

Abstract
Introduction:
Cystic fibrosis (CF) is a genetic disease characterized by progressive lung disease. Most CF therapies focus on treating secondary pulmonary complications rather than addressing the underlying processes inducing airway remodeling and ineffective response to infection. Transforming growth factor beta (TGFβ) is a cytokine involved in fibrosis, inflammation, and injury response as well as a genetic modifier and biomarker of CF lung disease. Targeting the TGFβ pathway has been pursued in other diseases, but the mechanism of TGFB effects in CF is less well understood.
Areas Covered:
In this review, we discuss CF lung disease pathogenesis with a focus on potential links to TGFβ. TGFβ signaling in lung health and disease is reviewed. Recent studies investigating TGFβ’s impact in CF airway epithelial cells are highlighted. Finally, an overview of potential therapies to target TGFβ signaling relevant to CF are addressed.
Expert Opinion:
The broad impact of TGFβ signaling on numerous cellular processes in homeostasis and disease is both a strength and a challenge to developing TGFβ dependent therapeutics in CF. We discuss the challenges inherent in developing TGFβ-targeted therapy, identifying appropriate patient populations, and questions regarding the timing of treatment. Future directions for research into TGFβ focused therapeutics are discussed.
Keywords: Cystic Fibrosis, cystic fibrosis transmembrane conductance regulator, genetic modifier, Transforming Growth Factor beta
1. Cystic Fibrosis
Cystic fibrosis (CF) is a recessive genetic disease common in the Caucasian population; as of 2015, CF affects approximately 70,000 individuals worldwide, including nearly 30,000 in the United States, with a predicted median survival of 41.7 years [1, 2]. Mutations in the cystic fibrosis transmembrane conductance regulator protein (CFTR) disrupt bicarbonate and chloride transport in epithelia throughout the body, leading to progressive multi-organ disease [3]. Lung disease is the most significant source of morbidity and mortality for patients with CF. More advanced CF disease is characterized by acquisition of chronic pulmonary infections (i.e. Pseudomonas aeruginosa or Burkholderia cepacia), recurrent bronchopneumonia, progressive lung injury, and loss of lung function typically measured by decline in the forced expiratory volume in one second (FEV1) [3]. As we better understand the course of early CF lung disease, it is apparent that FEV1 decline and recurrent pulmonary exacerbations are a late marker of lung disease; a third of children under age 3 years have airway damage (bronchiectasis) visible on chest CT [4]. Pulmonary inflammation and infection are also demonstrable early in life, leading to the proposal that very early childhood is a critical window to intervene with new therapies and change the trajectory of future CF lung disease [5]. New therapies that restore function to mutated CFTR, CFTR modulators, have been shown to benefit patients with certain CFTR mutations but are not curative [6, 7]. Thus, it is critical to understand contributors to CF lung disease and identify other novel therapeutic targets that might improve or prevent CF lung disease.
1.1. CF Lung Disease Pathogenesis
Loss of CFTR function produces a cascade of pathologic changes in the lung. Altered ion transport results from CFTR malfunction, loss of CFTR-dependent chloride and bicarbonate transport, and loss of the normal CFTR regulation of the epithelial sodium channel (ENaC) [8–10]. This results in airway surface liquid (ASL) dehydration, increasing mucus solid concentration and compression of the periciliary compartment [11–13]. Epithelial goblet cell and submucosal gland hyperplasia is also seen in CF, with a higher mucin content within goblet cells [14]. Reduction of pH and altered bicarbonate transport due to absent CFTR function results in reduced bacterial killing and thickened, abnormally adherent mucus [15–18]. These defects result in airway obstruction, poor mucociliary clearance, and increased inflammation and infection. Airways in CF are primed for a more aggressive inflammatory response to infectious and obstructive stimuli, with increased cytokine release resulting in chronic neutrophilic inflammation [19, 20]. Lungs of CF patients are thus vulnerable to infection and the severe downstream responses to the resulting inflammation and lung injury. Although later in life the morbidity associated with CF lung disease is quite apparent, the disease starts early in life when it is often asymptomatic; indeed, significant inflammation and ventilatory defects are detectable in the majority of children with CF [4]. The causative and temporal relationships between inflammation and infection in CF are unclear, although it is recognized that both occur early, prior to the onset of clinically identified respiratory symptoms [21].
Multiple signaling pathways (intra and extracellular) are implicated in CF lung disease pathogenesis, such as activation of hypoxic response pathways, NFkB, IL-1, and IL-8 [5, 22, 23]. Free neutrophil elastase in young infants (as early as 3 months old, typically prior to recognized bronchopulmonary pneumonias) has been found to be significantly associated with bronchiectasis by age 3 years, indicating that abnormal early and potentially sterile inflammation sets the stage for CF lung disease progression [4]. A number of investigators have observed abnormal microRNA (miRNA, small non-coding RNAs involved in gene expression regulation) expression in cystic fibrosis lungs, with downregulation of miR-126, miR-31, and miR-93 potentially allowing increased expression of their targets that cause inflammatory dysregulation, including Target of Myb1, cathepsin S, and IL-8, respectively [24]. In addition to effects on inflammation, a number of miRNAs downregulate CFTR expression, thus exacerbating the underlying etiology that drives cystic fibrosis [25].
1.2. Current Therapies for CF
Until the recent availability of CFTR modulators, CF pulmonary therapy had focused on alleviating secondary symptoms: thinning and mobilizing mucus, counteracting inflammation, and treating infection. Hypertonic saline (a mucus hydrator) and dornase alfa (a mucolytic) are two commonly used pulmonary therapies in CF, and both are associated with decreased pulmonary exacerbations and small improvements in FEV1 [26, 27]. Multiple modalities of airway clearance are used in CF patients, including postural drainage and percussion, PEP devices, percussion vests, and variable pressure (e.g.: flutter) devices. Little evidence exists regarding the relative benefits of the different types of airway clearance, or indeed as to the precise benefit of airway clearance in general; however, these tools are strongly recommended as part of the daily routine for CF patients given the high theoretical benefit of mobilizing the thick, inspissated lung secretions typical in CF [28]. Anti-inflammatory therapies such as macrolide antibiotics and ibuprofen have also been shown in several studies to improve lung function and decrease exacerbation frequency, and remain an area of intense drug development [29, 30]. Several studies have demonstrated that acute and chronic antibiotic therapies targeting important CF pathogens including Pseudomonas aeruginosa and methicillin-resistant Staphyloccus aureus are beneficial to CF patients [31–34]. Together, these symptom-based treatments have led to steady improvements in CF outcomes over the past several decades, including lung function and patient longevity.
CFTR modulators, which improve the function of defective, disease-causing CFTR mutations, are an exciting new avenue of CF therapies. Although more modulators are in the pipeline, currently only two exist that are approved for a subset of CF patients: ivacaftor, approved for patients age 2 years and above with certain gating mutations and a number of partially functional CFTR mutations, and ivacaftor in combination with lumacaftor, approved for patients age 6 years and up homozygous for the F508del mutation [6, 7]. The clinical benefits observed in CF patients treated with CFTR modulators (and indeed all therapies) vary, and understanding the factors responsible for variable responses are an area of growing interest in CF pharmacotherapy that strives for personalized and precise treatment regimens [35, 36].
With the combined improvements in patient diagnosis, care, and treatment, the course of cystic fibrosis has shifted dramatically. Today, the majority of patients with cystic fibrosis are adults (51.6% > 18 years) [1]. Our challenge now is to develop agents that modify and alleviate the fundamental defects in CF, with a focus on early and aggressively treatment, before secondary manifestations and irreversible organ damage occur.
2. TGFβ in CF Lung Disease
CF lung disease severity is not predicted by CFTR genotype alone. Numerous factors beyond CFTR genotype contribute to disease heterogeneity, including socioeconomic factors, adherence, environment, and other non-CFTR genetic modifiers [37–40]. Among these contributors, identification of genetic modifiers of cystic fibrosis has become an important goal for both disease forecasting and identification of novel therapeutic targets [37, 41]. Transforming Growth Factor β (TGFβ), a cytokine that drives diverse, crucial, and context dependent cellular activities has emerged as a master regulator of pulmonary health and disease that may offer insights into new approaches towards our understanding and novel treatment of CF lung disease [42].
2.1. TGFβ Signaling
The TGFβ super-family is vast, encompassing over 30 members including the TGFβ isoforms, anti-Mullerian hormone, and the bone morphogenic proteins (BMPs); for the purpose of this review, we will focus specifically on TGFβ [42–44]. There are three mammalian isoforms of TGFβ: TGFβ1, TGFβ2, and TGFβ3. These isoforms have similar bioactivities and share some function while also fulfilling some unique roles. They are important regulators of lung development, inflammation, injury, and repair, but their roles depend largely on the context of their expression and other parallel cellular processes. TGFβ1 is involved in pulmonary branching during development; overexpression arrests lung morphogenesis [45]. Knockout mice survive in utero but die early after birth secondary to pneumonitis and systemic inflammation [46]. Both TGFβ2 and TGFβ3 are expressed in the bronchiolar epithelium, and knockout of either results in perinatal death secondary to pulmonary defects, likely due to abnormal structural development and failure of appropriate epithelial-mesenchymal interactions [47–49].
