

Abstract
Backgrounds
Apis mellifera scutellata and A.m. capensis (the Cape honey bee) are western honey bee subspecies indigenous to the Republic of South Africa (RSA). Both bees are important for biological and economic reasons. First, A.m. scutellata is the invasive “African honey bee” of the Americas and exhibits a number of traits that beekeepers consider undesirable. They swarm excessively, are prone to absconding (vacating the nest entirely), usurp other honey bee colonies, and exhibit heightened defensiveness. Second, Cape honey bees are socially parasitic bees; the workers can reproduce thelytokously. Both bees are indistinguishable visually. Therefore, we employed Genotyping-by-Sequencing (GBS), wing geometry and standard morphometric approaches to assess the genetic diversity and population structure of these bees to search for diagnostic markers that can be employed to distinguish between the two subspecies.
Results
Apis mellifera scutellata possessed the highest mean number of polymorphic SNPs (among 2449 informative SNPs) with minor allele frequencies > 0.05 (Np = 88%). The RSA honey bees generated a high level of expected heterozygosity (Hexp = 0.24). The mean genetic differentiation (FST; 6.5%) among the RSA honey bees revealed that approximately 93% of the genetic variation was accounted for within individuals of these subspecies. Two genetically distinct clusters (K = 2) corresponding to both subspecies were detected by Model-based Bayesian clustering and supported by Principal Coordinates Analysis (PCoA) inferences. Selected highly divergent loci (n = 83) further reinforced a distinctive clustering of two subspecies across geographical origins, accounting for approximately 83% of the total variation in the PCoA plot. The significant correlation of allele frequencies at divergent loci with environmental variables suggested that these populations are adapted to local conditions. Only 17 of 48 wing geometry and standard morphometric parameters were useful for clustering A.m. capensis, A.m. scutellata, and hybrid individuals.
Conclusions
We produced a minimal set of 83 SNP loci and 17 wing geometry and standard morphometric parameters useful for identifying the two RSA honey bee subspecies by genotype and phenotype. We found that genes involved in neurology/behavior and development/growth are the most prominent heritable traits evolved in the functional evolution of honey bee populations in RSA. These findings provide a starting point for understanding the functional basis of morphological differentiations and ecological adaptations of the two honey bee subspecies in RSA.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4998-x) contains supplementary material, which is available to authorized users.
Keywords: Apis mellifera capensis, Apis mellifera scutellata, Genetic differentiation, Population structure, GBS-SNP, Wing geometry, Standard morphometrics
Background
Within the insect family Apidae, western honey bees, Apis mellifera L. (Hymenoptera: Apidae), are cosmopolitan eusocial insects that play an important role in the cultivation of various crops and maintenance of healthy ecosystems globally [1, 2]. The genus Apis, which is comprised of nine honey bee species, is believed to have an evolutionary origin in Asia [3]. From there, honey bees have adapted to a diverse range of ecological conditions globally, diverging into eight Asian species and a ninth species, the western honey bee, which is endemic to Europe, Africa and the Middle East [4, 5]. Six evolutionary groups composed of about 25–30 subspecies of A. mellifera have been identified: (A) African subspecies, (M) northern and western European subspecies, (C) North Mediterranean subspecies, (O and Z) Middle Eastern subspecies, and (Y) in Ethiopia [4, 6–8].
Africa, specifically, is home to at least 11 A. mellifera subspecies distributed across the continent with substantial geographical variability among the areas in which the lineages are endemic [9]. It has been suggested that selective adaptation of honey bees to the huge variance of biotopes where they occur is the primary mechanism driving subspecies differentiation in Africa [10]. Two subspecies of African honey bees, A.m. scutellata from the Savannah areas of central and southern Africa, and A.m. capensis from the southern part of the Western and Eastern Cape of the Republic of South Africa (RSA), are of particular interest due to the behavioral characteristics they present [11].
Outside of its endemic range, A.m. scutellata is referred to as the “African,” “Africanized”, or “killer” bee of the Americas where it is considered invasive. A.m. scutellata and its hybrid populations have spread throughout South America, Central America and the southern parts of North America [12–14]. Apis mellifera scutellata exhibits several behaviors that beekeepers consider undesirable, but that are biologically important to the bee. These include excessive swarming, absconding, aggressive usurpation and heightened defensiveness [9, 12, 15]. Additionally, this bee competes for limited resources against, and hybridizes with, European-derived honey bees. These traits negatively impact beekeepers, bee colonies, and general public opinion of the honey bees. Furthermore, the ecological impact of A.m. scutellata in the Americas has not been quantified but is likely significant.
Apis mellifera capensis is a facultative social parasite that can reproduce thelytokously (unfertilized eggs can develop into diploid females). These bees are characterized by a unique set of genetic, behavioral and physiological traits expressed by the worker bees [9, 16–19]. The workers can develop into pseudoqueens (female bees that are neither queens nor workers, but possess qualities of both [20, 21]. Furthermore, they may have high number of ovarioles [22], well-developed spermathecae [23], shorter latency periods [24], and the ability to produce queen-like pheromones if a colony loses their queen [25, 26]. These traits facilitate the socially parasitic nature of some A.m. capensis workers (i.e. worker females can invade non-A.m. capensis honey bee colonies and become the resident reproductive female) [27, 28]. This led to the “capensis calamity” in the RSA when A.m. capensis colonies were moved by beekeepers to areas where A.m. scutellata were indigenous. Once there, A.m. capensis workers drifted into A.m. scutellata colonies and became social parasites of these colonies, thus leading to widespread colony collapse and the recognition of the threat A.m. capensis pose to non-capensis colonies [17].
The behavioral and ecological diversity of A.m. scutellata and A.m. capensis makes them ideal model organisms to investigate the genetic variability and population structure of African honey bee subspecies. Wild honey bees in sub-Saharan Africa are believed to have low levels of genetic differentiation which may be due to a high degree of panmixia and large dispersal capacity of colonies [11, 29]. The RSA subspecies are two exceptional cases, as they are reported to be structured genetically despite the lack of regional physical barriers existing between them [30]. Additionally, an intermediate zone between the distributions of A.m. scutellata and A.m. capensis is occupied by hybrids of the two subspecies [9]. Predictably, the hybrid bees have a mixed gene pool [17].
Knowledge concerning the population structure of A.m. capensis and A.m. scutellata and the stability of the hybrid zone over time is still incipient. Recent progress in the development of next-generation sequencing (NGS) platforms has enabled scientists to genotype large groups of individuals using a genotyping-by-sequencing (GBS) approach [31]. GBS is a highly multiplexed, high-throughput, low-cost method and is one of the simplest reduced representation genome approaches developed thus far [31, 32]. The large numbers of SNPs obtained with the GBS method result in an accurate assessment of genetic diversity and population structure and simplify the detection of adaptive putative loci associated with environmental pressure [33–35].
Herein, we investigated genetic differentiation and population structure within 464 A.m. capensis, A.m. scutellata and hybrid honey bees collected from 69 different apiaries, representing 28 geographical regions across the natural distribution of honey bees in the RSA. We also determined if allele frequencies at divergent loci were significantly correlated with environmental variables, in an effort to identify regions of the genome under natural selection. We further measured wing geometry and standard morphometric parameters to evaluate the differentiation pattern between A.m. capensis and A.m. scutellata, given that morphometrics is the current tool utilized to separate the subspecies [36] and the possibility that wing geometry could offer a quicker identification method with comparable accuracy [37, 38]. The resulting GBS and morphometric data provide information critically needed for designing diagnostic markers to differentiate between A.m. capensis and A.m. scutellata bees.
Methods
Honey bee collections
We collected samples of worker honey bees from 1000+ managed colonies across the RSA in 2013 and 2014. These collections spanned the native geographical distribution of A.m. capensis, A.m. scutellata and hybrids of the two in the RSA. The number of bees examined per region/apiary and geographic information are reported in Table 1, Fig. 1. We analyzed between one and 21 bees per apiary, from 69 different apiaries, representing 28 geographical regions in and 464 bees from the RSA. Ten European-derived A. mellifera samples collected from honey bee colonies located at Honey Bee research Extension Laboratory, University of Florida were included in the genetic analysis for reference purposes. The RSA honey bees were collected into 50 ml vials containing absolute ethanol, imported into the US per USDA APHIS protocol and approval, and stored at − 20 °C prior to morphological and molecular analyses.
Table 1.
Summary information for honey bee samples collected in the Republic of South Africa
| No. | Geographic region - apiary | Apiary identifier | N | Geographical coordinates |
|---|---|---|---|---|
| 1 | Bloemfontein – A | BL – A | 5 | 29.24°S – 26.95°E |
| 2 | Bloemfontein – B | BL – B | 6 | 29.20°S – 27.20°E |
| 3 | Bloemfontein – C | BL – C | 8 | 29.24°S – 26.94°E |
| 4 | Kroonstad – A | KR – A | 6 | 27.58°S – 27.30°E |
| 5 | Kroonstad – B | KR – B | 1 | 27.33°S – 27.50°E |
| 6 | Kroonstad – C | KR – C | 10 | 27.27°S – 27.50°E |
| 7 | Pretoria – A | PT – A | 3 | 25.74°S – 28.26°E |
| 8 | Pretoria – B | PT – B | 6 | 25.70°S – 28.10°E |
| 9 | Springbok – B | SP – B | 4 | 29.71°S – 17.78°E |
| 10 | Springbok – C | SP – C | 5 | 29.67°S – 17.81°E |
| 11 | Upington – A | UP – A | 5 | 28.48°S – 21.18°E |
| 12 | Upington – B | UP – B | 5 | 28.72°S – 20.98°E |
| 13 | Upington – C | UP – C | 9 | 28.52°S – 21.24°E |
| 14 | Bredasdorp – A | BD – A | 6 | 34.50°S – 20.35°E |
| 15 | Citrusdaal – A | CD – A | 8 | 32.86°S – 19.21°E |
| 16 | Citrusdaal – B | CD – B | 8 | 32.84°S – 19.24°E |
| 17 | Citrusdaal – C | CD – C | 11 | 32.67°S – 19.06°E |
| 18 | Cape Town – A | CT – A | 5 | 33.80°S – 18.36°E |
| 19 | Cape Town – B | CT – B | 3 | 33.97°S – 18.51°E |
| 20 | Cape Town – C | CT – C | 12 | 33.96°S – 18.45°E |
| 21 | George – A | GE – A | 9 | 33.90°S – 22.33°E |
| 22 | George – B | GE – B | 8 | 33.95°S – 22.75°E |
| 23 | George – C | GE – C | 8 | 33.98°S – 22.47°E |
| 24 | Grahamstown – A | GT – A | 9 | 33.31°S – 26.49°E |
| 25 | Grahamstown – B | GT – B | 6 | 33.37°S – 26.42°E |
| 26 | Knysna – A | KN – A | 13 | 34.05°S – 22.99°E |
| 27 | Knysna – B | KN – B | 19 | 34.02°S – 22.97°E |
| 28 | Langebaan – A | LA – A | 10 | 33.04°S – 18.09°E |
| 29 | Langebaan – B | LA – B | 6 | 33.00°S – 18.31°E |
| 30 | Langebaan – C | LA – C | 6 | 33.03°S – 18.10°E |
| 31 | Laingsburg – A | LB – A | 4 | 33.27°S – 20.85°E |
| 32 | Laingsburg – B | LB – B | 6 | 33.28°S – 20.97°E |
| 33 | Moorreesburg – A | MB – A | 2 | 33.10°S – 18.74°E |
| 34 | Moorreesburg – B | MB – B | 3 | 33.11°S – 18.56°E |
| 35 | Moorreesburg – C | MB – C | 2 | 33.02°S – 18.85°E |
| 36 | Modderfontein – A | MF – A | 15 | 33.18°S – 25.80°E |
| 37 | Oudtshoorn – A | OD – A | 1 | 33.50°S – 22.51°E |
| 38 | Oudtshoorn – B | OD – B | 5 | 33.53°S – 22.54°E |
| 39 | Oudtshoorn – C | OD – C | 2 | 33.58°S – 22.49°E |
| 40 | Plettenburg Bay – A | PB – A | 3 | 34.05°S – 23.36°E |
| 41 | Plettenburg Bay – B | PB – B | 12 | 34.09°S – 23.34°E |
| 42 | Port Elizabeth – A | PE – A | 21 | 33.87°S – 25.39°E |
| 43 | Riversdale – A | RD – A | 9 | 34.31°S – 21.50°E |
| 44 | Riversdale – B | RD – B | 3 | 34.23°S – 21.58°E |
| 45 | Riversdale – C | RD – C | 14 | 34.10°S – 21.20°E |
| 46 | St. Francis – A | SF – A | 6 | 34.17°S – 24.81°E |
| 47 | Stellenbosch – A | ST – A | 13 | 33.89°S – 18.89°E |
| 48 | Stellenbosch – B | ST – B | 14 | 33.91°S – 18.81°E |
| 49 | Stellenbosch – C | ST – C | 10 | 33.85°S – 18.82°E |
| 50 | Swellendam – A | SW – A | 4 | 34.05°S – 20.65°E |
| 51 | Swellendam – B | SW – B | 7 | 34.40°S – 20.60°E |
| 52 | Swellendam – C | SW – C | 8 | 34.19°S – 20.30°E |
| 53 | Touwsrivier – A | TR – A | 2 | 33.15°S – 20.47°E |
| 54 | Touwsrivier – C | TR – C | 5 | 33.17°S – 20.26°E |
| 55 | Worcester – A | WD – A | 2 | 33.59°S – 19.45°E |
| 56 | Worcester – B | WD – B | 7 | 33.52°S – 19.49°E |
| 57 | Worcester – C | WD – C | 10 | 33.62°S – 19.69°E |
| 58 | Beaufort West – A | BW – A | 6 | 32.34°S – 22.64°E |
| 59 | Beaufort West – B | BW – B | 4 | 32.34°S – 22.64°E |
| 60 | Beaufort West – C | BW – C | 2 | 32.34°S – 22.62°E |
| 61 | East London – A | EL – A | 3 | 33.04°S – 27.86°E |
| 62 | East London – B | EL – B | 1 | 32.94°S – 27.97°E |
| 63 | East London – C | EL – C | 2 | 32.97°S – 27.90°E |
| 64 | Graaff-Reinet – A | GR – A | 7 | 32.25°S – 24.53°E |
| 65 | Graaff-Reinet – B | GR – B | 6 | 32.17°S – 24.56°E |
| 66 | Graaff-Reinet – C | GR – C | 8 | 32.26°S – 24.54°E |
| 67 | Klawer – A | KL – A | 1 | 32.02°S – 18.78°E |
| 68 | Klawer – B | KL – B | 8 | 32.17°S – 18.51°E |
| 69 | Klawer – C | KL – C | 6 | 32.10°S – 18.84°E |
| 70 | European Apis mellifera | AM | 10 | |
| Total | 474 |
Bees were sampled from 69 apiaries (no.). The apiaries are identified by their geographic region (the city closest to all apiaries in the region) and apiary in/around that city (A – C sampled apiaries in a region). This was coded into an apiary identifier which includes a two-letter city abbreviation and apiary letter. N represents the number of honey bees examined from each apiary. The GPS location of each apiary is reported in the final column
Fig. 1.

