

Abstract
A next-generation reagent-controlled approach for the synthesis of 2,6-dideoxy and 2,3,6-trideoxy sugar donors in good yield and high β-selectivity is reported. The use of p-toluenesulfonyl chloride and potassium hexa-methyldisilazide (KHMDS) greatly simplifies deoxy-sugar glycoside construction, and can be used for gram-scale glycosylation reactions. The development of this approach and its application to the construction of β-linked deoxy-sugar oligosaccharides are described.
Keywords: carbohydrates, diastereoselectivity, glycosylation, oligosaccharides, synthetic methods
The field of glycoscience has received considerable attention over the past decade, due in large part to the recognition of the important roles carbohydrates play in biology and medicine. This has, in turn, led to calls for the development of new methods for the synthesis of oligosaccharides, with the intent to make the field more accessible to the boarder biomedical research community.[1] Among glycosidic linkages, the construction of β-linked 2-deoxy-sugars remains particularly challenging.[2] The development of diastereoselective synthetic approaches to these molecules is of interest owing to their prevalence in many bioactive natural products. Over 3400 bacterial natural products are glycosylated,[3] many of which possess oligosaccharides composed of β-linked 2,6-deoxy-hexopyranoses (Figure 1).[4] Furthermore, altering the composition of these oligosaccharides has been shown to dramatically affect the bio-activities of these natural products, including mitigating toxicity.[5] Thus, methods for the stereoselective construction of β-linked 2-deoxy-sugars have the potential to aid the development of next-generation therapeutics.[6]
Figure 1.
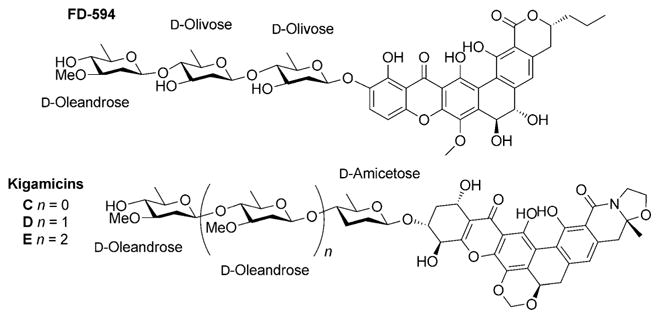
Naturally occurring 2,6-deoxy and 2,3,6-deoxy sugars.
The major challenge in the construction of β-linked 2,6-dideoxy sugars is the lack of functionality at the C-2 position, which precludes the use of neighboring group participation to control selectivity in glycosylation reactions.[7] Without such assistance, glycosylation reactions with deoxy-sugar donors frequently provide products as a mixture of α,β-anomers.[8] This lack of control has led many researchers to develop elegant approaches for the stereospecific installation of β-linked 2-deoxy glycosyl-linkages.[9] Indirect methods and de novo[10] approaches can provide excellent levels of selectivity, however, both approaches require post-glycosylation modification of the products to afford native structures. This drawback has led to renewed interest in developing methods for the direct synthesis of β-linked 2-deoxy-sugars where selectivity is independent of the nature of the coupling partners.[11] Despite these efforts, a universal selective glycosylation method that uses inexpensive and easy-to-handle reagents has yet to emerge.
As part of an ongoing program to develop a toolkit for selective reagent-controlled methods for 2-deoxy-sugar synthesis,[12] we have studied the utility of glycosyl sulfonates for the stereospecific construction of β-linkage targets.[13] Specifically, we have found that by matching the sulfonate leaving group ability with the 2-deoxy sugars reactivity, it is possible to generate species that react selectively through an SN2-like manifold.[14] During our attempts to apply our chemistry to oligo-saccharide synthesis, we encountered several limitations in our previous approaches. Specifically, the instability of reagents such as sulfonic anhydrides[15] led to problems with reproducibility and scalability. To address this issue, we initiated efforts to establish a method for glycosyl sulfonate generation that is more robust and reliable. To that end, we saw potential in examining the use of the inexpensive and easy to handle p-toluenesulfonyl chloride (TsCl) as a promoter for large-scale β-selective glycosylation reactions with 2-deoxy-sugars. Herein, we describe the evolution of this third-generation promotor system for β-selective glycosylation reactions and report its application to oligosaccharide synthesis.
