ng/μL cmRNA using Amicon Ultra centrifugal filters (EMD Millipore).
Table 1.
Composition of LNP Organic Phase
| Function | Common Name | Full Name | Molar Ratio |
|---|---|---|---|
| Ionizable lipid | D-Lin-MC3-DMA | dilinoleylmethyl-4-dimethylaminobutyratea | 50 |
| Structural lipid | DSPC | distearoylphosphatidlycholineb | 10 |
| Structural lipid | cholesterol | cholesterolc | 38.5 |
| Coating lipid | DMG-PEG2k | 1,2-dimyristoyl-rac-glycerol, methoxypolyethylene glycold | 1.5 |
Biofine International
Avanti Polar Lipids
MP Biomedicals
Sunbright GM-020, NOF America
Particle Characterization
LNPs were characterized using single-particle tracking (ViewSizer 3000, MANTA Instruments) and DLS (Zetasizer Nano ZSP, Malvern). cmRNA encapsulation efficiency was evaluated using a modified Quanti-iT RiboGreen RNA reagent assay (Life Technologies) as described previously.110
Cell Culture
CF cells (RRID: CVCL_HL93), which are derived from a patient homozygous for the F508del mutation, were used as an in vitro model system for cystic fibrosis. To study normal CFTR behavior, we used CF cells stably transfected with an Epstein-Barr virus (EBV)-based episomal expression vector, pCEP4β (CF-WT).75 All cell lines were kind gifts from J.P. Clancy and Dieter Gruenert. Cells were grown in minimal essential medium supplemented with 10% regular fetal bovine serum (Corning) and 5% penicillin/streptomycin/glutamate (Corning) at 37°C, supplemented with 5% CO2, and cultured between 20%–80% confluency on 10-cm culture dishes (Corning). Air-liquid interface cultures were seeded onto permeable supports (TransWell, Corning) at a density of 20,000 cells per support. Cells were grown to confluency, as determined by tri-weekly transepithelial electrical resistance (TEER) measurements (Millicell ERS-2 with an adjustable electrode set, EMD Millipore). When TEER measurements stabilized, air-liquid interface culture was initiated by removing medium supplementation from the top of the support. A 16-day polarization period followed, during which the airway-interfacing side of the cells was rinsed with PBS, and the medium was replaced triweekly.
Luciferase Assay
Cells were plated 24 h prior to transfection. LNP-cmFLuc was exposed to cells, and after 24 hr, luciferase expression (One-Glo Luciferase Assay, Promega) and cell viability (CellTiter Fluor Cell Viability Assay, Promega), were measured as described previously.110
mRNA Uptake Study
Cells were plated 24 h prior to transfection. Cy5-cmEGFP (TriLink Biotechnologies) was added to cultures, and after 36 hr, cells were washed three times in PBS and fixed using 4% paraformaldehyde (15 min, room temperature), and nuclei were stained using Hoechst 33342 (Life Technologies). Cells were imaged at 20× magnification with a 455 μm × 455 μm field size, 10 fields per well (Thermo Scientific Cellnsight NXT High Content Screening Platform, Thermo Scientific HCS Studio: Cellomics Scan v.6.4.4 2013). Uptake was calculated as mean intracellular Cy5 intensity divided by the nucleus count.
Western Blot Analysis
Samples
CF cells were plated at 40,000 cells per well and transfected after 36 hr with 1 μg/well cmRNA. After 36 hr exposure to LNP-cmCFTR, CF cells were washed three times in PBS and lysed by adding Tm-PER (FIVEphoton Biochemicals) containing 1× HALT protease inhibitor (Thermo Scientific) directly to plated cells. Cells were lysed on ice for 90 min.
Migration Standards
CF-WT and an identical sister cell line expressing CFTR-F508del were plated at 40,000 cells per well and harvested after 60 hr. Lysates were obtained using the method described for CF cells. Total protein was quantified by Micro BCA Protein Assay Kit according to the manufacturer’s instructions (Thermo Scientific). Samples and migration standards were prepared under reducing conditions and heated at 37°C for 30 min prior to loading. Wells were loaded at 400 ng (standards) or 40 μg total protein/well (lysates) into a Novex WedgeWell 8% Tris-glycine gel (Invitrogen) and separated in Tris-glycine SDS running buffer (Novex). Transfer to a polyvinylidene fluoride (PVDF) membrane (Novex) was performed in Tris-glycine transfer buffer (12 μM Tris base, 100 mM glycine, 10% methanol) at 20 V for 90 min (Mini Blot Module, Invitrogen).
Blocking
For blocking, we used 5% Carnation instant dry milk in 1× Tris-buffered saline with Tween-20 (TBS-T; Novex). Antigens were detected singly with stripping (Restore PLUS Western Blot Stripping Buffer, Thermo Scientific) between detections.
Primary Antibodies
Antibodies used were Ms-anti-CFTR Ab596 (University of North Carolina), 1:2,000, and Ms-anti-β-actin (Abcam), 1:10,000. The secondary antibody used was horseradish peroxidase (HRP)-Gt-anti-Ms, 1:5,000 (Abcam).
Detection and Imaging
For detection and imaging, we used SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and a myECL Imager (model 62236X, Thermo Scientific).