TGFβ is secreted in a latent form, which is then activated by one of a number of mechanisms including interactions with integrins or matrix metalloproteinases, acidification, and/or reactive oxygen species [43]. Active TGFβ then binds to a complex consisting of two Type I (TGFβRl) and two Type II TGFβ (TGFβR2) receptors, both Ser/Thr kinases. From here, two general downstream cellular pathways are activated (Figure 1).
Figure 1. Schematic demonstrating simplified TGFβ signaling and downstream pathways.
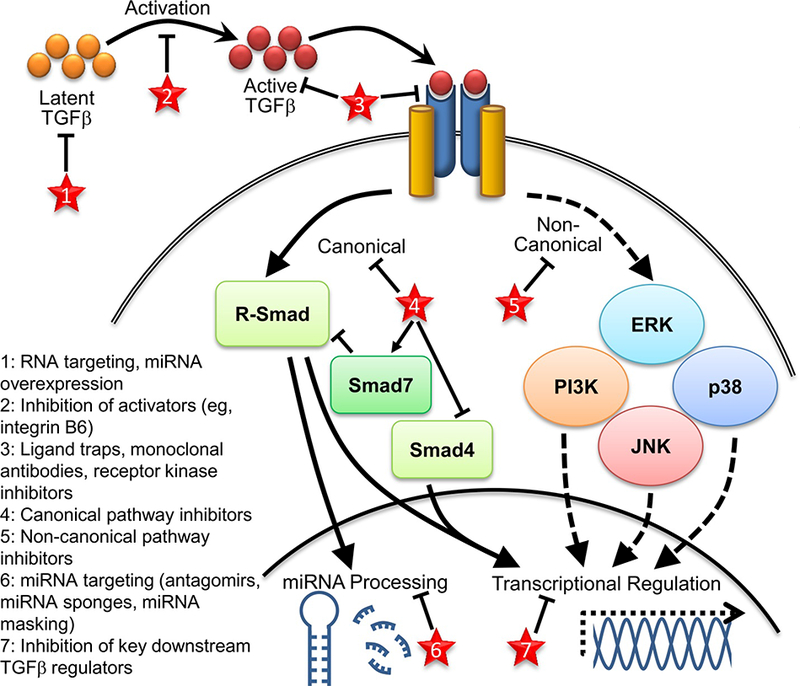
Potential sites of therapeutic targeting discussed in text are indicated by red stars.
Canonical TGFβ signaling operates through phosphorylation of Smad transcription factors. While the majority of Smad proteins are activating, Smads 6 and 7 function to inhibit TGFβ activities. TGFβ signaling via this pathway mainly functions through phosphorylation of Smad2 and Smad3 (termed receptor Smads or R-Smads), which interact with Smad4, the shared R-Smad partner, to regulate transcription of several downstream genes [43]. Activated Smads translocate to the nucleus, interacting with co-factors to facilitate binding to specific DNA sequences [42].
Non-canonical TGFβ signaling through non-Smad dependent pathways is perhaps less well understood and may function to either complement or counteract Smad activity. TGFβR2 can directly phosphorylate proteins such as PAR6, leading to altered cell morphology and differentiation [50]. TGFβ signaling can also activate the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways, such as Erk, p38, and c-Jun N-terminal Kinase (JNK), although the timing of activation is variable and dependent upon cellular context [42, 43, 51]. The balance of canonical to non-canonical signaling likely functions to modify the cellular response to TGFβ, along with the availability of other co-factors and signaling pathway partners.
TGFβ signaling is also intertwined, upstream and downstream, with miRNAs. miRNAs both modify TGFβ signaling and regulate downstream TGFβ effects; some miRNAs function in both capacities. A multitude of miRNAs are predicted to target TGFβ signaling pathway member mRNAs, but only a subset have been verified experimentally. miRNA regulators of TGFβ include the miR-200 family, which targets TGFβ2, TGFβR1, and Smad2 in a variety of cell types; the miR20 family, which targets TGFβR2 as shown in lung cancer tissues; miR-1343, which targets TGFβR1 and R2 in lung epithelial cells; and miR-21, which targets the inhibitory Smad7, thereby allowing increased canonical pathway activation and driving pro-fibrogeni( activity in pulmonary fibroblasts [52, 53].
TGFβ signaling also alters miRNA expression levels; activated R-Smads interact with the Drosha miRNA processing complex to promote maturation of discreet miRNAs [54, 55]. TGFβ influences the levels of some of the regulatory miRNAs described above, causing downregulation of the miR-200 family and upregulation of miR-21, allowing for greater TGFβ pathway activation [52]. TGFβ decreases miR-203, which normally functions to inhibit epithelial mesenchymal transition (EMT) and metastasis [56]. miR-155 is upregulated after TGFβtreatment of mammary cells in vitro, and knockdown of miR-155 suppresses EMT and cell migration [57]. The intimate interactions between miRNAs and TGFβ signaling have only begun to be described; miRNAs appear to be important modifiers of TGFβ induced pathology.
TGFβ broadly functions to influence injury responses, inflammation, and differentiation [42]. As described above, studies in transgenic mice show that the expression of TGFβ plays a critical role in pulmonary development and regulation of autoimmunity. TGFβ can also drive inflammation in the appropriate context and causes a shift towards Th2-type responses [58]. Also dependent upon the setting, TGFβ can be both pro- and anti-proliferative; in general, TGFβ promotes a mesenchymal phenotype and cell proliferation while inducing extracellular matrix production [59]. Although TGFβ is essential for wound healing, dysregulation of the pathway can drive remodeling and fibrosis, leading to end organ disease. As many pulmonary diseases are characterized by a cycle of pathologic lung inflammation/injury followed by aberrant repair mechanisms, TGFβ is positioned to be a master regulator of lung disease in a variety of disorders.
2.2. TGFβ in Pulmonary Disease
In humans, TGFβ isoforms continue to be expressed in the healthy lung through adulthood. TGFβ expression has been described in human airway epithelium, alveolar macrophages, and airway smooth muscle cells [60–62]. In various pulmonary diseases, TGFβ signaling has been reported to be increased in the airway epithelium, fibroblasts, macrophages, and smooth muscle cells [60, 63]. Reviewing the proposed role of TGFβ in non-CF pulmonary diseases provides a useful framework for understanding and potentially targeting the TGFβ pathway to impact CF lung disease.
Idiopathic pulmonary fibrosis (IPF) is a devastating interstitial lung disease characterized by injury, remodeling, and ultimately progressive fibrosis; median survival is 2.5–3.5 years after diagnosis [64]. A fundamental driver of fibrosis is TGFβ, and levels are elevated in airway epithelium and fibroblasts from IPF patients [60, 63, 65, 66]. TGFβ also drives myofibroblast differentiation from fibroblasts, a cell type which is proposed to drive the cycle of increased epithelial injury (especially alveolar type II cells) and aberrant injury response that promotes continued fibrosis in IPF [67–70]. In mice, inhibiting TGFβ signaling results in protection in fibrotic disease models [71, 72]. These findings led to a search for therapeutics targeting the TGFβ pathway that are effective in IPF. The first such drug to receive FDA approval for IPF was pirfenidone, an orally available small molecule inhibitor of TGFβ (as well as other signaling pathways including PDGF and TNF-α) with anti-fibrotic and anti-inflammatory effects; the exact mechanism of pirfenidone action is unknown [73]. A 2014 phase 3 study of pirfenidone in IPF showed a significant reduction in the proportion of patients with decline of forced vital capacity (FVC) of 10% or more in the active treatment group versus placebo (16.5% versus 31.8% respectively, p<0.001) [74]. Subsequent meta-analysis also supported the therapeutic benefit of pirfenidone in IPF, with pirfenidone use associated with reduction in lung function decline and improved survival over the course of a year [75].
TGFβ signaling has also been implicated in the pathogenesis of asthma and chronic obstructive pulmonary disease (COPD). Both diseases are characterized by airway obstruction (typically considered reversible in asthma but irreversible in COPD), inflammation, and remodeling. TGFβ1 levels were elevated in bronchoalveolar lavage (BAL) fluid obtained from asthmatic patients and in the airway and alveolar epithelium of patients with COPD [76, 77]. Higher producing polymorphisms in TGFβ1 (especially a C-509T variant in the TGFβ1 promotor) are associated with worsening asthma severity [78]. Mechanistically, it is hypothesized that TGFβ produces pathologic effects in these diseases by promoting goblet cell hyperplasia, subepithelial fibrosis, epithelial damage, and airway smooth muscle hypertrophy [79–85]. It is important to note, however, that the effects of TGFβ are not universally clear or consistently detrimental. In fact, TGFβ1 underexpressing mice have a more severe phenotype in the ovalbumin induced murine model of asthma, and overexpression of TGFβ1 in Th2 cells reduces disease severity in this model [86, 87]. These studies illustrate the complex and context dependent importance of TGFβ signaling. While TGFβ has been discussed and studied as a potential therapeutic target in asthma for over a decade, no approved medications targeting TGFβ in asthma have advanced to the clinic [88, 89].