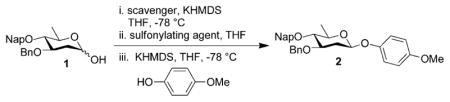
Initially, we were concerned that the chloride counterion generated during activation could react with the glycosyl tosylate intermediate, leading to the formation of an unreactive glycosyl chloride.[12a] To assess the utility of the reagent for glycoside construction, we carried out side-by-side glycosylations comparing p-toluenesulfonic anhydride (Ts2O) and TsCl in the reaction between donor 1 and p-methoxyphenol. Pleasingly, both reagents provided the β-glycoside 2 as a single diastereomer in 54 % and 56 % yield, respectively (Table 1). Increasing the scale of the reaction led to an increase in yield. Furthermore, we also found that superior results are obtained if the TsCl is recrystallized prior to use (Table 1, entry 3 vs. 4). Further studies revealed that the reaction could be performed on gram scale in the presence of either 2,4,6-tri-tert-butylpyrimidine (TTBP) or β-pinene[16] (Table 1 entries 8–10).
Table 1.
p -Toluenesulfonyl chloride screening with 1 and p-methoxyphenol.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Scavenger | Donor [g] | Acceptor [g] | Sulfonylating Agent | Yield [%][a] | β:α |
| 1[b] | TTBP | 0.14 | 0.03 | Ts2O | 54 | β-only |
| 2[b] | TTBP | 0.14 | 0.03 | TsCl | 56 | β-only |
| 3[b] | TTBP | 0.42 | 0.09 | TsCl | 70 | β-only |
| 4 | TTBP | 0.42 | 0.09 | TsCl | 90 | β-only |
| 5 | TTBP | 1 | 0.22 | TsCl | 93 | β-only |
| 6 | TTBP | 2.3 | 0.5 | TsCl | 97 | β-only |
| 7 | TTBP | 4.5 | 1 | TsCl | 96 | β-only |
| 8 | β-pinene | 0.14 | 0.03 | TsCl | 99 | β-only |
| 9 | β-pinene | 2.3 | 0.5 | TsCl | 97 | β-only |
| 10 | β-pinene | 4.5 | 1 | TsCl | 96 | β-only |
Yield of isolated product.
Sulfonylating agent used without recrystallization.
Nap = 2-naphthylmethyl; TTBP = 2,4,6-tri-tert-butylpyrimidine; Ts2O = p-toluenesulfonic anhydride; TsCl = p-toluenesulfonyl chloride.
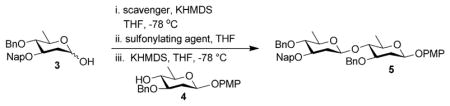
Having established that we could generate aryl glycosides with the reaction, we turned our attention to disaccharide formation. To this end, we evaluated the reaction between olivo-sides 3 and 4 (Table 2). On small scale, both TsCl and Ts2O (Table 2, entries 1 and 2) afforded products with excellent β-selectivity. Upon scale up, however, the yield dropped precipitously when Ts2O was used as the promoter (see the Supporting Information, Table S2.2.1). Increasing the activation time to 1 h, when TsCl was used as the promoter, led to a modest improvement in yield (Table 2, entry 2 vs. 3). On the other hand, increasing the reaction concentration resulted in an improved yield of the product, albeit with slight diminished β-selectivity (Table 2, entry 4). We also considered alternative proton scavengers, 2,6-di-tert-butylpyridine (DTBP), and β-pinene. Both species increased the reaction yield, but eroded the selectivity further (Table 2, entries 5–6). This screening also revealed that a proton scavenger was necessary, as there was a marked decrease in selectivity when the reaction was carried out in the absence of one (Table 2, entry 7). Pleasingly, we found that pre-forming the reaction on close to gram scale afforded 5 in good yield and selectivity, demonstrating the advantages of the current protocol over our previous generation-approach (Table 2, entry 8).