Geographic distribution of apiaries (N = 69) from which honey bees were collected in the Republic of South Africa. Adjacent apiaries are clustered into single geographical regions (N = 28) and assigned an abbreviation corresponding to the nearest town (corresponds to Table 1). The pie charts represent the composition of the three genetic clusters from each geographical region (shown as orange, blue, and dark blue). The colors indicate the different proportion of allele frequencies assigned to each region
Dissection and collection of morphometric data
Lateral images of the thorax and hairs on abdominal tergite 6 (A6) were taken of each bee prior to dissection. Four different parts of each bee’s body (right forewings, right hindwings, tergite A3 and sternite A4) were dissected to facilitate imaging of the morphometric characteristics. Forewings and hindwings were removed from the thorax using forceps. The sternites and tergites were removed by tearing the connective tissue between them. The dissected sternite A4 was cleaned with a paint brush and soaked in KOH to remove any remaining tissue. It also was stained with Bioquip double stain (6379B) and dried on a Kim-tech wipe. Excess stain was removed with ethanol. All body parts were dried on a Kim-tech wipe and mounted on slides using Euparal mounting medium. Mounts were made on 25 × 75 mm Fisher glass slides (S17466A) and cover glass (12–518-105H, Thermo Fisher Co.), warmed at 60 °C for 3 days on a slide warmer (Premiere xh-2004 or C.S. & E. 26,020), and imaged individually using Leica M205 light microscope equipped with a Leica MC170 camera (1600 × 1200 pixels) with its related software.
Ten standard morphometric characters were chosen based on information published by [9, 22]. These characters included: 1) forewing length, 2) the length of cover hair on abdominal tergite A6, 3) transverse width (TW) of the wax plate on abdominal sternite A4, 4) transverse length (TL) of the wax plate on abdominal sternite A4, 5) pigmentation of abdominal tergite A3, 6) number of ovarioles, 7) pigmentation of the scutellum, 8) pigmentation of the scutellar plate, 9) forewing angle N23, and 10) forewing angle O26. The pigmentations of the abdominal tergite A3, scutellum and scutellar plates were determined per the standard color ranks established by Ruttner [4]. Tergites and scutellar plates were ranked from 0 (fully pigmented black) to 9 (no black pigmentation). The scutellum color was ranked from 0 (fully pigmented black) to 5 (no black pigmentation). To avoid bias and improve accuracy, pigmentation was assessed by three different observers for each sample, and the resulting average used for analysis.
Forewing geometric landmarks were chosen based on the information published by Francoy et al. [39]. These included 19 landmarks of right forewings (venation intersections). Ten additional landmarks of the right hindwings were also included (Fig. 2). The two-dimensional x, y Cartesian coordinates of the identified landmarks were recorded using custom-built, assistive measuring software (unpublished licensed, software development project by Honey Bee Research and Extension Laboratory, University of Florida).
Fig. 2.

Location of the geometric landmarks on the honey bee wing. a 10 landmarks on the right hindwing; b 19 landmarks on the right forewing
DNA extraction
After morphometric analysis, total DNA was extracted from the dissected honey bee thoraces in accordance with the protocol outlined in [40]. DNA quality was assessed using a 1% agarose gel and quantified using Qubit® 3.0 Fluorometer per manufacturer’s guidelines (Thermo Scientific Inc., USA). The extracted DNAs were submitted for sequencing at the Genomic Diversity Facility of Cornell University. The DNA concentration was normalized (< 10 ng/ul) prior to sequencing. Samples with failed extractions were excluded from further analysis.
Genotyping-by-sequencing (GBS), sequence alignments and quality control
We constructed a GBS library containing 474 honey bee DNA samples (5 × 96 plate), including a negative control (no DNA) in accordance with the methods outlined by [31, 41]. Each DNA sample was digested with methylation-sensitive EcoT22I, a type II restriction endonuclease which recognizes a degenerate 6 bp sequence (ATGCAT) (New England Biolabs, Ipswich, MA), by incubation at 37 °C for 2 h. The digested DNAs were ligated with an equal amount of a different barcode-containing adapter and the same common adapter. The 474 barcode sequences were pooled (5 μl each) and purified with QIAquick PCR Purification Kit (Qiagen, Valencia, CA). The sequence of barcodes used for EcoT22I GBS library construction was published by [31, 41]. The pooled library was amplified by PCR per the cycling conditions outlined in [31]. The amplified genomic fragments were purified and quantified using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE) based on [31]. The constructed pooled EcoT22I library was sequenced on an Illumina HiSeq 2000 (Illumina Inc., San Diego, CA) in one sequencing flowcell lane (100 bp single-end sequencing) at the Cornell University Life Sciences Core Facility.
The raw Illumina DNA sequence reads for the EcoT22I library were quality-filtered by removing adapter sequences and enzyme recognition sites, followed by trimming by quality score utilizing the GBS analysis pipeline as implemented in TASSEL v3.0 [42]. We retained only the highest quality first 64 bp of each sequence to minimize the errors associated with base calling. To determine genomic SNP coordinates, we aligned sequence reads to the A. mellifera reference genome [43] using the Burrows-Wheeler alignment tool (BWA) version 0.7.8- r455 [44]. We further filtered the resulting genotypes by minor allele frequency (MAF) > 0.01, and missing data per site < 0.1. The filtered EcoT22I library reads produced the average individual depth of 38.75 (SD ± 6.76; median 38.61) with the average site depth of 27.88 (SD ±41.8; median 7.3) across all genotypes. All submitted samples generated sufficient genotypes for analysis and the effectiveness of the GBS method using EcoT22I digestion genomic library was previously shown by [45].
Post SNP filtering pipelines
Prior to the population genetic analyses, stringent filtering strategies were performed to obtain the most informative SNPs using VCFtools [46]. We applied eight different filters using the following parameters: (1) minimum read depth > 6 (−-min-meanDP 6), (2) MAF > 5% (−-MAF 0.05), (3) missing data at no more than 5% of samples (−max-missing 0.95), (4) average read depth < 100 (−max-meanDP 100), (5) missingness on per individual (−-missing-indv), (6) remove indel between reads (−-remove-indels), (7) include only bi-allelic sites (−-min-alleles 2 --max-alleles 2), and (8) remove SNPs in linkage disequilibrium (LD) (−-exclude-positions). After applying all filtering pipelines, we identified 2449 biallelic loci for further analysis of the 474 individuals.
Detecting SNP loci under selection based on FST outlier tests
Two different coalescent-based simulations were used to detect SNP loci deviating from neutrality. With these approaches, we expected to detect low levels of genetic differentiation under balancing selection (neutral loci) and high levels of differentiation under directional selection (divergent loci). We used two different FST-based methods including FDIST [47] and hierarchical [48]. FDIST was performed using the program LOGISTAN [49]. LOGISTAN calculates the neutral distribution of FST values with significant P-value for each locus. This method computes the distribution between FST and expected heterozygosity (HE) using two options “neutral mean Fst” and “force mean Fst” to detect genes under selection. A total number of 2449 SNPs were analyzed based on the following parameters: 50,000 simulations, confidence interval of 0.95, false discovery rate of 0.1, attempted FST ≥ 0.9, and mutation model of infinite alleles. We considered the FST values higher than expected neutral distribution as directional selection and FST values lower than expected neutral distribution as balancing selection.
The hierarchical method is a modification of the FDIST approach performed using an Arlequin package ver. 3.5.1.2 [48]. We used a hierarchical island model with 50,000 simulations to calculate the relationships between FST and heterozygosity. Loci with FST values above the 0.99 limits of neutral distribution were considered as putative outliers under the divergent selection [50]. The remaining loci with non-significant FST values were considered as neutral SNPs. All procedures reduced the bias and kept the highly diverged loci between individuals of subspecies.
Gene ontology (GO) analysis
The closest gene to each of the 83 divergent SNPs was determined using bedops v 2.4.22 in vcf2bed and closest features [51], relative to the gene annotations (gff3 file) which was available from NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCF_000002195.4/). The DAVID gene accession conversion tool was used to identify homologous genes from Apis mellifera and D. melanogaster as listed in BeeBase and FlyBase [52]. Functional enrichment of these gene IDs was conducted in the GeneMania and g:Profiler platforms [53, 54].
Environmental data
Nineteen bioclimatic variables were obtained from the WorldClim database (acquired in January 2016 at http://www.worldclim.org/). These bioclimatic variables (Additional file 1: Table S1) at a resolution of 2.5 arc-minutes derived from basic monthly climatic variables generated through interpolation of average monthly climate data from weather stations on a 0.5 arc-minute resolution grid [55]. These derived bioclimatic variables could better reflect biologically meaningful information instead of raw precipitation and temperature variables [56, 57]. We acquired these data for the 69 apiaries using each apiary’s geographic coordinates.
Statistical analysis
Morphometric analysis
The wing images from each bee were scaled, rotated and aligned using a Generalized Procrustes Alignment analysis (GPA) [58]. GPA analysis is a standard method to align landmark coordinate data [59]. Both the wing geometry and standard morphometric data were analyzed to determine which variables can best discriminate the subspecies using linear stepwise discrimination function analysis (a form of multivariate analysis of variance) (JMP ver. 12, SAS Institute, Cary, NC). Cross-validation (JMP ver. 12, SAS Institute, Cary, NC) was applied to determine the cutoff value and confirm accuracy for each wing geometry and standard morphometric parameter. A One-way Analysis of Variance (ANOVA) was applied to determine the average for each wing geometry and standard morphometric parameter (JMP ver. 12, SAS Institute, Cary, NC). A Tukey multiple comparisons test was used to compare mean values of each parameter at α ≤ 0.05). A dendrogram showing the relationships between individuals based on wing shape and morphometric characteristics was made using the unweighted pair group method with arithmetic means (UPGRMA). The analysis was done with 1000 as the bootstrap value and based on the discriminant values of each bee. Our stepwise discrimination function analysis to differentiate subspecies was based on using a combination of wing geometry/standard morphometric data. Once stepwise analysis had determined the best characteristics to use, standard loadings were calculated in R v.3.4.2. The subspecies groups were assigned based on the historical geographical distribution shown by [9]. The northernmost and southernmost bees were considered A.m. scutellata and A.m. capensis respectively. Bees falling between the two geographical regions were identified as hybrids. We repeated the analysis, grouping together samples by region instead of subspecies.
Population genetics analyses
We computed the pairwise evolutionary divergence among regions using MEGA7 [60] based on the p-distance model with 1000 bootstrap value across entire SNP loci. Population genetic diversity indices such as observed heterozygosity (Hobs), expected heterozygosity (Hexp), proportion of polymorphic SNP (Np), and inbreeding coefficient (FIS) were calculated for SNP loci using the statistical package R. Departure from Hardy-Weinberg equilibrium (HWE) was assessed by an exact test using Genepop 4.2 [61] based on the following Markov chain Monte Carlo simulation parameters: dememorization = 5000, batches = 5000, and iterations per batch = 1000 [62]. Analysis of molecular variance (AMOVA) was performed to determine the proportion of genetic variation within and between regions as implemented in Arlequin 3.5.1.2 [48] with 1000 permutations. The populations were structured by aligning Bayesian clustering pattern with the historical geographic distribution of the bees [9]. The overall and pairwise values of population differentiation statistic (FST) [63] were determined among regions and within subspecies using the SNP loci as calculated by Arlequin ver. 3.5.1.2 [48]. We permuted 1000 iterations to calculate the p-values for the mean and pairwise FST values. FST varies from 0 (lack of genetic structure and no sign of population subdivision) to 1 (distinct population structure or extreme segregation), with FST of up to 0.05 indicating a moderate genetic differentiation [64].
Association of environmental variables with SNP loci
We examined the association of divergent SNP loci with environmental characteristics using an individual-based spatial analysis as implemented in Samβada program [65] (available at lasig.epfl.ch/sambada). Samβada examines the associations between all environmental variables and allele frequencies of divergent SNP loci across sampling locations by a logistic regression approach. Models are selected through the examination of the significance of regression coefficient across environmental variables. The significance value of each model was evaluated with likelihood G and Wald scores. The Bonferroni-corrected threshold was considered as α = 0.05, indicating a significant association between loci and the environmental variables.
Bayesian population structure and principal coordinates analysis (PCoA)
We applied a model-based Bayesian clustering approach to characterize the existence of distinct genetic clusters among regions of both subspecies as implemented in STRUCTURE 2.3.4 [66]. The genome of each bee positioned into a predefined number of components (K) with variable proportions of allele frequency of the ancestral population. This approach allows the characterization of an ancestral population using admixed bees [66]. We ran STRUCTURE for n = 2449 loci and q subset of divergent loci (n = 83) using an admixture model and by applying a putative number of clusters (K) varying from 1 to 10. The analysis was performed without prior information of population identity by a simulation of 50,000 pre-burn steps and 100,000 post-burn iterations of MCMC algorithm for each run. We performed 10 independent runs for each K to estimate the most reliable number of distinct genetic clusters (K) using the likelihood of the posterior probability (LnP (N/K)) [67] and ad hoc quantity DK for each K partition. Posterior probability changes with respect to K between different runs are assigned as a method for determination of the true K value [68]. The most likely value for K was identified based on average log likelihood, Ln P (D) using Evanno’s ∆K method [68] from the web-based software STRUCTURE HARVESTER [69]. The population structure barplots were visualized using the CLUMPAK program as implemented in CLUMPP ver. 1.1.2 [70].
A principal coordinate analysis (PCoA) was performed with both sets of SNPs and the entire and divergent SNP loci to visualize the divergence pattern among individuals between two subspecies using TASSEL v3.0 and JMP Pro v12 (SAS Institute, Cary, NC). The PCoA analysis enables us to visualize the relatedness of individual honey bees across individuals/regions in multidimension scales.
