

SUMMARY
Herpes simplex virus-1 (HSV-1) establishes infections in humans and mice but some non-human primates exhibit resistance via unknown mechanisms. Innate immune recognition pathways are highly conserved but are pivotal in determining susceptibility to DNA virus infections. We report that variation of a single amino acid residue in the innate immune sensor cGAS determines species-specific inactivation by HSV-1. The HSV-1 UL37 tegument protein deamidates human and mouse cGAS. Deamidation impairs the ability of cGAS to catalyze cGAMP synthesis, which activates innate immunity. HSV-1 with deamidase-deficient UL37 promotes robust antiviral responses and is attenuated in mice, in a cGAS- and STING-dependent manner. Mutational analyses identified a single asparagine in human and mouse cGAS that is not conserved in many non-human primates. This residue underpins UL37-mediated cGAS deamidation and species permissiveness of HSV-1. Thus, HSV-1 mediates cGAS deamidation for immune evasion and exploits species sequence variation to disarm host defenses.
In Brief
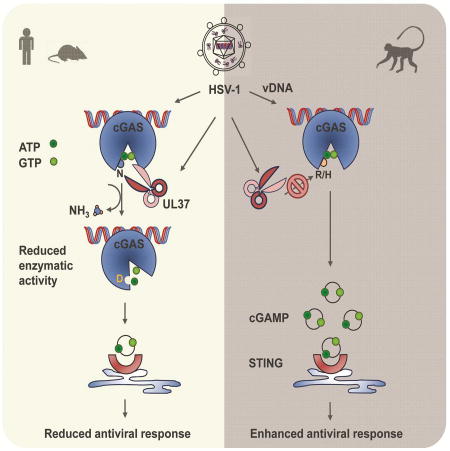
Natural sequence variations in immune factors may enable viral pathogens to evade or dismantle host defenses. Zhang et al report herpes simplex virus-1 exploits species-specific variation of a single amino acid residue in cGAS for deamidation, thereby inactivating innate defense in susceptible hosts.
INTRODUCTION
Upon infection, eukaryotic cells immediately respond with innate immune activation to defeat the invading pathogen. Along with other DNA sensors, cyclic GMP-AMP (cGAMP) synthase (cGAS) plays a pivotal role to detect double-stranded (ds) DNA in the cytosol (Li et al., 2013; Sun et al., 2013). Upon binding dsDNA, cGAS catalyzes the synthesis of cGAMP (Wu et al., 2013) that induces the dimerization and activation of the ER-anchored STING (also known as MITA) (Ishikawa and Barber, 2008; Zhong et al., 2008). STING then activates IκB kinase (IKK kinase) and TANK-binding kinase 1 (TBK-1) that further activate the transcription factors nuclear factor κB (NF-κB) and interferon regulatory factor 3 (IRF3) to induce inflammatory cytokines and interferons (IFN).
Though key components of innate immune pathways are conserved and species-specific difference has been linked to various RNA viruses (Aguirre et al., 2012; Meyerson et al., 2017; Patel et al., 2012), the importance of species-specific variation with regard to DNA viruses remains largely unexplored. As one family of the most successful pathogens, herpesviruses have evolved numerous intricate strategies to manipulate, evade and exploit host immune response (Ma and Damania, 2016). Coevolving with their hosts, herpesviruses are expected to leave footprint on host immune system via an arms race between host immune defense and viral evasion thereof. We have characterized herpesvirus tegument proteins that function as either authentic deamidase or pseudo-enzymes to deamidate RIG-I, thus evading innate immune activation induced by double-stranded RNA (dsRNA) (He et al., 2015; Zhao et al., 2016b). Considering the pivotal role of cGAS in sensing viral DNA, we hypothesize that cGAS might be targeted by viral evasion mechanism.
Despite the fact that cGAS is primarily regulated by dsDNA, additional regulatory mechanisms must exist to ensure physiological homeostasis, e.g., responding to DNA pathogens effectively while winding down in a timely manner to prevent collateral damage. Surprisingly, our understanding in cGAS regulation is in its infancy at best. Previous work has shown that post-translational modifications, including phosphorylation, sumolyation and glutamylation, modulate cGAS enzyme activity directly or indirectly (Cui et al., 2017; Hu et al., 2016; Seo et al., 2015; Xia et al., 2016). Investigating the evasion mechanism of herpes simplex virus 1 (HSV-1) conveyed by the UL37 protein deamidase, we discovered that human and mouse cGAS proteins, but not nonhuman primate cGAS, are targeted for deamidation by UL37. Biochemical and mutational analysis identified a single asparagine residue within the activation loop of cGAS that endowed the deamidation of mouse and human cGAS by UL37 to facilitate HSV-1 lytic replication. Consequently, the deamidase-deficient HSV-1 more robustly induced antiviral cytokines and was highly attenuated in mice compared to wild-type HSV-1, in a cGAS- and STING-dependent manner. These findings uncover an intricate immune evasion mechanism whereby a viral pathogen exploits natural sequence variation of innate immune components to dismantle host defense.
RESULTS
The UL37 deamidase antagonizes the cGAS-STING pathway in mice and primary macrophages
We previously reported that the UL37 tegument protein of HSV-1 deamidates RIG-I to avoid dsRNA-induced innate immune activation (Zhao et al., 2016b). Recombinant HSV-1 carrying the deamidase-inactivating UL37C819S mutation (HSV-1 UL37C819S) more robustly induced antiviral cytokines than HSV-1 containing UL37 wild-type (HSV-1 UL37WT), while they replicated comparatively in VERO cells that are defective in type I interferon production (Figure S1A). To determine whether the deamidase activity of UL37 is important for HSV-1 infection in vivo, we infected BL6 mice with HSV-1. Approximately 50% mice developed ataxia and paralysis and succumbed to HSV-1 UL37WT, while all mice survived from HSV-1 UL37C819S infection (Figure 1A). We also found that HSV-1 UL37C819S induced significantly elevated levels of antiviral cytokines (including IFN-α, IFN-β, IL-6 and CCL5) in the serum than HSV-1 UL37WT (Figure 1B). Conversely, the viral load of HSV-1 UL37C819S in the brain was ~3% of that of HSV-1 UL37WT (Figure 1C). To determine the role of cGAS in innate immune response, we infected cGAS-deficient mice with HSV-1. Remarkably, cGAS−/− mice succumbed to both viruses with nearly identical kinetics (Figure 1D). Consistent with this result, equally high loads of HSV-1 UL37WT and HSV-1 UL37C819S were detected in the brain of infected mice at 3 days post-infection (Figure 1E). Loss of cGAS greatly reduced the cytokine production triggered by HSV-1 and both viruses induced similar levels of residual cytokines, including IFN-β, IFN-α, IL-6 and CCL5 (Figure 1F, compared to Figure 1B). Furthermore, STING−/− mice infected with both HSV-1 demonstrated the same survival kinetics (Figure S1B). Cytokine production and lytic replication of both viruses in the brain of STING−/− mice essentially phenocopied those of cGAS−/− mice (Figure S1C and S1D).
