

Abstract
Background
This double-blind, randomized, 78-week study evaluated the efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics of PF-06410293, a candidate adalimumab biosimilar, versus adalimumab reference product (Humira®) sourced from the EU (adalimumab-EU) in biologic-naïve patients with active rheumatoid arthritis (RA) despite methotrexate (MTX) (10–25 mg/week). We report results for the first 26 weeks of treatment.
Methods
Patients with active RA (N = 597) were randomly assigned (1:1) to PF-06410293 or adalimumab-EU, while continuing with MTX treatment. The primary endpoint was American College of Rheumatology 20% improvement (ACR20) at week 12. Therapeutic equivalence was concluded if the two-sided 95% confidence interval (CI) for the ACR20 difference between the two arms was entirely contained within the symmetric equivalence margin (±14%). Additionally, a two-sided 90% CI was calculated by using an asymmetric equivalence margin (−12%, 15%). Secondary efficacy endpoints to week 26 included ACR20/50/70, change from baseline Disease Activity Score based on high-sensitivity C-reactive protein [DAS28–4(CRP)], European League Against Rheumatism (EULAR) response, DAS28–4(CRP) of less than 2.6, and ACR/EULAR remission. QuantiFERON-TB testing was performed at screening and week 26.
Results
Patients (78.7% of whom were female and whose mean age was 52.5 years) had a mean baseline RA duration of 6.8 years. The mean baseline DAS28–4(CRP) values were 5.9 (PF-06410293) and 6.1 (adalimumab-EU). The observed week-12 ACR20 values were 68.7% (PF-06410293) and 72.7% (adalimumab-EU) in the intention-to-treat population. With non-responder imputation, the treatment difference in week-12 ACR20 was −2.98% and corresponding CIs—95% CI (−10.38%, 4.44%) and 90% CI (−9.25%, 3.28%)—were entirely contained within the equivalence margins (symmetric and asymmetric, respectively). The secondary efficacy endpoints were similar between arms. Over 26 weeks, injection-site reactions occurred in 1.7% versus 2.0%, hypersensitivity events in 4.4% versus 8.4%, pneumonia in 0.7% versus 2.0%, and opportunistic infections in 2.4% versus 1.7% in the PF-06410293 and adalimumab-EU arms, respectively. One death due to myocardial infarction occurred (adalimumab-EU arm). Rates of anti-drug antibody incidence were 44.4% (PF-06410293) and 50.5% (adalimumab-EU).
Conclusions
The study results demonstrate that efficacy, safety, and immunogenicity of PF-06410293 and adalimumab-EU were similar during the first 26 weeks of treatment in patients with active RA on background MTX.
Trial registration
ClinicalTrials.gov Identifier: NCT02480153. First posted on June 24, 2015; EU Clinical Trials Register EudraCT number: 2014-000352-29. Start date: October 27, 2014.
Electronic supplementary material
The online version of this article (10.1186/s13075-018-1676-y) contains supplementary material, which is available to authorized users.
Keywords: Rheumatoid arthritis, Adalimumab, Biosimilar, Comparative clinical study
Background
The introduction of biologic disease-modifying anti-rheumatic drugs (bDMARDs) has been a major advance in the treatment of patients with rheumatoid arthritis (RA), providing an important addition to the previously available therapy options [1]. Adalimumab, a recombinant fully human immunoglobulin G1 monoclonal antibody, inhibits the interaction of tumor necrosis factor (TNF) with surface TNF receptors by specifically binding to TNF-α and has been shown to reduce clinical symptoms and inhibit radiographic progression in patients with RA [2–4]. Adalimumab is approved for multiple indications in addition to RA [5, 6].
The US Food and Drug Administration (FDA) defines a biosimilar as “a biopharmaceutical that is highly similar to an already licensed biologic product (the reference product), notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences in purity, potency, and safety between the two products” [7]. The European Medicines Agency requires that a biosimilar show “similarity to the reference biologic with respect to quality, biologic activity, safety, and efficacy” [8]. Biosimilars may expand patient access to bDMARDs because of potentially lower drug prices as a result of price competition within the product market, resulting in savings for health-care systems and patients [9–11].
PF-06410293 is in development as a candidate adalimumab biosimilar. Peptide mapping data demonstrate that PF-06410293 has a primary amino acid sequence identical to that of adalimumab reference product and is similar in comparative analytical, functional, and binding assessments [12]. Pharmacokinetic (PK) similarity was demonstrated following single-dose administration of PF-06410293 and adalimumab to healthy volunteers (Pfizer unpublished observation) [13]. The current comparative clinical study compared the efficacy, safety, immunogenicity, PK, and pharmacodynamics (PD) of PF-06410293 with adalimumab reference product (Humira®) sourced from the EU (adalimumab-EU) in patients with active RA and an inadequate response to methotrexate (MTX).
Methods
Study population
Patients with active RA and an inadequate response to MTX represent a sensitive and appropriate population for biosimilar comparability trials. Adults (at least 18 years old) with a diagnosis of active RA at least 4 months, based on the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) criteria [14], were eligible for inclusion. Active RA was defined as at least six tender and at least six swollen joints (at screening and baseline) with a high-sensitivity C-reactive protein (hs-CRP) of at least 8 mg/L at screening (Additional file 1).
Patients were ineligible if they met any of the following criteria: prior treatment with adalimumab, lymphocyte-depleting therapy, or more than two doses of one biologic therapy; inadequate washout of any second DMARD, pregnancy or breastfeeding, clinically significant laboratory abnormalities, current infection, congestive heart failure (New York Heart Association grade 3/4), untreated or inadequately treated latent or active tuberculosis (TB), malignancy within the previous 5 years, or a positive test for human immunodeficiency virus or hepatitis B or C virus (Additional file 2).
Study design and treatments
This was a multinational, two-arm, double-blind, randomized, comparative clinical study in patients with active RA and was conducted at 173 centers in Australia, Brazil, Bulgaria, Colombia, the Czech Republic, Estonia, Georgia, Germany, Hungary, Japan, Lithuania, Mexico, New Zealand, Peru, Poland, the Republic of Korea, Serbia, South Africa, Spain, Taiwan, Ukraine, the Russian Federation, UK, and the US. Patients were randomly assigned (1:1) on day 1 (stratified by geographic region) to receive either PF-06410293 or adalimumab-EU. There were three 26-week treatment periods and a 16-week follow-up after last dose of study drug (Fig. 1). Prior to dosing at week 26, patients in the adalimumab-EU arm were blindly re-randomized (1:1) to continue on adalimumab-EU or switch to PF-06410293. At week 52, all patients remaining on adalimumab-EU were switched to PF-06410293 for open-label treatment during the third treatment period. Herein, we report data from the first 26 weeks of the study. The number of tender (68) and swollen (66) joints was determined by an independent blinded joint assessor.
Fig. 1.