Table 2.
Reaction optimization for disaccharide 5.
| |||||||
|---|---|---|---|---|---|---|---|
| Scavenger | Sulfonylating Agent | Activation time [h] | [Activation] [°] | [Glycosylation] [°] | Yield [%][a] | β:α[b] | |
| 1 | TTBP | Ts2O | 0.5 | 0.051 | 0.040 | 76 | β-only |
| 2 | TTBP | TsCl | 0.5 | 0.050 | 0.040 | 50 | β-only |
| 3 | TTBP | TsCl | 1 | 0.053 | 0.041 | 64 | β-only |
| 4 | TTBP | TsCl | 1 | 0.074 | 0.053 | 74 | 18:1 |
| 5 | β-pinene | TsCl | 1 | 0.074 | 0.053 | 78 | 15:1 |
| 6 | DTBP | TsCl | 1 | 0.074 | 0.053 | 71 | 16:1 |
| 7 | — | TsCl | 1 | 0.053 | 0.041 | 80 | 6.4:1 |
| 8[c] | TTBP | TsCl | 1 | 0.075 | 0.054 | 60 | β-only |
Yield of isolated product.
The β:α ratio of the product after isolation.
820 mg donor scale and 500 mg acceptor scale.
DTBP = 2,6-di-tert-butylpyridine; [Activation] = Donor concentration after addition of sulfonylating agent; [Glycosylation] = Donor concentration after addition of metalated acceptor.
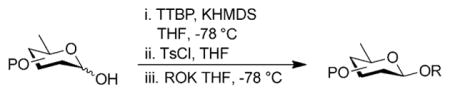
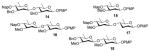
With optimized conditions in hand, we next evaluated the scope of the reaction with a variety of donors and acceptors (Figure 2). Glycosylation of 1 with 4 under optimal conditions afforded disaccharide 14 as a single isomer in 75 % yield (Table 3). Furthermore, by shortening the activation time to 15 min and increasing the donor to acceptor ratio from 1.5:1 to 2:1 we were able to obtain β-oleandrosides 16, 17, and 18 in moderate to good yields (Table 3, entries 3–5). Again, the reactions could be scaled up without any loss in selectivity (Table 3, entries 6–8) Finally, it is interesting to note that the acceptor can be treated with base after addition to the activated donor without appreciable loss of selectivity, further illustrating the user-friendly nature of the approach (Table 3, entry 9 and 10).
Figure 2.
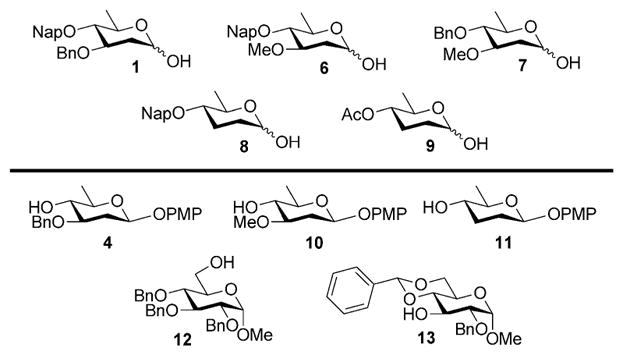
Donors and acceptors used for substrate screening.
Table 3.