Figure 1. HSV-1 carrying deamidase-deficient UL37C718S more robustly induces cytokine production than wild-type HSV-1 in vivo.

(A–C) Age- and gender-matched BL6 mice were infected with HSV-1 UL37 wild-type (WT) or HSV-1 UL37C819S (C819S) (5 × 107 PFU). (A) Mouse survival was recorded and shown in percentage over time (n=9). (B) Blood was collected at 8 hours post-infection (hpi) and indicated cytokines in sera were determined by ELISA (n=5). (C) Mice were sacrificed at 3 days post-infection (dpi) and the viral genome copy number in the brain was determined by real-time PCR analysis (n=5).
(D–F) cGAS−/− mice were infected with HSV-1 UL37 wild-type (WT) or HSV-1 UL37C819S (UL37C819S) (5 × 107 PFU). (D) Mouse survival was recorded over time (n=10). (E) Virus titer in the brain (at 3 dpi) and (F) cytokines in the sera (at 8 hpi) were determined (n=5) by plaque assay and ELISA, respectively.
Each value is the mean + s.d. of the results of three independent experiments (B, C, E, F). Statistical analysis was performed by log-rank test for (A, D) and unpaired t-test for (B, C, E, F); *P<0.05, **P<0.01, ***P<0.005. Data are representative of at least two independent experiments (A and D).
See also Figure S1.
We previously reported that UL37 deamidates RIG-I to avoid dsRNA-induced innate immune activation (Zhao et al., 2016b). To discern the contribution of RIG-I and cGAS to host defense against HSV-1, we generated primary bone marrow-derived macrophages (BMDMs) from WT, MAVS−/− and cGAS−/− mice and examined innate immune response upon infection. We observed that HSV-1 UL37C819S infection induced much higher levels of expression of immune genes (Ifnb1, Isg56, Ccl5 and Cxcl10) in WT BMDMs than HSV-1 UL37WT (Figure S1E–S1I). Consistent with previous studies (Chiang et al., 2018; Zhao et al., 2016b), MAVS deficiency reduced the expression of immune genes induced by HSV-1. Remarkably, loss of cGAS abolished HSV-1-induced antiviral gene expression in BMDMs. MAVS- and cGAS-deficiency restored HSV-1 UL37C819S replication to comparable levels of HSV-I UL37WT in BMDMs (Figure S1J). These results collectively support the conclusion that the UL37 deamidase antagonizes both the RIG-I- and cGAS-mediated nucleic acid sensing pathways.
The UL37 deamidase antagonizes cGAS in monocytes
To delineate the role of UL37 in evading innate immune defense, we analyzed host gene expression of human monocyte THP-1 cells infected with HSV-1 UL37WT or HSV-1 UL37C819S by RNA sequencing. Both viruses up-regulated the expression of a broad spectrum of host defense genes in THP-1 cells, with more pronounced effect by HSV-1 UL37C819S (Figure 2A). HSV-1 UL37WT and HSV-1 UL37C819S induced the expression of 1198 and 2701 genes, respectively (Figure 2A). Among the 1198 genes induced by HSV-1 UL37WT, 1122 were up-regulated by HSV-1 UL37C819S. Comparative analysis involving gene ontology and ingenuity pathway classified these overlapped set of genes to host response, particularly innate immune response entailing interferon and cytokine signaling (Figure 2B and S2A–S2D). Those genes that were up-regulated only by HSV-1 UL37C819S also belong to host innate immune response pathways, indicating the specificity of UL37 in targeting host inflammatory pathways. To validate these results, we performed real-time PCR analysis of representative antiviral effectors (including IFNB1, ISG56, CXCL10, MX1, IFIT3 and IL6) and found that HSV-1 UL37C819S virus induced ~5–10-fold higher expression of these genes than HSV-1 UL37WT in THP-1 cells (Figure 2C and S3A). Moreover, HSV-1 UL37C819S induced significantly higher levels of secreted IFN-β than HSV-1 UL37WT in THP-1 cells (Figure 2D). Furthermore, HSV-1 UL37C819S more robustly activated the IRF-IFN pathway than HSV-1 UL37WT in THP-1 cells, as evidenced by the phosphorylation (and activation) of TBK-1 (Ser172) and IRF3 (Ser396) (Figure S3B). Thus, HSV-1 UL37C819S more robustly induces innate immune activation than HSV-1 UL37WT in THP-1 cells.
Figure 2. The deamidase activity of UL37 is required to prevent innate immune activation by HSV-1.

(A, B) Human THP-1 monocytes were infected with HSV-1 UL37WT or HSV-1 UL37C819S at MOI=5. Total RNA was extracted and subjected to RNA sequencing analysis. (A) Venn diagram indicates the number of genes that are commonly and differentially upregulated by HSV-1 UL37WT and HSV-1 UL37C819S (FDR <= 0.05, fold change >= 2). (B) Representative genes of the interferon pathway were shown.
(C, D) Human THP-1 cells were infected as described in (A). The expression of inflammatory cytokines was determined by real-time PCR at the indicated time points after infection (C). Medium was collected at 16 hours post-infection (hpi) and IFN-β was determined by ELISA (D).
(E) cGAS wild-type and knockout L929 mouse fibroblasts were infected with HSV-1 WT and HSV-1 UL37C819S at MOI=5. L929 cells were harvested at 6 hpi and the expression of indicated cytokine genes was analyzed by real-time PCR.
(F) THP-1 monocytes were infected as described in (A). cGAMP was extracted and quantified using permeabilized THP-1 reporter cells.
Each value is the mean + s.d. of the results of three independent experiments (C–F). Statistical analysis was performed by unpaired t-test for (C–F); *P<0.05, ***P<0.005. Data are representative of two independent experiments (A and B).
See also Figure S2 and S3.