Study design. Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, EOT end of treatment
PF-06410293 or adalimumab-EU was administered as a subcutaneous injection (40 mg every other week using a prefilled syringe) in addition to a stable background dose of oral or intramuscular MTX (10–25 mg/week) and oral folic/folinic acid; lower doses of MTX (6 mg/week) were allowed if indicated in local guidance or standards of care. Patients could receive concomitant low-dose oral corticosteroids (≤10 mg prednisone or equivalent per day), one non-steroidal anti-inflammatory drug, and non-opioid or specific opioid analgesics or both. Treatment could be delayed by up to 24 h prior to the next injection for illness or scheduling issues. Dosing could be temporarily held at the discretion of the investigator for an adverse event (AE) and resumed after the AE resolved, unless the patient missed three sequential injections.
Primary study endpoint
The primary efficacy endpoint was the proportion of patients achieving an ACR20 response [15] at week 12. Week 12 is considered the beginning of the plateau of the time-response curve for ACR20 and, as such, is a more sensitive time point for the assessment of rapidity of response in biosimilar comparability trials in RA as suggested by regulatory authorities. Therefore, this trial evaluated the primary endpoint at week 12 rather than week 26, as used in the historical registration trials for adalimumab in patients with RA.
Secondary endpoints and assessments
Secondary efficacy endpoints through week 26 included ACR20 (at time points in addition to week 12), ACR50, ACR70, change from baseline in Disease Activity Score 28 joints: four components based on hs-CRP [DAS28–4(CRP)], EULAR response, DAS28–4(CRP) of less than 2.6, ACR/EULAR remission, and change from baseline in individual ACR components, including Health Assessment Questionnaire Disability Index (HAQ-DI). The sponsor selected DAS28–4(CRP) rather than DAS28–4(erythrocyte sedimentation rate [ESR]) to determine clinical response, as CRP is performed in a central laboratory. A cutoff of less than 2.6 was used to define DAS28(CRP) “remission” and not more than 3.2 as “low disease activity” rather than the lower numbers that have been shown to best correlate with the DAS28(ESR) formula [16].
Safety endpoints included type, incidence, severity, timing, seriousness, and investigator-determined relatedness of AEs—using the National Cancer Institute Common Terminology Criteria for Adverse Events (Version 4.03)—and laboratory abnormalities (Covance, Indianapolis, IN, USA). Safety evaluations during study treatment included physical examinations, electrocardiograms, and QuantiFERON-TB Gold testing (at screening and week 26).
Prespecified treatment-emergent adverse events (TEAEs) of special interest were injection-site reactions (ISRs), opportunistic infections (defined for this study to include zoster, cytomegalovirus, latent/active TB, atypical mycobacteria, systemic fungal infections and oral thrush, pneumocystis, legionella, salmonellosis, shigellosis, vibrio, and other infections), and anaphylaxis/angioedema/urticaria. Additional prespecified TEAE categories of interest included blood and lymphatic events, cardiovascular events, demyelinating conditions, gastric/hepatic events, hypersensitivity events, infections and infestations, and neoplasms.
Anti-drug antibodies (ADAs) and neutralizing antibodies (NAbs) were tested at baseline and weeks 2, 6, 12, and 26. Serum samples were analyzed by using a tiered approach of laboratory screening, confirmation, and titer determination. Serum samples were analyzed for ADA at QPS, LLC (Newark, DE, USA) by using a single validated electrochemiluminescent immunoassay. ADA-positive samples were then tested for neutralizing activity with a validated cell-based assay using PF-06410293 as the capture agent.
PK serum samples were obtained at baseline and weeks 1, 2, 6, 12, and 26 and evaluated for PF-06410293 or adalimumab-EU concentrations by using a validated, sensitive, and specific enzyme-linked immunosorbent assay with a lower limit of quantification of 250 ng/mL (QPS). The prespecified PD marker was hs-CRP.
Statistical methods
With the assumption of a week-12 ACR20 response rate of 60% for both PF-06410293 and adalimumab-EU, a sample size of 560 patients was determined to provide about 85% power to demonstrate therapeutic equivalence between the treatment arms, and the symmetric margin of ±14% was used for the primary endpoint. This equivalence margin was derived from a meta-analysis of published data from registration studies for adalimumab in patients with RA [2, 3, 17, 18] and was endorsed by both the European Medicines Agency and the Pharmaceuticals and Medical Devices Agency. Exact methods were used to calculate the confidence interval (CI) for the treatment difference in primary efficacy endpoint of week-12 ACR20, using non-responder imputation (NRI) for missing data and for patients with permanent discontinuation of study drug prior to week 12. Therapeutic equivalence was concluded if the two-sided 95% CI for the treatment difference was entirely contained within ±14% margin and additionally if the two-sided 90% CI for the same treatment difference was within the asymmetric margin of −12% to 15% (as requested by the FDA).
The intention-to-treat (ITT) population, defined as all randomly assigned patients, was the primary analysis population. Sensitivity analyses of the primary and secondary endpoints used the per protocol (PP) population, defined as all patients who received study treatment up to week 12, had a week-12 evaluation, and had no major protocol deviations. The DAS28–4(CRP) change from baseline was analyzed by using an analysis of covariance for repeated-measures data approach.
Safety and immunogenicity analyses were performed for the safety population (defined as randomly assigned patients who received any study treatment) on the prespecified TEAEs of special interest and categories of special interest with risk differences (RDs) and 95% CIs by using the asymptotic approach of Miettinen and Nurminen [19]. Transient ADA response after treatment (including the follow-up period) was defined as either a single positive ADA result or two positive sampling time points where the first and last ADA-positive samples (irrespective of any negative samples in between) were separated by less than 16 weeks and the patient’s last ADA sampling time result was negative [20]. PK analysis was conducted for all dosed patients who provided at least one post-dose drug concentration measurement and was summarized by treatment and ADA status by using descriptive statistics (mean, standard deviation [SD], median, and minimum and maximum). PD analysis using hs-CRP concentration over time was summarized by descriptive statistics according to treatment.
Results
Patient disposition and demographics
In total, 1231 patients were screened and 597 eligible patients—297 to PF-06410293 and 300 to adalimumab-EU—were randomly assigned to receive study treatment (Additional file 3). Low hs-CRP level was the main reason for screen failure. The safety population included 596 patients, and one adalimumab-EU patient was randomly assigned and not dosed. In both treatment arms, the median duration of study treatment was 24.1 weeks. The first treatment period to week 26 was completed by 286 (96.3%) out of 297 patients in the PF-06410293 arm and 273 (91.0%) out of 300 in the adalimumab-EU arm. Overall, 30 (10.1%) out of 297 patients in the PF-06410293 arm and 46 (15.3%) out of 300 in the adalimumab-EU arm were excluded from the PP population. In most cases, exclusion was due to incomplete study drug dosing up to week 12 for 16 (5.4%) out of 297 patients in the PF-06410293 arm and 34 (11.3%) out of 300 in the adalimumab-EU arm. In the PF-06410293 arm, 29 (9.8%) out of 297 patients, compared with 51 (17.1%) out of 299 in the adalimumab-EU arm, missed one or more doses. This included 18 (6.1%) out of 297 and 34 (11.4%) out of 299 patients who missed one or more doses because of an AE in the PF-06410293 and adalimumab-EU arms, respectively.