Results
Wing geometry and standard morphometrics
The 19 forewing landmarks created 38 Cartesian coordinates for each specimen and the ten hindwing landmarks generated 20 Cartesian coordinates for each specimen. At the subspecies level, the linear stepwise discriminant function analysis of combined wing geometry and standard morphometric variables incorporated six out of ten variables (ovariole number, abdomen hair, scutellar plate, angle O, forewing length and tergite color), 11 out of 38 forewing coordinates (F4X, F5X, F6X, F6Y, F7X, F8X, F15Y, F16X, F16Y, F18Y, F19Y), and ten out of 20 hindwing coordinates (H2X, H2Y, H3Y, H5X, H7X, H7Y, H8X, H9X, H9Y, H10Y) in the statistically significant classification model for the honey bee populations (P < 0.05). The linear stepwise discriminant function analysis based on all examined variables placed all individual bees into the three groups with a high percentage of classification (87%). The cross-validation test misclassified 61 (13.11%) out of 464 bees. At the regional level, a linear stepwise discriminant function analysis of all wing geometry and standard morphometric variables included six out of ten standard variables (ovariole number, abdomen hair, scutellar plate, scutellum, sternite H, angle N, forewing length and tergite color), 12 out of 38 forewing coordinates (F1Y, F2Y, F4Y, F6Y, F7Y, F8Y, F10Y, F12Y, F13Y, F16Y, F18Y, F19Y), and seven out of 20 hindwing coordinates (H1X, H1Y, H2X, H2Y, H3Y, H10X, H10Y) in the significant classification model for the honey bee populations (P < 0.05). A linear stepwise discriminant function analysis based on all examined variables placed all individual bees into the three groups with an average percentage of classification (53%). The cross- validation test misclassified 217 (47%) out of 464 bees.
The discriminant function analysis of all 464 honey bees showed that the clusters created by the three groups overlapped in Canonical space. Canonical Vector 1 (CV1) explained 88% of the variance and Canonical Vector 2 (CV2) explained 12% (Fig. 3). Twenty-seven of 39 wing geometry and standard morphometric parameters have variations in the positive and negative factor-loading axis onto CV1 after normalizing the data. The ovariole count (− 0.45) and scutellar plate color (− 0.42) were significant characters in the first canonical function and the hindwing landmark coordinate 7X (0.59) and forewing coordinate 7X (0.55) in second canonical function. The least influential characters were the forewing landmark coordinate 7X (− 0.02) and hindwing coordinate 2X (0.03) in the first canonical function, and forewing coordinate Y15 (− 0.03) and forewing length (− 0.03) in the second canonical function (Additional file 2: Figure S1).
Fig. 3.
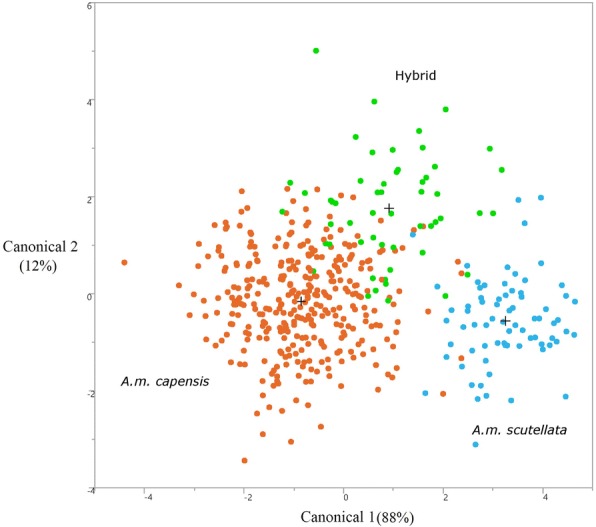
Morphological clustering pattern based on the final model from a stepwise discriminant analysis using 48 wing geometry and standard morphometric parameters. Three distinct morphological groups are shown by different colors, of which orange (N = 337), blue (N = 73) and green (N = 54) colors reflect Apis mellifera capensis, A.m. scutellata and hybrid honey bees respectively
An analysis of the honey bees grouped into the 28 geographical regions where they were collected revealed an incongruent and overlapped clustering pattern in the Canonical scatter plot of CV 1 (33%) and CV 2 (12%). The ovariole count (0.54) and abdomen hair length (− 0.47) were significant characteristics in the first canonical function and the forewing landmark coordinate 5X (0.76) and forewing angle O (0.51) in the second canonical function. The least influential characteristics were forewing coordinate X7 (0.007) and hindwing coordinate X2 (0.007) in the first canonical function, and forewing coordinate X7 (0.03) in the second canonical function (Additional file 3: Figure S2).
A dendrogram constructed by hierarchical cluster analysis of the squared Euclidian distances across all individuals revealed six major morphological groups (Additional file 4: Figure S3). Each group was composed of bees clustering across all three major groups (A.m. capensis, A.m. scutellate and hybrids). The cluster analysis showed a consistent pattern of overlap with hybrids located between the two subspecies.
The mean values of wing geometry and standard morphometric variables for three groups (A.m. scutellata, A.m. capensis and hybrids) are presented in Table 2. F values generated significant differences among all three groups except for characteristics 4, 7, 8, 11, 12, 14, 20, 21, 22, 24 and 25 as depicted in Table 2. Scutellar plate and tergite color show the highest between-group variability and statistical differences among mean values (Table 2). In contrast hindwing characteristic 5X, hindwing characteristic 9X, hindwing characteristic 8X, and forewing characteristic 16X have the lowest variability and are not significantly different from other parameters (Table 2).
Table 2.
The mean values (SD) of wing geometry and standard morphometric characters calculated for Apis mellifera capensis, A.m. scutellata, and hybrids of the two
| Characters | A.m. scutellata | A.m. capensis | Hybrid | F-value | P-value |
|---|---|---|---|---|---|
| number of ovarioles | 5.12a (0.69) | 12.76c (0.32) | 9.24b (0.8) | 53.61 | < 0.01 |
| abdomen hair (mm) | 0.0002a (0.00) | 0.0001c (0.00) | 0.0001b (0.00) | 84.09 | < 0.01 |
| scutellar plate (0–9 scale) | 7.86a (0.29) | 2.42c (0.13) | 5.01b (0.34) | 149.57 | < 0.01 |
| angle O (degrees) | 1.01a (0.007) | 1a (0.003) | 1a (0.008) | 0.21 | 0.81 |
| forewing length (mm) | 0.008a (0.00) | 0.008b (0.00) | 0.008b (0.00) | 13.73 | < 0.01 |
| tergite color (0–9 scale) | 8.45a (0.18) | 5.37c (0.08) | 6.94b (0.21) | 129.62 | < 0.01 |
| forewing 4X | 705.38a (5.18) | 716.41a (2.4) | 706.22a (6.02) | 2.69 | 0.06 |
| forewing 5X | 749.32a (5.44) | 758.56a (2.52) | 748.79a (6.32) | 1.91 | 0.14 |
| forewing 6X | 775.10b (4.99) | 788.60a (2.32) | 786.57ab (5.81) | 3 | 0.05 |
| forewing 6Y | 655.17b (7.53) | 703.39a (3.5) | 688.55a (8.75) | 17.08 | < 0.01 |
| forewing 7X | 794.09a (4.98) | 806.24a (2.31) | 803.11a (5.8) | 2.45 | 0.08 |
| forewing 8X | 797.61a (5.01) | 808.47a (2.32) | 802.33a (5.82) | 2.16 | 0.11 |
| forewing 15Y | 445.63b (7.03) | 491.01a (3.26) | 468.22b (8.17) | 18.56 | < 0.01 |
| forewing 16X | 1009.21a (4.97) | 1015.71a (2.31) | 1015.14a (5.78) | 0.71 | 0.49 |
| forewing 16Y | 629.11b (7.03) | 669.71a (3.26) | 649.61ab (8.18) | 14.79 | < 0.01 |
| forewing 18Y | 547.41b (6.97) | 587.36a (3.24) | 564.68b (8.11) | 15.13 | < 0.01 |
| forewing 19Y | 437.97b (7.07) | 477.39a (3.29) | 450.20b (8.23) | 15.41 | < 0.01 |
| hindwing 2X | 561.13ab (4.77) | 570.35a (2.22) | 554.38b (5.55) | 4.47 | 0.01 |
| hindwing 2Y | 506.04b (7.2) | 546.63a (3.34) | 557.79a (8.37) | 15.19 | < 0.01 |
| hindwing 3X | 852.26a (4.85) | 857.30a (2.25) | 848.77a (5.63) | 1.25 | 0.28 |
| hindwing 5X | 1096.02a (4.98) | 1096.60a (2.31) | 1092.79a (5.79) | 0.19 | 0.83 |
| hindwing 7X | 734.21a (5.51) | 745.44a (2.56) | 736.75a (6.4) | 2.19 | 0.11 |
| hindwing 7Y | 664.52b (7.14) | 701.16a (3.32) | 709.20a (8.31) | 12.16 | < 0.01 |
| hindwing 8X | 1003.83a (5.04) | 1008.5a (2.34) | 1005.01a (5.86) | 0.43 | 0.64 |
| hindwing 9X | 1104.13a (4.95) | 1103.73a (2.3) | 1098.92a (5.76) | 0.32 | 0.72 |
| hindwing 9Y | 549.09b (7.13) | 585.29a (3.31) | 589.01a (8.29) | 11.25 | < 0.01 |
| hindwing 10Y | 581.94b (7.19) | 616.71a (3.34) | 614.33a (8.36) | 9.7 | < 0.01 |
Row means with the same superscript letters are not significantly different from one another (Tukey’s test, P ≤ 0.05
At the regional level, forewing characteristics 1Y (F = 6.84), 2Y (F = 6.9) and 4Y (F = 6.39) show the highest between group variability and statistical difference among mean values (P ≤ 0.05), while angle N (F = 1.85) and hindwing characteristic 10X (F = 1.31) have the lowest variability and are not significantly different from other parameters (P > 0.05) (data not shown).
Genotypic data, genetic diversity and divergence
A total of 3,103,730 reads of paired-end sequencing data were generated with 474 individual bees using the GBS method. An average of 65% were uniquely aligned to the honey bee reference genome (GCF_000002195.4_Amel_4.5_genomic.fna.gz), resulting in 2,028,130 reads with 98,134 SNPs. After evaluating the dataset with informative pipelines (MAF overall populations > 0.01, missing data per site < 90%), we filtered 70,475 SNPs with a mean individual depth of 38.7. By applying stringent additional post filtering SNP criteria, we kept 2449 high quality SNPs out of 70,475 total to analyze among the 474 individual bees in the final data set. A test for HWE departure indicated that all SNPs and regions are in HWE after the sequential Bonferoni correction. At the regional level, ten of 29 regions were in HWE (BL, CD, GE, GR, KN, LA, PE, RD, ST and SW – see Table 1 for abbreviation location) but the rest showed a significant departure from HWE. Genetic diversity estimates for mean value of allelic richness for A.m. capensis, A.m. scutellata and hybrids were 1.53, 1.52 and 1.52 respectively. A similar level of mean observed and expected heterozygosity was found for A.m. capensis, A.m. scutellata, and hybrids (Table 3). The observed heterozygosity (Hobs) was the highest across all regions (Hobs = 0.26) except for the PT region (Hobs = 0.21). The inbreeding coefficient (FIS) was negative in BD, MF, OD and TR regions for A.m. capensis and in the KR region for A.m. scutellata. Within the hybrids, two regions, BW and KL, generated negative values that indicate an outbreeding outcome in these regions. The FIS value estimated for the 2449 SNP loci revealed the inbreeding outcome occurred across the regions. The percentage of polymorphic SNPs (Np) ranged from 60.27 to 95.51% among regions. The mean percent of polymorphic SNPs was higher in A.m. scutellata than in A.m. capensis and hybrid populations (Table 3). The population genetic diversity indices across 28 geographical regions is depicted in Table 3. Fifteen regions of A.m. capensis, two regions of A.m. scutellata and two regions of hybrids showed the maximum amount of genetic divergence (0.08). The minimum value was observed in two regions of A.m. capensis (BD and TR), and one region of A.m. scutellata (KR) (0.06). A net pair-wise evolutionary divergence among regions ranged from 0.01 to 0.08, with an average of 0.09 (Additional file 5: Table S2).
Table 3.