2.3. TGFβ as a Genetic Modifier and Biomarker of CF Lung Disease
CF lung disease severity cannot be predicted simply by CFTR genotype [37, 41]; over 50% of the variation in lung disease severity is attributable to non-CFTR genetic contributors [90]. Two TGFβ1 polymorphisms (C-509T polymorphism in the promotor region and T29C polymorphism in codon 10) have been found in genome-wide association studies to be linked to more severe CF lung disease [41, 91, 92]. These polymorphisms have been linked in vitro and in non-CF populations to higher levels of gene expression and secretion. Transfection of the T29C polymorphism in HeLa cells causes a 2.8-fold increase in TGFβ1 secretion, and having at least a single copy of the T29C allele was associated with increased serum TGFβ1 protein levels in a study of Caucasian and African-American normotensive and hypertensive adults and in groups of healthy or osteoporotic Japanese women [93–95]. The C-509T promotor region polymorphism is associated with higher transcriptional activity of TGFβ1 in vitro and higher plasma TGFβ1 levels in a group of female UK subjects [96, 97]. However, the relationship between these polymorphisms and blood or BAL levels of TGFβ1 has not been clearly defined in the CF population.
In addition to TGFβ1’s role as a genetic modifier of CF lung disease, it is also a biomarker of increased disease severity. Increased TGFβ1 blood (plasma) and BAL levels in CF patients have been associated with pulmonary exacerbations, severity of lung disease, increased neutrophilic inflammation in BAL, and infection with Pseudomonas aeruginosa [98, 99]. Taken together, these studies implicate TGFβ1 as a prominent potential therapeutic target in CF. In a separate study, serum concentrations of TGFβ2 were demonstrated to be elevated in young CF patients compared to healthy controls, although linkage to disease severity is unclear [100].
2.4. Mechanism of TGFβ Action in CF
Airway remodeling in cystic fibrosis is complex and progressive. Early disease is characterized by inflammation, mucus obstruction, chronic infection, and goblet cell hyperplasia with a relative preservation of FEV1; later, bronchiectasis and fibrosis result from the cycles of infection and inflammation, chronic infection with dominant organism(s) may take hold, and FEV1 ultimately declines [101, 102]. TGFβ signaling is involved in different disease mechanisms at different stages of the disease, and TGFβ pathways should not be considered universally detrimental in CF.
One possible mechanism through which TGFβ could negatively affect lung health in CF is through direct downregulation of chloride transport. Several groups have shown that TGFβ1 exposure in human airway epithelial cells in vitro causes downregulated expression and function of CFTR and an alternative chloride transport pathway, Calcium activated Chloride Conductance (CaCC) [103–105]. TGFβ1 exposure also negates the beneficial effects of treatment with VX- 809, a CFTR corrector, on F508del CFTR function [103, 104]. One potential mechanism for this downregulation is through microRNAs, small (~22 nucleotides) non-coding RNAs that bind to specific mRNAs to decrease expression of target genes. miR-145 is a microRNA upregulated by TGFβ1 which downregulates CFTR expression; preliminary evidence suggests that the TGFβ1 induced decrease in CFTR is mediated at least in part by miR-145 upregulation [106, 107]. Increased TGFβ signaling in CF is predicted to induce other pathologies downstream of miRNA alterations. miR-155, an miRNA upregulated by TGFβ as described in section 1.2, is upregulated in CF epithelial cell lines and increases both IL-8 and Smad phosphorylation through inhibition of regulatory associated protein of mTOR, complex 1 (RPTOR) [108].
TGFβ1 may also promote networks of gene expression that drive pathologic airway remodeling. Goblet cell hyperplasia and increased mucin secretion is a well described feature of CF lung disease [109]. Inhibition of TGFβ1 signaling, specifically through Smad dependent pathways, in mouse models of allergen induced rhinitis and airway remodeling suppresses goblet cell hyperplasia [79, 80]. TGFβ1 has recently been shown to drive airway smooth muscle shortening and hyperresponsiveness in vitro via Smad signaling [110]. As described above, TGFβ is linked to pulmonary fibrosis in multiple animal models as well is in IPF and drives myofibroblast differentiation. Studies of CF patients have identified activation of TGFβ signaling associated with areas of fibrosis and myofibroblast differentiation, and, furthermore, that constrictive bronchiolitis in lung biopsies was associated with myofibroblast differentiation potentially induced by TGFβ [111, 112].
TGFβ signaling is involved in driving inflammatory responses that may augment injurious inflammatory stimuli in CF, both in young patients prior to acquiring chronic pulmonary infections and in older patients with more progressed lung disease and established infection [113]. Neutrophilic inflammation is a hallmark of CF lung disease, and TGFβ is a potent neutrophil chemoattractant [114]. Pro-inflammatory cytokines including IL-1β, IL-6, and IL-8 are higher in CF BAL, while the anti-inflammatory cytokine IL-10 is reduced [115]. Human neutrophil elastase (NE) is also elevated even in young children with CF and is predictive of bronchiectasis and low BMI [4, 116]. TGFβ signaling has mixed effects on these inflammatory regulators and is likely context dependent. NE appears to induce TGFβ1 secretion and activation, and mice lacking NE are resistant to bleomycin induced lung injury and have a lack of active TGFβ in lung tissue [117, 118]. IL-6 and TGFβ synergistically drive differentiation of proinflammatory T helper 17 cells, while TGFβ and IL-1β work together to enhance IL-6 and IL-8 production in retinal epithelial cells [119, 120]. TGFβ1 inhibits IL-10 expression in immune cells, thus reducing modulation of the immune response by this anti-inflammatory cytokine [121]. Human airway epithelial cells exposed in vitro to TGFβ demonstrate suppression of IL-8 production, but in other cell types such as renal epithelial cells and cancer cell lines TGFβ induces IL-8; the role of TGFβ in vivo on IL-8 production in CF is unclear [122–124].
Aberrant secondary inflammatory responses may be another mechanism of TGFβ mediated pulmonary pathology in CF. TGFβ is expressed by a variety of immune cells, including alveolar macrophages, eosinophils, and T lymphocytes, and is a potent neutrophil chemoattractant [61, 125, 126]. Recent studies have shown that TGFβ1 suppresses innate immune responses and alters host responses to infection, with reduced clearance and augmented inflammation, and promotes viral replication in several epithelial cell models [127–129]. TGFβ signaling may thus enhance susceptibility to viral infections while promoting inflammatory responses that lead to lung injury and damage.
Alterations in TGFβ expression are induced by P. aeruginosa quorum-sensing (QS) systems, an intercellular density dependent communication linked to biofilm formation and virulence, and TGFβ may in turn lessen the effectiveness of the resulting inflammatory response [130]. In a murine model of burn infection, QS-capable P. aeruginosa induced TGFβ1 expression while a QS-defective mutant did not [131]. Similarly, in a rat model of pulmonary P. aeroginosa biofilm infection, wild type P. aeruginosa but not QS mutant strains induced pulmonary TGFβ1 expression; the authors hypothesized that the cytokines induced by the QS system may upregulate regulatory T cells and thus decrease the effectiveness of the resulting immune response [132]. Adaptations of quorum signaling occur during the transition of initial to chronic infection in cystic fibrosis patients, and it is unknown how TGFβ production and function is altered during this complex process [133, 134].
TGFβ has a multifactorial and complex role in promoting lung disease in CF, as summarized in Table 1. While the time course of TGFβ dependent effects is unclear, it is possible that early effects and responses to TGFβ1 signaling contribute to early events in CF lung disease pathology. In this scenario, increased TGFβ signaling in young CF patients may drive goblet cell hyperplasia, immune system dysregulation, and counterproductive responses to infection and fibrosis that ultimately contribute to lung function decline.
Table 1.
CF relevant effects and potential mechanisms of pathologic TGFβ actions in CF.
| Effect | Suggested Mechanism | Relevance to CF | Refs |
|---|---|---|---|
| Downregulation of epithelial chloride transport | Downregulated CFTR and CaCC expression and function, potentially microRNA mediated | Exacerbates already dysregulated ion transport in epithelia throughout the body in CF | 94, 95, 96 |
| Driving goblet cell hyperplasia | Potentially via Smad dependent pathways based upon mouse models of allergen induced airway remodeling | Goblet cell hyperplasia and mucin secretion are pathologic features of CF lung disease | 72, 73, 79 |
| Aberrant inflammatory responses | Suppression of innate immune responses with augmented inflammation | CF lungs are known to be primed for a more aggressive inflammatory response, inability to clear chronic infection, and dysregulated innate immunity | 104, 105, 106 |
| Enhanced fibrosis | Driving myofibroblast differentiation and aberrant injury response that promotes fibrosis | After cycles of infection and inflammation, fibrotic lung disease significantly contributes to pulmonary decline in CF patients | 56, 100, 101 |
CaCC, Calcium activated Chloride Conductance; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulation.
3. Targeting TGFβ in CF
Due to its broad relevance across multiple organ systems and diseases, targeting TGFβ and its downstream pathways is an area of intense research and development interest. However, the complex network of pathways activated downstream of TGFβ, as well as TGFβ’s diverse involvement in critical cellular functions, makes developing an effective but safe therapy challenging. There are several potential strategies for targeting TGFβ and its signaling pathways: broad TGFβ pathway inhibition at the receptor or ligand level, directed blocking of downstream signaling components, targeting microRNAs, or focusing on modifiers and downstream products (Figure 1, Table 2).