Patient demographic and baseline RA characteristics were similar between the treatment arms (Table 1). At baseline, patients had a mean age of 52.5 years, 78.7% were female, and the mean RA duration was 6.8 years. Mean baseline swollen joint counts were 15.4 versus 17.0 and tender joint counts were 24.3 versus 26.7 in the PF-06410293 and adalimumab-EU arms, respectively. Mean baseline DAS28–4(CRP) values were 5.9 (PF-06410293) and 6.1 (adalimumab-EU). Across the two arms, the mean MTX dose was 15.2 mg/week and 55.9% of patients were receiving oral corticosteroids (Table 1).
Table 1.
Baseline patient demographic and clinical characteristics (ITT population)
| PF-06410293 n = 297 |
Adalimumab-EU n = 300 |
|
|---|---|---|
| Demographicsa | ||
| Gender, n (%) | ||
| Female | 241 (81.1) | 229 (76.3) |
| Male | 56 (18.9) | 71 (23.7) |
| Age, mean (SD), years | 51.5 (13.6) | 53.5 (12.9) |
| Weight, mean (SD), kg | 74.7 (17.5) | 76.2 (20.8) |
| Body mass index, mean (SD), kg/m2 | 27.5 (6.1) | 28.1 (7.3) |
| Race, n (%) | ||
| White | 261 (87.9) | 256 (85.3) |
| Black | 6 (2.0) | 9 (3.0) |
| Asian | 16 (5.4) | 17 (5.7) |
| Other | 14 (4.7) | 18 (6.0) |
| Ethnicity, n (%) | ||
| Hispanic/Latino | 25 (8.4) | 29 (9.7) |
| Not Hispanic/Latino | 272 (91.6) | 271 (90.3) |
| Clinical characteristics | ||
| RA duration, mean (SD), years | 6.8 (7.2) | 6.8 (6.9) |
| Positive RF or anti-CCP antibody or both, n (%) | 242 (81.5) | 245 (81.7) |
| Swollen joint count, mean (SD) | 15.4 (7.8) | 17.0 (9.8) |
| Tender joint count, mean (SD) | 24.3 (12.3) | 26.7 (14.8) |
| hs-CRP, mg/L | ||
| Mean (SD) | 21.3 (22.7) | 22.8 (25.2) |
| Median (range) | 14.7 (0.2–169) | 16.0 (0.2–192) |
| DAS28–4(CRP), mean (SD) | 5.9 (0.9) | 6.1 (0.9) |
| HAQ-DI, mean (SD) | 1.5 (0.6) | 1.7 (0.6) |
| Prior use of one biologic drug, n (%) | 8 (2.7) | 5 (1.7) |
| Number of prior and current non-biologic DMARDs (in addition to MTX), mean (SD) | 1.5 (0.9) | 1.5 (0.9) |
| MTX dose, mean (SD), mg/week | 15.2 (4.4) | 15.2 (4.5) |
| Corticosteroid use, n (%) | 164 (55.2) | 170 (56.7) |
Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, CCP cyclic citrullinated peptide, DAS28–4(CRP) Disease Activity Score-28: four components based on high-sensitivity C-reactive protein, DMARD disease-modifying anti-rheumatic drug, HAQ-DI Health Assessment Questionnaire Disability Index, hs-CRP high-sensitivity C-reactive protein, ITT intention-to-treat, MTX methotrexate, n number of patients in each category, RA rheumatoid arthritis, RF rheumatoid factor, SD standard deviation
aRandomization stratified by geographic region (North America and Western Europe; Japan; Republic of Korea and Taiwan; Latin America; rest of world)
Efficacy
Primary endpoint
Based on the primary efficacy endpoint of ACR20 response rate at week 12, therapeutic equivalence between PF-06410293 and adalimumab-EU was demonstrated by using both prespecified equivalence margins. With observed data in the ITT population, 204 (68.7%) out of 297 patients in the PF-06410293 arm and 218 (72.7%) out of 300 in the adalimumab-EU arm achieved an ACR20 response at week 12, and treatment difference was −3.98%. For the ITT population, response was imputed as non-responder in 19 patients, the treatment difference was −2.98%, based on ACR20 response in 203 (68.4%) out of 297 patients in the PF-06410293 arm and 214 (71.3%) out of 300 in the adalimumab-EU arm, and the 95% CI (−10.38%, 4.44%) was entirely contained within the symmetric margin (Fig. 2a) and 90% CI (−9.25%, 3.28%) was entirely contained within the asymmetric margin (Fig. 2b).
Fig. 2.
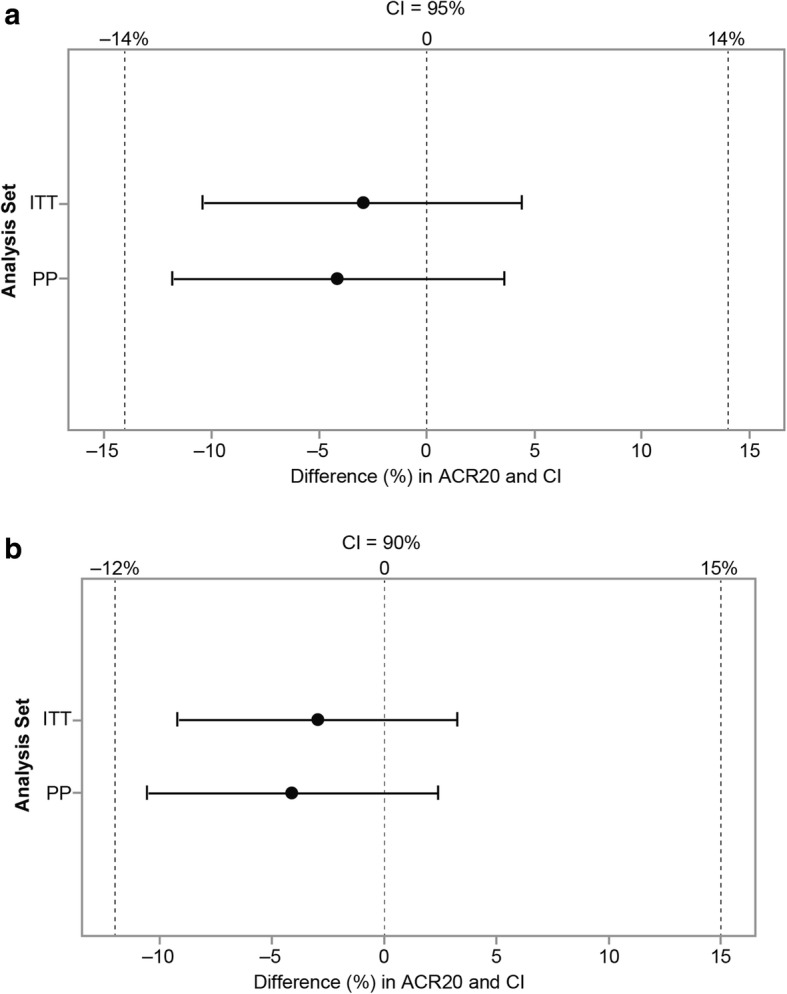
Primary efficacy endpoint of ACR20 at week 12 (with non-responder imputation). a Difference (95% CI) between PF-06410293 and adalimumab-EU using a symmetric equivalence margin. b Difference (90% CI) between PF-06410293 and adalimumab-EU using an asymmetric equivalence margin. Abbreviations: ACR20 American College of Rheumatology 20% improvement, Adalimumab-EU adalimumab sourced from the European Union, CI confidence interval, ITT intention-to-treat, PP per protocol
For the PP population sensitivity analysis, 189 (71.1%) out of 266 patients in the PF-06410293 arm and 191 (75.2%) out of 254 in the adalimumab-EU arm achieved an ACR20 response at week 12. The treatment difference was −4.14%, and the corresponding 95% (−11.79%, 3.61%) and 90% (−10.60%, 2.38%) CIs were entirely contained within the symmetric (±14%) and asymmetric (−12%, 15%) equivalence margins, respectively. Other sensitivity analyses of the primary endpoint, including an analysis adjusting for the stratification variable of geographic region and a multiple imputation-based tipping point analysis for missing data, were consistent with the primary result of therapeutic equivalence between PF-06410293 and adalimumab-EU (Additional file 4).