Given the pivotal role of cGAS in DNA sensing, we assessed whether cGAS is required for elevated immune response against HSV-1 UL37C819S. We infected wild-type and cGAS−/− L929 fibroblasts with HSV-1 and determined antiviral gene expression. In wild-type L929 fibroblasts, HSV-1 UL37C819S more robustly induced Isg56 and Cxcl10 expression than HSV-1 UL37WT, recapitulating the phenotype that was observed in human THP-1 monocytes (Figure 2E) and in mice (Figure 1B). Remarkably, loss of cGAS abolished Isg56 and Cxcl10 expression in response to both viruses (Figure 2E). Next, we determined intracellular cGAMP concentrations using the THP-1/Lucia reporter cell line as previously described (Wu et al., 2015). With known concentrations of cGAMP, we established a standard that demonstrated a high correlation between cGAMP concentration and luciferase activity within the range of 0–30 ng/ml of cGAMP (Figure S3C and S3D). We found that HSV-1 UL37WT induced ~0.33 ng of cGAMP per ten million THP-1 cells, while HSV-1 UL37C819S increased cGAMP production to ~1 ng per ten million THP-1 cells (Figure 2F). Together, these data indicate that the UL37 deamidase activity is crucial to dampen cGAS-mediated response.
UL37 is sufficient to antagonize cGAS-mediated immune response
To determine whether UL37 is sufficient to inhibit cGAS-mediated immune response, we established a THP-1 cell line stably expressing UL37 by lentiviral transduction (Figure 3A). Upon stimulation with herring testis DNA (HT-DNA), ISD-DNA and HSV60 that activate cGAS, UL37 expression in THP-1 cells reduced IFNB1 expression by ~60% (Figure 3B). A similar level of reduction in secreted IFN-β by THP-1 cells was also observed upon UL37 expression (Figure 3C). Interestingly, the induction of IL6 and IL8 by HT-DNA transfection was not affected by UL37 expression (Figure S4A). Moreover, CXCL10 and ISG56 expression induced by LPS was significantly increased by UL37 expression (Figure S4B). These results are consistent with a previous report that UL37 activates NF-κB (Liu et al., 2008).
Figure 3. UL37 targets cGAS to inhibit innate immune activation in human THP-1 cells.

(A) THP-1 cells infected with control (Vector) or UL37-expressing lentivirus were selected with puromycin and whole cell lysates (WCLs) were analyzed by immunoblotting with indicated antibodies.
(B) THP-1 stable cells as described in (A) were transfected with HT-DNA (2 µg/ml), ISD-DNA (1 µg/ml) or HSV60 (1 µg/ml). Cells were harvested at 6 hours post-transfection and the expression of IFNB1 was analyzed by real-time PCR.
(C) Medium was harvested at 16 hpi and IFN-β was determined by ELISA.
(D) THP-1 stable cells were transfected with cGAMP (2 µg/ml). The expression of IFNB1 and ISG56 was analyzed by real-time PCR at 6 hours post-transfection.
(E) Control (V) or UL37-expressing (U) THP-1 cells were mock-transfected (NT), or transfected with HT-DNA (2 µg/ml) or cGAMP (2 µg/ml). Cells were harvested at 6 hours post-transfection and whole cell lysates were analyzed by immunoblotting with the indicated antibodies.
(F) WCLs of the indicated THP-1 stable cells were analyzed by immunoblotting with indicated antibodies.
(G) THP-1 stable cells as described in (F) were transfected with HT-DNA (2 µg/ml). Cells were harvested at 6 h post-transfection. The expression of IFNB1 and ISG56 was determined by real-time PCR.
(H) Cells were harvested and intracellular cGAMP was determined by a reporter assay.
Each value is the mean + s.d. of the results of three independent experiments (B–D, G, H). Statistical analysis was performed by unpaired t-test for (B–D, G, H); *P<0.05, **P<0.01, ***P<0.005. Data are representative of three independent experiments (A, E, F).
See also Figure S4.
To determine the point of inhibition by UL37, we assessed IFNB1 and ISG56 gene expression in THP-1 cells upon cGAMP transfection that bypasses cGAS. Remarkably, UL37 expression did not significantly affect either the expression of IFNB1 and ISG56 (Figure 3D), nor the secretion of IFN-β (Figure S4C). These results suggest that UL37 targets cGAS to evade innate immune activation, leaving the downstream signaling events intact. To further test this hypothesis, we established wild-type and cGAS−/− L929 cells with stable UL37 expression (Figure S4D) and examined DNA-induced innate immune signaling. UL37 expression reduced the mRNA levels of Ifnb1 and Isg56 in L929 cells transfected with HT-DNA (Figure S4E). Consistent with previous reports (Sun et al., 2013), loss of cGAS abolished Ifnb1 and Isg56 expression induced by HT-DNA in L929 cells (Figure S4E). UL37 expression in THP-1 cells inhibited the phosphorylation of TBK-1 and IRF3 induced by HT-DNA, but not by cGAMP (Figure 3E). Similar results were obtained in L929 cells (Figure S4F). Interestingly, transfected HT-DNA induced the phosphorylation of TBK-1 and IRF3 in cGAS−/− L929 cells, albeit to a lower level than those in cGAS wild-type L929 cells. The HT-DNA-induced, cGAS-independent phosphorylation of TBK-1 and IRF3 was not affected by UL37 expression (Figure S4F). Similar to what was observed in THP-1 cells, UL37 expression didn’t affect the induction of Ifnb1 and Isg56 by cGAMP in wild-type and cGAS−/− L929 cells (Figure S4G). cGAMP-induced expression of Isg56 in cGAS−/− L929 cells was significantly lower than that in wild-type L929 cells, suggesting that cGAS may contribute to the induction of Isg56 (Figure S4G). Immunoblotting analyses further showed that UL37 did not affect the phosphorylation of TBK-1 and IRF3 induced by cGAMP (Figure S4H). These results collectively support the conclusion that UL37 antagonizes cGAS to inhibit DNAinduced innate immune response.
Given that HSV-1 UL37C819S more robustly induced antiviral cytokine production in mice (Figure 1) and THP-1 cells (Figure 2), we sought to determine whether the deamidase activity of UL37 is necessary to antagonize cGAS in THP-1 monocytes using the deamidase-deficient UL37C819S. Expression of UL37WT, but not UL37C819S (Figure 3F), reduced the expression of IFNB1 and ISG56 in THP-1 cells transfected with HT-DNA (Figure 3G). Similar reduction in CXCL10 expression was observed upon UL37WT expression, while UL37C819S increased CXCL10 expression in THP-1 cells (Figure S4I). Furthermore, transfected HT-DNA induced ~6 ng of cGAMP per ten million THP-1 cells (equivalent to 0.5 million molecules of cGAMP per cell), while UL37WT expression reduced cGAMP to ~3.1 ng per ten million THP-1 cells (Figure 3H). UL37C819S expression increased cGAMP production to ~8.5 ng per ten million THP-1 cells. These results demonstrate that the deamidase activity of UL37 is required to suppress cGAMP synthesis catalyzed by cGAS.