Population genetic characteristics, determined using 2449 SNP loci, of the honey bees sampled from 28 geographical regions in the Republic of South Africa
| Geographic region | N a | Ar | H obs | H exp | F IS | Np (%) |
|---|---|---|---|---|---|---|
| Apis mellifera scutellata, N = 73 bees | ||||||
| BL | 1.88 | 1.52 | 0.24 | 0.23 | 0.04 | 88.18 |
| KR | 1.87 | 1.53 | 0.25 | 0.23 | −0.02 | 87.03 |
| PT | 1.71 | 1.48 | 0.21 | 0.21 | 0.003 | 71.8 |
| SP | 1.73 | 1.51 | 0.22 | 0.22 | 0.001 | 73.81 |
| UP | 1.88 | 1.54 | 0.25 | 0.23 | 0.01 | 88.1 |
| Mean | 1.81 | 1.52 | 0.24 | 0.22 | 0.006 | 87.78 |
| Apis mellifera capensis, N = 337 bees | ||||||
| BD | 1.6 | 1.45 | 0.22 | 0.19 | −0.04 | 60.27 |
| CD | 1.93 | 1.55 | 0.24 | 0.24 | 0.02 | 93.12 |
| CT | 1.92 | 1.55 | 0.25 | 0.24 | 0.03 | 92.46 |
| GE | 1.94 | 1.55 | 0.23 | 0.24 | 0.06 | 94.48 |
| GT | 1.86 | 1.53 | 0.23 | 0.23 | 0.02 | 87 |
| KN | 1.95 | 1.55 | 0.24 | 0.24 | 0.04 | 95 |
| LA | 1.92 | 1.55 | 0.23 | 0.24 | 0.04 | 92.26 |
| LB | 1.82 | 1.54 | 0.24 | 0.23 | 0.02 | 82.37 |
| MB | 1.72 | 1.52 | 0.23 | 0.22 | 0.003 | 72.33 |
| MF | 1.86 | 1.55 | 0.26 | 0.24 | −0.01 | 86.66 |
| OD | 1.76 | 1.52 | 0.23 | 0.22 | − 0.02 | 76.5 |
| PB | 1.85 | 1.53 | 0.24 | 0.23 | 0.02 | 85.6 |
| PE | 1.9 | 1.54 | 0.23 | 0.24 | 0.03 | 90.4 |
| RD | 1.94 | 1.55 | 0.24 | 0.24 | 0.04 | 94.03 |
| SF | 1.69 | 1.5 | 0.23 | 0.22 | 0.03 | 69 |
| ST | 1.95 | 1.54 | 0.23 | 0.24 | 0.04 | 95.51 |
| SW | 1.87 | 1.53 | 0.24 | 0.23 | 0.03 | 87.64 |
| TR | 1.66 | 1.48 | 0.23 | 0.21 | −0.04 | 66.07 |
| WD | 1.91 | 1.54 | 0.25 | 0.23 | 0.03 | 91.23 |
| Mean | 1.84 | 1.53 | 0.24 | 0.23 | 0.02 | 84.83 |
| Hybrid bees, N = 54 bees | ||||||
| BW | 1.81 | 1.52 | 0.25 | 0.23 | −0.02 | 81.22 |
| EL | 1.69 | 1.5 | 0.23 | 0.21 | 0.02 | 69.41 |
| GR | 1.9 | 1.54 | 0.23 | 0.23 | 0.02 | 90.77 |
| KL | 1.83 | 1.52 | 0.24 | 0.23 | −0.001 | 83.07 |
| Mean | 1.81 | 1.52 | 0.24 | 0.22 | 0.004 | 81.12 |
| European A. mellifera reference, N = 10 bees | ||||||
| AM | 1.36 | 1.25 | 0.12 | 0.11 | −0.05 | 35.73 |
Geographical abbreviations are defined in Table 1. Abbreviations: Na, mean number of observed alleles per locus; Ar, mean allelic richness; Hobs, observed heterozygosity; Hexp, expected heterozygosity; FIS; Fixation index and NP (%), percent mean number of private alleles per region. Mean values are calculated by pooling correspondent regions of each subspecies
Detection of divergent SNP loci
When we considered all 474 honey bees including A.m. scutellate, A.m. capensis and the European A. mellifera, the Arlequin hierarchical method revealed 90 candidate SNPs for divergent selection at the 1% significance level (Fig. 4a). Based on the same data set, the LOGISTAN method detected 103 divergent loci with evidence of divergent selection (4b). These approaches concurred on 83 divergent SNPs at the α= 0.01 significance level. The list of these 83 divergent SNP loci, together with their locations in the honey bee genome and information on functional gene annotation, are listed in Table 4.
Fig. 4a, b.

Identification of putative divergent SNP loci under directional selection based on FST outlier approaches. a Hierarchical structure model using Arlequin 3.5. FST: locus –specific genetic divergence among the populations; Heterozygosity: measure of heterozygosity per locus. The significant loci are shown with red dots (P < 0.01). b Finite island model (fdist) by LOGISTAN. Loci under positive selection above 99% percentile are shown in the red area. Loci in the gray area are neutral loci. Those in the yellow area are under balancing selection
Table 4.
List of 83 divergent SNPs with their functions obtained by outlier tests as putative markers and used for their association with environmental variables using Samβada package
| SNPID | SNP Position | Gene product | Gene ID (BEEBASE) | Gene ID (FLYBASE) |
|---|---|---|---|---|
| S1_4,550,788 | 4,550,788 | LIM/homeobox protein Awh | GB53183 | FBgn0013751 |
| S1_6,686,923 | 6,686,923 | Uncharacterized LOC725682, transcript variant X1 | GB40830 | FBgn0260997 |
| S1_12,811,137 | 12,811,137 | Protein prickle-like, transcript variant X1 | GB44796 | FBgn0003090 |
| S1_15,039,694 | 15,039,694 | Krueppel-like factor 6, transcript variant X1 | GB52133 | FBgn0040765 |
| S1_17,699,574 | 17,699,574 | Protein-serine O-palmitoleoyltransferase porcupine | GB47458 | FBgn0004957 |
| S1_18,694,215 | 18,694,215 | Protein eva-1, transcript variant X1 | GB51947 | FBgn0259821 |
| S1_33386163 | 3,492,655 | Unknown | Unknown | Unknown |
| S1_50443612 | 5,000,737 | Dumpy | GB55781 | FBgn0053196 |
| S1_52257404 | 6,814,529 | Unknown | Unknown | Unknown |
| S1_52257424 | 6,814,549 | Unknown | Unknown | Unknown |
| S1_62957554 | 4,280,238 | Sestrin homolog, transcript variant X1 | GB49567 | FBgn0034897 |
| S1_69083742 | 10,406,426 | Protein 4.1 homolog | GB44175 | FBgn0010434 |
| S1_74943997 | 3,548,247 | Hemicentin-2, transcript variant X1 | GB51391 | Unknown |
| S1_77638337 | 6,242,587 | Solute carrier family 35 member F5, transcript variant X1 | GB46790 | FBgn0034032 |
| S1_77638361 | 6,242,611 | Solute carrier family 35 member F5, transcript variant X1 | GB46790 | FBgn0034032 |
| S1_86109011 | 349,889 | Eye-specific diacylglycerol kinase, transcript variant X1 | GB51219 | FBgn0261549 |
| S1_86386875 | 627,753 | Lateral signaling target protein 2 homolog | GB51210 | FBgn0039492 |
| S1_88925416 | 3,166,294 | Charged multivesicular body protein 7 | GB54484 | FBgn0027565 |
| S1_88925462 | 3,166,340 | UDP-glucuronosyltransferase 1–3-like | GB54485 | Unknown |
| S1_89224631 | 3,465,509 | Jazigo, transcript variant X1 | GB43054 | FBgn0261259 |
| S1_90545383 | 4,786,261 | GRAM domain-containing protein 3-like, transcript variant X1 | GB52157 | Unknown |
| S1_96460901 | 10,701,779 | Guanylate cyclase, soluble, beta 1 | GB52953 | FBgn0013973 |
| S1_103184872 | 17,425,750 | Potassium channel subfamily T member 2 | GB45474 | FBgn0261698 |
| S1_104349915 | 117,756 | Zinc finger protein GLI4-like, transcript variant X1 | GB40273 | FBgn0039039 |
| S1_108444799 | 4,212,640 | Protein tincar, transcript variant X1 | GB49246 | FBgn0261649 |
| S1_108537914 | 4,305,755 | Tachykinin | GB49248 | Unknown |
| S1_108537929 | 4,305,770 | Tachykinin | GB49248 | Unknown |
| S1_108682550 | 4,450,391 | Unknown | Unknown | Unknown |
| S1_108724951 | 4,492,792 | Uncharacterized LOC725260 | GB54634 | FBgn0034808 |
| S1_108786716 | 4,554,557 | Uncharacterized LOC725260 | GB54634 | FBgn0034808 |
| S1_110307882 | 6,075,723 | Thyrotropin-releasing hormone-degrading ectoenzyme-like, transcript variant X1 | GB43314 | Unknown |
| S1_110854546 | 6,622,387 | CCR4-NOT transcription complex subunit 6-like, transcript variant X1 | GB48300 | FBgn0011725 |
| S1_110950229 | 6,718,070 | CCR4-NOT transcription complex subunit 6-like, transcript variant X1 | GB48300 | FBgn0011725 |
| S1_110951124 | 6,718,965 | CCR4-NOT transcription complex subunit 6-like, transcript variant X1 | GB48300 | FBgn0011725 |
| S1_110951168 | 6,719,009 | CCR4-NOT transcription complex subunit 6-like, transcript variant X1 | GB48300 | FBgn0011725 |
| S1_123838315 | 6,386,711 | Plasma membrane calcium-transporting ATPase 2 | GB43909 | FBgn0259214 |
| S1_125133095 | 7,681,491 | Uncharacterized LOC411277, transcript variant X1 | GB41894 | FBgn0023531 |
| S1_126922494 | 9,470,890 | Probable G-protein coupled receptor 158 | GB52840 | FBgn0085401 |
| S1_127479850 | 10,028,246 | Ecdysone receptor, transcript variant A | GB48059 | FBgn0000546 |
| S1_130971599 | 13,519,995 | Uncharacterized LOC725485 | GB51646 | FBgn0259927 |
| S1_132727867 | 1,729,619 | Prefoldin subunit 5 | GB43750 | FBgn0038976 |
| S1_134667854 | 3,669,606 | Fasciclin-1 | Unknown | Unknown |
| S1_141112580 | 10,114,332 | Sortilin-related receptor | GB53341 | Unknown |
| S1_143133972 | 1,015,171 | Uncharacterized LOC102654435 | Unknown | Unknown |
| S1_143743719 | 1,624,918 | Unknown | Unknown | Unknown |
| S1_150101379 | 7,982,578 | Beta-1-syntrophin, transcript variant X1 | GB54295 | FBgn0037130 |
| S1_153385191 | 11,266,390 | Uncharacterized protein CG43867 | GB51041 | FBgn0259100 |
| S1_153549815 | 11,431,014 | Brachyury protein, transcript variant X1 | GB51013 | FBgn0011723 |
| S1_153549857 | 11,431,056 | Brachyury protein, transcript variant X1 | GB51013 | FBgn0011723 |
| S1_155093443 | 8589 | Unknown | Unknown | Unknown |
| S1_155468419 | 383,565 | Uncharacterized protein C1orf112 homolog | GB46620 | FBgn0050424 |
| S1_162084123 | 6,999,269 | Protein FAM49B, transcript variant X1 | GB53506 | FBgn0052066 |
| S1_162216249 | 7,131,395 | tRNA dimethylallyltransferase, mitochondrial-like | GB53505 | Unknown |
| S1_164267544 | 9,182,690 | Carbonic anhydrase-related protein 10, transcript variant X1 | GB45092 | FBgn0029962 |
| S1_167336698 | 12,251,844 | Tyrosine-protein kinase transmembrane receptor Ror2, transcript variant X1 | GB45194 | FBgn0020391 |
| S1_168107630 | 13,022,776 | Fringe glycosyltransferase | GB44913 | FBgn0011591 |
| S1_176503904 | 6,692,394 | Disks large 1 tumor suppressor protein | GB40648 | FBgn0001624 |
| S1_182557882 | 843,618 | Unknown | Unknown | Unknown |
| S1_192304686 | 301,823 | Inactive rhomboid protein 1 | GB49046 | FBgn0041723 |
| S1_193370056 | 1,367,193 | Unknown | GB53054 | Unknown |
| S1_197495828 | 5,492,965 | Negative elongation factor E-like, transcript variant X1 | Unknown | Unknown |
| S1_200092025 | 8,089,162 | Chromatin assembly factor 1 subunit A-B, transcript variant X1 | GB52767 | FBgn0025833 |
| S1_202452286 | 195,668 | MAX dimerization protein | Unknown | Unknown |
| S1_209285909 | 7,029,291 | Uncharacterized LOC100576529 | GB50066 | Unknown |
| S1_212445471 | 21,524 | Scavenger receptor class B member 1-like, transcript variant X1 | Unknown | Unknown |
| S1_217211608 | 4,787,661 | Unknown | Unknown | Unknown |
| S1_218446595 | 6,022,648 | Ankyrin repeat domain-containing protein 39-like, transcript variant X1 | GB45961 | FBgn0031674 |
| S1_220569872 | 317,721 | Histone-lysine N-methyltransferase SETMAR-like | Unknown | Unknown |
| S1_226482382 | 27,314 | Histone-lysine N-methyltransferase SETMAR-like | Unknown | Unknown |
| S1_226482388 | 27,320 | Histone-lysine N-methyltransferase SETMAR-like | Unknown | Unknown |
| S1_227605662 | 117,306 | Unknown | Unknown | Unknown |
| S1_228624508 | 85,274 | Unknown | Unknown | Unknown |
| S1_228990350 | 24,441 | Unknown | Unknown | Unknown |
| S1_229285085 | 47,268 | Cytohesin-1-like | GB55123 | FBgn0086779 |
| S1_229285093 | 47,276 | Cytohesin-1-like | GB55123 | FBgn0086779 |
| S1_229818309 | 42,135 | Unknown | Unknown | Unknown |
| S1_231880268 | 35,546 | Lactosylceramide 4-alpha-galactosyltransferase, transcript variant X1 | GB46832 | FBgn0039378 |
| S1_231894656 | 49,934 | Unknown | Unknown | Unknown |
| S1_231918135 | 9237 | Putative phosphatidate phosphatase, transcript variant X1 | GB49207 | FBgn0016078 |
| S1_233321962 | 34,497 | Homogentisate 1,2-dioxygenase | GB53288 | FBgn0040211 |
| S1_233858922 | 67,499 | Zinc finger and BTB domain-containing protein 24-like | GB51241 | Unknown |
| S1_234352063 | 52,465 | Unknown | Unknown | Unknown |
| S1_241871073 | 419 | Unknown | Unknown | Unknown |
The Gene IDs mapped with Apis mellifera and Drosophila melanogaster genomes are listed for each SNP
In order to examine environmental correlations within subspecies, we also tested loci under selection within each of the sub-populations. Using the 2449 SNPs within A.m. capensis, the Arlequin hierarchical method revealed 54 divergent SNPs (α = 0.01) and LOGISTAN identified 132 divergent SNPs (Additional file 6: Figure S4 A, B). Both approaches revealed 47 divergent SNPs under directional selection (Table 5). For A.m. scutellata, the Arlequin hierarchical method revealed 31 divergent SNPs (α= 0.01) and LOGISTAN identified 45 divergent SNPs (Additional file 7: Figure S5 A, B) with 21 divergent SNPs in common between the two approaches (Table 6).
Table 5.