The ACR20 rates at week 12 for subgroups were numerically higher in ADA-negative (70.9% and 77.2%) compared with ADA-positive (63.7% and 65.7%) patients for the PF-06410293 and adalimumab-EU arms, respectively, defined as subjects with a positive ADA test in the first 26 weeks. ACR20 rates for NAb-negative patients were also numerically higher (70.9% and 74.0%) as compared with NAb-positive patients (50.0% and 64.0%) for the PF-06410293 and adalimumab-EU arms, respectively.
Secondary endpoints
The ACR20/50/70 response rates through week 26 were similar between the PF-06410293 and adalimumab-EU arms (Fig. 3a). Mean changes from baseline in DAS28–4(CRP) were similar between treatment arms at each study visit, and the changes from baseline at week 26 were −2.7 for the PF-06410293 arm and −2.8 for the adalimumab-EU arm (Fig. 3b). At week 26, 162 (54.5%) out of 297 and 147 (49.0%) out of 300 of patients had a good EULAR response in the PF-06410293 and adalimumab-EU arms, respectively (Additional file 5). In the PF-06410293 arm, 87 (29.3%) out of 297 patients achieved DAS28–4(CRP) of less than 2.6 at week 26 compared with 99 (33.0%) out of 300 in the adalimumab-EU arm (Additional file 6). A total of 38 (12.8%) out of 297 patients in the PF-06410293 arm and 44 (14.7%) out of 300 in the adalimumab-EU arm achieved ACR/EULAR remission at week 26, including 26 (8.8%) out of 297 and 27 (9.0%) out of 300 using only the Boolean definition (Additional file 6). At week 26, mean HAQ-DI decreased from baseline by 0.654 in the PF-06410293 arm and by 0.674 in the adalimumab-EU arm (Additional file 7).
Fig. 3.
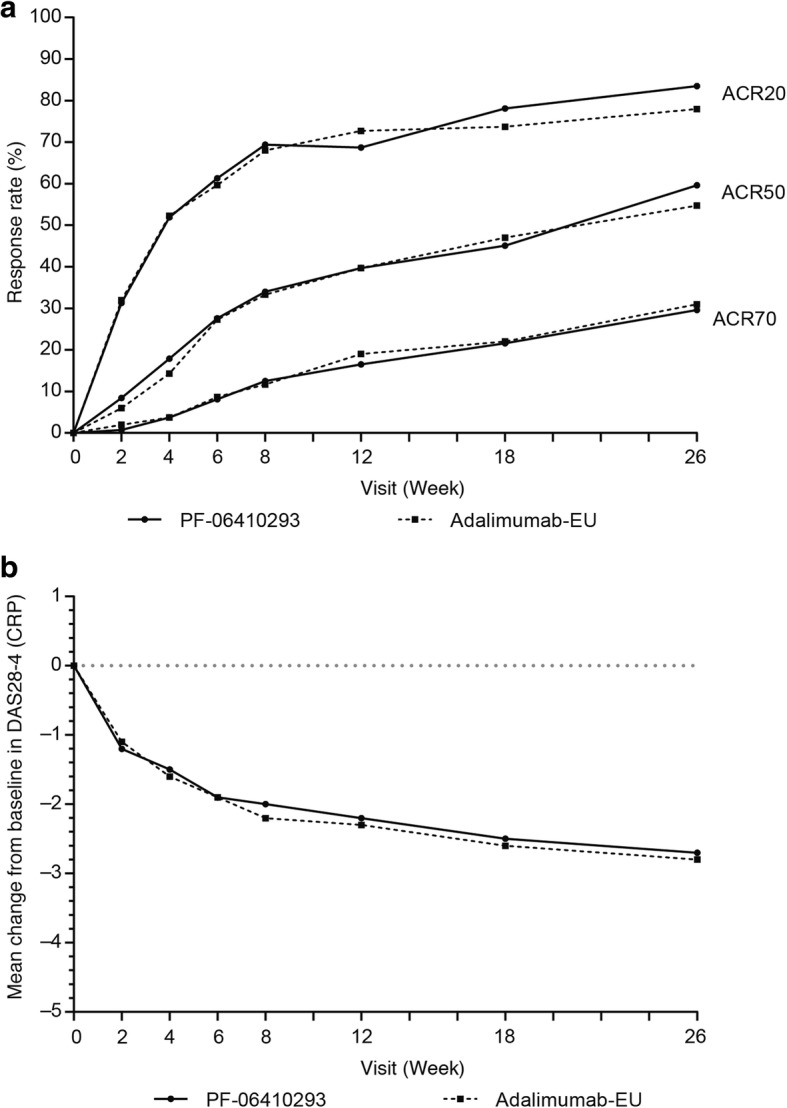
Secondary efficacy endpoints (intention-to-treat population). a ACR20/50/70 response rates by study visit. b Mean change from baseline in DAS28–4(CRP) by study visit. Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, ACR20/50/70 American College of Rheumatology 20%/50%/70% improvement, DAS28–4(CRP) Disease Activity Score-28: four components based on high-sensitivity C-reactive protein
Safety
A total of 143 (48.1%) out of 297 patients in the PF-06410293 arm and 143 (47.8%) out of 299 in the adalimumab-EU arm reported one or more TEAEs. The System Organ Classes (SOCs) with the highest proportion of patients with AEs were infections and infestations in 24.9% and 25.1%, musculoskeletal and connective tissue disorders in 10.4% and 8.7%, and investigations in 8.8% and 7.7% for the PF-06410293 and adalimumab-EU patients, respectively. The number of patients who permanently discontinued treatment because of TEAEs was 11 (3.7%) versus 14 (4.7%) and the number of patients who temporarily discontinued treatment because of TEAEs was 17 (5.7%) versus 29 (9.7%) in the PF-06410293 and adalimumab-EU arms, respectively.
Serious adverse events (SAEs) were reported by 4.0% (PF-06410293) and 4.3% (adalimumab-EU) of patients (Table 2). This included one death due to myocardial infarction in the adalimumab-EU arm. The SOC with the highest proportion of patients with SAEs was infections and infestations, occurring in three patients in each treatment arm.
Table 2.