UL37 deamidates cGAS in cells and in vitro
The observation that the deamidase activity of UL37 is crucial to antagonize cGAS suggests that UL37 deamidates cGAS. To test that, we first determined whether UL37 interacts with cGAS in HSV-1-infected cells. Using recombinant HSV-1 carrying FLAG-tagged UL37 (Zhao et al., 2016b), we found that cGAS was readily precipitated by UL37 in HSV-1-infected THP-1 cells (Figure 4A). Mutational analysis showed that the Mab21 enzyme domain (residue 162–522) of cGAS interacted with UL37, and both the N- and C-terminal domains of UL37 were sufficient for binding cGAS (Figure S5A and S5B).
Figure 4. UL37 deamidates cGAS in cells and in vitro.

(A) Human THP-1 monocytes were infected with HSV-1 carrying FLAG-tagged UL37 at MOI=0.5. At 16 hours post-infection (hpi), cells were harvested and whole cell lysates (WCLs) were precipitated with anti-FLAG (UL37) or mouse IgG as a control. Precipitated proteins and WCLs were analyzed by immunoblotting with antibody against cGAS or FLAG (UL37).
(B) 293T cells stably expressing FLAG-tagged cGAS were transfected with a vector or that containing UL37. At 30 hours post-transfection, WCLs were prepared and analyzed by two-dimensional gel electrophoresis (2-DGE) and immunoblotting with the indicated antibodies.
(C) Primary bone marrow-derived macrophages were infected with HSV-1 (MOI=10). At 4 hpi, cells were harvested and WCLs were analyzed by 2-DGE and immunoblotting with anti-cGAS antibody.
(D) The structure of a cGAS dimer with the four deamidated residues highlighted in yellow (N210 and N389) and red (Q451 and Q454) (PDB:4K9B).
(E, F) Purified cGAS or cGAS-DDEE (as fusion constructs with maltose-binding protein from E.coli), UL37 and its deamidase-deficient UL37C819S mutant were analyzed by silver staining (E). In vitro deamidation reactions were analyzed by 2-DGE and immunoblotting (F).
Data are representative of three independent experiments (A–C, E, F).
See also Figure S5.
To assess whether UL37 induces cGAS deamidation, we analyzed the charge status of cGAS without or with UL37 expression by two-dimensional gel electrophoresis. UL37 expression shifted cGAS toward the positive side of the gel strip, indicative of an increase in negative charge potentially due to deamidation (Figure 4B). Furthermore, HSV-1 UL37WT, but not HSV-1 UL37C819S, increased the negative charge of endogenous cGAS of BMDMs, (Figure 4C), indicating the essential role of UL37 in deamidating cGAS during HSV-1 infection. To probe the contribution of phosphorylation to cGAS charge change, we purified cGAS from HSV-1-infected cells and treated with phosphatase. The result showed that a minor fraction of cGAS was shifted by phosphatase-treatment (Figure S5C).
To identify sites of deamidation in cGAS, we purified cGAS in 293T cells without or with UL37 expression and analyzed cGAS deamidation by tandem mass spectrometry. Additionally, we performed in vitro deamidation assays to augment the protein coverage of cGAS analyzed by tandem mass spectrometry. The analysis identified a total of four sites of deamidation, all located within the Mab21 enzyme domain of mouse cGAS (mcGAS): N196, N377, Q436 and Q439 (corresponding to N210, N389, Q451 and Q454 of hcGAS) (Figure 4D and S5D) (PDB: 4K9B). To validate the deamidation sites of cGAS, we generated a cGAS mutant containing all four deamidated residues, designated cGAS-DDEE. UL37 expression shifted cGAS-WT toward the positive side of the strip and to the position that was identical to cGAS-DDEE (Figure S5E). UL37 failed to shift cGAS-DDEE, implying that there are no other deamidation sites. There was a residual amount of cGAS-WT and cGAS-DDEE that was not shifted by UL37. This small portion may represent deamidated cGAS, particularly that of the cGAS-DDEE, with other post-translational modifications that offset the charge change of deamidation.
To determine whether UL37 is sufficient to deamidate cGAS in vitro, we purified cGAS, UL37WT and UL37C819S to high homogeneity (Figure 4E). There were two major species of the purified cGAS as analyzed by two-dimensional gel electrophoresis, likely due to intrinsic deamidation or other modifications (Figure 4F). Nevertheless, two-dimensional gel electrophoresis showed that UL37WT engendered three additional species, in addition to the original two species. All three additional species migrated toward the positive end of the gel strip to a position of the deamidated cGAS-DDEE mutant, while the deamidase-deficient UL37C819S failed to do so (Figure 4F). Furthermore, UL37WT did not further shift cGAS-DDEE, indicating the lack of other deamidation sites. Taken together, UL37 is a bona fide protein deamidase that deamidates cGAS in cells and in vitro.
Deamidated cGAS fails to sense dsDNA and restrict HSV-1 infection
To probe the role of protein deamidation in cGAS-mediated immune response, we first performed reporter assays to analyze the ability of various deamidated cGAS mutants in activating IFN-β and NF-κB promoters. In addition to the four deamidation sites (N210, N389, Q451 and Q454), we have also generated deamidation mutations of all N (>D) and Q (>E) residues within the Mab21 enzyme domain that are conserved between hcGAS and mcGAS (Figure S6A). Reporter assays showed that N210D mutation reduced cGAS-mediated expression of the IFN-β promoter by 50%, while all other individual deamidation mutations did not significantly impair cGAS to activate the IFN-β promoter (Figure S6A and S6B). However, combining the three mutations in NQQ389,451,454DEE modestly reduced cGAS-induced IFN-β expression (Figure S6A and S6C). The cGAS-DDEE mutant failed to activate the IFN-β and NF-κB promoters (Figure 5A and 5B). These results conclude that deamidation negatively regulates cGAS-induced innate immune activation.
Figure 5. Deamidated cGAS fails to respond to DNA stimulation.

(A, B) 293T cells were transfected with an IFN-β (A) or NF-κB (B) reporter plasmid cocktail with plasmids containing cGAS wild-type or the deamidated cGAS-DDEE mutant. IFN-β or NF-κB activation was determined by luciferase assay (top panels), while whole cell lysates (WCLs) were analyzed by immunoblotting (bottom panels).