Immunocytochemistry
After 36 hr exposure to LNP-cmCFTR, cells were washed three times in PBS. Cells or populated Transwell membranes were fixed in 4% paraformaldehyde, washed, blocked, and treated with CFTR primary antibody (Ms-anti-CFTR Ab596, 1:250, University of North Carolina). Alexa Fluor 647-Gt-anti-Ms secondary antibody (1:2,000, Thermo Fisher Scientific) was used for detection. Cells were counterstained for actin (ActinGreen 488, Molecular Probes) and the nucleus (Hoechst 33258, Life Technologies). Imaging was performed using an AiryScan LSM-880 laser-scanning confocal microscope (Zeiss).
In Vitro Chloride Efflux Assay
Chloride efflux was measured using the chloride-quenched fluorescent dye MQAE. Treated and untreated cells were incubated in medium containing MQAE and NucRed Live 647 nuclear stain (Molecular Probes) for 3 hr under standard incubation conditions. Dye-containing medium was removed, and cells were perfused with chloride-containing Ringer’s solution (135 mM NaCl, 3 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 20 mM HEPES, 1 mM KH2PO4, and 11 mM glucose [pH 7.4]) containing 5 μM forskolin and 100 μM 3-isobutyl-1-methylxanthine, facilitating chloride uptake and quenching of intracellular MQAE. Chloride efflux was measured upon replacing the chloride-containing perfusate with an isotonic 0Cl− solution containing 5 μM forskolin and 100 μM 3-isobutyl-1-methylxanthine, in which chloride is fully substituted by nitrate (0Cl− solution: 135 mM NaNO3, 3 mM KNO3, 1.8 mM Ca(NO3)2, 0.8 mM MgSO4, 20 mM HEPES, 1 mM KH2PO4, and 11 mM glucose [pH 7.4]).72, 119 Fluorescence data were collected using an EVOS FL Auto microscopy system (Life Technologies). MQAE fluorescence images were collected every 15 s for 10 m (DAPI channel). Change in fluorescence was calculated as the mean fluorescence across 25 cells at a given time normalized to the mean at the time of the change in perfusate (Ft/F0). Data were transformed to account for logarithmic decay in fluorescent signal, which was determined on a per-sample basis from the signal bleaching during the window from 300–600 s.
Animals
All animal studies were conducted at Oregon Health and Sciences University and approved by the Institutional Animal Care and Use Committee (IACUC). Female BALB/c mice (Charles River Laboratories) were sedated using isoflurane (2%), and LNP-cmFluc was pipetted onto the nostrils for spontaneous inhalation into the lungs (0.6 mg/kg). Bioluminescent imaging was performed on isolated organs using the IVIS Lumina XRMS imaging system (PerkinElmer) following intraperitoneal injection of 200 μL of D-luciferin substrate (PerkinElmer, 150 mg/kg body weight). Image acquisition and analysis were conducted using IVIS Living Image software (PerkinElmer). Cftr−/−tm1Unc Tg(FABPCFTR)1 Jaw/J double-transgenic CFKO mice were obtained from The Jackson Laboratory (JAX 002364). Mice were produced with full knockout of the endogenous mouse gene Cftr. To avoid complications in the intestines associated with Cftr knockout, the human CFTR (hCFTR) transgene was complimented back using fatty acid binding protein 2 (FABP2) as a promoter, resulting in localized hCFTR expression in the gastrointestinal tract.76 For particle administration, mice were anesthetized with an intraperitoneal injection of a mixture of ketamine and xylazine (100 μg/10 μg/kg body weight). LNPs were administered on two consecutive days to a single nostril (2 μL/application, 10 applications over 20 min, 0.1 mg/kg/day).
Nasal Brushing and PCR Amplification
Nasal brushings were taken 1 day after particle administration was completed. Mice were sacrificed by anesthesia overdose by intraperitoneal injection of a mixture of ketamine and xylazine (300 μg/30 μg per kg body weight). Mice were exsanguinated, and the nares (left and right) were brushed twice each to a depth of 3–5 mm using a 0.4 mm interdental brush (TePe, Sweden). Brushings underwent 1 freeze/thaw cycle and 2 passages through a 30G needle. RNA was isolated from lysates or 40 μL LNP-cmCFTR (process control) using the RNEasy Plus Micro Kit (QIAGEN), and cDNA libraries were generated by RT-PCR using the SuperScript VILO cDNA Synthesis Kit (Invitrogen) according to the provided protocols. To demonstrate the presence of LNP-delivered hCFTR, primers were designed to amplify exon 11 (forward: 5′-AAC TGG AGC CTT CAG AGG GT-3′; reverse: 5′-TTG GCA TGC TTT GAT GAC GC-3′) using GAPDH as a loading control (forward: 5′-ACC ACA GTC CAT GCC ATC AC-3′; reverse: 5′-TCC ACC CTG TTG CTG TA-3′). All primers were purchased from Integrated DNA Technologies. PCR was carried out using SuperScript (Invitrogen) in a 30-cycle reaction with a 10-min, 95°C polymerase activation step. Each repeating cycle consisted of two steps: 15 s at 95°C and 1 min at 60°C. PCR products were analyzed on a 2% agarose gel stained with ethidium bromide.