All-causality treatment-emergent adverse events (safety population)
| PF-06410293 n = 297 |
Adalimumab-EU n = 299 |
|
|---|---|---|
| Number of AEs | 343 | 379 |
| Patients with events, n (%) | ||
| AEs | 143 (48.1) | 143 (47.8) |
| SAEs | 12 (4.0) | 13 (4.3) |
| Grade 3 AEs | 15 (5.1) | 16 (5.4)a |
| Grade 4 AEs | 2 (0.7) | 4 (1.3) |
| Grade 5 AEs | 0 | 1 (0.3) |
| Patients with temporary treatment discontinuation due to AEs, n (%) | 17 (5.7) | 29 (9.7) |
| Patients discontinued from treatment due to AEsc, n (%) | 11 (3.7)b | 14 (4.7) |
| Patients discontinued from the study due to AEs, n (%) | 8 (2.7) | 9 (3.0) |
AEs were graded in accordance with National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. Grade 1–5 AEs are defined as mild, moderate, severe, life-threatening AEs, and death related to AE, respectively.
Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, AE adverse event, SAE serious adverse event
aOne patient had an AE of neutropenia incorrectly recorded as grade 2; the correct severity was grade 3 (not corrected in this table)
bOne patient was incorrectly recorded as treatment discontinuation due to an AE; the correct reason was insufficient clinical response (not corrected in this table)
cThe System Organ Class with the highest proportion of subjects who had AEs leading to permanent treatment discontinuation was infections and infestations (8 [2.7%] subjects on PF-06410293 and 3 [1.0%] subjects on adalimumab-EU)
In total, 5.7% of patients in the PF-06410293 arm and 7.0% in the adalimumab-EU arm reported TEAEs of grade 3 or higher. All-causality grade 4 TEAEs were reported in two patients in the PF-06410293 arm (intentional self-injury, and hemorrhoids with rectal hemorrhage and resulting anemia) and four patients in the adalimumab-EU arm (atrial fibrillation, ileus secondary to colon cancer, gastroenteritis, and papillary thyroid cancer).
The most frequently reported TEAEs occurring in at least 2% of patients in any treatment arm were viral upper respiratory tract infections, increased alanine aminotransferase, hypertension, and headaches (Additional file 8).
Of the TEAEs of special interest, ISRs were reported by five (1.7%) and six (2.0%) patients in the PF-06410293 and adalimumab-EU arms, respectively (Table 3). The primary symptom was redness (three patients in the PF-06410293 arm and two in the adalimumab-EU arm). In addition, one patient in each arm reported pain and swelling. No patients discontinued treatment because of an ISR. For one patient in the PF-06410293 arm and two patients in the adalimumab-EU arm, the ISR occurred on or after the date the patient first tested positive for ADA.
Table 3.
All-causality treatment-emergent adverse events of special interest with risk difference (safety population)
| Event of special interest | PF-06410293 n = 297 n (%) |
Adalimumab-EU n = 299 n (%) |
Risk difference (95% CI) (%) |
|---|---|---|---|
| Injection-site reactions | 5 (1.7) | 6 (2.0) | −0.32 (−2.84, 2.12) |
| Opportunistic infections | 7 (2.4) | 5 (1.7) | 0.69 (−1.80, 3.32) |
| Herpes zoster | 1 (0.3) | 3 (1.0) | −0.67 (−2.61, 0.97) |
| Latent tuberculosis | 5 (1.7) | 1 (0.3) | 1.35 (−0.35, 3.59) |
| Confirmed active tuberculosis | 0 | 0 | 0 (NA) |
| Oral candidiasis | 0 | 1 (0.3) | −0.33 (−1.87, 0.95) |
| Pneumocystis jirovecii pneumonia | 1 (0.3) | 0 | 0.34 (−0.94, 1.88) |
| Urticaria, angioedema, anaphylactic reactiona | 0 | 2 (0.7) | −0.67 (−2.41, 0.61) |
Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, CI confidence interval, NA not applicable
aOnly urticaria reported
Overall infection rates were similar at 24.9% and 25.1% for the PF-06410293 and adalimumab-EU arms, respectively. Opportunistic infections (predefined in the study as including latent TB) were reported by seven (2.4%) in the PF-06410293 arm and five (1.7%) patients in the adalimumab-EU arm (Table 3). One case of herpes zoster was reported in the PF-06410293 arm and three cases in the adalimumab-EU arm. Five and one cases of seroconversion with a subsequent diagnosis of latent TB (based on specialist consultation following a positive week-26 QuantiFERON-TB Gold test result) were reported in the PF-06410293 and adalimumab-EU arms, respectively. The RD for latent TB (1.35, 95% CI −0.35, 3.59) was not statistically significant. In total, 5.6% of patients in the PF-06410293 arm and 4.9% in the adalimumab-EU arm had a negative QuantiFERON-TB test at screening and a positive test at week 26. There were no cases of active TB in any patient in either treatment arm. One case of oral candidiasis (adalimumab-EU) and one case of Pneumocystis jirovecii pneumonia (PF-06410293) were reported. Overall, pneumonia was reported by 0.7% and 2.0% of the PF-06410293 and adalimumab-EU arms, respectively. There were no cases of anaphylaxis or angioedema in either treatment arm; two cases of urticaria were reported in the adalimumab-EU arm.
A total of 39.1% of patients in each arm reported 183 (PF-06410293) and 202 (adalimumab-EU) AEs in one or more prespecified TEAE categories of interest (Table 4). Hypersensitivity TEAEs were reported in 13 (4.4%) out of 297 patients in the PF-06410293 arm compared with 25 (8.4%) out of 299 in the adalimumab-EU arm (RD −3.98, 95% CI −8.15, −0.06). The most frequently reported hypersensitivity TEAEs were cough (5 versus 3), erythema (4 versus 1), and rash (1 versus 3) in the PF-06410293 and adalimumab-EU arms, respectively. Hypersensitivity TEAEs occurring on or after the date a patient first tested positive for ADA included six AEs reported by five patients in the PF-06410293 arm and nine AEs reported by seven patients in the adalimumab-EU arm. Two patients in the PF-06410293 arm reported grade 3 hypersensitivity SAEs, including interstitial lung disease and toxic skin eruption. The rate of blood and lymphatic system events was numerically higher in the PF-06410293 versus adalimumab-EU arms—22 (7.4%) out of 297 versus 14 (4.7%) out of 299—but this was not statistically significant (RD 2.73, 95% CI −1.15, 6.79). The reported malignancies included one (basal cell carcinoma) and two (adenocarcinoma of the colon and papillary thyroid cancer) patients in the PF-06410293 and adalimumab-EU arms, respectively.
Table 4.