Glycosylation reactions with various glycosyl donors and acceptors.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Donor | R′OK | Activation time [h] | Product | Yield [%][b] | β:α |
| 1 | 1 | 4 | 1 | 14 | 75 | β-only |
| 2 | 6 | PMP | 0.5 | 15 | 87 | β-only |
| 3[c] | 6 | 10 | 0.25 | 16 | 75 | β-only |
| 4[c] | 6 | 11 | 0.25 | 17 | 55 | β-only |
| 5 | 7 | 10 | 1 | 18 | 70 | β-only |
| 6[c,d] | 1 | 4 | 1 | 14 | 66 | β-only |
| 7[d] | 6 | 10 | 0.25 | 16 | 62 | β-only |
| 8[d] | 6 | 11 | 0.4 | 17 | 50 | β-only |
| 9[e] | 6 | 10 | 0.25 | 16 | 66 | β-only |
| 10[f] | 6 | 10 | 0.25 | 16 | 48 | β-only |
| ||||||
Donor:acceptor ratio 2:1 unless otherwise noted.
Yield of isolated product.
Donor:acceptor ratio 1.5:1.
500 mg acceptor scale.
Added KHMDS immediately after addition of acceptor.
Added KHMDS 3 h after addition of acceptor.
PMP = p-methoxyphenyl.
We next turned our attention to the more reactive 2,3,6-tri-deoxy donor, 8. Under our optimized conditions, the reaction proceeded in high yield, but with attenuated β-selectivity. Interestingly, scaling the reaction up to 3 g scale proceeded in much better selectivity (16:1 β:α, Table 4, entries 1 and 2). Reasoning that a bulkier and less reactive sulfonate would improve selectivity through the generation of a more stable intermediate, we next examined triisopropylbenzenesulfonyl,[17] chloride (TrisylCl) as a promoter. However, this reagent afforded the product with diminished selectivity (Table 4, entry 3). We next examined donor 9 in the reaction to see if a less armed sugar would improve selectivity. Contrary to our expectations, the activation of disarmed donor 9 with TsCl resulted in a reversal of selectivity (1:2.2 β:α) (Table 4, entry 4). In an effort to improve α-selectivity, we next examined using TrisylCl to activate 9, as this promoter decreased β-selectivity with 8. Indeed, activation of 9 with TrisylCl led to the production of 20 as a single α-anomer in 84 % yield (Table 4, entry 5). These latter conditions could also be used to produce disaccharides in moderate yields (Table 4, entries 6 and 7).
Table 4.
Glycosylation of 2,3,6-trideoxy sugars 8 and 9 with various acceptors.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Sulfonylating Agent | Product | Yield [%][a] | β:α[b] |
| 1 | 8 | PMP | TsCl | 19 | 80 | 8:1 |
| 2 | 8 | PMP | TrisylCl | 19 | 42 | 5:1 |
| 3 | 9 | PMP | TsCl | 20 | 60 | 1:2.2 |
| 4 | 9 | PMP | TrisylCl | 20 | 84 | α-only |
| 5 | 9 | 12 | TrisylCl | 21 | 27 | α-only |
| 6 | 9 | 13 | TrisylCl | 22 | 41 | α-only |
|
| ||||||
Yield of isolated product.
Selectivity determined by 1H NMR spectroscopy.
3 g donor scale and 930 mg acceptor scale.
TrisylCl = triiso-propylbenzenesulfonyl chloride.