The list of divergent SNP loci for Apis mellifera capensis using FST outlier and environmental correlation tests
| No. | SNP loci | SNP Position | Heterozygosity | FST statistic | Environmental correlation |
|---|---|---|---|---|---|
| 1 | S1_13,998,280 | 13,998,280 | 0.25 | 0.1 | V1, V6, V10, V11 |
| 2 | S1_14,309,089 | 14,309,089 | 0.13 | 0.32 | |
| 3 | S1_19,198,024 | 19,198,024 | 0.1 | 0.13 | |
| 4 | S1_33680990 | 3,787,482 | 0.19 | 0.26 | |
| 5 | S1_34025116 | 4,131,608 | 0.14 | 0.18 | |
| 6 | S1_38187276 | 8,293,768 | 0.24 | 0.11 | |
| 7 | S1_39180700 | 9,287,192 | 0.25 | 0.11 | V15 |
| 8 | S1_47120892 | 1,678,017 | 0.47 | 0.16 | V13, V16, V19 |
| 9 | S1_50443500 | 5,000,625 | 0.39 | 0.2 | |
| 10 | S1_52257404 | 6,814,529 | 0.46 | 0.21 | V8, V15, V19, Longitude |
| 11 | S1_52257424 | 6,814,549 | 0.5 | 0.27 | V15, Longitude |
| 12 | S1_58794304 | 116,988 | 0.12 | 0.19 | |
| 13 | S1_71833698 | 437,948 | 0.5 | 0.2 | |
| 14 | S1_72542859 | 1,147,109 | 0.2 | 0.13 | |
| 15 | S1_84555306 | 13,159,556 | 0.22 | 0.17 | V13, V16, V19 |
| 16 | S1_84555310 | 13,159,560 | 0.21 | 0.13 | V15, Latitude |
| 17 | S1_94849186 | 9,090,064 | 0.32 | 0.15 | |
| 18 | S1_96460917 | 10,701,795 | 0.42 | 0.15 | |
| 19 | S1_100472193 | 14,713,071 | 0.29 | 0.13 | |
| 20 | S1_105727753 | 1,495,594 | 0.13 | 0.11 | |
| 21 | S1_108033805 | 3,801,646 | 0.29 | 0.11 | |
| 22 | S1_108684700 | 4,452,541 | 0.14 | 0.16 | V4, V5, V7, V14, V15, V17, V18, Longitude |
| 23 | S1_108684756 | 4,452,597 | 0.15 | 0.16 | V4, V5, V7, V14, V15, V17, V18, Longitude |
| 24 | S1_108786716 | 4,554,557 | 0.45 | 0.42 | V9 |
| 25 | S1_109332327 | 5,100,168 | 0.21 | 0.25 | V3, V4, V5, V7,V14, V15, V17, V18, Longitude |
| 26 | S1_110951124 | 6,718,965 | 0.18 | 0.28 | V4, V5, V7, V14, V17, V18, Longitude |
| 27 | S1_110951168 | 6,719,009 | 0.18 | 0.28 | V4, V5, V7, V14, V17, V18, Longitude |
| 28 | S1_110951197 | 6,719,038 | 0.19 | 0.27 | V3, V4, V5, V7, V14, V15, V17, V18, Longitude |
| 29 | S1_111171339 | 6,939,180 | 0.18 | 0.24 | V3, V4, V5, V7, V14, V15, V17, V18, Longitude |
| 30 | S1_111171425 | 6,939,266 | 0.18 | 0.24 | V3, V4, V5, V7, V14, V15, V17, V18, Longitude |
| 31 | S1_111285658 | 7,053,499 | 0.18 | 0.23 | V4, V14, V15, V17, V18, Longitude |
| 32 | S1_118855642 | 1,404,038 | 0.48 | 0.14 | |
| 33 | S1_120132551 | 2,680,947 | 0.2 | 0.13 | Longitude |
| 34 | S1_132727867 | 1,729,619 | 0.2 | 0.25 | V3, V9, Longitude |
| 35 | S1_132761070 | 1,762,822 | 0.5 | 0.38 | V3, V9, Longitude |
| 36 | S1_133026966 | 2,028,718 | 0.23 | 0.18 | Longitude |
| 37 | S1_146906109 | 4,787,308 | 0.16 | 0.21 | |
| 38 | S1_152157525 | 10,038,724 | 0.43 | 0.14 | |
| 39 | S1_158767923 | 3,683,069 | 0.44 | 0.11 | |
| 40 | S1_177861915 | 8,050,405 | 0.2 | 0.2 | V1, V5, V8, V10 |
| 41 | S1_192304686 | 301,823 | 0.16 | 0.36 | V8 |
| 42 | S1_207842400 | 5,585,782 | 0.07 | 0.13 | |
| 43 | S1_210484669 | 8,228,051 | 0.3 | 0.1 | |
| 44 | S1_212486639 | 62,692 | 0.5 | 0.17 | Longitude |
| 45 | S1_238491065 | 11,486 | 0.16 | 0.15 | |
| 46 | S1_238491098 | 11,519 | 0.36 | 0.15 | V13, V15, V16, V19 |
| 47 | S1_238498932 | 1677 | 0.47 | 0.11 |
The significant SNP genotypes correlated with environmental variables for both the likelihood ratio (G) and Wald tests at the Bonferroni-corrected 0.05 alpha level are shown. The variable numbers are explained in Additional file 1: Table S1
Table 6.
The list of divergent SNP loci for Apis mellifera scutellata using FST outlier and environmental correlation tests
| No. | SNP loci | SNP Position | Heterozygosity | FST statistic | Environmental correlation |
|---|---|---|---|---|---|
| 1 | S1_22,206,418 | 22,206,418 | 0.21 | 0.1 | |
| 2 | S1_39180699 | 9,287,191 | 0.1 | 0.17 | |
| 3 | S1_75314820 | 3,919,070 | 0.17 | 0.08 | |
| 4 | S1_88966222 | 3,207,100 | 0.48 | 0.1 | Latitude |
| 5 | S1_89371176 | 3,612,054 | 0.3 | 0.11 | |
| 6 | S1_90412686 | 4,653,564 | 0.13 | 0.12 | |
| 7 | S1_90545383 | 4,786,261 | 0.2 | 0.4 | |
| 8 | S1_92199236 | 6,440,114 | 0.1 | 0.12 | |
| 9 | S1_98916246 | 13,157,124 | 0.22 | 0.1 | Latitude |
| 10 | S1_109332316 | 5,100,157 | 0.16 | 0.14 | |
| 11 | S1_109332327 | 5,100,168 | 0.21 | 0.25 | |
| 12 | S1_110307739 | 6,075,580 | 0.21 | 0.18 | |
| 13 | S1_122235324 | 4,783,720 | 0.25 | 0.13 | |
| 14 | S1_131785203 | 786,955 | 0.3 | 0.11 | |
| 15 | S1_166680858 | 11,596,004 | 0.14 | 0.1 | |
| 16 | S1_174926422 | 5,114,912 | 0.2 | 0.04 | |
| 17 | S1_190284134 | 8,569,870 | 0.35 | 0.1 | |
| 18 | S1_190284138 | 8,569,874 | 0.16 | 0.06 | |
| 19 | S1_238491065 | 11,486 | 0.16 | 0.15 | |
| 20 | S1_238491098 | 11,519 | 0.36 | 0.15 | V1, V10, V12, V13, V16, V18, Longitude |
| 21 | S1_241084639 | 2280 | 0.38 | 0.11 | V12, V13, V16, V18, Longitude |
The significant SNP genotypes correlated with environmental variables for both the likelihood ratio (G) and Wald tests at the Bonferroni-corrected 0.05 alpha levels are shown. The variable numbers are explained in Additional file 1: Table S1
Gene ontology annotation of genes at divergent loci
We retrieved 52 non-redundant BeeBase Genes IDs for known gene products most proximal to each of the 83 divergent SNPS, as well as gene names and functions, as listed in Table 4. Given the more extensive gene annotation available for D. melanogaster, we converted these 52 genes to homologs, resulting in 38 recognized genes examined in the GeneMania tool for functional enrichment. The eleven significantly enriched functional categories are listed in Table 7. Utilizing the same D. melanogaster gene list, g:Profiler highlighted three genes participating in one significant Biological Process: chitin-based embryonic cuticle biosynthetic process (GO:0008362, P = 3.69e-02).
Table 7.
Functional enrichment for the genes closest to the divergent SNP loci between A.m. capensis and A.m. scutellata
| GO Functional Category | FDR | # of genes in network | # of genes in genome |
|---|---|---|---|
| Plasma membrane region | 0.00722 | 6 | 70 |
| Regulation of growth | 0.0252 | 8 | 215 |
| Embryo development ending in birth or egg hatching | 0.0341 | 7 | 181 |
| Embryonic development via the syncytial blastoderm | 0.0341 | 7 | 170 |
| Membrane region | 0.0341 | 6 | 120 |
| Regulation of synaptic growth at neuromuscular junction | 0.0344 | 5 | 73 |
| Morphogenesis of an epithelium | 0.0360 | 8 | 273 |
| Regulation of synapse assembly | 0.0360 | 5 | 77 |
| Regulation of nervous system development | 0.0401 | 7 | 204 |
| Regulation of synapse organization | 0.0541 | 5 | 89 |
| Apicolateral plasma membrane | 0.0613 | 3 | 17 |
Associations between genetic and environmental parameters
Analysis of allele frequency variation of 47 divergent SNPs within A.m. capensis (337 individuals and 15,564 non-missing genotypes) generated a total of 2961 models, within which 25 SNPs were significantly correlated (Bonferroni-corrected P < 0.05) with one or more environmental parameters (191 significant associations, 6.5% of the total) (Table 5). Of those, 16 loci were significantly related to the longitude of the apiary where the sample was collected; one was significantly related to latitude of the same. Six of 25 loci were significantly associated with isothermality and one with annual mean temperature. Furthermore, six of the 25 loci were exclusively correlated with temperature, and six with precipitation. The strong locus-environment associations were observed in eight SNP loci including S1_108684700, S1_108684756, S1_109332327, S1_110951124, S1_110951168, S1_110951197, S1_111171339, S1_111171425 and S1_111285658 (Table 5).
Within Apis mellifera scutellata (73 individuals and 21 divergent loci) generated 1533 non-missing genotypes with a total of 1323 models, of which four SNPs were significantly correlated (Bonferroni-corrected P < 0.05) with one or more environmental parameters (21 significant associations, 1.6% of the total) (Table 6). Two loci were significantly related to longitude and two with latitude. The strongest locus-environment association was observed in two SNPs: S1_238491098 and S1_241084639. The highest contribution by percentage among the environmental parameters was to temperature and precipitation in the warmest seasons.
Model-based Bayesian population structure and genetic differentiation between A.m. capensis and A.m. scutellata
The model-based Bayesian population structure was determined for the two sets of data, all 2449 SNPs, and the 83 divergent SNPs. Using 2449 SNP loci, the delta K method suggested three genetic groups (K = 3) with inclusion of European subspecies of A. mellifera (Fig. 1, Additional file 8: Table S3). When we excluded European A. mellifera (n = 10), two genetic groups were observed according to the delta K calculation as determined by the Harvester method (K = 2) (Fig. 5). Apis mellifera scutellata constituted a distinct genetic group with genetic homogeneity across the regions (Fig. 5). In A.m. capensis, nine regions (CD, CT, GE, KN, LA, PE, RD, ST and WD) confirmed the occurrence of distinct genetic groups, and variable degrees of genetic heterogeneity were observed across its natural distribution in the Cape region (Fig. 5). Hybrid regions showed a mixture pattern of genotypes, with a variable percent of K membership in each region (Fig. 5). When we examined individual membership across K populations, A.m. scutellate possessed, on average, 5% of K1, 93% of K2, and 2% of K3 ancestry. In contrast, A.m. capensis was assigned primarily to K1 (78%), with lesser contributions from K2 (21%) and K3 (K3). The hybrid population appropriately contained roughly equivalent proportions of the two primary Ks: 44% K1, 55% K2 and 1% K3. The PCoA analysis confirmed these results, positioning A.m. scutellata and A.m. capensis into two clusters with hybrids placed at intermediate positions (Fig. 6). European A. mellifera clustered into a single distant group. The first and second component accounted for 23.29% and 33.34% of the variance, respectively. With European A. mellifera excluded, 20% of the individual A.m. scutellata and hybrids overlapped in the PCoA plot, 44.5% of A.m. capensis and hybrids overlapped, 4% of A.m. capensis and A.m. scutellata overlapped, and 4% overlapped between all three groups.
Fig. 5.

A model-based Bayesian population structure of 474 honey bees from the Republic of South Africa using admixture pattern as included in STRUCTURE program. The Y-axis in each plot indicates the estimated membership coefficients for each individual. Each individual’s genotype is represented by a single vertical line, which is partitioned into colored segments corresponding to the estimated membership in the two or three groups. AM in each plot represents European Apis mellifera. a Population clustering based on 2449 SNP loci with European AM. b Population clustering based on 2449 SNP loci without European AM. c Population clustering based on 83 divergent SNP loci with European AM. d Population clustering based on 83 divergent SNP loci without European AM
Fig. 6.

Clustering pattern obtained by discriminant analysis of principal coordinates using 2449 SNP loci with 474 honey bees collected from 28 geographical regions in the Republic of South Africa. Each group is shown by different colors and the colored circle dots in the figures represent individuals of each subspecies. European Apis mellifera are indicated with a star. a Clustering plot of all individuals with European A. mellifera and b without European A. mellifera
Using the reduced set of just 83 divergent SNP loci and including European A. mellifera in the analysis, A.m. scutellata and A.m. capensis revealed a similar delta K value consistent with the pattern produced with the 2449 SNP set, supporting three distinctive genetic clusters (K = 3) across individuals (Fig. 5). Two genetic groups were defined (K = 2), when European A. mellifera were excluded. In contrast, the PCoA analysis displayed increased resolution with the reduced marker set, capturing 14.95% and 67.96% of the variance in the first and second components respectively (Fig. 7). With European A. mellifera excluded, 37% of the individual A.m. scutellata and hybrids overlapped in the PCoA plot, 69% of the A.m. capensis and hybrids overlapped, 0.2% of the A.m. capensis and A.m. scutellata overlapped, and 9% of all three groups overlapped.
Fig. 7.

Clustering pattern obtained by discriminant analysis of principal coordinate using 83 divergent SNP loci with collected from 28 geographical regions in the Republic of South Africa. Each group is shown by different colors and the colored circle dots in the figures represent individuals of each subspecies. European Apis mellifera are indicated with a star. a Clustering plot of all individuals with European A. mellifera and b without European A. mellifera
Using the set of 2449 SNP loci, a low level of population differentiation (FST) was observed within each group, estimated at 0.035 for A.m. capensis and 0.04 for A.m. scutellata.
We observed the highest significant pairwise genetic differentiation between BD and all other regions (0.07 to 0.12, P < 0.05). The lowest FST values observed among other pairs of regions ranged from 0.02 to 0.09, P < 0.05 (Table 8). Apis mellifera capensis and A.m. scutellata revealed a lower value of genetic differentiation using all 2449 SNP loci, although they are distinctly clustered in the structure plots. AMOVA results revealed that most of the total genetic variation occurred within populations (93%, P < 0.001), while only 3% was attributed across populations (P < 0.001).