(C) cGAS−/− L929 cells were infected with control (Vector) lentivirus or that containing cGAS wild-type (WT) or the deamidated cGAS-DDEE mutant (DDEE). WCLs were analyzed by immunoblotting.
(D) “Reconstituted” cGAS stable cells as described in (C) were transfected with HT-DNA (2 µg/ml). Cells were harvested at 6 h post-transfection and the expression of the indicated inflammatory genes was analyzed by real-time PCR.
(E) “Reconstituted” cGAS stable cells were infected with HSV-1 UL37WT or HSV-1 UL37C819S at MOI=5, cells were harvested at 6 h post-infection. The relative mRNA quantity of the indicated genes was determined by real-time PCR.
(F) “Reconstituted” cGAS stable cells were infected with murine herpesvirus 68 (MHV68) (MOI=0.05). Viral titers in the medium at the indicated time points were determined by plaque assays.
Each value is the mean + s.d. of the results of three independent experiments (A, B, D–F).
Statistical analysis was performed by unpaired t-test for (D–F); **P<0.01, ***P<0.005. Data are representative of three independent experiments (A–C).
See also Figure S6.
To assess the effect of deamidation on cGAS-mediated innate immune response, we “reconstituted” cGAS−/− L929 cells with cGAS wild-type or the deamidated cGAS-DDEE mutant (Figure 5C). In response to HD-DNA transfection, L929 cells “reconstituted” with cGAS wild-type up-regulated the expression of Ifnb, Isg56, Cxcl10 and Ifit3 genes (Figure 5D and S6D). In contrast, L929 cells “reconstituted” with cGAS-DDEE failed to induce the expression of these immune genes upon HD-DNA transfection. We further “reconstituted” cGAS−/− L929 cells with cGAS carrying individual deamidated residues (Figure S6E). When these cells were transfected with HT-DNA, we found that cytokine gene expression was significantly and most reduced in L929 cells “reconstituted” with cGAS-D210 compared to L929 “reconstituted” with cGAS wild-type (Figure S6F). D389 and E454 also consistently reduced the expression of cytokines, including Ifnb1 and Cxcl10. E451 had a minor effect on the expression of Ifnb1, but no effect on other cytokines. These results show that the deamidation of N210, N389 and N454 reduces the activity of cGAS in dsDNA-induced host defense
To assess the biological significance of cGAS deamidation, we examined cellular immune response and viral production in these “reconstituted” L929 cell lines. When infected with HSV-1 UL37WT or HSV-1 UL37C819S, L929 cells “reconstituted” with cGAS wild-type up-regulated the expression of inflammatory genes as potent as wild-type L929 cells (data not shown). cGAS-deficient L929 cells “reconstituted” with cGAS-DDEE essentially behaved like cGAS−/− L929 cells, demonstrating no induction of immune gene expression upon HSV-1 infection (Figure 5E). Moreover, the antiviral gene expression elevated by HSV-1 UL37C819S was completely abolished in cGAS−/− L929 cells or those “reconstituted” with cGAS-DDEE (Figure 5E). We further tested the ability of cGAS wild-type and cGAS-DDEE in restricting murine gamma herpesvirus 68 (MHV68), a DNA virus lacking a homologue of UL37. We found that cGAS wild-type significantly reduced the replication of MHV68 at all time points analyzed (Figure 5F), whereas cGAS-DDEE had no effect on MHV68 productive infection in cGAS−/− L929 cells. These results demonstrate that deamidated cGAS fails to induce innate immune defense that restricts DNA virus infection.
Deamidation specifically impairs the cGAMP-synthesizing activity of cGAS
Upon binding to dsDNA, cGAS forms dimer that coordinates the cGAMP synthesis to induce innate immune activation. Structural analyses also indicate that N376 and N377 of mcGAS (corresponding to N388 and N389 of hcGAS) extend into the minor groove of the dsDNA helix (Figure S7A) (Gao et al., 2013), suggesting that deamidation of these residues potentially interferes cGAS ability to sense dsDNA. However, precipitation of biotinylated interferon-stimulating DNA (ISD) demonstrated that neither UL37-WT, nor UL37C819S diminished cGAS co-precipitated with ISD (Figure S7B). In fact, UL37WT, but not UL37C819S, increased the interaction between cGAS and ISD by ~50% (Figure S7C). We then probed the interaction between cGAS-DDEE and ISD. The result showed that the deamidated cGAS-DDEE demonstrated slightly enhanced interaction with ISD compared to cGAS wild-type (Figure 6A). Thus, these results show that deamidation does not impair cGAS to interact with dsDNA.
Figure 6. cGAS deamidation abolished its enzymatic activity.

(A) WCLs were prepared from 293T/cGAS-FLAG or 293T/cGAS-DDEE-FLAG stable cells. Increasing amount of biotinylated intereferon-stimulating DNA (Biotin-ISD) was used to pull down cGAS wild-type or the DDEE mutant in vitro and precipitated proteins were analyzed by immunoblotting.
(B) 293T stable cells expressing both cGAS-FLAG and cGAS-V5 were transfected with empty vector, plasmids encoding UL37 or UL37C819S. At 30 h post-transfection, WCLs were incubated with anti-FLAG agarose. Precipitated proteins and WCLs were analyzed by immunoblotting.
(C) WCLs were prepared from 293T-cGAS-FLAG or 293T-cGAS-DDEE-FLAG stable cell lines. Immunoprecipitation was performed with ATP-agarose, GTP-agarose or control agarose. Input (WCL) and precipitated proteins were analyzed by immunoblotting.
(D) The N196 residue locates in proximity to the catalytic active site, consisting of E211, D213 and D307 (PDB: 4K9B). cGAMP, in relation to E211, D213 and D307, is also shown.
(E) In vitro cGAMP synthesis was measured with purified hcGAS or its deamidated mutants, including N210D and DDEE, with or without HSV60 DNA. Reactions were analyzed by thin layer chromatography (left panel).
(F) In vitro cGAMP activity assay was performed in the presence of 1 µg of UL37 or UL37C819S (left panel). BSA was included as a negative control. The relative intensity of cGAMP was determined by densitometry analysis (right panel).
Each value is the mean + s.d. of the results of three independent experiments (F). Statistical analysis was performed by unpaired t-test for (F); ***P<0.005. Data are representative of three independent experiments (A–C, E, F).
See also Figure S7.