Table 2.
Potential approaches to normalize TGFβ signaling in CF.
| Target | Drug Type or Approach |
|---|---|
| Broad TGFβ pathway inhibition | Monoclonal antibodies targeting ligands or receptors Ligand traps Small molecule inhibitors |
| TGFβ ligand specific inhibition | Monoclonal antibodies Gene silencing, e. g. siRNAs and antisense oligonucleotides |
| Smad dependent signaling blockade | Small molecule inhibitors Receptor kinase inhibitors Smad7 upregulation |
| Non-canonical signaling inhibition | Small molecule inhibitors of relevant pathways, e. g. PI3K and MAPK inhibitors |
| Pathologic miRNA s | Antagomirs |
| Modifiers of TGFβ effects: | |
| Co-regulator inhibition | Small molecule inhibitors, antibodies targeting EGFR pathway |
| Promotion of Th1 inflammatory response | Upregulation of IFN-γ to balance Th2 response driven by TGFβ |
| Blockade of TGFβ activation | Monoclonal antibodies targeting integrin β6 |
IFN-γ, interferon gamma; MAPK, mitogen-activated protein kinase; miRNA, microRNA; PI3K, phosphoinositide 3-kinase; siRNA, small interfering RNA
As with all CF therapies, drug delivery poses a unique challenge. Pulmonary disease remains the largest source of morbidity and mortality for CF patients, so inhalation is a logical method of delivery. As TGFβ in BALF is a biomarker of more advance CF lung disease, blockade of TGFβ signaling via an inhaled drug may be sufficient to ameliorate TGFβ induced pathology [98]. However, CF pulmonary disease presents unique barriers to effective inhaled drug delivery, including impaired drug deposition in diseased airways, permeation through thickened mucus, and an environment with sustained infection and inflammation [135]. In addition, evidence is gathering that dysfunction related to loss of CFTR not only affects epithelia throughout the body but also the immune system, smooth muscle cells, and fibroblasts [3, 136, 137]. If TGFβ does further downregulate CFTR in CF, systemic drug delivery may provide enough CFTR restoration in certain CF genotypes to lessen CF induced pancreatic and intestinal dysfunction [103]. While systemic drug delivery, either enteral or parenteral, has the benefit of modulating TGFβ signaling throughout the body, it also exposes the patient to more potential toxicity and off-target effects.
While many types of pharmacologic strategies have been investigated for other disease processes, including neutralizing antibodies, small molecule inhibitors, antisense RNAs, ligand traps, and gene therapy, no TGFβ targeting drugs are currently approved for use in CF. There are currently no reports from clinical trials targeting TGFβ signaling in CF, so strategies to modulate TGFβ signaling to treat CF must be extrapolated from animal or cell studies and patient data from other disease processes. What follows is not a complete list of therapeutics that target the TGFβ pathway; rather, in the remaining sections of this review we seek to identify feasible and/or rational approaches based on the scientific literature.
3.1. Existing Medications with Known TGFβ Effects
Several approved medications have known effects on TGFβ signaling pathways, although the exact mechanism of these effects is often not well understood. Pirfenidone, currently approved for IPF, blocks TGFβ signaling and production by an unclear mechanism; TGFβ1- induced airway surface liquid (ASL) dehydration in CF bronchial epithelial cells has been shown to be abrogated by treatment with pirfenidone in vitro [74, 138]. Losartan, an angiotensin II type 1 receptor inhibitor, reduces TGFβ signaling and also reduces mucociliary dysfunction in CF bronchial epithelial cells after TGFβ treatment [139]. Corticosteroids have been demonstrated to inhibit TGFβ1 expression in a mouse model of asthma, yet these results have not been replicated in humans, and the utility of using corticosteroids to reduce TGFβ signaling is unclear [88]. Simvastatin, used primarily for its lipid-lowering effects, also inhibits TGFβ1-induced myofibroblast production [140]. Finally, mepacrine, an anti-malarial drug, inhibits TGFβ1 expression and subepithelial fibrosis in a mouse model of asthma [141]. As these medications have known side effect profiles and are approved for other indications in humans, they represent low-hanging fruit in the search for TGFβ modifying medications in CF. However, their utility and direct translation to CF may be limited by off target effects and unclear mechanisms of action.
3.2. Broad TGFβ Pathway Inhibition at Ligands/Receptor Level
TGFβsignaling can be broadly inhibited by targeting TGFβ ligands or receptors. A panoply of monoclonal antibodies have been developed, both ligand-specific and capable of targeting multiple TGFβ ligands [51, 127]. Many of these antibodies have been humanized and proceeded through at least the early stages of preclinical and clinical trials in non-CF diseases where TGFβ ligands are overexpressed. The benefit of these monoclonal antibodies includes the possibility of specificity for select TGFβ ligands (while retaining TGFβ signaling via other ligands) and their pharmacokinetic stability. The TGFβ1 isoform has the most evidence for involvement in CF lung disease, and therefore is a logical initial target for consideration. Animals studies have provided evidence of successful TGFβ pathology inhibition via antibodies. In rodents, anti-TGFβ antibodies have been used to halt nephropathy and suppress tumor metastasis; such antibodies are undergoing clinical development for a variety of disease including cancers and scleroderma [52].
Soluble receptor constructs are another strategy to sequester excess TGFβ ligands. Ligand traps using modified or mimetic TGFβII and TGFβRIII constructs have been developed; these pharmacologics have prevented tumor metastasis in animal models and shown promise in suppression of cell growth of human breast and colon cancer in vitro [52]. These therapeutics have not moved far in clinical development, and there may be challenges to systemic drug delivery [51].
Another method of TGFβ pathway inhibition is RNA targeting. Gene silencing techniques such as antisense oligonucleotides, short interfering RNAs (siRNAs), and promoter targeting pyrrole-imidazole polyamides inhibit mRNA translation and have been successfully used against TGFβ ligands in non-CF cell and animal models, but difficulties with drug delivery and stability persist [51, 88]. Antisense oligonucleotides against TGFβ family members have been used to make antisense-modified tumor cell vaccines to promote antitumor immunity; such techniques have unclear applications in CF [142].
Beyond inhibition of ligands, receptors are also appealing targets; monoclonal antibodies against TGFβR1 and R2 have been developed [51]. These antibodies have been shown to have effects on both canonical and noncanonical signaling and would presumably provide the greatest universal, ligand and TGFβ isoform-independent downregulation of TGFβ signaling. Intracellular signal transduction can be blocked by TGFβ1 receptor kinase inhibitors, which would also provide broad TGFβ pathway inhibition. In vitro data for such inhibitors are promising, with inhibition of TGFβ induced EMT and fibrosis, and clinical development of some such pharmaceutics for cancers such as hepatocellular carcinoma and glioblastoma is advanced [142].
3.3. Targeting Downstream TGFβ Signaling
Several small molecule inhibitors targeting TGFβR1 to inhibit Smad 2/3 phosphorylation have also been developed and have the benefit of oral availability, but may have off target effects on other activin-like kinases (ALKs) [51]. Upregulation of the inhibitory Smad 7 can also be a tool to decrease Smad dependent signaling [88]. However, it is unclear in CF if the primary pathology downstream of TGFβ is driven by the Smad-dependent canonical pathways and/or the non-canonical pathways. In other words, the necessary balance of canonical and non-canonical signaling has not been established to date. Indeed, TGFβ1 induced reduction of CFTR expression and function appears to be via a p38 MAPK mechanism rather than Smad dependent signaling, indicating that Smad inhibition would, at best, only partially relieve TGFβ induced pathology [103, 104].
Non-canonical pathways are also potential targets for inhibition. In oncology and other diseases, small molecule inhibitors of the Erk pathway have been trialed, but their use is limited by variable responses and development of resistance [143]. JNK and p38 MAPK inhibitors have been investigated for use in IPF and beyond, but side effects and toxicity are continuing challenges [144, 145]. The PI3K pathway is also felt to contribute to dysregulated inflammatory responses, airway hyperresponsiveness, and mucin production [146]. Small molecule inhibitors of this pathway are promising, but further work is needed to avoid dangerous immunosuppression in CF (where chronic infection is common), off target effects, unacceptable side effects; indeed, certain inhibitors of the pathway, especially macrolide mTOR inhibitors such as sirolimus, carry a risk of interstitial pneumonitis [146].
3.4. MicroRNA as Targets
A number of studies have implicated miRNAs as altered in CF and potentially playing a role in mediating cystic fibrosis pathophysiology related to TGFβ signaling [147]. These may be amenable to anti-miR targeting, or in the case of protective miRNAs downregulated by TGFβ or miRNAs that target TGFβ pathway members, overexpression. As outlined above, TGFβ signaling can directly promote maturation of specific microRNAs through interaction with the microRNA processing complex [54, 55]. There is extensive crosstalk between TGFβ signaling and microRNAs, and a number of miRNAs are altered by TGFβ [52]. miR-145 specifically has been shown to be upregulated in response to TGFβ signaling and to decrease CFTR expression and function [106, 107, 148].