Table 8.
Pairwise FST values computed using 2449 SNPs from honey bees collected from 28 geographical regions in the Republic of South Africa. European A. mellifera (AM) was included as a single region in the dataset
| BL | KR | PT | SP | UP | OD | PB | PE | RD | SF | ST | SW | TR | WD | MF | MB | LB | LA | KN | GT | GE | CT | CD | BD | GR | BW | EL | KL | AM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL | 0 | ||||||||||||||||||||||||||||
| KR | 0.03 | 0.00 | |||||||||||||||||||||||||||
| PT | 0.04 | 0.04 | 0.00 | ||||||||||||||||||||||||||
| SP | 0.04 | 0.05 | 0.06 | 0.00 | |||||||||||||||||||||||||
| UP | 0.03 | 0.04 | 0.05 | 0.03 | 0.00 | ||||||||||||||||||||||||
| OD | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.00 | |||||||||||||||||||||||
| PB | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.03 | 0.00 | ||||||||||||||||||||||
| PE | 0.04 | 0.04 | 0.05 | 0.06 | 0.04 | 0.03 | 0.04 | 0.00 | |||||||||||||||||||||
| RD | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.03 | 0.04 | 0.04 | 0.00 | ||||||||||||||||||||
| SF | 0.04 | 0.04 | 0.05 | 0.05 | 0.04 | 0.04 | 0.03 | 0.03 | 0.03 | 0.00 | |||||||||||||||||||
| ST | 0.05 | 0.05 | 0.06 | 0.07 | 0.06 | 0.03 | 0.04 | 0.04 | 0.03 | 0.04 | 0.00 | ||||||||||||||||||
| SW | 0.05 | 0.06 | 0.06 | 0.07 | 0.06 | 0.03 | 0.04 | 0.04 | 0.03 | 0.04 | 0.03 | 0.00 | |||||||||||||||||
| TR | 0.07 | 0.07 | 0.09 | 0.08 | 0.07 | 0.05 | 0.06 | 0.06 | 0.06 | 0.06 | 0.05 | 0.05 | 0.00 | ||||||||||||||||
| WD | 0.06 | 0.06 | 0.07 | 0.07 | 0.06 | 0.03 | 0.04 | 0.04 | 0.03 | 0.04 | 0.03 | 0.03 | 0.06 | 0.00 | |||||||||||||||
| MF | 0.04 | 0.04 | 0.05 | 0.06 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.03 | 0.04 | 0.04 | 0.06 | 0.05 | 0.00 | ||||||||||||||
| MB | 0.05 | 0.05 | 0.07 | 0.06 | 0.05 | 0.04 | 0.04 | 0.04 | 0.03 | 0.04 | 0.03 | 0.04 | 0.06 | 0.03 | 0.04 | 0.00 | |||||||||||||
| LB | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.03 | 0.03 | 0.04 | 0.03 | 0.04 | 0.02 | 0.03 | 0.06 | 0.03 | 0.04 | 0.03 | 0.00 | ||||||||||||
| LA | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.03 | 0.03 | 0.04 | 0.03 | 0.03 | 0.03 | 0.03 | 0.05 | 0.03 | 0.04 | 0.03 | 0.03 | 0.00 | |||||||||||
| KN | 0.04 | 0.05 | 0.05 | 0.06 | 0.05 | 0.03 | 0.03 | 0.04 | 0.03 | 0.03 | 0.03 | 0.03 | 0.06 | 0.03 | 0.03 | 0.04 | 0.03 | 0.03 | 0.00 | ||||||||||
| GT | 0.04 | 0.04 | 0.05 | 0.06 | 0.04 | 0.03 | 0.04 | 0.03 | 0.03 | 0.03 | 0.04 | 0.04 | 0.06 | 0.04 | 0.03 | 0.03 | 0.03 | 0.03 | 0.04 | 0.00 | |||||||||
| GE | 0.04 | 0.04 | 0.05 | 0.06 | 0.04 | 0.03 | 0.03 | 0.04 | 0.02 | 0.03 | 0.03 | 0.03 | 0.05 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.02 | 0.03 | 0.00 | ||||||||
| CT | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.03 | 0.03 | 0.04 | 0.03 | 0.03 | 0.02 | 0.03 | 0.05 | 0.03 | 0.04 | 0.03 | 0.03 | 0.02 | 0.03 | 0.03 | 0.02 | 0.00 | |||||||
| CD | 0.04 | 0.05 | 0.06 | 0.05 | 0.05 | 0.03 | 0.04 | 0.04 | 0.03 | 0.03 | 0.03 | 0.03 | 0.05 | 0.03 | 0.04 | 0.03 | 0.03 | 0.02 | 0.03 | 0.03 | 0.03 | 0.02 | 0.00 | ||||||
| BD | 0.10 | 0.10 | 0.12 | 0.12 | 0.10 | 0.08 | 0.08 | 0.08 | 0.07 | 0.09 | 0.07 | 0.07 | 0.11 | 0.07 | 0.08 | 0.08 | 0.08 | 0.07 | 0.08 | 0.08 | 0.07 | 0.08 | 0.07 | 0.00 | |||||
| GR | 0.03 | 0.04 | 0.05 | 0.05 | 0.04 | 0.03 | 0.04 | 0.03 | 0.03 | 0.03 | 0.04 | 0.04 | 0.06 | 0.04 | 0.03 | 0.04 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.08 | 0.00 | ||||
| BW | 0.04 | 0.05 | 0.05 | 0.05 | 0.04 | 0.05 | 0.05 | 0.04 | 0.05 | 0.04 | 0.05 | 0.05 | 0.07 | 0.05 | 0.04 | 0.05 | 0.05 | 0.04 | 0.05 | 0.04 | 0.04 | 0.04 | 0.04 | 0.09 | 0.04 | 0.00 | |||
| EL | 0.04 | 0.04 | 0.05 | 0.06 | 0.04 | 0.03 | 0.04 | 0.04 | 0.04 | 0.03 | 0.04 | 0.04 | 0.07 | 0.04 | 0.03 | 0.04 | 0.04 | 0.04 | 0.03 | 0.03 | 0.03 | 0.04 | 0.04 | 0.09 | 0.03 | 0.04 | 0.00 | ||
| KL | 0.05 | 0.05 | 0.06 | 0.06 | 0.05 | 0.04 | 0.05 | 0.05 | 0.04 | 0.04 | 0.04 | 0.05 | 0.06 | 0.05 | 0.05 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.09 | 0.04 | 0.05 | 0.05 | 0.00 | |
| AM | 0.23 | 0.22 | 0.26 | 0.27 | 0.23 | 0.27 | 0.25 | 0.23 | 0.22 | 0.29 | 0.23 | 0.25 | 0.30 | 0.24 | 0.24 | 0.28 | 0.26 | 0.24 | 0.22 | 0.24 | 0.22 | 0.24 | 0.22 | 0.35 | 0.22 | 0.25 | 0.28 | 0.26 | 0.00 |
The geographical abbreviations are shown in Table 1. The significant values of all pairwise comparisons are based on Bonferroni correction (P < 0.05)
Discussion
Morphological studies revealed two distinct morphoclusters of honey bee colonies in RSA [9, 71, 72]. Apis mellifera capensis became distinct from other subspecies in Africa due to three phenotypic traits (thelytoky, spermatheca size, and ovariole number) [23]. We found some degree of overlap in the clustering patterns of the two subspecies in RSA, as is observed among other African honey bee subspecies [36]. Our wing geometry, standard morphometric and genomic data supported a clear differentiation between A.m. capensis and A.m. scutellata, though there were no individual variables that alone predicted this separation. Our study demonstrated that 17 wing geometry and standard morphometric parameters can be used to separate the bees into three clusters coinciding with their subspecies and hybrid distributions. We found that honey bee populations from several regions fell outside of the confidence ellipses and instead positioned at the intermediate regions, as expected for hybrids [4, 9].
With the aid of high-throughput sequencing technologies, it has become possible to genotype a large number of samples economically, this in order to determine genetic diversity, population structure and degree of introgression in different honey bee populations [73, 74]. Here, we utilized GBS technology to infer genetic diversity and population structure in a large collection of honey bees from the Republic of South Africa. To our knowledge, this is the first attempt to use GBS, in comparison with wing geometry and standard morphometric parameters, to characterize the population structure and identify ancestry informative markers that can be applied to distinguish between A.m. capensis and A.m. scutellata in research and production settings. GBS provided a total of 2449 highly informative SNP markers based on very stringent quality criteria. We found the maximum evolutionary divergence between the African subspecies of honey bees and the European honey bees we tested.
The majority of the SNPs identified in the examined honey bees exhibited a high degree of polymorphism. The average level of polymorphic SNP markers observed in A.m. capensis, A.m. scutellata and hybrids (Np = >80%) was higher than that previously reported for each subspecies of A. mellifera (Np = 40%) [75]. The average number of SNP polymorphisms across the honey bees we collected in the RSA was higher than those observed in four of the evolutionary groups (A, M, C and O) of honey bees (Np = 84.5% vs 30%) [75]. The large SNP variation we detected could be due to number of SNPs tested, the number of honey bees genotyped and geological history of honey bees in Africa.
Genetic diversity based on expected heterozygosity was the highest in almost all regions of A.m. capensis, A.m. scutellata and hybrids, with an average of Hexp = 0.23. The highest level of heterozygosity previously reported in the honey bee literature was observed in African bees (Hobs = 0.12) [73]. This is consistent with previous studies, suggesting that African honey bee subspecies exhibit high genetic diversity, most likely due to their large effective population size, low level of inbreeding between lineages, and lack of population bottlenecks incurred during quaternary ice ages [75–79]. The SNP heterozygosity values reported across regions in our study were lower than those obtained using microsatellite markers [8, 29]. These differences between the two techniques could reflect the multi-allelic nature of microsatellite markers [80]. In one study conducted by Fuller et al. [79], the heterozygosity level observed in the Forkhead Box Protein O (Foxo, GB48301) gene was greater in savannah honey bees (likely A.m. scutellata) than in desert honey bee populations (likely A.m. yemenitica and A.m. simensis) in Kenya.
The clustering pattern from the STRUCTURE analysis of 2449 SNPs illustrated shared ancestry correlating to the two known subspecies in the RSA, and was mirrored by the PCoA. These methods distinguished between A.m. capensis and A.m. scutellata, supporting the idea of two subspecies with distinctive physiological and behavioral differences [9, 79]. In contrast, a set of ancestry informative markers distinguishing these two subspecies could not be found in several recent studies examining both genomic and whole mitochondrial genomes [3, 81–83]. Recently, the effects of Africanization on the genome diversity of 32 Africanized honey bees from Brazil was determined [84]. In that study, signals of positive selection on chromosome 11 indicative of adaptive evolution in the Africanized honey bee population were identified. The authors concluded that African Brazilian honey bee populations are indistinguishable from African ancestry because these two populations have not sufficiently diverged.
Our success in genetically identifying the two subspecies is likely due to the sizable number of individuals we analyzed, and the use of SNP markers specifically derived from the target populations [3, 81, 82]. Our findings are consistent with a study conducted on 11 honey bees collected from four distinct ecological regions (savannah, coast, desert and mountain) in Kenya. Those authors concluded that A.m. scutellata in savannah regions can be distinguished from other honey bees based on the phylogenetic analysis of complete mitochondrial genome sequences [79].
The performance of outlier approaches to differentiate honey bee populations has been investigated by others [74, 82]. Here, the outlier analysis suggested that 83 SNPs were potential candidates for use to differentiate A.m. capensis and A.m. scutellata. Our STRUCTURE and PCoA analyses using 83 divergent loci enhanced the resolution power of SNPs used to discriminate the two subspecies. We suggest that the accuracy and robustness of these markers should be determined on randomly collected samples from RSA. This could validate the discriminatory power of the divergent loci.
The detected signature of admixture within A.m. scutellata could be due to the fact that A.m. scutellata differs in genetic characteristics from A.m. capensis. Unique clustering of A.m. scutellata was also observed in phylogenetic analysis based on whole genome data of honey bee populations in Kenya [79]. We found several genomic regions under selective pressure within A.m. capensis, allowing reliable assignment of individuals to the population of origin and providing effective tools to identify pure A.m. capensis colonies in RSA. We propose that these 83 markers may be utilized effectively for the identification of both subspecies, a critical application for both research and agricultural efforts. These 83 divergent loci may be under natural selection for physiological and behavioral characters adaptive to the native environments of both subspecies.
A functional analysis highlighted processes involved in neurology/behavior and growth/development which were among the most rapidly evolving genes identified in the two subspecies. Apis mellifera scutellata and A.m. capensis are the two divergent honey bee subspecies that exhibit distinct biological functions in RSA [9, 16–19]. Indeed, these subspecies differ in several aspects of behavior maturation in a presumably adaptive way, including foraging activity and defensive behaviors [9, 12, 15, 17, 27, 28]. The defensive behavior and colony usurpation tendencies of A.m. scutellata and thelytoky in A.m. capensis are the most heritable traits supporting the functional behavior in this study [12, 15, 17]. Foraging, which encodes a cyclic G-dependent protein kinase, affects feeding and food gathering–related activities in both honey bees and Drosophila melanogaster [85, 86].
Honey bees exhibit a suite of diverse behaviors in social environment and several hundred genes have been closely associated with brain function and physiological behaviors in bees [86]. Several genes have been found to regulate neuronal function and behavior, for example the gene metabotropic GABA-B receptor subtype 1 [87]. Chemical signaling is used to coordinate the behavior and physiology of colony members. Changes in the protein-coding sequence are possibly related to the evolution of the chemical communication system found in honey bees [88].
We found several genes of the divergent loci shown to impact embryonic development and growth. Eusocial insects have remarkably diverse exocrine gland functions and produce many novel glandular secretions, including pheromones, brood food, and antimicrobial compounds [89, 90].
Genes involved in caste differentiation, worker development and reproduction in both subspecies are the most prominent examples of gene families gaining diverse functions through the social interactions [91]. Our results provide an avenue for linking specific genetic changes to the functional evolution in these bees. Major challenges in this attempt include determining the genes associated with morphological and behavioral differences between the subspecies and furthering our understanding of how changes in the gene function affect a biological process in these subspecies. However, additional work to measure LD length and haplotype blocks encompassing these divergent loci, and considerable improvement in functional annotation of the reference genome, are needed before conclusive work on selective sweeps can be accomplished in these subspecies.