An interesting observation is that all four deamidation sites reside in regions proximal to the dimerization interface of cGAS (Figure 4D). Thus, we sought to determine whether UL37 influences cGAS self-dimerization that is induced by DNA-binding. With 293T cells that stably express FLAG-cGAS and cGAS-V5, we found that DNA transfection induced robust dimerization of cGAS, and that the expression of UL37WT or UL37C819S did not alter cGAS dimerization by Co-IP assay (Figure 6B). We further assessed the binding of cGAS to nucleotide substrates (ATP and GTP), and found that cGAS and cGAS-DDEE bound comparatively to these nucleotides (Figure 6C). Collectively, these results support the conclusion that deamidation does not impair the dsDNA-binding, self-dimerizing or nucleotide-binding activity of cGAS.
Previous structural studies revealed an active site, known as a catalytic triad, of cGAS that catalyzes cGAMP synthesis. The catalytic triad consists of E211, D213 and D307 that are donated from two core anti-parallel β-sheets, i.e., β2 and β7 (PDB: 4K9B) (Figure 6D) (Gao et al., 2013). β1 and β6 sheets sandwich the two core β-sheets. Moreover, an important sequence known as the activation loop, consisting of nonpolar or uncharged amino acids with small side chain, provides free rotation to coordinate the catalytic triad (Zhang et al., 2014). Locating within the activation loop, N196 (corresponding to N210 of hcGAS) is also proximal to residues, particularly D307 and D213, of the catalytic triad (Figure 6D). Together, these suggest that deamidation may impinge on the catalytic triad responsible for cGAMP synthesis. We thus assessed the effect of the deamidation on the enzyme activity of cGAS. Indeed, HSV60 DNA stimulated cGAMP synthesis of cGAS, but had no significant effect on that of cGAS-N210D or cGAS-DDEE (Figure 6E). Furthermore, addition of UL37 to the cGAS-catalyzed reaction reduced cGAMP production nearly to basal level, while the deamidase-deficient UL37C819S mutant had less inhibition on cGAMP production (Figure 6F). These results collectively show that UL37-mediated deamidation specifically inhibits the cGAMP synthase activity of cGAS.
Nonhuman primate cGAS proteins are resistant to UL37-mediated deamidation and more potently restrict HSV-1 lytic replication than human and mouse cGAS
Deamidation of N210 of hcGAS had the most significant effect on cGAS activity. In order to determine if this residue is evolutionarily conserved, we aligned protein sequences of cGAS from human, mouse, and several nonhuman primate species. Interestingly, among the four deamidation sites by UL37, cGAS homologues differ only at residue 210, while the other three residues are identical in all cGASs (Figure 7A). Human, mouse and gorilla cGAS encode asparagine (N) at residue 210, while cGAS from other nonhuman primates, including black mangabey, patas monkey, rhesus macaque and orangutan, instead encode either arginine (R) or histidine (H). It is well known that immune effectors targeted by viral evasion mechanisms can sometimes experience positive selection in favor of non-synonymous nucleotide substitutions, changing the protein sequence in the region targeted by a viral antagonist (Duggal and Emerman, 2012). Therefore, we hypothesized that cGAS could be evolving under natural selection at the sites targeted by HSV-1 for deamidation. Natural selection is inferred by detecting an increased rate of non-synonymous mutations (dN) in comparison to synonymous mutations (dS) in DNA sequences (dN/dS>1) (Hurst, 2002). In line with a previous study (Hancks et al., 2015), a commonly used model of selection (implemented in PAML (Yang, 2007)), revealed that cGAS was under strong positive selection, with several codons bearing a signature of dN/dS > 1 (Figure 7B). Importantly, our analysis identified N210 of hcGAS (N196 of mcGAS) as one of the residues that is positively selected (Figure 7B). Codon 210 has a dN/dS = 2.74, which is significantly greater than 1 (Naïve Empirical Bayes posterior probability = 0.64). This indicates that selection is acting to change the residue encoded at this site.
Figure 7. Species-specific deamidation of cGAS promotes HSV-1 lytic replication.

(A) cGAS proteins of human and 5 different nonhuman primate species were aligned, with sequences flanking the four deamidation sites (asterisks) shown.
(B) Starting with an alignment of cGAS from 29 primate species, dN/dS values of each amino acid of cGAS were calculated. The dN/dS value (Y axis) is shown for each codon along the length of the cGAS gene (X axis). Amino acid sites with statistically significant elevation of dN/dS > 1 (PP > 0.5) values are indicated by red lines. The asterisk denotes N210 of human cGAS.
(C–E) cGAS−/− L929 cells were “reconstituted” with human and nonhuman primate cGASs. (C) Whole cell lysates (WCLs) were analyzed by immunoblotting. (D) Cells were infected with HSV-1 (MOI=0.01), virus titer in the medium at 24 hours post-infection (hpi) was determined by plaque assay. (E) the expression of Isg56 was analyzed by real-time PCR.
(F) Plasmids containing cGAS of human and nonhuman primates, without or with that containing UL37, were transfected into 293T cells. WCLs were analyzed by two-dimensional gel electrophoresis and immunoblotting.
(G) Transfection of 293T cells with plasmids containing black mangabey (BM) or orangutan (OR) cGAS with the H210N or R209N mutation and that containing UL37, two-dimensional gel electrophoresis and immunoblotting were carried out as in (F).
(H–J) cGAS−/− L929 cells were “reconstituted” with black mangabey (BM) or orangutan (OR) cGAS or that containing H210N or R209N. (H) WCLs were analyzed by immunoblotting. (I) Cells were infected with HSV-1 (MOI=0.01), virus titer in the medium was determined. (J) Cells were infected with HSV-1 at MOI=5. The expression of Isg56 was analyzed by real-time PCR.
Each value is the mean + s.d. of the results of three independent experiments (D, E, I, J). Statistical analysis was performed by unpaired t-test for (D, E, I, J); **P<0.01, ***P<0.005. Data are representative of three independent experiments (C, F, G, H).
See also Figure S8 and S9.