We next turned to variable-temperature (VT) NMR to study the mechanism of the reaction. Activating 6 in [D8]THF with TsCl at −78 °C, led to the formation of a species with an anomeric proton signal at 6.0 ppm indicative of an α-glycosyl sulfonate[12a,e,13a,18] (see Supporting Information, Section S5.1). The 1H–13C heteronuclear single-quantum correlation (HSQC) NMR spectrum correlates the 1H NMR spectrum singlet at 6.0 ppm to a 13C NMR signal at 101.5 ppm, which is again consistent with an anomeric sulfonate. The tosylate species was stable at temperature up to −50 °C, above which it readily decomposed to the glycal (see Supporting Information, Section S5.2). This supports a mechanism where the donor is converted into an α-glycosyl sulfonate intermediate, which reacts predominantly through an SN2-like manifold.[12e]
Having established the utility of the reaction in disaccharide construction, we turned our attention to using the chemistry for production of larger targets. We first examined the synthesis of the trisaccharide of FD-594 (Figure 1). To this end, selective removal of the naphthylmethyl ether on 14 afforded disaccharide 23 (Scheme 1). In the subsequent glycosylation, we found that we had to increase the reaction concentration to 0.07 °, (see Supporting Information) however, we were pleased to find that the reaction between 6 and 23, afforded trisaccharide 24 as a single β-anomer in 75 %.
Scheme 1.
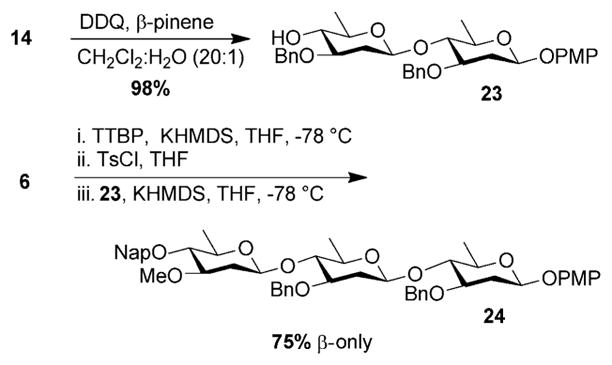
Synthesis of FD-594 trisaccharide 24.
We next focused on the construction of the tetrasaccharide fragment of kigamicin E, since this molecule also contains a labile β-linked 2,3,6-trideoxy-sugar (Figure 1). Initially, we examined a convergent [2+2] assembly of kigamicin E. To this end, disaccharide 16 was cleanly converted to donor 25 by removing the anomeric PMP group with cerium ammonium nitrate (CAN).[19] Similarly, acceptor 26 was generated from 17 using a combination of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and β-pinene (Scheme 2). Much to our disappointment, convergent glycosylation of 25 with 26 afforded 27 as a mixture of anomeric products in 50 % yield.[20] Reasoning that the loss in selectivity may be due to the diminished reactivity of the larger donor, we next examined a linear approach to the target. This would involve first synthesizing the trisaccharide of kigamicin D. This latter species could then be elongated to afford the tetrasaccharide of kigamicin E (Scheme 3). In the synthetic direction, activating donor 6 with p-toluenesulfonyl chloride followed by treatment with acceptor 26 afforded 28 as a single β-anomer in 58 % yield (Scheme 3). Removal of the naphthylmethyl ether from 28 was uneventful, revealing 29 in good yield. Finally, activation of 6 with potassium hexamethyl-disilazide (KHMDS) and TsCl followed by treatment with the potassium salt of 29 afforded tetrasaccharide 27 as a single isomer in 50 % yield (Scheme 3).
Scheme 2.
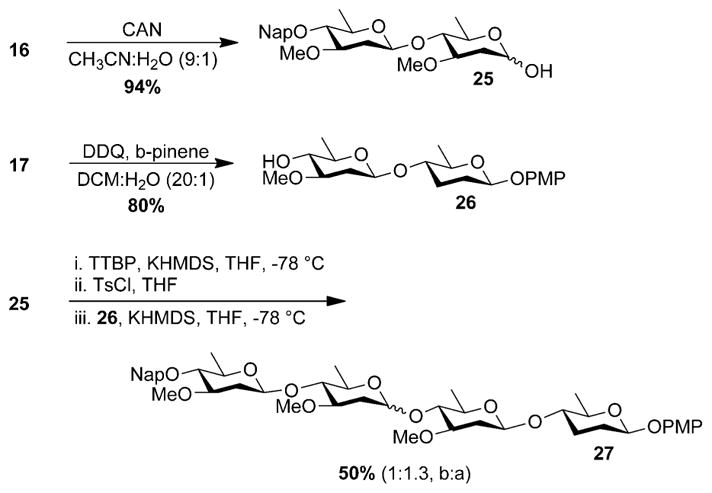
Convergent synthesis of kigamicin E tetrasaccharide 27.