The distribution and clear differentiation of the two subspecies suggests that they may have been separated by a permanent barrier historically, a barrier likely influenced by environmental conditions such as temperature and precipitation. This is consistent with the literature, which suggests that A.m. scutellata prefers warm and dry climates while A.m. capensis prefers cooler wetter ones [9, 15]. Indeed, we found significant associations of several divergent SNP loci with environmental parameters, most notably temperature for A.m. scutellata SNPs and precipitation for A.m. capensis ones. These findings explain the population distribution of the two subspecies along the west-east axis, and supports the occurrence of adaptive divergence related to environmental parameters. Such signals of local adaptation to environmental variables were previously observed in Iberian honey bee populations [92]. We believe that temperature and precipitation could be two important parameters maintaining the population structure of honey bees in RSA. Another factor contributing to genetic divergence of these two species could be isolating differences in the behaviors of A.m. capensis and A.m. scutellata as demonstrated by Hepburn and Radloff [9], Jaffé et al. [11] and Onions [16].
The low level of differentiation between these two subspecies in our study (FST = 0.06) could be related to a substantial level of gene flow between populations [93]. A second possible reason for low FST values could be attributed to the large population size of A.m. capensis and A.m. scutellata [9]. The FST values observed in this study for A.m. capensis (0.035), A.m. scutellata (0.04) and hybrids (0.04), is lower than previously reported for honey bees in Africa [29] and higher than values observed between savannah and desert honey bees in Kenya [79]. We suggest that the lack of any physical barrier between the indigenous ranges of the two subspecies and exchanging of queens and colonies between beekeepers in both areas are contributing factors supporting the admixture pattern within A.m. capensis [94, 95]. It was previously noted that honey bee colonies pollinating in hot/dry regions (e.g. A.m. scutellata) migrate over large distances to environments with more resources in order to withstand reduced nutrient intake during winter seasons better [79, 84].
We identified 47 and 21 divergent loci for A.m. capensis and A.m. scutellata, respectively. These results provide evidence for signatures of natural selection in RSA honey bee populations. We demonstrated the relevance of environmental heterogeneity in driving locally adaptive genetic variation within these candidate loci. For divergent loci, temperature and precipitation variables were significantly associated with SNP variants within signatures of selection, highlighting the importance of these environmental factors in adaptation to local conditions [9, 15, 79]. The present findings provide some testable hypotheses for additional experimental analyses of the functional role these genes play in ecological adaptation.
Conclusions
Considerable genetic diversity is retained within indigenous honey bee populations in RSA. Principal coordinate and population structure analyses clearly differentiated A.m. capensis and A.m. scutellata populations, and quantified ancestry in hybrid bees, as expected based on their behavioral and ecological characteristics. The differentiation pattern describes the genetic distinctiveness of A.m. scutellata and A.m. capensis populations. The regional admixture observed in A.m. capensis populations represents a unique genetic resource, and an unexploited opportunity, that necessitates initiatives for the sustainable conservation of this subspecies. The significant identification of divergent SNP loci by environmental variables suggests adaptive selection occurring within the RSA honey bee subspecies. The wing geometry and standard morphometric analyses supported grouping the bees into two subspecies, which was consistent with genetic structure. We believe the 83 divergent SNPs discovered here enable distinguishing between A.m. scutellata and A.m. capensis with improved efficiency and accuracy.
Additional files
Table S1. Bioclimatic variables used in the distribution model analysis for the two honey bee subspecies from the Republic of South Africa. (DOC 127 kb)
Figure S1. Canonical variance analysis factor loadings of wing geometry and standard morphometric measurements onto Canonical Vector 1 (CV1) (A) and Canonical Vector 2 (CV2) (B), based on subspecies classifications, for 464 measured honey bees collected from the Republic of South Africa. CV1 and CV2 factor loadings all generated the varied sign and contributed in positive and negative values. (TIF 8470 kb)
Figure S2. Canonical variance analysis factor loadings of wing geometry and standard morphometric measurements onto Canonical Vector 1 (CV1) (A) and Canonical Vector 2 (CV2) (B), based on regional classifications, for 464 measured honey bees collected from the Republic of South Africa. CV1 and CV2 factor loadings all generated the varied sign and contributed in positive and negative values. (TIF 8470 kb)
Figure S3. The hierarchical clustering structure of 464 honey bees collected from 28 geographical regions in the Republic of South Africa. The colors indicate different subspecies: blue = Apis mellifera scutellata (N = 73), red = A.m. capensis (N = 337) and green = hybrids (N = 54). (TIF 622 kb)
{kind=link}
Table S2. A pair-wise evolutionary divergence matrix based on a corrected p-distance nucleotide model among honey bees in 29 geographical regions in the Republic of South Africa and a reference European Apis mellifera. The geographical abbreviations are explained in Table 1. (JPG 270 kb)
{kind=link}
Figure S4 A, B. Identification of putative divergent SNP loci under directional selection for Apis mellifera capensis based on FST outlier approaches. (A) Hierarchical structure model using Arlequin 3.5. FST: locus –specific genetic divergence among the populations; Heterozygosity: measure of heterozygosity per locus. The significant loci are shown with red dots (P < 0.01). (B) Finite island model (fdist) by LOGISTAN. Loci under positive selection above 99% percentile are shown in the red area. Loci shown in the gray area are neutral loci and those in the yellow area are under balancing selection. (JPG 592 kb)
{kind=link}
Figure S5 A, B. Identification of putative divergent SNP loci under directional selection for Apis mellifera scutellata based on FST outlier approaches. (A) Hierarchical structure model using Arlequin 3.5. FST: locus –specific genetic divergence among the populations; Heterozygosity: measure of heterozygosity per locus. The significant loci are shown with red dots (P < 0.01). (B) Finite island model (fdist) by LOGISTAN. Loci under positive selection above 99% percentile are shown in the red area. Loci shown in the gray area are neutral loci and those in the yellow area are under balancing selection. (JPG 272 kb)
{kind=link}
Table S3. Estimated posterior probabilities and delta K for each K partition. (JPG 571 kb)
Acknowledgements
The authors would like to thank the current and former members of the University of Florida Honey Bee Research and Extension Laboratory who collected honey bee samples across the Republic of South Africa. This includes Mark Dykes, Ashley Mortensen, and Daniel Schmehl. We graciously acknowledge Mike Allsopp (ARC-Plant Protection Research Institute, South Africa), Christian Pirk (University of Pretoria, South Africa), Garth Cambray (beekeeper), and Eddie Hart (beekeeper) for the assistance they provided in coordinating field sample collections and/or providing samples. We acknowledge Heather Marie Holl and Laura Patterson-Rosa in Department of Animal Sciences, University of Florida for their technical assistance and proof-reading support. We also thank the South African beekeepers who allowed us to sample their colonies. We also acknowledge the Cornell University, Institute of Biotechnology (Genomic facilities) for performing Genotyping-By-Sequencing of 474 honey bees and bioinformatic service regarding the association analyses of the project. The authors acknowledge Tao Zhang from Department of Biology, University of Florida for providing the bioclimatic variables of two subspecies from the WorldClim database.
Funding
This project was financed through a cooperative agreement provided by the United States Department of Agriculture, Animal and Plant Health Inspection Service (USDA-APHIS, Projects 14–8130-0414-CA and 16–8130-0414-CA) and by the Florida Department of Agriculture and Consumer Services through the guidance of the Honey Bee Technical Council.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available through Dryad Digital Repository 10.5061/dryad.98jh446.
Authors’ contributions
AE and JDE designed and coordinated the research. TB and JDE collected the samples. AE conducted library construction and sequencing. AE, SAB and TB analyzed the data. AE drafted the paper with the input of JDE and SAB. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Amin Eimanifar, Phone: 352 273 3958, Email: amineimanifar1979@gmail.com.
Samantha A. Brooks, Email: samantha.brooks@ufl.edu
Tomas Bustamante, Email: g4spar@ufl.edu.
James D. Ellis, Email: jdellis@ufl.edu
References
- 1.Haddad N, Mahmud Batainh A, Suleiman Migdadi O, Saini D, Krishnamurthy V, Parameswaran S, et al. Next generation sequencing of Apis mellifera syriaca identifies genes for Varroa resistance and beneficial bee keeping traits. Insect science. 2016;23(4):579–590. doi: 10.1111/1744-7917.12205. [DOI] [PubMed] [Google Scholar]
- 2.Steffan-Dewenter I, Tscharntke T. Effects of habitat isolation on pollinator communities and seed set. Oecologia. 1999;121(3):432–440. doi: 10.1007/s004420050949. [DOI] [PubMed] [Google Scholar]
- 3.Wallberg A, Han F, Wellhagen G, Dahle B, Kawata M, Haddad N, et al. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat Genet. 2014;46(10):1081. doi: 10.1038/ng.3077. [DOI] [PubMed] [Google Scholar]
- 4.Ruttner F. Biogeography and taxonomy of honeybees. Springer Science & Business Media. 1988;
- 5.Al-Ghamdi AA, Nuru A, Khanbash MS, Smith DR. Geographical distribution and population variation of Apis mellifera jemenitica Ruttner. J Apic Res. 2013;52(3):124–133. [Google Scholar]
- 6.Garnery L, CORNUET JM, Solignac M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol Ecol. 1992;1(3):145–154. doi: 10.1111/j.1365-294x.1992.tb00170.x. [DOI] [PubMed] [Google Scholar]
- 7.Franck P, Garnery L, Solignac M, Cornuet JM. Molecular confirmation of a fourth lineage in honeybees from the near east. Apidologie. 2000;31(2):167–180. [Google Scholar]
- 8.Alburaki M, Bertrand B, Legout H, Moulin S, Alburaki A, Sheppard WS, et al. A fifth major genetic group among honeybees revealed in Syria. BMC Genet. 2013;14(1):117. doi: 10.1186/1471-2156-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hepburn HR, Radloff SE. Honeybees of Africa: Springer Science & Business Media; 1998.
- 10.Fletcher DJ. The African bee, Apis mellifera adansonii, in Africa. Annu Rev Entomol. 1978;23(1):151–171. [Google Scholar]
- 11.Jaffé R, Dietemann V, Crewe RM, Moritz RF. Temporal variation in the genetic structure of a drone congregation area: an insight into the population dynamics of wild African honeybees (Apis mellifera scutellata) Mol Ecol. 2009;18(7):1511–1522. doi: 10.1111/j.1365-294X.2009.04143.x. [DOI] [PubMed] [Google Scholar]
- 12.Rinderer TE, Oldroyd BP, Sheppard WS. Africanized bees in the US. Sci Am. 1993;269(6):84–90. [Google Scholar]
- 13.Spivak M, Fletcher DJ, Breed MD. The African honey bee. USA: Westview press; boulder; 1991. [Google Scholar]
- 14.Kono Y, Kohn JR. Range and frequency of africanized honey bees in California (USA) PLoS One. 2015;10(9):e0137407. doi: 10.1371/journal.pone.0137407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seeley TD. Honeybee ecology: a study of adaptation in social life: Princeton University Press; 2014.
- 16.Onions GW. South African fertile-worker bees. Agricultural Journal of the Union of South Africa. 1912;3(5):720. [Google Scholar]
- 17.Neumann P, Moritz R. The cape honeybee phenomenon: the sympatric evolution of a social parasite in real time? Behav Ecol Sociobiol. 2002;52(4):271–281. [Google Scholar]
- 18.Hepburn HR, Crewe RM. Portrait of the cape honeybee, Apis mellifera capensis. Apidologie. 1991;22(6):567–580. [Google Scholar]
- 19.Hepburn HR, Allsopp MH. Reproductive conflict between honeybees: usurpation of Apis mellifera scutellata colonies by Apis mellifera capensis. S Afr J Sci. 1994;90(4):247–249. [Google Scholar]
- 20.Wossler TC. Pheromone mimicry by Apis mellifera capensis social parasites leads to reproductive anarchy in host Apis mellifera scutellata colonies. Apidologie. 2002;33(2):139–163. [Google Scholar]
- 21.Moritz RF, Lattorff HM, Crewe RM. Honeybee workers (Apis mellifera capensis) compete for producing queen ‘like pheromone signals. Proc R Soc Lond B Biol Sci. 2004;271(3):S98–100. doi: 10.1098/rsbl.2003.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phiancharoen M, Pirk CW, Radloff SE, Hepburn R. Clinal nature of the frequencies of ovarioles and spermathecae in cape worker honeybees, Apis mellifera capensis. Apidologie. 2010;41(2):129–134. [Google Scholar]
- 23.Ruttner F. The cape bee: a biological curiosity. Proc Apimondia Symp Afr Bees. 1977:127–31.