To validate this analysis, we first determined whether nonhuman primate cGASs more potently inhibit HSV-1 replication using cGAS−/− L929 cells. Specifically, we complemented cells with cGAS from four primate species that encoded an amino acid at position 210 that is different from the human asparagine (N), namely black mangabey, patas monkey, rhesus macaque, and orangutan to establish stable cell lines. In all cases, these cGASs more potently reduced HSV-1 replication in comparison to human or gorilla cGAS (Figure 7C and 7D). Conversely, these primate cGASs more effectively induced the expression of immune effectors, e.g., Isg56 and Cxcl10, than human and gorilla cGAS upon HSV-1 infection (Figure 7E and S8A). Upon HT-DNA stimulation, cGASs from all primate species robustly upregulated cytokine gene expression to similar levels (Figure S8B). Analysis by two-dimensional gel electrophoresis showed that black mangabey, patas monkey, rhesus macaque, and orangutan cGASs were resistant to UL37-mediated deamidation, while human and gorilla cGAS were deamidated by UL37 (Figure 7F and S8C). To test whether residue 210 is sufficient to confer sensitivity to UL37, this site was mutated in both black mangabey (H210N) and orangutan (R209N) cGASs and UL37-mediated deamidation was assessed. This showed that replacing H of black mangabey cGAS or R of orangutan cGAS with N resulted in UL37-mediated deamidation (Figure 7G). Furthermore, “reconstituted” expression of cGAS wild-type of black mangabey and orangutan (Figure 7H) more robustly induced Isg56 and Cxcl10 expression than that of their deamidation-sensitive mutants (H210N or R209N) upon HSV-1 infection (Figure 7I and S8D). Consistently, wild-type of black mangabey and orangutan cGAS more effectively reduced HSV-1 replication than their corresponding mutants (H210N and R209N) (Figure 7J). The expression of wild-type and deamidation mutants of black mangabey and orangutan cGASs led to comparable innate immune activation when stimulated with HT-DNA (Figure S8E). To test whether N210R and N210H confer hcGAS the resistance to UL37-mediated immune evasion, hcGAS mutants containing N210R or N210H were generated and used for two-dimensional gel electrophoresis analysis. Indeed, cGAS-R210 and cGAS-H210 were minimally, if any, shifted by UL37 expression as analyzed by two-dimensional gel electrophoresis (Figure S9A), indicative of their resistance to UL37-mediated deamidation. Consistent with this, the expression of hcGAS-R210 and hcGAS-H210 in cGAS−/− L929 cells (Figure S9B) more potently upregulated Isg56 and Cxcl10 expression than hcGAS wild-type upon HSV-1 infection (Figure S9C). Consequently, hcGAS-R210 and hcGAS-H210 more robustly inhibited HSV-1 replication than hcGAS wild-type (Figure S9D). In response to transfected HT-DNA, hcGAS-R210 and hcGAS-H210 induced levels of immune gene expression similar to hcGAS wild-type (Figure S9E). These two hcGAS mutants behaved indistinguishably from hcGAS wild-type in restricting VSV replication and activating the IFN-β promoter by reporter assay (Figure S9F and S9G). Taken together, these results show that the N210 of hcGAS is necessary and sufficient for UL37-mediated deamidation, defining a species-specific deamidation of cGAS by human HSV-1.
DISCUSSION
To investigate the in vivo roles of the deamidase activity of UL37, we discovered that UL37 antagonizes the cGAS-STING immune pathway in mice. In wild-type BL6 mice, HSV-1 carrying the deamidase-deficient UL37 is highly attenuated in lytic replication and pathogenesis compared to HSV-1 wild-type, indicating the pivotal role of the UL37 deamidase activity in HSV-1 infection in vivo. Surprisingly, loss of cGAS and STING in mice rendered HSV-1 UL37C819S as potent as HSV-1 UL37WT in replication and pathogenesis. When host innate immune response was examined, HSV-1 UL37C819S more robustly induced antiviral cytokine production in mice and in monocytes ex vivo. Furthermore, loss of cGAS in mice and in L929 cells resulted in basal and indistinguishable level of cytokine production induced by HSV-1 UL37WT and HSV-1 UL37C819S. These findings are consistent with previous studies (Ishikawa and Barber, 2008; Li et al., 2013), and also provide genetic credentials to support the corollary that UL37 counteracts the cGAS-STING pathway during HSV-1 infection. Remarkably, deamidation of cGAS by UL37 specifically abolishes the cGAMP-synthesizing activity, without impairing the DNA-binding, self-dimerizing and nucleotide-binding activities of cGAS. The negative effect of deamidation on cGAS-mediated signaling is evident by the reduced cGAMP synthesis of deamidated cGAS mutants and the deamidase-dependent inhibition mediated by UL37. The inability to activate innate immune response and restrict DNA virus replication of the deamidated cGAS-DDEE mutant further supports the critical role of deamidation in inactivating cGAS antiviral activity.
Among all four deamidation sites, N210 locates within the activation loop that impinges on the cGAS-catalyzed cGAMP synthesis. Mutational analysis indicates that deamidation of N210 of hcGAS significantly reduced cGAS-dependent cGAMP production and innate immune signaling induced by dsDNA, while the other three deamidations had marginal effect alone, or further diminished cGAS enzyme activity when combined with N210D mutation. Evolution analysis of cGAS amino acid sequence suggests that selection has acted on N210 of hcGAS. In contrast to human and mouse cGAS, most of nonhuman primate species encode cGAS molecules containing an R or H at the position corresponding to hcGAS N210. In fact, mutational analysis swapping the N210 of hcGAS with nonhuman primate R or H or vice versa shows that the N210 is necessary and sufficient to endow UL37-dependent cGAS deamidation, despite the fact that the other three deamidation sites are conserved among mouse, human and nonhuman primate cGAS proteins. The prerequisite of N210 deamidation for subsequent deamidation of the other three sites calls for further investigation, and echoes the priming mechanism of NFAT phosphorylation by protein kinase A (PKA) in inactivating NFAT and suppressing T cell activation (Sheridan et al., 2002). Nevertheless, these results uncover an immune evasion strategy that herpes simplex virus exploits species-specific variation of immune components to inactivate host innate defense. Previous studies have extensively documented species-specific host factors in restricting HIV replication (Mariani et al., 2003; Stremlau et al., 2004). Recent work has also extended this theme into the cross-species restriction of flaviviruses, herpesviruses (Aguirre et al., 2012; Lou et al., 2016; Patel et al., 2012) and influenza virus (Long et al., 2016; Meyerson et al., 2017). These studies provide critical mechanistic insight into the barrier of cross-species transmission of viruses. Our study that cGAS inhibition by UL37-mediated deamidation constitutes a cross-species barrier for HSV-1 replication defines protein deamidation, a post-translational modification, as a restriction mechanism in cross-species transmission of viruses.