Scheme 3.
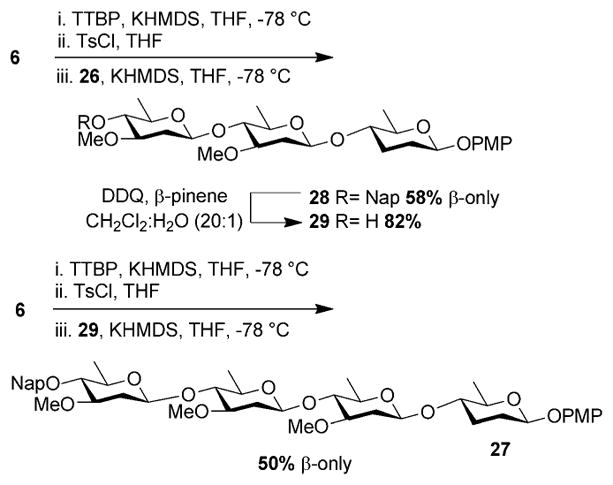
Linear synthesis of kigamicin D trisaccharide 28 and E tetraaccharide 27.
In summary, we have developed an improved third-generation reagent-controlled approach for deoxy-sugar oligosaccharide synthesis. Using p-toluenesulfonyl chloride to activate di-deoxy-sugar donors results in the formation of a species that reacts with acceptors in excellent to near perfect β-selectivity. Furthermore, highly sensitive 2,3,6-trideoxy-sugar substrates are also viable coupling partners using this current protocol. These latter species can be activated to react in either a highly α- or β-selective fashion, depending on the protecting group at C-4 and promoter (TrisylCl vs. TsCl) used in the reaction. Importantly, the chemistry is also readily scalable, and can be carried out using multi-gram quantities of donor. VT NMR studies revealed that the reaction proceeds through the formation of an α-glycosyl tosylate, which presumably reacts through an SN2-like manifold with nucleophiles to afford β-linked products. The utility and robustness of the protocol was demonstrated in the stereospecific construction of tri- and tetrasaccharides found in the bioactive natural products FD-594 and kigamicin D and E. The application of this chemistry to more complex systems is currently under investigation in our laboratory.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (NSF 1300334, CHE-1566233) and National Institutes of Health (R01-GM115779 and U01-GM120414) for generous financial support.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/chem.201800736.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.National Research Council of the National Academies. Transforming Glycoscience: a Roadmap for the Future. National Academies Press (US); Washington (DC): 2012. [PubMed] [Google Scholar]
- 2.Bennett CS. Selective Glycosylations. Wiley-VCH; Weinheim: 2017. [Google Scholar]
- 3.Elshahawi SI, Shaaban KA, Kharel MK, Thorson JS. Chem Soc Rev. 2015;44:7591–7697. doi: 10.1039/c4cs00426d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Henkel T, Rohr J, Beale JM, Schwenen L. J Antibiot. 1990;43:492–503. doi: 10.7164/antibiotics.43.492. [DOI] [PubMed] [Google Scholar]; b) Kondo K, Eguchi T, Kakinuma K, Mizoue K, Qiao YF. J Antibiot. 1998;51:288–295. doi: 10.7164/antibiotics.51.288. [DOI] [PubMed] [Google Scholar]; c) Kunimoto S, Lu J, Esumi H, Yamazaki Y, Kinoshita N, Honma Y, Hamada M, Ohsono M, Ishizuka M, Takeuchi T. J Antibiot. 2003;56:1004– 1011. doi: 10.7164/antibiotics.56.1004. [DOI] [PubMed] [Google Scholar]