- 24.Okosun OO, Yusuf AA, Crewe RM, Pirk CW. Effects of age and reproductive status on Tergal gland secretions in Queenless honey bee workers, Apis mellifera scutellata and A. m. capensis. J Chem Ecol. 2015;41(10):896–903. doi: 10.1007/s10886-015-0630-6. [DOI] [PubMed] [Google Scholar]
- 25.Crewe RM, Velthuis HH. False queens: a consequence of mandibular gland signals in worker honeybees. Naturwissenschaften. 1980;67(9):467–469. [Google Scholar]
- 26.Velthuis HH, Ruttner F, Crewe RM. Differentiation in reproductive physiology and behaviour during the development of laying worker honey bees. Social Insects. 1990;9:231–243. [Google Scholar]
- 27.Härtel S, Neumann P, Kryger P, Von Der Heide C, Moltzer GJ, Crewe RM, et al. Infestation levels of Apis mellifera scutellata swarms by socially parasitic cape honeybee workers (Apis mellifera capensis) Apidologie. 2006;37(4):462–470. [Google Scholar]
- 28.Martin SJ, Beekman M, Wossler TC, Ratnieks FL. Parasitic cape honeybee workers, Apis mellifera capensis, evade policing. Nature. 2002;415(6868):163. doi: 10.1038/415163a. [DOI] [PubMed] [Google Scholar]
- 29.Franck P, Garnery L, Loiseau A, Oldroyd BP, Hepburn HR, Solignac M, et al. Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity. 2001;86(4):420–430. doi: 10.1046/j.1365-2540.2001.00842.x. [DOI] [PubMed] [Google Scholar]
- 30.Neumann P, Härtel S, Kryger P, Crewe RM, Moritz RF. Reproductive division of labour and thelytoky result in sympatric barriers to gene flow in honeybees (Apis mellifera L.) J Evol Biol. 2011;24(2):286–294. doi: 10.1111/j.1420-9101.2010.02167.x. [DOI] [PubMed] [Google Scholar]
- 31.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12(7):499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- 33.Milano I, Babbucci M, Cariani A, Atanassova M, Bekkevold D, Carvalho GR, et al. Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius) Mol Ecol. 2014;23(1):118–135. doi: 10.1111/mec.12568. [DOI] [PubMed] [Google Scholar]
- 34.Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G, Lowry DB, et al. Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am Nat. 2016;188(4):379–397. doi: 10.1086/688018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taranto F, D’Agostino N, Greco B, Cardi T, Tripodi P. Genome-wide SNP discovery and population structure analysis in pepper (Capsicum annuum) using genotyping by sequencing. BMC Genomics. 2016;17(1):943. doi: 10.1186/s12864-016-3297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kandemir İ, Özkan A, Fuchs S. Reevaluation of honeybee (Apis mellifera) microtaxonomy: a geometric morphometric approach. Apidologie. 2011;42(5):618. [Google Scholar]
- 37.Slice DE. 2007. Geometric morphometrics. Annu. Rev. Anthropol. 2007;36:261–281. [Google Scholar]
- 38.Meixner MD, Pinto MA, Bouga M, Kryger P, Ivanova E, Fuchs S. Standard methods for characterising subspecies and ecotypes of Apis mellifera. J Apic Res. 2013;52(4):1–28. [Google Scholar]
- 39.Francoy TM, Wittmann D, Drauschke M, Müller S, Steinhage V, Bezerra-Laure MA, et al. Identification of Africanized honey bees through wing morphometrics: two fast and efficient procedures. Apidologie. 2008;39(5):488–494. [Google Scholar]
- 40.Eimanifar AT, Kimball RL, Braun E, Ellis JD. The complete mitochondrial genome of the cape honey bee, Apis mellifera capensis Esch. (Insecta: hymenoptera: apidae) Mitochondrial DNA Part B. 2016;1(1):817–819. doi: 10.1080/23802359.2016.1241682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Donato M, Peters SO, Mitchell SE, Hussain T, Imumorin IG. Genotyping-by-sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One. 2013;8(5):e62137. doi: 10.1371/journal.pone.0062137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 43.Elsik CG, Worley KC, Bennett AK, Beye M, Camara F, Childers CP, et al. Finding the missing honey bee genes: lessons learned from a genome upgrade. BMC Genomics. 2014;15(1):86. doi: 10.1186/1471-2164-15-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson JL, Wittgenstein H, Mitchell SE, Hyma KE, Temnykh SV, Kharlamova AV, et al. Genotyping-by-sequencing (GBS) detects genetic structure and confirms behavioral QTL in tame and aggressive foxes (Vulpes vulpes) PLoS One. 2015;10(6):e0127013. doi: 10.1371/journal.pone.0127013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B. 1996;263(1377):1619–1626. [Google Scholar]
- 48.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10(3):564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 49.Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G. LOSITAN: a workbench to detect molecular adaptation based on a F ST-outlier method. BMC Bioinformatics. 2008;9(1):323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acheré V, Favre JM, Besnard G, Jeandroz S. Genomic organization of molecular differentiation in Norway spruce (Picea abies) Mol Ecol. 2005;14(10):3191–3201. doi: 10.1111/j.1365-294X.2005.02646.x. [DOI] [PubMed] [Google Scholar]
- 51.Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, et al. BEDOPS: high-performance genomic feature operations. Bioinformatics. 2012;28(14):1919–1920. doi: 10.1093/bioinformatics/bts277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 53.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(2):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, et al. G:profiler—a web server for functional interpretation of gene lists. Nucleic Acids Res. 2016;44(1):W83–W89. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005;25(15):1965–1978. [Google Scholar]
- 56.Hijmans RJ, Graham CH. The ability of climate envelope models to predict the effect of climate change on species distributions. Glob Chang Biol. 2006;12(12):2272–2281. [Google Scholar]
- 57.Booth TH, Nix HA, Busby JR, Hutchinson MF. BIOCLIM: the first species distribution modelling package, its early applications and relevance to most current MAXENT studies. Divers Distrib. 2014;20(1):1–9. [Google Scholar]
- 58.Rohlf FJ. Shape statistics: Procrustes superimpositions and tangent spaces. J Classif. 1999;16(2):197–223. [Google Scholar]
- 59.Viscosi V, Cardini A. Leaf morphology, taxonomy and geometric morphometrics: a simplified protocol for beginners. PLoS One. 2011;6(10):e25630. doi: 10.1371/journal.pone.0025630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raymond M, Rousset F. GENEPOP: population genetics software for exact tests and ecumenism. J Hered. 1995;86:248–249. [Google Scholar]
- 62.Guo SW, Thompson EA. Performing the exact test of hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–372. [PubMed] [Google Scholar]
- 63.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38(6):1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 64.Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol. 2002;11(2):155–165. doi: 10.1046/j.0962-1083.2001.01436.x. [DOI] [PubMed] [Google Scholar]
- 65.Joost S, Bonin A, Bruford MW, Després L, Conord C, Erhardt G, et al. A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol. 2007;16(18):3955–3969. doi: 10.1111/j.1365-294X.2007.03442.x. [DOI] [PubMed] [Google Scholar]
- 66.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Resour. 2007;7(4):574–578. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 69.Earl DA. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–361. [Google Scholar]
- 70.Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 71.Maa TC. An enquiry into the systematics of the tribus Apidini or honey bees (Hymenoptera) Treubia. 1953;21:525–640. [Google Scholar]
- 72.DuPraw EJ. Non-Linnean taxonomy and the systematics of honeybees. Syst Zool. 1965;14(1):1–24. [Google Scholar]
- 73.Harpur BA, Minaei S, Kent CF, Zayed A. Management increases genetic diversity of honey bees via admixture. Mol Ecol. 2012;21(18):4414–4421. doi: 10.1111/j.1365-294X.2012.05614.x. [DOI] [PubMed] [Google Scholar]
- 74.Muñoz I, Henriques D, Johnston JS, Chávez-Galarza J, Kryger P, Pinto MA. Reduced SNP panels for genetic identification and introgression analysis in the dark honey bee (Apis mellifera mellifera) PLoS One. 2015;10(4):e0124365. doi: 10.1371/journal.pone.0124365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han F, Wallberg A, Webster MT. From where did the western honeybee (Apis mellifera) originate? Ecology and Evolution. 2012;2(8):1949–1957. doi: 10.1002/ece3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Estoup A, Garnery L, Solignac M, Cornuet JM. Microsatellite variation in honey bee (Apis mellifera L.) populations: hierarchical genetic structure and test of the infinite allele and stepwise mutation models. Genetics. 1995;140(2):679–695. doi: 10.1093/genetics/140.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McMichael M, Hall HG. DNA RFLPs at a highly polymorphic locus distinguish European and African subspecies of the honey bee Apis mellifera L. and suggest geographical origins of New World honey bees. Mol Ecol. 1996;5(3):403–416. [Google Scholar]
- 78.Franck P, Garnery L, Solignac M, Cornuet JM. The origin of west European subspecies of honeybees (Apis mellifera): new insights from microsatellite and mitochondrial data. Evolution. 1998;52(4):1119–1134. doi: 10.1111/j.1558-5646.1998.tb01839.x. [DOI] [PubMed] [Google Scholar]
- 79.Fuller ZL, Niño EL, Patch HM, Bedoya-Reina OC, Baumgarten T, Muli E, et al. Genome-wide analysis of signatures of selection in populations of African honey bees (Apis mellifera) using new web-based tools. BMC Genomics. 2015;16(1):518. doi: 10.1186/s12864-015-1712-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nietlisbach P, Keller LF, Postma E. Genetic variance components and heritability of multiallelic heterozygosity under inbreeding. Heredity. 2016;116(1):1. doi: 10.1038/hdy.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chapman NC, Harpur BA, Lim J, Rinderer TE, Allsopp MH, Zayed A, et al. A SNP test to identify Africanized honeybees via proportion of ‘African’ ancestry. Mol Ecol Resour. 2015;15(6):1346–1355. doi: 10.1111/1755-0998.12411. [DOI] [PubMed] [Google Scholar]
- 82.Harpur BA, Chapman NC, Krimus L, Maciukiewicz P, Sandhu V, Sood K, et al. Assessing patterns of admixture and ancestry in Canadian honey bees. Insect Soc. 2015;62(4):479–489. [Google Scholar]
- 83.Eimanifar A, Kimball RT, Braun EL, Ellis JD. Mitochondrial genome diversity and population structure of two western honey bee subspecies in the Republic of South Africa. Sci Rep. 2018;228(1):1333. doi: 10.1038/s41598-018-19759-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nelson RM, Wallberg A, Simões ZL, Lawson DJ, Webster MT. Genome-wide analysis of admixture and adaptation in the Africanized honeybee. Mol Ecol. 2017;26(14):3603–3617. doi: 10.1111/mec.14122. [DOI] [PubMed] [Google Scholar]
- 85.Kent CF, Daskalchuk T, Cook L, Sokolowski MB, Greenspan RJ. The Drosophila foraging gene mediates adult plasticity and gene-environment interactions in behaviour, metabolites, and gene expression in response to food deprivation. PLoS Genet. 2009;5:e1000609. doi: 10.1371/journal.pgen.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ben-Shahar Y, Leung H-T, Pak WL, Sokolowski MB, Robinson GE. cGMP-dependent changes in phototaxis: a possible role for the foraging gene in honey bee division of labor. J Exp Biol. 2003;206:2507–2515. doi: 10.1242/jeb.00442. [DOI] [PubMed] [Google Scholar]
- 87.Root CM, Masuyama K, Green DS, Enell LE, Nässel DR, Lee CH, et al. A presynaptic gain control mechanism fine-tunes olfactory behavior. Neuron. 2008;59(2):311–321. doi: 10.1016/j.neuron.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woodard SH, Fischman BJ, Venkat A, Hudson ME, Varala K, Cameron SA, et al. Clark AG, Robinson GE. Genes involved in convergent evolution of eusociality in bees. Proc Natl Acad Sci. 2011;108(18):7472–7477. doi: 10.1073/pnas.1103457108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wilson EO. The insect societies. Press, Cambridge, MA: Harvard Univ; 1971. [Google Scholar]
- 90.Roubik DW. Ecology and natural history of tropical bees. Press, Cambridge, UK: Cambridge Univ; 1992. [Google Scholar]
- 91.Honeybee Genome Sequencing Consortium Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443(7114):931. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chávez-Galarza J, Henriques D, Johnston JS, Azevedo JC, Patton JC, Muñoz I, et al. Signatures of selection in the Iberian honey bee (Apis mellifera iberiensis) revealed by a genome scan analysis of single nucleotide polymorphisms. Mol Ecol. 2013;22(23):5890–5907. doi: 10.1111/mec.12537. [DOI] [PubMed] [Google Scholar]
- 93.Otis GW, Winston ML, Taylor OR., Jr Engorgement and dispersal of Africanized honeybee swarms. J Apic Res. 1981;20(1):3–12. [Google Scholar]
- 94.Gupta RK, Reybroeck W, van Veen JW, Gupta A. Beekeeping for poverty alleviation and livelihood security. 2014. [Google Scholar]
- 95.Techer MA, Clémencet J, Turpin P, Volbert N, Reynaud B, Delatte H. Genetic characterization of the honeybee (Apis mellifera) population of Rodrigues Island, based on microsatellite and mitochondrial DNA. Apidologie. 2015;46(4):445–454. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Bioclimatic variables used in the distribution model analysis for the two honey bee subspecies from the Republic of South Africa. (DOC 127 kb)
Figure S1. Canonical variance analysis factor loadings of wing geometry and standard morphometric measurements onto Canonical Vector 1 (CV1) (A) and Canonical Vector 2 (CV2) (B), based on subspecies classifications, for 464 measured honey bees collected from the Republic of South Africa. CV1 and CV2 factor loadings all generated the varied sign and contributed in positive and negative values. (TIF 8470 kb)
Figure S2. Canonical variance analysis factor loadings of wing geometry and standard morphometric measurements onto Canonical Vector 1 (CV1) (A) and Canonical Vector 2 (CV2) (B), based on regional classifications, for 464 measured honey bees collected from the Republic of South Africa. CV1 and CV2 factor loadings all generated the varied sign and contributed in positive and negative values. (TIF 8470 kb)
Figure S3. The hierarchical clustering structure of 464 honey bees collected from 28 geographical regions in the Republic of South Africa. The colors indicate different subspecies: blue = Apis mellifera scutellata (N = 73), red = A.m. capensis (N = 337) and green = hybrids (N = 54). (TIF 622 kb)
Table S2. A pair-wise evolutionary divergence matrix based on a corrected p-distance nucleotide model among honey bees in 29 geographical regions in the Republic of South Africa and a reference European Apis mellifera. The geographical abbreviations are explained in Table 1. (JPG 270 kb)
Figure S4 A, B. Identification of putative divergent SNP loci under directional selection for Apis mellifera capensis based on FST outlier approaches. (A) Hierarchical structure model using Arlequin 3.5. FST: locus –specific genetic divergence among the populations; Heterozygosity: measure of heterozygosity per locus. The significant loci are shown with red dots (P < 0.01). (B) Finite island model (fdist) by LOGISTAN. Loci under positive selection above 99% percentile are shown in the red area. Loci shown in the gray area are neutral loci and those in the yellow area are under balancing selection. (JPG 592 kb)
Figure S5 A, B. Identification of putative divergent SNP loci under directional selection for Apis mellifera scutellata based on FST outlier approaches. (A) Hierarchical structure model using Arlequin 3.5. FST: locus –specific genetic divergence among the populations; Heterozygosity: measure of heterozygosity per locus. The significant loci are shown with red dots (P < 0.01). (B) Finite island model (fdist) by LOGISTAN. Loci under positive selection above 99% percentile are shown in the red area. Loci shown in the gray area are neutral loci and those in the yellow area are under balancing selection. (JPG 272 kb)
Table S3. Estimated posterior probabilities and delta K for each K partition. (JPG 571 kb)
Data Availability Statement
The datasets generated and/or analyzed during the current study are available through Dryad Digital Repository 10.5061/dryad.98jh446.