NPD Assay
NPD was measured using a modification of the previously described methods and using a previously described circuit.77, 120 Briefly, mice were positioned at a 15° head-down tilt, and a high-impedance voltmeter (World Precision Instruments, Sarasota, FL) attached to chloride pellet electrodes was used to measure the potential difference between an exploring nasal bridge and subcutaneous reference probe. A syringe pump continuously perfused solution into the nostril through a polyethylene tube stretched to approximately 0.5 mm in diameter (PE10, 0.28 mm inner diameter [ID]; Clay-Adams, BD, Sparks, MD). Solutions were pumped into the nasal cavity sequentially (Table 2). DMSO (0.5%) was added to the CFTRInh-172 solution to improve solubility; this has been shown previously not to affect NPD measurements.
Table 2.
Perfusion Solutions Used in NPD Studies
| Ringer’s | Amiloride | 0Cl−/iso | CFTRInh-172 | ATP | |
|---|---|---|---|---|---|
| NaCl (mM) | 135 | 135 | – | – | – |
| Na gluconate (mM) | – | – | 135 | 135 | 135 |
| CaCl2 ⋅ 2H2O (mM) | 2.25 | 2.25 | – | – | – |
| Ca gluconate (mM) | – | – | 2.25 | 2.25 | 2.25 |
| MgCl2 ⋅ 6H2O (mM) | 1.2 | 1.2 | – | – | – |
| MgSO4 ⋅ 7H2O (mM) | – | – | 1.2 | 1.2 | 1.2 |
| K2HPO4 (mM) | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 |
| KH2PO4 (mM) | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 |
| Amiloride (μM) | – | 100 | 100 | 100 | 100 |
| Isoproterenol (μM) | – | – | 100 | 100 | 100 |
| CFTRInh-172 (μM) | – | – | – | 20 | – |
| ATP (μM) | – | – | – | – | 100 |
Statistical Analysis
Significance was determined using Student’s t test for all analyses.
Author Contributions
G.S. directed the project and conceived ideas to conduct studies. E.R. was involved in conducting all experiments. E.R. and S.P. formulated and characterized LNPs with cmRNA. E.R. performed western blot analysis, confocal microscopy, polarized cell culture, and the MQAE assay with assistance from K.S. and M.M. K.D.M. oversaw all in vivo work, maintained the animal colony, and performed the NPD assay with assistance from E.R. C.S. provided ideas for in vivo lung biodistribution studies. G.S., K.D.M., and E.R. analyzed all data and wrote the paper. All authors refined the manuscript.
Conflicts of Interest
The authors have no conflict of interest.
Acknowledgments
This project was supported by funding from the Cystic Fibrosis Foundation (to G.S. and K.D.M.), OSU College of Pharmacy startup funding (to G.S.), the National Institute of Biomedical Imaging and Bioengineering (1R15EB021581-01 to G.S.), and the National Institute of General Medical Sciences (NIGMS) (1R35GM119839-01 to C.S.). We thank Ajay Sapre for assistance with collecting single-particle tracking data and Emily Pulliam for maintenance of the CFKO mouse colony.
References
- 1.Cystic Fibrosis Foundation (2018) About Cystic Fibrosis. https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/.
- 2.Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 3.Welsh M.J. Abnormal regulation of ion channels in cystic fibrosis epithelia. FASEB J. 1990;4:2718–2725. doi: 10.1096/fasebj.4.10.1695593. [DOI] [PubMed] [Google Scholar]
- 4.Chu C.S., Trapnell B.C., Curristin S., Cutting G.R., Crystal R.G. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat. Genet. 1993;3:151–156. doi: 10.1038/ng0293-151. [DOI] [PubMed] [Google Scholar]
- 5.Welsh M.J., Anderson M.P., Rich D.P., Berger H.A., Denning G.M., Ostedgaard L.S., Sheppard D.N., Cheng S.H., Gregory R.J., Smith A.E. Cystic fibrosis transmembrane conductance regulator: a chloride channel with novel regulation. Neuron. 1992;8:821–829. doi: 10.1016/0896-6273(92)90196-k. [DOI] [PubMed] [Google Scholar]
- 6.Clinical and Functional Translation of CFTR (2018). CFTR2 Variant List History. https://cftr2.org/.
- 7.Boucher R.C. Pathogenesis of cystic fibrosis airways disease. Trans. Am. Clin. Climatol. Assoc. 2001;112:99–107. [PMC free article] [PubMed] [Google Scholar]
- 8.Boucher R.C. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 9.Stoltz D.A., Meyerholz D.K., Welsh M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015;372:351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong D.S., Grimwood K., Carlin J.B., Carzino R., Gutièrrez J.P., Hull J., Olinsky A., Phelan E.M., Robertson C.F., Phelan P.D. Lower airway inflammation in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1997;156:1197–1204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- 11.National Jewish Health (2018) Cystic Fibrosis: Life Expectancy. https://www.nationaljewish.org/conditions/cystic-fibrosis-cf/life-expectancy.