Development of therapeutics targeting miRNAs has been an enticing goal, especially in oncologic diseases, with some therapeutics reaching clinical development for non-CF diseases [149]. The most common method of miRNA suppression is utilizing an antisense oligonucleotide (antagomir) that bind miRNA targets to prevent miRNA-mRNA interactions [150]. To target knockdown of a network of miRNAs, miRNA sponges are being developed that contain multiple binding sites for related miRNA targets [52]. A third technique, miRNA masking, involves utilizing a single-stranded oligoribonucleotide that is complementary to the miRNA binding site on the target mRNA to hinder miRNA binding [52]. For miRNAs that might target and downregulate TGFβ pathway members, therapies include miRNA overexpression or development of artificial double-stranded miRNA mimic [149].
There are several challenges involved in developing miRNA therapeutics. With systemic delivery, there is the potential for cytotoxic effects (related to either the miRNA or the vector) and immunogenic reactions; local treatment may be more feasible, with intranasal delivery previously described [52]. Furthermore, difficulties with drug delivery due to size and charge in the setting of pulmonary disease have slowed durg development, although studies are ongoing [147].
In addition to inhibiting TGFβ-upregulated miRNAs and increasing TGFβ-downregulated miRNAs, antagonizing or expressing miRNAs that influence relevant TGFβ target genes may be a viable strategy. Further research would be needed both to identify miRNA targets and validate their utility in blocking TGFβ induced pathology in CF.
3.5. Modulating TGFβ Signaling through Modifiers, Co-Regulators, and Downstream Products
Given the complex network of pathways and genes influenced by and interacting with TGFβ in a context dependent manner, there are a vast number of targets outside the direct TGFβ signaling pathways that may be amenable to therapeutic modulation. The EGFR pathway has been implicated as a positive regulator of TGFβ induced pulmonary fibrosis and functions through activation of non-canonical pathways including PI3K and ERK [151]. Inhibition EGFR signaling in lung diseases has been an area of intense research, but a clinical benefit has only been seen in lung cancer therapy; difficulties with clinical efficacy and toxicities limit therapeutic development [152]. Interferon-gamma (IFN-γ) is a cytokine that promotes a Th1 type response, perhaps balancing the Th2 cytokine production driven by TGFβ, and also downregulates TGFβ2 [88]. Monoclonal antibodies against integrin β6, a key activator of latent TGFβ, prevent activation of TGFβ and have undergone studies in non-CF patients [51]. Key downstream regulators of TGFβ such as protein tyrosine phosphatase α (PTP-α), which mediates profibrotic signaling, are also potentially amenable to inhibition [42]. The principle challenges involved in inhibiting regulators and downstream products involved in transmitted TGFβ effects include difficulty in target selection and potentially narrow windows of clinical efficacy.
4. Conclusion
TGFβ signaling in certain contexts drives a number of processes involved in CF lung disease pathophysiology, including fibrosis, goblet cell hyperplasia, abnormal inflammatory responses, and dysregulated ion transport. TGFβ1 in particular has been shown to be both a genetic modifier and a biomarker of CF lung disease severity. The complexity and context dependence of TGFβ signaling, however, make it a challenging therapeutic target, as exemplified by the difficulties in developing TGFβ-targeting therapies for diseases such as IPF where TGFβ has long been identified as a driving force of lung disease. Given the building evidence that TGFβ signaling is involved in CF lung disease and can directly drive further decreases in CFTR expression and function, we postulate that it is time to re-evaluate TGFβ pathways and regulators as potential therapeutic targets in CF. Furthermore, as TGFβ signaling may impact the efficacy of emerging and expensive CFTR modulators that are becoming available for certain CF patients, understanding the implications of TGFβ signaling may allow clinicians to make more educated and personalized choices to better treat CF patients.
5. Expert Opinion
Modulating TGFβ signaling in CF presents a unique opportunity and challenge, as it is a critical regulator of lung development and homeostasis, as well as a tumor suppressor and antiinflammatory cytokine depending on its cellular context. Broad TGFβ suppression may well have undesirable effects upon cellular homeostasis, inflammation, and tumor suppression. Efficacy in CF patients y to be variable and depend upon genetic variations in patients, stage of lung disease, and personal disease characteristics including the pulmonary microbiome. Other co-therapies, such as CFTR modulators, will likely impact the response to TGFβ-targeted therapies, including which aspects of CF pathology are affected. A better understanding of the role of TGFβ in CF lung disease will help researchers test novel therapies that impact rational signaling components and ultimately help clinicians select appropriate drugs to administer to the right patient at the right stage of disease.
5.1. Targeting Appropriate Components of TGFβ Signaling
As described above, the TGFβ signaling network is complex and context-dependent. Pan-inhibition of TGFβ signaling would potentially disrupt a number of critical cellular processes. Choosing appropriate targets will be necessary to developing efficacious, safe therapies in CF. Preliminary work has identified that TGFβ-induced p38 signaling contributes to CFTR downregulation in human airway epithelial cells (AECs), but it is unclear if this is a significant contributor to lung pathology in vivo [103, 105]. Furthermore, miR-145 has been identified as an important mediator of CFTR downregulation by TGFβ1, but the relationship between this mechanism and p38 signaling is currently undefined [106, 148, 153]. TGFβ’s influence on goblet cell hyperplasia appears to be through Smad-dependent mechanisms, while profibrotic effects appear to include both canonical and noncanonical signaling pathways. Thus, there may be no single target downstream of TGFβ that will abrogate all, or even the majority, of potential detrimental effects. Further studies are needed to define the role of TGFβ in CF lung disease severity, describe the downstream pathways involved, validate potential targets identified using simple systems in translational models, and elucidate the time course of TGFβ effects.
Drug delivery is another translational challenge. Systemic therapies increase the potential for off target effects in unaffected tissues. Direct intrapulmonary delivery may allow higher pulmonary drug exposures and avoid systemic effects, but penetrance and transepithelial transport of drugs is variable and depends on drug geometry, size, and charge. Furthermore, as is the case with other inhaled therapeutics, delivery may be poor in those pulmonary regions with greater obstruction and mucous plugging, which may ironically be where TGFβ targeting has the greatest potential impact.
5.2. Determining the Right Patient
Other co-therapies may impact the efficacy of TGFβ modulation in CF, or TGΕβ inhibitors may improve the efficacy of existing drugs. In vitro studies have shown that TGFβ treatment of human AECs negates the beneficial effects of VX-809 on F508del CFTR; it stands to reason that certain patients may see more benefit from VX-809 with concurrent TGFβ targeted therapy where excessive TGFβ signaling is manifest. This logic may extend to other CFTR modulators, but preclinical support is currently lacking. Clarifying these important interactions in the context of complex CF treatment regimens will be important, but not straightforward.
A patient with one of the previously described polymorphisms in TGFβ linked to development of more severe lung disease may experience more clinical benefit from therapy targeting TGFβ, but this too is currently only theoretical. Beyond CFTR-mutation specific modulator therapy, we are currently far from effectively harnessing our growing knowledge of CF genetic modifiers to inform treatment plans. As a first step, using patient derived samples to identify excessive TGFβ signaling and to test in vitro responses may help guide the selection of patients for clinical trials. With the growing concept of personalized medicine in the CF community, we feel this general approach may become a valuable tool to choose the most appropriate patient to receive next generation therapies.
5.3. Timing of TGFβ Targeting Therapies
It is becoming clear that effective treatment of CF lung disease needs to begin early in life. Bronchiectasis is already present in many toddlers with CF, and studies in pigs suggest that host-defense defects are present at birth [4, 154]. As TGFβ is a genetic modifier of CF, it is possible that it has an early impact on processes such as innate immune response, airway remodeling, inflammation, and fibrosis. However, we currently do not have an understanding of the expression and role of TGFβ in very young children with CF. It is possible, perhaps even likely, that the later elevation of TGFβ in the BAL and blood of CF patients with more advanced lung disease is not only causative but also a marker of lungs with great infectious, inflammatory, and fibrotic burdens. Further studies are needed to define the pattern of TGFβ expression and correlation with the development of lung disease in children with CF.
Although early therapy has the most potential to change long term CF lung disease trajectories, targeting young (and potentially asymptomatic) children for therapy brings certain ethical and medical considerations. Long term toxicity and impact on lung development must be thoroughly evaluated before these medications could be considered in this vulnerable population. It is highly likely that approval would first be sought for anti-TGFβ therapeutics in adults, and then extended towards younger age groups as efficacy and safety are shown. This requirement of drug development could potentially be a barrier, as the impact of TGFβ-targeted therapies may vary substantially based on the stage of lung disease. Thus, findings in older patients with established disease may not predict or reflect effects in young patients with less lung damage.
5.4. Future Directions in Developing Therapies Directed at the TGFβPathway
As TGFβ inhibiting medications are already approved for other disease processes (i.e.: pirfenidone in IPF, losartan in Marfan syndrome), these would be logical initial therapies to trial in CF based on supportive evidence from preclinical and translational model systems. These preclinical studies are essential to understand how these therapies may influence CF lung disease and to select appropriate biomarkers to demonstrate drug bioactivity in the context of CF. Further study is needed, however, to better understand the mechanism(s) of TGFβ induced pathology in CF, the timeline of these effects, the downstream mediators involved, and the connection between discreet TGFβ signaling pathways and pulmonary pathology. We will also need to consider the personal disease characteristics in our CF patients that may impact the efficacy of TGFβ targeting, including TGFβ polymorphisms, CFTR genotype, CF co-therapies, and disease stage. There is a risk of failing to prove efficacy or provoking unacceptable side effects if patient and drug selection are not carefully considered.