Prespecified treatment-emergent adverse event categories of interest with risk difference (safety population)
| Category | PF-06410293 n = 297 n (%) |
Adalimumab-EU n = 299 n (%) |
Risk difference (95% CI) (%) |
|---|---|---|---|
| Blood and lymphatic system events | 22 (7.4) | 14 (4.7) | 2.73 (−1.15, 6.79) |
| Cardiovascular events | 9 (3.0) | 16 (5.4) | −2.32 (−5.84, 0.96) |
| Demyelinating conditions | 0 | 0 | 0 (NA) |
| Gastric/hepatic events | 11 (3.7) | 14 (4.7) | −0.98 (−4.44, 2.39) |
| Hypersensitivitya | 13 (4.4) | 25 (8.4) | −3.98 (−8.15, −0.06) |
| Infections and infestations | 74 (24.9) | 75 (25.1) | −0.17 (−7.13, 6.80) |
| Neoplasms | 5 (1.7) | 5 (1.7) | 0.01 (−2.38, 2.42) |
| Otherb | 11 (3.7) | 10 (3.3) | 0.36 (−2.80, 3.57) |
Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, CI confidence interval, MedDRA Medical Dictionary for Regulatory Activities, NA not applicable
aHypersensitivity events identified by Hypersensitivity Standardized MedDRA Query (broad and narrow), Anaphylactic reactions Standardized MedDRA Query (broad and narrow), and High-Level Group Terms Immunology and allergy investigations
bOther events identified by High-Level Group Terms Skin Vascular Abnormalities, Central Nervous System Vascular Abnormalities and Medication Errors; Higher Level Terms Connective Tissue Disorders, Vasculitides (not elsewhere classified), Rashes, Eruptions and Exanthems (not elsewhere classified); Lower Level Teams Seizure and Convulsions; and Preferred Terms Lupus-like Syndrome, Headache and Migraine
Immunogenicity, PK, and PD
Overall, 44.4% and 50.5% of patients in the PF-06410293 and adalimumab-EU treatment arms, respectively, had at least one post-dose sample that tested positive for ADA (Fig. 4; Additional file 9). ADAs were transient in 11.4% of patients in the PF-06410293 arm and in 6.0% in the adalimumab-EU arm. Of the ADA-positive patients, 31.1% in the PF-06410293 and 27.8% in the adalimumab-EU treatment arms tested positive for NAb.
Fig. 4.
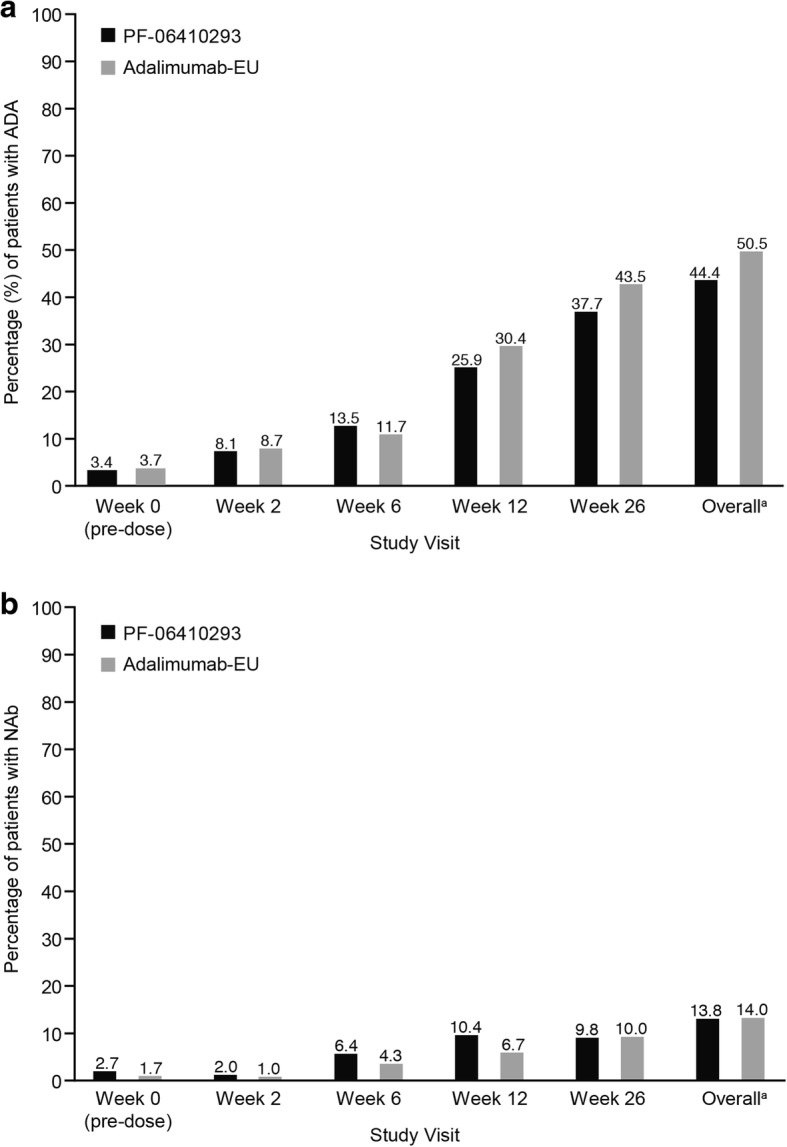
ADA and NAb incidence by study visit (safety population). a ADA incidence. b NAb incidence. The percentage of NAb-positive patients is based on the total number of patients in each treatment group. a“Overall” includes data from week 2, week 6, week 12, week 26, end-of-treatment/early termination, follow-up, and unplanned visits in treatment period 1. Abbreviations: ADA anti-drug antibody, Adalimumab-EU adalimumab sourced from the European Union, NAb neutralizing antibody
The mean serum drug trough concentrations at week 26 were 8244 and 7190 ng/mL in the PF-06410293 and adalimumab-EU arms, respectively. Mean serum concentrations of both drugs were lower in ADA-positive patients (4683 and 4041 ng/mL) compared with ADA-negative patients (11,090 and 10,460 ng/mL) for the PF-06410293 and adalimumab-EU arms, respectively. The mean hs-CRP concentrations (prespecified PD marker) were decreased at week 26 in both arms (ITT population); the changes from baseline to week 26 were −11.1 (PF-06410293) and −13.6 mg/L (adalimumab-EU).
Discussion
This comparative clinical study was conducted to evaluate the biosimilarity of PF-06410293 and adalimumab-EU. The primary goal of the study was met by demonstrating therapeutic equivalence of PF-06410293 and adalimumab-EU using the symmetric and asymmetric margins for the week-12 ACR20 primary endpoint comparison. Sensitivity analyses of the primary endpoint supported a conclusion of therapeutic equivalence. As expected, ACR20 response rates at week 12 were numerically higher in ADA-negative patients compared with ADA-positive in both treatment arms. Secondary endpoints reported up to week 26, including ACR50, ACR70, change from baseline in DAS28–4(CRP), EULAR response, DAS28–4(CRP) of less than 2.6, ACR/EULAR remission, and HAQ-DI all supported therapeutic equivalence.
The safety profiles of PF-06410293 and adalimumab-EU to week 26 were comparable, including similar findings with respect to the number of AEs, SAEs, and prespecified TEAEs and TEAE categories of special interest. The only statistically significant safety difference was a lower rate of hypersensitivity events observed in the PF-06410293 arm compared with the adalimumab-EU arm; however, no correction for multiplicity was performed in this study. As might be expected in a single clinical trial, numerical differences were observed between the treatment arms, including an imbalance in the development of latent TB (more common in the PF-06410293 group) and hypersensitivity events (more common in the adalimumab-EU group). Of note, the diagnosis of latent TB was based on investigator judgment, local practice, and consultation with a pulmonary or infectious disease specialist and after a protocol-mandated QuantiFERON-TB test was performed. The percentage of patients who converted to a QuantiFERON-TB test positive at week 26 was balanced between the treatment arms, suggesting that there was no clinically meaningful difference in the rate of QuantiFERON-TB test conversion between PF-06410293 and adalimumab-EU. The safety profile for both study drugs appears to be consistent with the known safety profile of reference adalimumab-EU.