- 12.Van Goor F., Hadida S., Grootenhuis P.D.J., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Accurso F.J., Rowe S.M., Clancy J.P., Boyle M.P., Dunitz J.M., Durie P.R., Sagel S.D., Hornick D.B., Konstan M.W., Donaldson S.H. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Goor F., Hadida S., Grootenhuis P.D.J., Burton B., Stack J.H., Straley K.S., Decker C.J., Miller M., McCartney J., Olson E.R. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowe S.M., Daines C., Ringshausen F.C., Kerem E., Wilson J., Tullis E., Nair N., Simard C., Han L., Ingenito E.P. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017;377:2024–2035. doi: 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren C.L., Morgan R.L., Oermann C., Resnick H.E., Brady C., Campbell A. Cystic Fibrosis Foundation Pulmonary Guidelines: Use of CFTR Modulator Therapy in Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2018;15:271–280. doi: 10.1513/AnnalsATS.201707-539OT. [DOI] [PubMed] [Google Scholar]
- 17.Cholon D.M., Quinney N.L., Fulcher M.L., Esther C.R., Jr., Das J., Dokholyan N.V., Randell S.H., Boucher R.C., Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014;6:246ra96. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veit G., Avramescu R.G., Perdomo D., Phuan P.-W., Bagdany M., Apaja P.M., Borot F., Szollosi D., Wu Y.S., Finkbeiner W.E. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci. Transl. Med. 2014;6:246ra97. doi: 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubert D., Chiron R., Camara B., Grenet D., Prévotat A., Bassinet L., Dominique S., Rault G., Macey J., Honoré I. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J. Cyst. Fibros. 2017;16:388–391. doi: 10.1016/j.jcf.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Jennings M.T., Dezube R., Paranjape S., West N.E., Hong G., Braun A., Grant J., Merlo C.A., Lechtzin N. An Observational Study of Outcomes and Tolerances in Patients with Cystic Fibrosis Initiated on Lumacaftor/Ivacaftor. Ann. Am. Thorac. Soc. 2017;14:1662–1666. doi: 10.1513/AnnalsATS.201701-058OC. [DOI] [PubMed] [Google Scholar]
- 21.Hajj K.A., Whitehead K.A. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017;2 natrevmats201756. [Google Scholar]
- 22.Zabner J., Fasbender A.J., Moninger T., Poellinger K.A., Welsh M.J. Cellular and molecular barriers to gene transfer by a cationic lipid. J. Biol. Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 23.Vaughan E.E., Dean D.A. Intracellular trafficking of plasmids during transfection is mediated by microtubules. Mol. Ther. 2006;13:422–428. doi: 10.1016/j.ymthe.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahin U., Karikó K., Türeci Ö. mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 25.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 27.Karikó K., Ni H., Capodici J., Lamphier M., Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 28.Karikó K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pardi N., Muramatsu H., Weissman D., Karikó K. In Vitro Transcription of Long RNA Containing Modified Nucleosides. In: Rabinovich P.M., editor. Synth. Messenger RNA Cell Metab. Modul. Humana Press; 2013. pp. 29–42. [DOI] [PubMed] [Google Scholar]
- 31.Warren L., Manos P.D., Ahfeldt T., Loh Y.-H., Li H., Lau F., Ebina W., Mandal P.K., Smith Z.D., Meissner A. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 33.Cullis P.R., Hope M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. J. Am. Soc. Gene Ther. 2017;25:1467–1475. doi: 10.1016/j.ymthe.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stöter M. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 35.Paunovska K., Sago C.D., Monaco C.M., Hudson W.H., Castro M.G., Rudoltz T.G., Kalathoor S., Vanover D.A., Santangelo P.J., Ahmed R. A Direct Comparison of in Vitro and in Vivo Nucleic Acid Delivery Mediated by Hundreds of Nanoparticles Reveals a Weak Correlation. Nano Lett. 2018;18:2148–2157. doi: 10.1021/acs.nanolett.8b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gindy M.E., DiFelice K., Kumar V., Prud’homme R.K., Celano R., Haas R.M., Smith J.S., Boardman D. Mechanism of macromolecular structure evolution in self-assembled lipid nanoparticles for siRNA delivery. Langmuir. 2014;30:4613–4622. doi: 10.1021/la500630h. [DOI] [PubMed] [Google Scholar]
- 37.Reichmuth A.M., Oberli M.A., Jeklenec A., Langer R., Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016;7:319–334. doi: 10.4155/tde-2016-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberli M.A., Reichmuth A.M., Dorkin J.R., Mitchell M.J., Fenton O.S., Jaklenec A., Anderson D.G., Langer R., Blankschtein D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017;17:1326–1335. doi: 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue H.Y., Guo P., Wen W.-C., Wong H.L. Lipid-Based Nanocarriers for RNA Delivery. Curr. Pharm. Des. 2015;21:3140–3147. doi: 10.2174/1381612821666150531164540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulkarni J.A., Darjuan M.M., Mercer J.E., Chen S., van der Meel R., Thewalt J.L., Tam Y.Y.C., Cullis P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano. 2018;12:4787–4795. doi: 10.1021/acsnano.8b01516. [DOI] [PubMed] [Google Scholar]