In summary, TGFβ is an attractive target for new CF therapeutics, based on its clear lung disease modifying association and relationship to numerous important and fundamental aspects of CF lung disease. The complexity of TGFβ signaling and the enormous reach of its influence across organ systems and diseases also make it a challenging target, and the development of therapies will require careful mechanistic studies to understand relevant signaling pathways and downstream impact. Accumulating research in CF suggests, however, that it could be an excellent therapeutic target with broad beneficial impact on ion transport, goblet cell hyperplasia/mucus production, inflammation, airway remodeling, and/or fibrosis. Our current research understanding supports studies to identify and advance these therapies in CF, with an eye to balancing the relative roles of TGFβ in both lung health and disease.
Article Highlights.
-
-
TGFβ is a genetic modifier of CF lung disease and a biomarker of more severe pulmonary pathology in patients.
-
-
TGFβ signaling drives airway remodeling, dysfunctional inflammatory responses and epithelial ion channel dysregulation in experimental models.
-
-
The mechanism of CF disease modification by TGFβ, and the utility of targeting its pathway, is unknown.
-
-
Therapeutics targeting TGFβ signaling are under development for a variety of pulmonary and non-pulmonary diseases but have not been used in CF.
-
-
Inhibiting the TGFβ pathway in CF may improve outcomes for certain patients and allow for better efficacy of new CFTR modulating pharmaceutics.
Acknowledgments
Funding
This work was supported by the Cystic Fibrosis Foundation (grant number CLANCY15R0) and the National Center for Advancing Translational Sciences, National Institutes of Health (grant number KL2TR001426).
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.North American Cystic Fibrosis Foundation. Patient Registry: Annual Data Report. 2015. Available from: https://www.cff.org/Our-Research/CF-Patient-Registry/2015-Patient-Registry-Annual-Data-Report.pdf
- 2.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001 [DOI] [PubMed] [Google Scholar]
- 4.•.Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368:1963–1970.Description of early lung disease in children with CF under age 3 years.
- 5.Ranganathan SC, Hall GL, Sly PD, et al. Early Lung Disease in Infants and Pre-school Children with Cystic Fibrosis: What Have We Learnt and What Should We Do About It? Am J Respir Crit Care Med.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:1783–1784 [DOI] [PubMed] [Google Scholar]
- 8.Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79:S215–255 [DOI] [PubMed] [Google Scholar]
- 9.Schweibert EM, Benos DJ, Egan ME, et al. CFTR Is a Conductance Regulator as well as a Chloride Channel Physiological Reviews. 79: The American Physiological Society; 1999. p. S145–S166 [DOI] [PubMed] [Google Scholar]
- 10.Anderson MP, Gregory RJ, Thompson S, et al. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205 [DOI] [PubMed] [Google Scholar]
- 11.Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med. 2007;261:5–16 [DOI] [PubMed] [Google Scholar]
- 12.Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–170 [DOI] [PubMed] [Google Scholar]
- 13.Button B, Cai LH, Ehre C, et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science. 2012;337:937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Walker NM, Ootani A, et al. Defective goblet cell exocytosis contributes to murine cystic fibrosis-associated intestinal disease. J Clin Invest. 2015;125:1056–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen JH, Stoltz DA, Karp PH, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. Epub 2010/12/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoegger MJ, Fischer AJ, McMenimen JD, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345:818–822. Epub 2014/08/16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinton PM. Role of epithelial HCOFormula transport in mucin secretion: lessons from cystic fibrosis. Am J Physiol Cell Physiol.299:C1222–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang N, Garcia MA, Quinton PM. Normal mucus formation requires cAMP-dependent HCO3- secretion and Ca2+-mediated mucin exocytosis. The Journal of physiology. 2013;591:4581–4593. Epub 2013/07/03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konstan MW, Berger M. Current understanding of the inflammatory process in cystic fibrosis: onset and etiology. Pediatric Pulmonology. 1997;24:137–142; discussion 159–161 [DOI] [PubMed] [Google Scholar]
- 20.Cohen-Cymberknoh M, Kerem E, Ferkol T, et al. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax. 2013;68:1157–1162. Epub 2013/05/25 [DOI] [PubMed] [Google Scholar]
- 21.Khan TZ, Wagener JS, Bost T, et al. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082 [DOI] [PubMed] [Google Scholar]
- 22.Bodas M, Vij N. The NF-kappaB signaling in cystic fibrosis lung disease: pathophysiology and therapeutic potential. Discov Med. 2010;9:346–356 [PMC free article] [PubMed] [Google Scholar]
- 23.Montgomery ST, Mall MA, Kicic A, et al. Hypoxia and sterile inflammation in cystic fibrosis airways: mechanisms and potential therapies. Eur Respir J. 2017;49. [DOI] [PubMed] [Google Scholar]
- 24.McKiernan PJ, Greene CM. MicroRNA Dysregulation in Cystic Fibrosis. Mediators Inflamm. 2015;2015:529642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gillen AE, Gosalia N, Leir SH, et al. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem J. 2011;438:25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–240 [DOI] [PubMed] [Google Scholar]
- 27.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637–642 [DOI] [PubMed] [Google Scholar]
- 28.Flume PA, Robinson KA, O’Sullivan BP, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54:522–537 [PubMed] [Google Scholar]
- 29.Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749–1756 [DOI] [PubMed] [Google Scholar]
- 30.Lands LC, Milner R, Cantin AM, et al. High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J Pediatr. 2007;151:249–254 [DOI] [PubMed] [Google Scholar]
- 31.Mayer-Hamblett N, Kloster M, Rosenfeld M, et al. Impact of Sustained Eradication of New Pseudomonas aeruginosa Infection on Long-term Outcomes in Cystic Fibrosis. Clin Infect Dis. 2015;61:707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–951 [DOI] [PubMed] [Google Scholar]
- 33.Dasenbrook EC, Merlo CA, Diener-West M, et al. Persistent methicillin-resistant Staphylococcus aureus and rate of FEV1 decline in cystic fibrosis. Am J Respir Crit Care Med. 2008;178:814–821 [DOI] [PubMed] [Google Scholar]
- 34.Kappler M, Nagel F, Feilcke M, et al. Eradication of methicillin resistant Staphylococcus aureus detected for the first time in cystic fibrosis: A single center observational study. Pediatr Pulmonol. 2016;51:1010–1019 [DOI] [PubMed] [Google Scholar]
- 35.Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nichols DP, Happoldt CL, Bratcher PE, et al. Impact of azithromycin on the clinical and antimicrobial effectiveness of tobramycin in the treatment of cystic fibrosis. J Cyst Fibros. 2017;16:358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.•.Corvol H, Blackman SM, Boelle PY, et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun. 2015;6:8382.Genome wide association study identifying genetic modifiers of CF
- 38.Collaco JM, Cutting GR. Update on gene modifiers in cystic fibrosis. Curr Opin Pulm Med. 2008;14:559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collaco JM, McGready J, Green DM, et al. Effect of temperature on cystic fibrosis lung disease and infections: a replicated cohort study. PLoS One. 2011;6:e27784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephenson AL, Sykes J, Stanojevic S, et al. Survival Comparison of Patients With Cystic Fibrosis in Canada and the United States: A Population-Based Cohort Study. Ann Intern Med. 2017;166:537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.••.Drumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–1453.Study of genetic modifiers of CF lung disease identifying TGFβ as an important modifier.
- 42.••.Aschner Y, Downey GP. Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. American journal of respiratory cell and molecular biology. 2016;54:647–655.Review of physiologic and pathologic roles of TGFβ in the lung.
- 43.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morty RE, Konigshoff M, Eickelberg O. Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:607–613 [DOI] [PubMed] [Google Scholar]
- 45.Zhou L, Dey CR, Wert SE, et al. Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-beta 1 chimeric gene. Developmental biology. 1996;175:227–238 [DOI] [PubMed] [Google Scholar]
- 46.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmid P, Cox D, Bilbe G, et al. Differential expression of TGF beta 1, beta 2 and beta 3 genes during mouse embryogenesis. Development. 1991;111:117–130 [DOI] [PubMed] [Google Scholar]
- 48.Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421 [DOI] [PubMed] [Google Scholar]
- 50.Yi JJ, Barnes AP, Hand R, et al. TGF-beta signaling specifies axons during brain development. Cell. 2010;142:144–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.•.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811.Review of pharmacologic approaches to normalize TGFβ signaling.