The immunogenicity profiles observed during the first 26 weeks of treatment were similar for the two treatment arms, and there was a somewhat lower incidence of patients testing positive for ADA in the PF-06410293 arm. Serum drug concentrations were numerically higher in the PF-06410293 arm; however, these differences were not considered clinically meaningful, as the clinical response was similar in the two treatment arms. The hs-CRP response as a PD biomarker supports this lack of clinical significance, as the decrease in hs-CRP was similar for the two arms over the first 26 weeks of treatment. As expected, the serum drug concentrations of PF-06410293 and adalimumab-EU were lower in both treatment arms for ADA-positive compared with ADA-negative patients.
Conclusions
Results from the first 26 weeks of dosing demonstrated no clinically meaningful differences in efficacy, safety, immunogenicity, PK, or PD between PF-06410293 and adalimumab-EU in patients with active RA. Upcoming data from the subsequent 6 months of the trial will provide additional efficacy, safety, and immunogenicity information, including data on patients after a blinded transition from adalimumab-EU to PF-06410293 and those who receive a total of 1 year of treatment with either PF-06410293 or adalimumab-EU.
Additional files
Inclusion criteria for study enrollment. (DOCX 48 kb)
Exclusion criteria. (DOCX 53 kb)
{kind=link}
Consolidated Standards of Reporting Trials (CONSORT) flow diagram of patient progress in the two treatment arms. aPatients may screen fail for more than one reason. bPatients may be excluded from the per-protocol population for more than one reason. Abbreviation: Adalimumab-EU adalimumab sourced from the European Union. (PNG 171 kb)
Multiple imputation-based tipping-point analysis. (DOCX 48 kb)
EULAR response by study visit (ITT population). Abbreviations: EULAR European League Against Rheumatism, ITT intention-to-treat. (DOCX 48 kb)
DAS28–4(CRP) of less than 2.6 and ACR/EULAR remission by study visit (ITT population). Abbreviations: ACR/EULAR American College of Rheumatology/European League Against Rheumatism, DAS28–4(CRP) Disease Activity Score 28 joints: four components based on high-sensitivity C-reactive protein, ITT intention-to-treat. (DOCX 50 kb)
{kind=link}
Mean change from baseline in HAQ-DI by visit (ITT population). Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, HAQ-DI health assessment questionnaire disability index, ITT intention-to-treat (PNG 84 kb)
All-causality treatment-emergent adverse events in at least 2% of patients in any treatment arm (safety population). (DOCX 49 kb)
ADA and NAb incidence by study visit (safety population). Abbreviations: ADA anti-drug antibody, NAb neutralizing antibody. (DOCX 52 kb)
Acknowledgments
Medical writing support was provided by Jacqui Oliver and Neel Misra of Engage Scientific Solutions and was funded by Pfizer Inc.
Funding
This study was sponsored by Pfizer Inc.
Availability of data and materials
The policies of Pfizer Inc. on the provision of clinical trial data are set out in http://www.pfizer.com/research/clinical_trials/trial_data_and_results. In addition to posting clinical trial results on the ClinicalTrials.gov registry, Pfizer will provide access to anonymized patient-level data in response to scientifically valid research protocols. Data from Pfizer-sponsored global interventional clinical studies are available from trials conducted for medicines, vaccines, and medical devices for indications that have been approved in the US or the European Union or both and from trials conducted for medicines, vaccines, and medical devices that have been terminated (that is, development for all indications has been discontinued). Data from these trials will be made available 24 months after study completion. Pfizer will make reasonable efforts to fulfill all data requests for legitimate research purposes, but there may be instances in which retrieval or delivery of data is not feasible (for example, if Pfizer does not have legal authority to provide the data or if costs of retrieval of older or pre-electronic data are prohibitive; see page 5 at the following link: https://www.pfizer.com/files/research/research_clinical_trials/A_Guide_to_Requesting_Pfizer_Patient-Level_Clinical_Trial_Data_2017.pdf). Further detail can be found at http://www.pfizer.com/research/clinical_trials/trial_data_and_results/data_requests. Pfizer’s practices adhere to the principles for responsible data sharing laid out by the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Pharmaceutical Research and Manufacturers of America (PhRMA): http://phrma.org/sites/default/files/pdf/PhRMAPrinciplesForResponsibleClinicalTrialDataSharing.pdf.
Abbreviations
- ACR
American College of Rheumatology
- ACR20
American College of Rheumatology 20% improvement
- ADA
Anti-drug antibody
- Adalimumab-EU
Adalimumab sourced from the European Union
- AE
Adverse event
- bDMARD
Biologic disease-modifying anti-rheumatic drug
- CI
Confidence interval
- DAS28–4(CRP)
Disease Activity Score 28 joints: four components based on high-sensitivity C-reactive protein
- DMARD
Disease-modifying anti-rheumatic drug
- ESR
Erythrocyte sedimentation rate
- EULAR
European League Against Rheumatism
- FDA
US Food and Drug Administration
- HAQ-DI
Health Assessment Questionnaire Disability Index
- hs-CRP
High-sensitivity C-reactive protein
- ISR
Injection-site reaction
- ITT
Intention-to-treat
- MTX
Methotrexate
- NAb
Neutralizing antibody
- PD
Pharmacodynamics
- PK
Pharmacokinetics
- PP
Per protocol
- RA
Rheumatoid arthritis
- RD
Risk difference
- SAE
Serious adverse event
- SOC
System Organ Class
- TB
Tuberculosis
- TEAE
Treatment-emergent adverse event
- TNF
Tumor necrosis factor
Authors’ contributions
KLS, DA, and AEB made substantial contributions to study conception and design. SYH, CC, KLS, and AEB analyzed the data and SYH provided statistical support. RMF, RA, and MP contributed to the acquisition of data. RMF reviewed for investigator approval all study data in the clinical study report written by KLS. All authors made substantial contributions to the interpretation of data, were involved in drafting the manuscript or revising it critically for important intellectual content or both, and read and approved the final manuscript for submission.
Ethics approval and consent to participate
This study was conducted in compliance with the ethical principles of the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines. The study protocol, all amendments, and informed consent documentation were reviewed and approved by the institutional review boards or independent ethics committees or both. In addition, all local regulatory requirements were followed. All patients provided informed consent before undergoing any screening procedures.
Consent for publication
Not applicable.
Competing interests
RMF has received research grants and consulting fees from Pfizer Inc. and AbbVie. RA has received research grants and honoraria from Pfizer Inc. MP declares that she has no competing interests. SYH was an employee of and held stock holdings or stock options (or both) from Pfizer Inc. at the time of the study. KL, CC, DA, AEB, and KLS are full-time employees of and declare stock holdings or stock options (or both) from Pfizer Inc.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Roy M. Fleischmann, Phone: 214-540-0645, Email: rfleischmann@arthdocs.com
Rieke Alten, Email: rieke.alten@schlosspark-klinik.de.