- 41.Semple S.C., Klimuk S.K., Harasym T.O., Dos Santos N., Ansell S.M., Wong K.F., Maurer N., Stark H., Cullis P.R., Hope M.J., Scherrer P. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta. 2001;1510:152–166. doi: 10.1016/s0005-2736(00)00343-6. [DOI] [PubMed] [Google Scholar]
- 42.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W., Stebbing D., Crosley E.J., Yaworski E. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 43.Viger-Gravel J., Schantz A., Pinon A.C., Rossini A.J., Schantz S., Emsley L. Structure of Lipid Nanoparticles Containing siRNA or mRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. J. Phys. Chem. B. 2018;122:2073–2081. doi: 10.1021/acs.jpcb.7b10795. [DOI] [PubMed] [Google Scholar]
- 44.Zelphati O., Szoka F.C., Jr. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. USA. 1996;93:11493–11498. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lechanteur A., Sanna V., Duchemin A., Evrard B., Mottet D., Piel G. Cationic Liposomes Carrying siRNA: Impact of Lipid Composition on Physicochemical Properties, Cytotoxicity and Endosomal Escape. Nanomater. Basel. 2018;8:E270. doi: 10.3390/nano8050270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018 doi: 10.1016/j.ymthe.2018.03.010. Published online March 14, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siegwart D.J., Whitehead K.A., Nuhn L., Sahay G., Cheng H., Jiang S., Ma M., Lytton-Jean A., Vegas A., Fenton P. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc. Natl. Acad. Sci. USA. 2011;108:12996–13001. doi: 10.1073/pnas.1106379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leung A.K.K., Hafez I.M., Baoukina S., Belliveau N.M., Zhigaltsev I.V., Afshinmanesh E., Tieleman D.P., Hansen C.L., Hope M.J., Cullis P.R. Lipid Nanoparticles Containing siRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core. J Phys Chem C Nanomater Interfaces. 2012;116:18440–18450. doi: 10.1021/jp303267y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crawford R., Dogdas B., Keough E., Haas R.M., Wepukhulu W., Krotzer S., Burke P.A., Sepp-Lorenzino L., Bagchi A., Howell B.J. Analysis of lipid nanoparticles by Cryo-EM for characterizing siRNA delivery vehicles. Int. J. Pharm. 2011;403:237–244. doi: 10.1016/j.ijpharm.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 50.Owens D.E., 3rd, Peppas N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 51.Storm G., Belliot S.O., Daemen T., Lasic D.D. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv. Drug Deliv. Rev. 1995;17:31–48. [Google Scholar]
- 52.Klibanov A.L., Maruyama K., Torchilin V.P., Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 53.ClinicalTrials.gov. (2018). Expanded Access Protocol of Patisiran for Patients With Hereditary ATTR Amyloidosis (hATTR). https://clinicaltrials.gov/ct2/show/NCT02939820.
- 54.Bahl K., Senn J.J., Yuzhakov O., Bulychev A., Brito L.A., Hassett K.J., Laska M.E., Smith M., Almarsson Ö., Thompson J. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017;25:1316–1327. doi: 10.1016/j.ymthe.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lutz J., Lazzaro S., Habbeddine M., Schmidt K.E., Baumhof P., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Heidenreich R., Fotin-Mleczek M. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines. 2017;2:29. doi: 10.1038/s41541-017-0032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai S.K., O’Hanlon D.E., Harrold S., Man S.T., Wang Y.-Y., Cone R., Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl. Acad. Sci. USA. 2007;104:1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y.-Y., Lai S.K., Suk J.S., Pace A., Cone R., Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew. Chem. Int. Ed. Engl. 2008;47:9726–9729. doi: 10.1002/anie.200803526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suk J.S., Lai S.K., Wang Y.-Y., Ensign L.M., Zeitlin P.L., Boyle M.P., Hanes J. The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles. Biomaterials. 2009;30:2591–2597. doi: 10.1016/j.biomaterials.2008.12.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ensign L.M., Schneider C., Suk J.S., Cone R., Hanes J. Mucus penetrating nanoparticles: biophysical tool and method of drug and gene delivery. Adv. Mater. 2012;24:3887–3894. doi: 10.1002/adma.201201800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schuster B.S., Suk J.S., Woodworth G.F., Hanes J. Nanoparticle diffusion in respiratory mucus from humans without lung disease. Biomaterials. 2013;34:3439–3446. doi: 10.1016/j.biomaterials.2013.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suk J.S., Xu Q., Kim N., Hanes J., Ensign L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016;99(Pt A):28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boylan N.J., Suk J.S., Lai S.K., Jelinek R., Boyle M.P., Cooper M.J., Hanes J. Highly compacted DNA nanoparticles with low MW PEG coatings: in vitro, ex vivo and in vivo evaluation. J. Control. Release. 