- 5.a) Langenhan JM, Peters NR, Guzei IA, Hoffmann FM, Thorson JS. Proc Natl Acad Sci USA. 2005;102:12305–12310. doi: 10.1073/pnas.0503270102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Langenhan JM, Griffith BR, Thorson JS. J Nat Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]; c) Daniel PT, Koert U, Schuppan J. Angew Chem Int Ed. 2006;45:872–893. doi: 10.1002/anie.200502698. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:886–908. [Google Scholar]; d) Iyer AKV, Zhou M, Azad N, Elbaz H, Wang L, Rogalsky DK, Rojanasakul Y, O’Doherty GA, Langenhan JM. ACS Med Chem Lett. 2010;1:326– 330. doi: 10.1021/ml1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Rohr J, Thiericke R. Nat Prod Rep. 1992;9:103–135. doi: 10.1039/np9920900103. [DOI] [PubMed] [Google Scholar]; b) Weymouth-Wilson AC. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]; c) McCranie EK, Bachmann BO. Nat Prod Rep. 2014;31:1026– 1042. doi: 10.1039/c3np70128j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Zhu X, Schmidt RR. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2009;121:1932–1967. [Google Scholar]; b) Yang Y, Zhang X, Yu B. Nat Prod Rep. 2015;32:1331– 1355. doi: 10.1039/c5np00033e. [DOI] [PubMed] [Google Scholar]
- 8.Cumpstey I. Org Biomol Chem. 2012;10:2503–2508. doi: 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]
- 9.For reviews see: Marzabadi CH, Franck RW. Tetrahedron. 2000;56:8385–8417.Hou D, Lowary TL. Carbohydr Res. 2009;344:1911–1940. doi: 10.1016/j.carres.2009.07.013.Li Z, Ding N, Zhang W, Wang P, Li M, Li Y. Chin J Org Chem. 2012;32:1812–1826.Borovika A, Nagorny P. J Carbohydr Chem. 2012;31:255–283.Zeng J, Xu Y, Wang H, Meng L, Wan Q. Sci China Chem. 2017;60:1162– 1179.
- 10.a) McDonald FE, Reddy KS. J Organomet Chem. 2001;617:444–452. [Google Scholar]; b) Zhou M, O’Doherty GA. Org Lett. 2008;10:2283– 2286. doi: 10.1021/ol800697k. [DOI] [PubMed] [Google Scholar]
- 11.a) Lam SN, Gervay-Hague J. Org Lett. 2003;5:4219–4222. doi: 10.1021/ol035705v. [DOI] [PubMed] [Google Scholar]; b) Kaneko M, Herzon SB. Org Lett. 2014;16:2776–2779. doi: 10.1021/ol501101f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhu D, Baryal KN, Adhikari S, Zhu J. J Am Chem Soc. 2014;136:3172–3175. doi: 10.1021/ja4116956. [DOI] [PubMed] [Google Scholar]; d) Baryal K, Zhu J. Synlett. 2014;25:308–312. [Google Scholar]; e) Ruei JH, Venukumar P, Ingle AB, Mong KKT. Chem Commun. 2015;51:5394–5397. doi: 10.1039/c4cc08465a. [DOI] [PubMed] [Google Scholar]; f) Zhang W, Luo X, Wang Z, Zhang J. J Carbohydr Chem. 2016;35:315–325. [Google Scholar]; g) D’Angelo KA, Taylor MS. J Am Chem Soc. 2016;138:11058–11066. doi: 10.1021/jacs.6b06943. [DOI] [PubMed] [Google Scholar]; h) Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN. Science. 