Margarita Pileckyte, Email: pmargarita1@gmail.com.
Kasia Lobello, Email: kasia.lobello@pfizer.com.
Steven Y. Hua, Email: steven26190@yahoo.com
Carol Cronenberger, Email: carol.cronenberger@pfizer.com.
Daniel Alvarez, Email: daniel.f.alvarez@pfizer.com.
Amy E. Bock, Email: amy.e.bock@pfizer.com
K. Lea Sewell, Email: lea.sewell@pfizer.com.
References
- 1.Curtis JR, Singh JA. Use of biologics in rheumatoid arthritis: current and emerging paradigms of care. Clin Ther. 2011;33:679–707. doi: 10.1016/j.clinthera.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48:35–45. doi: 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- 3.Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50:1400–1411. doi: 10.1002/art.20217. [DOI] [PubMed] [Google Scholar]
- 4.Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26–37. doi: 10.1002/art.21519. [DOI] [PubMed] [Google Scholar]
- 5.European Medicines Agency . HUMIRA (adalimumab) summary of product characteristics. 2017. [Google Scholar]
- 6.Abbott Laboratories . HUMIRA (adalimumab) package insert. 2003. [Google Scholar]
- 7.US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015. [Google Scholar]
- 8.European Medicines Agency . Guideline on similar biological medicinal products. 2014. [Google Scholar]
- 9.QuintilesIMS . The impact of biosimilar competition in Europe. 2017. [Google Scholar]
- 10.Mulcahy A, Predmore Z, Soeren M. The cost savings potential of biosimilar drugs in the United States. 2014. [Google Scholar]
- 11.Crespi-Lofton J, Skelton JB. The growing role of biologics and biosimilars in the United States: Perspectives from the APhA Biologics and Biosimilars Stakeholder Conference. J Am Pharm Assoc (2003) 2017;57:e15–e27. doi: 10.1016/j.japh.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 12.Derzi D, Ripp S, Ng C, Shoieb A, Finch G, Lorello L, et al. Society of Toxicology 53rd Annual Meeting and ToxExpo 2014; Phoenix, Arizona. 2014. Comparative nonclinical assessments of the potential biosimilar PF-06410293 and adalimumab [abstract no. 276] [Google Scholar]
- 13.ClinicalTrials.gov. A study of PF-06410293 (adalimumab-Pfizer) and adalimumab (Humira) In healthy subjects (REFLECTIONS B538–07)) (B538–07). 2014 (last update: 13 April 2015). https://clinicaltrials.gov/ct2/show/NCT02237729. Accessed 19 Feb. 2018.
- 14.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–2581. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 15.Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38:727–735. doi: 10.1002/art.1780380602. [DOI] [PubMed] [Google Scholar]
- 16.Fleischmann R, van der Heijde D, Koenig AS, Pedersen R, Szumski A, Marshall L, et al. How much does disease activity score in 28 joints ESR and CRP calculations underestimate disease activity compared with the simplified disease activity index? Ann Rheum Dis. 2015;74:1132–1137. doi: 10.1136/annrheumdis-2013-204920. [DOI] [PubMed] [Google Scholar]
- 17.Kim HY, Lee SK, Song YW, Yoo DH, Koh EM, Yoo B, et al. A randomized, double-blind, placebo-controlled, phase III study of the human anti-tumor necrosis factor antibody adalimumab administered as subcutaneous injections in Korean rheumatoid arthritis patients treated with methotrexate. APLAR J Rheumatol. 2007;10:9–16. doi: 10.1111/j.1479-8077.2007.00248.x. [DOI] [Google Scholar]
- 18.Chen DY, Chou SJ, Hsieh TY, Chen YH, Chen HH, Hsieh CW, et al. Randomized, double-blind, placebo-controlled, comparative study of human anti-TNF antibody adalimumab in combination with methotrexate and methotrexate alone in Taiwanese patients with active rheumatoid arthritis. J Formos Med Assoc. 2009;108:310–319. doi: 10.1016/S0929-6646(09)60071-1. [DOI] [PubMed] [Google Scholar]
- 19.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213–226. doi: 10.1002/sim.4780040211. [DOI] [PubMed] [Google Scholar]
- 20.Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16:658–673. doi: 10.1208/s12248-014-9599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inclusion criteria for study enrollment. (DOCX 48 kb)
Exclusion criteria. (DOCX 53 kb)
Consolidated Standards of Reporting Trials (CONSORT) flow diagram of patient progress in the two treatment arms. aPatients may screen fail for more than one reason. bPatients may be excluded from the per-protocol population for more than one reason. Abbreviation: Adalimumab-EU adalimumab sourced from the European Union. (PNG 171 kb)
Multiple imputation-based tipping-point analysis. (DOCX 48 kb)
EULAR response by study visit (ITT population). Abbreviations: EULAR European League Against Rheumatism, ITT intention-to-treat. (DOCX 48 kb)
DAS28–4(CRP) of less than 2.6 and ACR/EULAR remission by study visit (ITT population). Abbreviations: ACR/EULAR American College of Rheumatology/European League Against Rheumatism, DAS28–4(CRP) Disease Activity Score 28 joints: four components based on high-sensitivity C-reactive protein, ITT intention-to-treat. (DOCX 50 kb)
Mean change from baseline in HAQ-DI by visit (ITT population). Abbreviations: Adalimumab-EU adalimumab sourced from the European Union, HAQ-DI health assessment questionnaire disability index, ITT intention-to-treat (PNG 84 kb)
All-causality treatment-emergent adverse events in at least 2% of patients in any treatment arm (safety population). (DOCX 49 kb)
ADA and NAb incidence by study visit (safety population). Abbreviations: ADA anti-drug antibody, NAb neutralizing antibody. (DOCX 52 kb)
Data Availability Statement
The policies of Pfizer Inc. on the provision of clinical trial data are set out in http://www.pfizer.com/research/clinical_trials/trial_data_and_results. In addition to posting clinical trial results on the ClinicalTrials.gov registry, Pfizer will provide access to anonymized patient-level data in response to scientifically valid research protocols. Data from Pfizer-sponsored global interventional clinical studies are available from trials conducted for medicines, vaccines, and medical devices for indications that have been approved in the US or the European Union or both and from trials conducted for medicines, vaccines, and medical devices that have been terminated (that is, development for all indications has been discontinued). Data from these trials will be made available 24 months after study completion. Pfizer will make reasonable efforts to fulfill all data requests for legitimate research purposes, but there may be instances in which retrieval or delivery of data is not feasible (for example, if Pfizer does not have legal authority to provide the data or if costs of retrieval of older or pre-electronic data are prohibitive; see page 5 at the following link: https://www.pfizer.com/files/research/research_clinical_trials/A_Guide_to_Requesting_Pfizer_Patient-Level_Clinical_Trial_Data_2017.pdf). Further detail can be found at http://www.pfizer.com/research/clinical_trials/trial_data_and_results/data_requests. Pfizer’s practices adhere to the principles for responsible data sharing laid out by the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Pharmaceutical Research and Manufacturers of America (PhRMA): http://phrma.org/sites/default/files/pdf/PhRMAPrinciplesForResponsibleClinicalTrialDataSharing.pdf.