2012;157:72–79. doi: 10.1016/j.jconrel.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cholon D.M., O’Neal W.K., Randell S.H., Riordan J.R., Gentzsch M. Modulation of endocytic trafficking and apical stability of CFTR in primary human airway epithelial cultures. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;298:L304–L314. doi: 10.1152/ajplung.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farinha C.M., Canato S. From the endoplasmic reticulum to the plasma membrane: mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017;74:39–55. doi: 10.1007/s00018-016-2387-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farinha C.M., Miller E., McCarty N. Protein and lipid interactions – Modulating CFTR trafficking and rescue. J. Cyst. Fibros. 2018;17(2S):S9–S13. doi: 10.1016/j.jcf.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 66.Ernst W.L., Shome K., Wu C.C., Gong X., Frizzell R.A., Aridor M. VAMP-associated Proteins (VAP) as Receptors That Couple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Proteostasis with Lipid Homeostasis. J. Biol. Chem. 2016;291:5206–5220. doi: 10.1074/jbc.M115.692749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berdiev B.K., Qadri Y.J., Benos D.J. Assessment of the CFTR and ENaC association. Mol. Biosyst. 2009;5:123–127. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peters K.W., Okiyoneda T., Balch W.E., Braakman I., Brodsky J.L., Guggino W.B., Penland C.M., Pollard H.B., Sorscher E.J., Skach W.R. CFTR Folding Consortium: methods available for studies of CFTR folding and correction. Methods Mol. Biol. 2011;742:335–353. doi: 10.1007/978-1-61779-120-8_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Puchelle E., Gaillard D., Ploton D., Hinnrasky J., Fuchey C., Boutterin M.C., Jacquot J., Dreyer D., Pavirani A., Dalemans W. Differential localization of the cystic fibrosis transmembrane conductance regulator in normal and cystic fibrosis airway epithelium. Am. J. Respir. Cell Mol. Biol. 1992;7:485–491. doi: 10.1165/ajrcmb/7.5.485. [DOI] [PubMed] [Google Scholar]
- 70.Yang Y., Janich S., Cohn J.A., Wilson J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dosanjh A., Lencer W., Brown D., Ausiello D.A., Stow J.L. Heterologous expression of delta F508 CFTR results in decreased sialylation of membrane glycoconjugates. Am. J. Physiol. 1994;266:C360–C366. doi: 10.1152/ajpcell.1994.266.2.C360. [DOI] [PubMed] [Google Scholar]
- 72.McNeer N.A., Anandalingam K., Fields R.J., Caputo C., Kopic S., Gupta A., Quijano E., Polikoff L., Kong Y., Bahal R. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat. Commun. 2015;6:6952. doi: 10.1038/ncomms7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verkman A.S., Sellers M.C., Chao A.C., Leung T., Ketcham R. Synthesis and characterization of improved chloride-sensitive fluorescent indicators for biological applications. Anal. Biochem. 1989;178:355–361. doi: 10.1016/0003-2697(89)90652-0. [DOI] [PubMed] [Google Scholar]
- 74.West M.R., Molloy C.R. A microplate assay measuring chloride ion channel activity. Anal. Biochem. 1996;241:51–58. doi: 10.1006/abio.1996.0377. [DOI] [PubMed] [Google Scholar]
- 75.Illek B., Maurisse R., Wahler L., Kunzelmann K., Fischer H., Gruenert D.C. Cl transport in complemented CF bronchial epithelial cells correlates with CFTR mRNA expression levels. Cell. Physiol. Biochem. 2008;22:57–68. doi: 10.1159/000149783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou L., Dey C.R., Wert S.E., DuVall M.D., Frizzell R.A., Whitsett J.A. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]
- 77.MacDonald K.D., McKenzie K.R., Henderson M.J., Hawkins C.E., Vij N., Zeitlin P.L. Lubiprostone activates non-CFTR-dependent respiratory epithelial chloride secretion in cystic fibrosis mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;295:L933–L940. doi: 10.1152/ajplung.90221.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knowles M.R., Carson J.L., Collier A.M., Gatzy J.T., Boucher R.C. Measurements of nasal transepithelial electric potential differences in normal human subjects in vivo. Am. Rev. Respir. Dis. 1981;124:484–490. doi: 10.1164/arrd.1981.124.4.484. [DOI] [PubMed] [Google Scholar]
- 79.Grubb B.R., Pickles R.J., Ye H., Yankaskas J.R., Vick R.N., Engelhardt J.F., Wilson J.M., Johnson L.G., Boucher R.C. Inefficient gene transfer by adenovirus vector to cystic fibrosis airway epithelia of mice and humans. Nature. 1994;371:802–806. doi: 10.1038/371802a0. [DOI] [PubMed] [Google Scholar]