2017;355:162– 166. doi: 10.1126/science.aal1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Nogueira JM, Nguyen SH, Bennett CS. Org Lett. 2011;13:2814–2817. doi: 10.1021/ol200726v. [DOI] [PubMed] [Google Scholar]; b) Nogueira JM, Issa JP, Chu AHA, Sisel JA, Schum RS, Bennett CS. Eur J Org Chem. 2012:4927–4930. [Google Scholar]; c) Nogueira JM, Bylsma M, Bright DK, Bennett CS. Angew Chem Int Ed. 2016;55:10088–10092. doi: 10.1002/anie.201605091. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2016;128:10242–10246. [Google Scholar]; d) Issa JP, Lloyd D, Steliotes E, Bennett CS. Org Lett. 2013;15:4170–4173. doi: 10.1021/ol4018547. [DOI] [PubMed] [Google Scholar]; e) Issa JP, Bennett CS. J Am Chem Soc. 2014;136:5740– 5744. doi: 10.1021/ja500410c. [DOI] [PubMed] [Google Scholar]
- 13.For earlier studies on glycosyl sulfonates see: Eby R, Schuerch C. Carbohydr Res. 1974;34:79–90.Koto S, Hamada Y, Zen S. Chem Lett. 1975Maroussek V, Lucas TJ, Wheat PE, Schuerch C. Carbohydr Res. 1978;60:85–96.Srivastava VK, Schuerch C. Carbohydr Res. 1980;79:C13–C16.Koto S, Sato T, Morishima N, Zen S. Bull Chem Soc Jpn. 1980;53:1761–1762.Srivastava VK, Schuerch C. J Org Chem. 1981;46:1121–1126.Nguyen HM, Chen Y, Duron SG, Gin DY. J Am Chem Soc. 2001;123:8766–8772. doi: 10.1021/ja015968p.Boebel TA, Gin DY. Angew Chem Int Ed. 2003;42:5874–5877. doi: 10.1002/anie.200352761.Angew Chem. 2003;115:6054–6057.Boebel TA, Gin DY. J Org Chem. 2005;70:5818–5826. doi: 10.1021/jo050294c.Frihed TG, Bols M, Pedersen CM. Chem Rev. 2015;115:4963– 5013. doi: 10.1021/cr500434x.
- 14.Krumper JR, Salamant WA, Woerpel KA. Org Lett. 2008;10:4907–4910. doi: 10.1021/ol8019956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Field L, McFarland JW, Johnson WS, Hindersinn RR, Fisher AG. Org Synth. 1956;36:91–94. [Google Scholar]
- 16.a) Gu X, Chen L, Wang X, Liu X, You Q, Xi W, Gao L, Chen G, Chen YL, Xiong B, Shen J. J Org Chem. 2014;79:1100–1110. doi: 10.1021/jo402551x. [DOI] [PubMed] [Google Scholar]; b) Chen G, Yin Q, Yin J, Gu X, Liu X, You Q, Chen YL, Xiong B, Shen J. Org Biomol Chem. 2014;12:9781– 9785. doi: 10.1039/c4ob01807a. [DOI] [PubMed] [Google Scholar]
- 17.Paleos CM, Varveri FS, Gregoriou GA. J Org Chem. 1974;39:3594–3595. [Google Scholar]
- 18.a) Kitowski A, Jiménez-Moreno E, Salvadó M, Mestre J, Castill+n S, Jiménez-Osés G, Boutureira O, Bernardes GJL. Org Lett. 2017;19:5490–5493. doi: 10.1021/acs.orglett.7b02886. [DOI] [PubMed] [Google Scholar]; b) van der Vorm S, Overkleeft HS, van der Marel GA, Codée JDC. J Org Chem. 2017;82:4793– 4811. doi: 10.1021/acs.joc.7b00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cattaneo V, Oldrini D, Corrado A, Berti F, Adamo R. Org Chem Front. 2016;3:753–758. [Google Scholar]
- 20.Similar loss of selectivity was seen during a test glycosylation of 25 with acceptor 10 (see Supporting Information, Section S4).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.