- 52.Butz H, Racz K, Hunyady L, et al. Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharmacol Sci. 2012;33:382–393 [DOI] [PubMed] [Google Scholar]
- 53.Stolzenburg LR, Wachtel S, Dang H, et al. miR-1343 attenuates pathways of fibrosis by the TGF-beta receptors. Biochem J. 2016;473:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis BN, Hilyard AC, Lagna G, et al. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis BN, Hilyard AC, Nguyen PH, et al. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ding X, Park SI, McCauley LK, et al. Signaling between transforming growth factor beta (TGF-beta) and transcription factor SNAI2 represses expression of microRNA miR-203 to promote epithelial- mesenchymal transition and tumor metastasis. J Biol Chem. 2013;288:10241–10253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kong W, Yang H, He L, et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maeda H, Shiraishi A. TGF-beta contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearing mice. Journal of immunology. 1996;156:73–78 [PubMed] [Google Scholar]
- 59.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827 [DOI] [PubMed] [Google Scholar]
- 60.Khalil N, O’Connor RN, Flanders KC, et al. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. American journal of respiratory cell and molecular biology. 1996;14:131–138 [DOI] [PubMed] [Google Scholar]
- 61.Coker RK, Laurent GJ, Shahzeidi S, et al. Diverse cellular TGF-beta 1 and TGF-beta 3 gene expression in normal human and murine lung. Eur Respir J. 1996;9:2501–2507 [DOI] [PubMed] [Google Scholar]
- 62.Magnan A, Frachon I, Rain B, et al. Transforming growth factor beta in normal human lung: preferential location in bronchial epithelial cells. Thorax. 1994;49:789–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khalil N, O’Connor R, Unruh H, et al. Enhanced expression and immunohistochemical distribution of transforming growth factor-beta in idiopathic pulmonary fibrosis. Chest. 1991;99:65S–66S [DOI] [PubMed] [Google Scholar]
- 64.King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961 [DOI] [PubMed] [Google Scholar]
- 65.Bergeron A, Soler P, Kambouchner M, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003;22:69–76 [DOI] [PubMed] [Google Scholar]
- 66.Xu Y, Mizuno T, Sridharan A, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;l:e90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thannickal VJ, Lee DY, White ES, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389 [DOI] [PubMed] [Google Scholar]
- 68.White ES, Lazar MH, Thannickal VJ. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol. 2003;201:343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xia H, Bodempudi V, Benyumov A, et al. Identification of a cell-of-origin for fibroblasts comprising the fibrotic reticulum in idiopathic pulmonary fibrosis. Am J Pathol. 2014;184:1369–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim KK, Wei Y, Szekeres C, et al. Epithelial cell alpha3betal integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li M, Krishnaveni MS, Li C, et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121:277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proc Am Thorac Soc. 2012;9:130–136 [DOI] [PubMed] [Google Scholar]
- 73.Schaefer CJ, Ruhrmund DW, Pan L, et al. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.King TE Jr., Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092 [DOI] [PubMed] [Google Scholar]
- 75.Fleetwood K, McCool R, Glanville J, et al. Systematic Review and Network Meta-analysis of Idiopathic Pulmonary Fibrosis Treatments. J Manag Care Spec Pharm. 2017;23:S5–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Redington AE, Madden J, Frew AJ, et al. Transforming growth factor-beta 1 in asthr Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642–64 [DOI] [PubMed] [Google Scholar]
- 77.de Boer WI, van Schadewijk A, Sont JK, et al. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158:1951–1957 [DOI] [PubMed] [Google Scholar]
- 78.Pulleyn LJ, Newton R, Adcock IM, et al. TGFbeta1 allele association with asthma severity. Hum Genet. 2001;109:623–627 [DOI] [PubMed] [Google Scholar]
- 79.•.Ouyang Y, Miyata M, Hatsushika K, et al. TGF-beta signaling may play a role in the development of goblet cell hyperplasia in a mouse model of allergic rhinitis. Allergol Int. 2010;59:313–319.Study linking TGFβ signaling to goblet cell hyperplasia in a mouse model.
- 80.Le AV, Cho JY, Miller M, et al. Inhibition of allergen-induced airway remodeling in Smad 3- deficient mice. Journal of immunology. 2007;178:7310–7316 [DOI] [PubMed] [Google Scholar]
- 81.Chu HW, Balzar S, Seedorf GJ, et al. Transforming growth factor-beta2 induces bronchial epithelial mucin expression in asthma. Am J Pathol. 2004;165:1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. Journal of immunology. 2005;174:5774–5780 [DOI] [PubMed] [Google Scholar]
- 83.Undevia NS, Dorscheid DR, Marroquin BA, et al. Smad and p38-MAPK signaling mediates apoptotic effects of transforming growth factor-beta1 in human airway epithelial cells. American journal of physiology Lung cellular and molecular physiology. 2004;287:L515–524 [DOI] [PubMed] [Google Scholar]
- 84.Hashimoto S, Gon Y, Takeshita I, et al. Transforming growth Factor-beta1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med. 2001;163:152–157 [DOI] [PubMed] [Google Scholar]
- 85.Chen G, Khalil N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir Res. 2006;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scherf W, Burdach S, Hansen G. Reduced expression of transforming growth factor beta 1 exacerbates pathology in an experimental asthma model. Eur J Immunol. 2005;35:198–206 [DOI] [PubMed] [Google Scholar]
- 87.Hansen G, McIntire JJ, Yeung VP, et al. CD4(+) T helper cells engineered to produce latent TGF-beta1reverse allergen-induced airway hyperreactivity and inflammation. J Clin Invest. 2000;105:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Al-Alawi M, Hassan T, Chotirmall SH. Transforming growth factor beta and severe asthma: a perfect storm. Respir Med. 2014;108:1409–1423 [DOI] [PubMed] [Google Scholar]
- 89.Howell JE, McAnulty RJ. TGF-beta: its role in asthma and therapeutic potential. Curr Drug Targets. 2006;7:547–565 [DOI] [PubMed] [Google Scholar]
- 90.Vanscoy LL, Blackman SM, Collaco JM, et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:1036–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haston CK, Hudson TJ. Finding genetic modifiers of cystic fibrosis. N Engl J Med. 2005;353:1509–1511 [DOI] [PubMed] [Google Scholar]
- 92.••.Arkwright PD, Laurie S, Super M, et al. TGF-beta(l) genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax. 2000;55:459–462.Initial description linking TGFβ higher producing genotypes to accelerated lung function decline in CF.
- 93.Dunning AM, Ellis PD, McBride S, et al. A transforming growth factorbeta1 signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer Res. 2003;63:2610–2615 [PubMed] [Google Scholar]
- 94.Suthanthiran M, Li B, Song JO, et al. Transforming growth factor-beta1 hyperexpression in African-American hypertensives: A novel mediator of hypertension and/or target organ damage. Proc Natl Acad Sci USA. 2000;97:3479–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamada Y, Miyauchi A, Goto J, et al. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res. 1998;13:1569–1576 [DOI] [PubMed] [Google Scholar]
- 96.Grainger DJ, Heathcote K, Chiano M, et al. Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet. 1999;8:93–97 [DOI] [PubMed] [Google Scholar]
- 97.Luedecking EK, DeKosky ST, Mehdi H, et al. Analysis of genetic polymorphisms in the transforming growth factor-beta1 gene and the risk of Alzheimer’s disease. Hum Genet. 2000;106:565–569 [DOI] [PubMed] [Google Scholar]
- 98.Harris WT, Muhlebach MS, Oster RA, et al. Transforming growth factor-beta(l) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr Pulmonol. 2009;44:1057–1064 [DOI] [PubMed] [Google Scholar]
- 99.Harris WT, Muhlebach MS, Oster RA, et al. Plasma TGF-β1 in pediatric cystic fibrosis: potential biomarker of lung disease and response to therapy. Pediatr Pulmonol. 2011;46:688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bernardi DM, Ribeiro AF, Mazzola TN, et al. The impact of cystic fibrosis on the immunologic profile of pediatric patients. J Pediatr (Rio J). 2013;89:40–47 [DOI] [PubMed] [Google Scholar]
- 101.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23:146–158 [DOI] [PubMed] [Google Scholar]
- 103.Sun H, Harris WT, Kortyka S, et al. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS One. 2014;9–el06842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.••.Snodgrass SM, Cihil KM, Cornuet PK, et al. Tgf-β1inhibits Cftr biogenesis and prevents functional rescue of ΔF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS One. 2013;8:e63167.Description of TGFβ induced inhibition of CFTR biogenesis and impairment of VX-809 functional rescule of F508del CFTR in airway epithelial cells.
- 105.Howe KL, Wang A, Hunter MM, et al. TGFbeta down-regulation of the CFTR: a means to limit epithelial chloride secretion. Exp Cell Res. 2004;298:473–484 [DOI] [PubMed] [Google Scholar]
- 106.Farruk MLK, William TH. MicroRNA-145 Regulates TGF-?1 Mediated Down-Regulation of CFTR Expression in CF and Non-CF Airway Epithelia. A108 MICRO RNAS, RNA SEQ, LNCRNA: BIOLOGY AND FUNCTION. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2016. p. A2808–A2808 [Google Scholar]
- 107.Oglesby IK, Chotirmall SH, McElvaney NG, et al. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145, −223, and −494 is altered in ΔF508 cystic fibrosis airway epithelium. Journal of immunology. 2013;190:3354–3362 [DOI] [PubMed] [Google Scholar]
- 108.Tsuchiya M, Kalurupalle S, Kumar P, et al. RPTOR, a novel target of miR-155, elicits a fibrotic phenotype of cystic fibrosis lung epithelium by upregulating CTGF. RNA Biol. 2016;13:837–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kreda SM, Davis CW, Rose MC. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb Perspect Med. 2012;2:a009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ojiaku CA, Cao G, Zhu W, et al. TGF-beta1 Evokes Human Airway Smooth Muscle Cell Shortening and Hyperresponsiveness via Smad3. American journal of respiratory cell and molecular biology. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harris WT, Boyd JT, McPhail GL, et al. Constrictive Bronchiolitis in Cystic Fibrosis Adolescents with Refractory Pulmonary Decline. Ann Am Thorac Soc. 2016;13:2174–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.•.Harris WT, Kelly DR, Zhou Y, et al. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One. 2013;8:e70196.Description of TGFβ signaling associated with myofibroblast differentiation in CF lung tissue.