- 80.Rowe S.M., Clancy J.-P., Wilschanski M. Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol. Biol. 2011;741:69–86. doi: 10.1007/978-1-61779-117-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Livraghi-Butrico A., Kelly E.J., Wilkinson K.J., Rogers T.D., Gilmore R.C., Harkema J.R., Randell S.H., Boucher R.C., O’Neal W.K., Grubb B.R. Loss of Cftr function exacerbates the phenotype of Na(+) hyperabsorption in murine airways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013;304:L469–L480. doi: 10.1152/ajplung.00150.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Briel M., Greger R., Kunzelmann K. Cl- transport by cystic fibrosis transmembrane conductance regulator (CFTR) contributes to the inhibition of epithelial Na+ channels (ENaCs) in Xenopus oocytes co-expressing CFTR and ENaC. J. Physiol. 1998;508:825–836. doi: 10.1111/j.1469-7793.1998.825bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knowles M.R., Paradiso A.M., Boucher R.C. In vivo nasal potential difference: techniques and protocols for assessing efficacy of gene transfer in cystic fibrosis. Hum. Gene Ther. 1995;6:445–455. doi: 10.1089/hum.1995.6.4-445. [DOI] [PubMed] [Google Scholar]
- 84.Alton E.W.F.W., Boyd A.C., Porteous D.J., Davies G., Davies J.C., Griesenbach U., Higgins T.E., Gill D.R., Hyde S.C., Innes J.A., UK Cystic Fibrosis Gene Therapy Consortium * A Phase I/IIa Safety and Efficacy Study of Nebulized Liposome-mediated Gene Therapy for Cystic Fibrosis Supports a Multidose Trial. Am. J. Respir. Crit. Care Med. 2015;192:1389–1392. doi: 10.1164/rccm.201506-1193LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alton, E.W., Armstrong, D.K., Ashby, D., Bayfield, K.J., Bilton, D., Bloomfield, E.V., et al. (2016). A randomised, double-blind, placebo-controlled trial of repeated nebulisation of non-viral cystic fibrosis transmembrane conductance regulator (CFTR) gene therapy in patients with cystic fibrosis. NIHR Journals Library, Southampton (UK).https://www.ncbi.nlm.nih.gov/books/NBK373650/. [PubMed]
- 86.Caplen N.J., Alton E.W.F.W., Middleton P.G., Dorin J.R., Stevenson B.J., Gao X., Durham S.R., Jeffery P.K., Hodson M.E., Coutelle C. Liposome-mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Nat. Med. 1995;1:39–46. doi: 10.1038/nm0195-39. [DOI] [PubMed] [Google Scholar]
- 87.Drumm M.L., Pope H.A., Cliff W.H., Rommens J.M., Marvin S.A., Tsui L.-C., Collins F.S., Frizzell R.A., Wilson J.M. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990;62:1227–1233. doi: 10.1016/0092-8674(90)90398-x. [DOI] [PubMed] [Google Scholar]
- 88.Rommens J.M., Iannuzzi M.C., Kerem B., Drumm M.L., Melmer G., Dean M., Rozmahel R., Cole J.L., Kennedy D., Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 89.Goldman M.J., Lee P.S., Yang J.S., Wilson J.M. Lentiviral vectors for gene therapy of cystic fibrosis. Hum. Gene Ther. 1997;8:2261–2268. doi: 10.1089/hum.1997.8.18-2261. [DOI] [PubMed] [Google Scholar]
- 90.Crystal R.G., McElvaney N.G., Rosenfeld M.A., Chu C.-S., Mastrangeli A., Hay J.G., Brody S.L., Jaffe H.A., Eissa N.T., Danel C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet. 1994;8:42–51. doi: 10.1038/ng0994-42. [DOI] [PubMed] [Google Scholar]
- 91.Harvey B.-G., Leopold P.L., Hackett N.R., Grasso T.M., Williams P.M., Tucker A.L., Kaner R.J., Ferris B., Gonda I., Sweeney T.D. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J. Clin. Invest. 1999;104:1245–1255. doi: 10.1172/JCI7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cooney A.L., Abou Alaiwa M.H., Shah V.S., Bouzek D.C., Stroik M.R., Powers L.S., Gansemer N.D., Meyerholz D.K., Welsh M.J., Stoltz D.A. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight. 2016;1:e88730. doi: 10.1172/jci.insight.88730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baum C., von Kalle C., Staal F.J.T., Li Z., Fehse B., Schmidt M., Weerkamp F., Karlsson S., Wagemaker G., Williams D.A. Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol. Ther. 2004;9:5–13. doi: 10.1016/j.ymthe.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 94.Guggino W.B., Cebotaru L. Adeno-Associated Virus (AAV) gene therapy for cystic fibrosis: current barriers and recent developments. Expert Opin. Biol. Ther. 2017;17:1265–1273. doi: 10.1080/14712598.2017.1347630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang C., Akita G.Y., Colledge W.H., Ratcliff R.A., Evans M.J., Hehir K.M., St George J.A., Wadsworth S.C., Cheng S.H. Increased contact time improves adenovirus-mediated CFTR gene transfer to nasal epithelium of CF mice. Hum. Gene Ther. 1997;8:671–680. doi: 10.1089/hum.1997.8.6-671. [DOI] [PubMed] [Google Scholar]
- 96.Aitken M.L., Moss R.B., Waltz D.A., Dovey M.E., Tonelli M.R., McNamara S.C., Gibson R.L., Ramsey B.W., Carter B.J., Reynolds T.C. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther. 2001;12:1907–1916. doi: 10.1089/104303401753153956. [DOI] [PubMed] [Google Scholar]
- 97.Moss R.B., Milla C., Colombo J., Accurso F., Zeitlin P.L., Clancy J.P., Spencer L.T., Pilewski J., Waltz D.A., Dorkin H.L. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007;18:726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- 98.Alton E.W.F.W., Armstrong D.K., Ashby D., Bayfield K.J., Bilton D., Bloomfield E.V., Boyd A.C., Brand J., Buchan R., Calcedo R., UK Cystic Fibrosis Gene Therapy Consortium Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015;3:684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leung A.K.K., Tam Y.Y.C., Chen S., Hafez I.M., Cullis P.R. Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J. Phys. Chem. B. 2015;119:8698–8706. doi: 10.1021/acs.jpcb.5b02891. [DOI] [PubMed] [Google Scholar]
- 100.Guimarães Sá Correia M., Briuglia M.L., Niosi F., Lamprou D.A. Microfluidic manufacturing of phospholipid nanoparticles: Stability, encapsulation efficacy, and drug release. Int. J. Pharm. 2017;516:91–99. doi: 10.1016/j.ijpharm.2016.11.025. [DOI] [PubMed] [Google Scholar]
- 101.Belliveau N.M., Huft J., Lin P.J., Chen S., Leung A.K., Leaver T.J., Wild A.W., Lee J.B., Taylor R.J., Tam Y.K. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids. 2012;1:e37. doi: 10.1038/mtna.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen D., Love K.T., Chen Y., Eltoukhy A.A., Kastrup C., Sahay G., Jeon A., Dong Y., Whitehead K.A., Anderson D.G. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc. 2012;134:6948–6951. doi: 10.1021/ja301621z. [DOI] [PubMed] [Google Scholar]
- 103.Di Gioia S., Conese M. Polyethylenimine-mediated gene delivery to the lung and therapeutic applications. Drug Des. Devel. Ther. 2009;2:163–188. doi: 10.2147/dddt.s2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oh P., Borgström P., Witkiewicz H., Li Y., Borgström B.J., Chrastina A., Iwata K., Zinn K.R., Baldwin R., Testa J.E., Schnitzer J.E. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotechnol. 2007;25:327–337. doi: 10.1038/nbt1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaczmarek J.C., Patel A.K., Kauffman K.J., Fenton O.S., Webber M.J., Heartlein M.W., DeRosa F., Anderson D.G. Polymer-Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. Int. Ed. Engl. 2016;55:13808–13812. doi: 10.1002/anie.201608450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sahay G., Querbes W., Alabi C., Eltoukhy A., Sarkar S., Zurenko C., Karagiannis E., Love K., Chen D., Zoncu R. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013;31:653–658. doi: 10.1038/nbt.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stewart M.P., Lorenz A., Dahlman J., Sahay G. Challenges in carrier-mediated intracellular delivery: moving beyond endosomal barriers. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016;8:465–478. doi: 10.1002/wnan.1377. [DOI] [PubMed] [Google Scholar]
- 108.Wittrup A., Ai A., Liu X., Hamar P., Trifonova R., Charisse K., Manoharan M., Kirchhausen T., Lieberman J. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015;33:870–876. doi: 10.1038/nbt.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dowdy S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017;35:222–229. doi: 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 110.Patel S., Ashwanikumar N., Robinson E., DuRoss A., Sun C., Murphy-Benenato K.E., Mihai C., Almarsson Ö., Sahay G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017;17:5711–5718. doi: 10.1021/acs.nanolett.7b02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Davis D.R. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res. 1995;23:5020–5026. doi: 10.1093/nar/23.24.5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haque A.K.M.A., Dewerth A., Antony J.S., Riethmüller J., Latifi N., Yasar H. Modified hCFTR mRNA restores normal lung function in a mouse model of cystic fibrosis. bioRxiv. 2017 doi: 10.1038/s41598-018-34960-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.ClinicalTrials.gov (2012). Safety Study of Ivacaftor in Subjects With Cystic Fibrosis. https://clinicaltrials.gov/ct2/show/NCT00457821.
- 114.Ramalho A.S., Beck S., Meyer M., Penque D., Cutting G.R., Amaral M.D. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2002;27:619–627. doi: 10.1165/rcmb.2001-0004OC. [DOI] [PubMed] [Google Scholar]
- 115.Hwang T.C., Wang F., Yang I.C., Reenstra W.W. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am. J. Physiol. 1997;273:C988–C998. doi: 10.1152/ajpcell.1997.273.3.C988. [DOI] [PubMed] [Google Scholar]
- 116.Van Goor F., Straley K.S., Cao D., González J., Hadida S., Hazlewood A., Joubran J., Knapp T., Makings L.R., Miller M. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 117.Villate-Beitia I., Zarate J., Puras G., Pedraz J.L. Gene delivery to the lungs: pulmonary gene therapy for cystic fibrosis. Drug Dev. Ind. Pharm. 2017;43:1071–1081. doi: 10.1080/03639045.2017.1298122. [DOI] [PubMed] [Google Scholar]
- 118.Hagemeijer M.C., Siegwart D.J., Strug L.J., Cebotaru L., Torres M.J., Sofoluwe A. Translational research to enable personalized treatment of cystic fibrosis. J. Cyst. Fibros. 2017;17:S46–S51. doi: 10.1016/j.jcf.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 119.Carbone A., Castellani S., Favia M., Diana A., Paracchini V., Di Gioia S., Seia M., Casavola V., Colombo C., Conese M. Correction of defective CFTR/ENaC function and tightness of cystic fibrosis airway epithelium by amniotic mesenchymal stromal (stem) cells. J. Cell. Mol. Med. 2014;18:1631–1643. doi: 10.1111/jcmm.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Das S., MacDonald K., Chang H.-Y.S., Mitzner W. A Simple Method of Mouse Lung Intubation. J. Vis. Exp. 2013;73:e50318. doi: 10.3791/50318. [DOI] [PMC free article] [PubMed] [Google Scholar]