- 113.Courtney JM, Ennis M, Elborn JS. Cytokines and inflammatory mediators in cystic fibrosis. J Cyst Fibros. 2004;3:223–231 [DOI] [PubMed] [Google Scholar]
- 114.Brandes ME, Mai UE, Ohura K, et al. Type I transforming growth factor-beta receptors on neutrophils mediate chemotaxis to transforming growth factor-beta. Journal of immunology. 1991;147:1600–1606 [PubMed] [Google Scholar]
- 115.Bonfield TL, Panuska JR, Konstan MW, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–2118 [DOI] [PubMed] [Google Scholar]
- 116.Ranganathan SC, Parsons F, Gangell C, et al. Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax. 2011;66:408–413 [DOI] [PubMed] [Google Scholar]
- 117.Lee KY, Ho SC, Lin HC, et al. Neutrophil-derived elastase induces TGF-beta1 secretion in human airway smooth muscle via NF-kappaB pathway. American journal of respiratory cell and molecular biology. 2006;35:407–414 [DOI] [PubMed] [Google Scholar]
- 118.Chua F, Dunsmore SE, Clingen PH, et al. Mice lacking neutrophil elastase are resistant to bleomycin-induced pulmonary fibrosis. Am J Pathol. 2007;170:65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sanjabi S, Zenewicz LA, Kamanaka M, et al. Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr Opin Pharmacol. 2009;9:447–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuppner MC, McKillop-Smith S, Forrester JV. TGF-beta and IL-1 beta act in synergy to enhance IL-6 and IL-8 mRNA levels and IL-6 production by human retinal pigment epithelial cells. Immunology. 1995;84:265–271 [PMC free article] [PubMed] [Google Scholar]
- 121.Reinhold D, Bank U, Buhling F, et al. Transforming growth factor beta 1 inhibits interleukin-10 mRNA expression and production in pokeweed mitogen-stimulated peripheral blood mononuclear cells and T cells. J Interferon Cytokine Res. 1995;15:685–690 [DOI] [PubMed] [Google Scholar]
- 122.Jagels MA, Hugli TE. Mixed effects of TGF-beta on human airway epithelial-cell chemokine responses. Immunopharmacology. 2000;48:17–26 [DOI] [PubMed] [Google Scholar]
- 123.Wang WM, Zhang HD, Jin YM, et al. PPAR-gamma agonists inhibit TGF-beta1-induced chemokine expression in human tubular epithelial cells. Acta Pharmacol Sin. 2009;30:107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cantelli G, Crosas-Molist E, Georgouli M, et al. TGFBeta-induced transcription in cancer. Semin Cancer Biol. 2017;42:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kelley J, Kovacs EJ, Nicholson K, et al. Transforming growth factor-beta production by lung macrophages and fibroblasts. Chest. 1991;99:85S–86S [PubMed] [Google Scholar]
- 126.Reibman J, Meixler S, Lee TC, et al. Transforming growth factor beta 1, a potent chemoattractant for human neutrophils, bypasses classic signal-transduction pathways. Proc Natl Acad Sci U S A. 1991;88:6805–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Thomas BJ, Kan OK, Loveland KL, et al. In the Shadow of Fibrosis: Innate Immune Suppression Mediated by Transforming Growth Factor-beta. American journal of respiratory cell and molecular biology. 2016;55:759–766 [DOI] [PubMed] [Google Scholar]
- 128.Thomas BJ, Lindsay M, Dagher H, et al. Transforming growth factor-beta enhances rhinovirus infection by diminishing early innate responses. American journal of respiratory cell and molecular biology. 2009;41:339–347 [DOI] [PubMed] [Google Scholar]
- 129.McCann KL, Imani F. Transforming growth factor beta enhances respiratory syncytial virus replication and tumor necrosis factor alpha induction in human epithelial cells. J Virol. 2007;81:2880–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee J, Zhang L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell. 2015;6:26–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rumbaugh KP, Hamood AN, Griswold JA. Cytokine induction by the P. aeruginosa quo sensing system during thermal injury. J Surg Res. 2004;116:137–144 [DOI] [PubMed] [Google Scholar]
- 132.Feng L, Xiang Q, Ai Q, et al. Effects of Quorum Sensing Systems on Regulatory T Cells i Related Pseudomonas aeruginosa Biofilm Infection Rat Models. Mediators Inflamm. 2016;2016:4012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Winstanley C, Fothergill JL. The role of quorum sensing in chronic cystic fibrosis Pseudomonas aeruginosa infections. FEMS Microbiol Lett. 2009;290:1–9 [DOI] [PubMed] [Google Scholar]
- 134.LaFayette SL, Houle D, Beaudoin T, et al. Cystic fibrosis-adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci Adv. 2015;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.d’Angelo I, Conte C, La Rotonda MI, et al. Improving the efficacy of inhaled drugs in cystic fibrosis: challenges and emerging drug delivery strategies. Adv Drug Deliv Rev. 2014;75:92–111 [DOI] [PubMed] [Google Scholar]
- 136.Bals R, Weiner DJ, Wilson JM. The innate immune system in cystic fibrosis lung disease. J Clin Invest. 1999;103:303–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huaux F, Noel S, Dhooghe B, et al. Dysregulated proinflammatory and fibrogenic phenotype of fibroblasts in cystic fibrosis. PLoS One. 2013;8:e64341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Manzanares D, Krick S, Baumlin N, et al. Airway Surface Dehydration by Transforming Growth Factor beta (TGF-beta) in Cystic Fibrosis Is Due to Decreased Function of a Voltage-dependent Potassium Channel and Can Be Rescued by the Drug Pirfenidone. J Biol Chem. 2015;290:25710–25716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Salathe M, Baumlin N, Kis A, et al. Losartan Rescues Cystic Fibrosis (CF)-Related Mucociliary Dysfunction in CF Cells In Vitro and in a Novel CF Sheep Model in Vivo. American Journal of Respiratory and Critical Care Medicine. 2016;193:A5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Park IH, Park SJ, Cho JS, et al. Effect of simvastatin on transforming growth factor beta-1-induced myofibroblast differentiation and collagen production in nasal polyp-derived fibroblasts. Am J Rhinol Allergy. 2012;26:7–11 [DOI] [PubMed] [Google Scholar]
- 141.Mabalirajan U, Aich J, Agrawal A, et al. Mepacrine inhibits subepithelial fibrosis by reducing the expression of arginase and TGF-beta1 in an extended subacute mouse model of allergic asthma. American journal of physiology Lung cellular and molecular physiology. 2009;297:L411–419 [DOI] [PubMed] [Google Scholar]
- 142.Neuzillet C, Tijeras-Raballand A, Cohen R, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31 [DOI] [PubMed] [Google Scholar]
- 143.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942 [DOI] [PubMed] [Google Scholar]
- 144.Yarza R, Vela S, Solas M, et al. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front Pharmacol. 2015;6:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chopra P, Kanoje V, Semwal A, et al. Therapeutic potential of inhaled p38 mitogen-activated protein kinase inhibitors for inflammatory pulmonary diseases. Expert Opin Investig Drugs. 2008;17:1411–1425 [DOI] [PubMed] [Google Scholar]
- 146.Medina-Tato DA, Ward SG, Watson ML. Phosphoinositide 3-kinase signalling in lung disease: leucocytes and beyond. Immunology. 2007;121:448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Greene CM. MicroRNA Expression in Cystic Fibrosis Airway Epithelium. Biomolecules. 2013;3:157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cihil KM, Winschel J, Ranganathan S, et al. TGF-beta1 Blocks delF508-CFTR Rescue via MicroRNA-145. Pediatr Pulmonol. 2016;51:20326716396 [Google Scholar]
- 149.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222 [DOI] [PubMed] [Google Scholar]
- 150.Christopher AF, Kaur RP, Kaur G, et al. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhou Y, Lee JY, Lee CM, et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J Biol Chem. 2012;287:41991–42000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vallath S, Hynds RE, Succony L, et al. Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur Respir J. 2014;44:513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kabir F, Halloran B, Mazur M, et al. MicroRNA-145 Mediates TGF-beta Downregulation of CFTR in Airway Epithelial Cells. Pediatr Pulmonol. 2016;51:21726677806 [Google Scholar]
- 154.Stoltz DA, Meyerholz DK, Pezzulo AA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]