Though reported more than half a century ago, protein deamidation is poorly understood and emerging as a key post-translational modification in regulating immune response (Zhao et al., 2016a). Among all post-translational modification events, deamidation is the simplest one and consumes no energy. However, this also permits the natural hydrolysis of asparaginyl and occasionally glutaminyl residues, shadowing the deamidation catalyzed by cellular and microbial enzymes. Our recent studies and other related work collectively demonstrate that protein deamidation can be a powerful regulatory mechanism governing fundamental biological processes, such as immune response (Cui et al., 2010; He et al., 2015; Sanada et al., 2012; Schmidt et al., 1997; Zhao et al., 2016b). The functional consequence of protein deamidation can be protein- and site-specific. Deamidation of RIG-I within the CARD domain induced by MHV68 vGAT promoted CARD-mediated oligomerization of RIG-I (He et al., 2015), whereas UL37-mediated deamidation within the helicase domain abolished RNA-binding of RIG-I (Zhao et al., 2016b). In this study, we show that deamidation of cGAS by UL37 specifically ablated the cGAMP-synthesizing activity of cGAS. Previous studies have shown that cGAS is subjected to diverse post-translational modifications, including sumoylation, glutamylation and phosphorylation (Cui et al., 2017; Hu et al., 2016; Seo et al., 2015; Xia et al., 2016), and direct antagonism by an array of microbial proteins (Aguirre et al., 2017; Wu et al., 2015; Zhang et al., 2016a). How deamidation, of RIG-I and cGAS, cross-talks with these regulatory mechanisms remain unclear and these potential cross-talks, if exist, will connect protein deamidation to other well-established signaling cascades and expand the functional repertoire of protein deamidation in high eukaryotes.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Viruses
Cell Culture
METHOD DETAILS
RNA Extraction and qRT-PCR
Luciferase Reporter Assay
Immunoprecipitation
Protein Purification
Two-dimensional Gel Electrophoresis
In Vitro Deamidation Assay
cGAMP Reporter Assay
In Vitro cGAMP Activity Assay
Stable Cell Line Generation
Deamidation Site Analysis by LC/MS/MS
Cytokine Measurement
Next Generation Sequencing and Analysis
Evolutionary analysis
QUANTIFICATION AND STATISTICAL ANALYSES
DATA AND SOFTWARE AVAILABILITY
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pinghui Feng (pinghui.feng@usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Ten to twelve-week old, gender-matched mice (male and female) were used for all experiments. All animal work was performed under strict accordance with the recommendation in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California. All mice were housed in a standard pathogen-free animal facility.
C57BL/6J and cGAS KO mice were purchased from the Jackson Laboratory. STING KO mice were kindly provided by Dr. Jae Jung. MAVS KO mice have been previously used and been maintained in the laboratory. All strains were confirmed by genotyping.
Viruses
Murine gamma herpesvirus 68 (MHV68) viruses were propagated in BHK21 cells (Zhang et al., 2015b). Herpes simplex virus 1 (HSV-1) WT and UL37C819S recombinant viruses were propagated using Vero cells. Vesicular stomatitis virus (VSV) was amplified using VERO cells (Minassian et al., 2015).
Cell Culture
HEK293T, VERO cells (ATCC) and BHK21 were cultured in DMEM (Corning) supplemented with 10% fetal calf serum (FCS), penicillin– streptomycin at 37 °C. THP -1 (ATCC) and THP1-Lucia ISG cells (InvivoGen) were cultured in RPMI-1640 (Corning) supplemented with 10% fetal calf serum (FCS), penicillin– streptomycin at 37 °C. L929 WT and cGAS KO cells (kindly provided by D r. James Chen) were cultured with DMEM (Corning) supplemented with 10% fetal calf serum (FCS). Primary bone marrow-derived macrophages (BMDM) were differentiated and maintained in DMEM supplemented with 30% L929 conditioned medium. Gender of the cell lines were not a consideration in this study. Cell lines were authenticated by genotyping, or obtained from authentic stocks (ATCC).
METHOD DETAILS
RNA Extraction and qRT-PCR
Human THP-1 monocytes, mouse BMDMs or mouse L929 fibroblasts were infected with HSV-1 (MOI = 5) or transfected with HT-DNA (2 µg/ml) or cGAMP (2 µg/ml) for 6 h, unless specifically indicated otherwise. Cells were washed with cold PBS, and total RNA was extracted using TRIzol Reagent (Invitrogen). RNA was digested with DNase I (New England Biolabs) to remove genomic DNA. One microgram of total RNA was used for reverse transcription with PrimeScript Reverse Transcriptase (Clontech) according to the manufacturer’s instruction. Approximately 0.5% of the cDNA was used as template in each quantitative real-time PCR (qRT-PCR) reaction with SYBR master mix (Applied biosystems).
Primers for q-PCR can be found in Table S1.
Luciferase Reporter Assay
HEK293T cells in 24-well plates were transfected with a reporter plasmid mixture containing 50 ng of the plasmid expressing IFN-β or NF-κB firefly luciferase reporter and 20 ng of the plasmid expressing TK-renilla luciferase reporter. At 30 h post-transfection, cells were harvested and cell lysates were prepared. Cell lysates were used for dual luciferase assay according to the manufacturer’s instruction (Promega).
Immunoprecipitation
Immunoprecipitation was carried out as previously described (Zhang et al., 2015a). Briefly, THP-1 cells were infected with HSV-1 FLAG-UL37 recombinant virus (MOI = 0.5) for 16 h. Cells were harvested and lysed with NP40 buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) supplemented with a protease inhibitor cocktail (Roche). Centrifuged cell lysates were pre-cleared with Sepharose 4B beads and incubated with FLAG-agarose at 4 °C for 4 h. Agaros e beads were washed three times with lysis buffer and precipitated proteins were released by boiling with 1×SDS sample buffer at 95 °C for 5 min. Precipitat ed proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
Protein Purification
Mouse maltose-binding protein (MBP)-cGAS fusion protein (141–507) (Seo et al., 2015) was expressed in BL21 (DE3) and bacteria were grown at 37 °C to an OD600 of 0.6. Then, cultures were cooled to 18 °C and pro tein expression was induced by adding 0.1 mM Isopropyl b-D-1-thiogalactopyranoside (IPTG) for 16 h. Cells were collected by centrifugation and lysed with Triton X-100 buffer (20 mM Tris-HCl [pH 7.4], 200 mM NaCl, 10% glycerol, 0.5% Triton X-100, 0.2 mg/ml lysozyme supplemented with protease inhibitor cocktail). Centrifuged lysates were mixed with amylose resin and incubated for 2 h at 4 °C. The resin was washed extensively with lysis buffer and recombinant proteins were eluted with 10 mM maltose.
For mass spectrometry analysis, purified MBP-mcGAS was digested with TEV protease overnight at 4 °C and MBP proteins were depleted by incubation with Ni-NTA agarose.
FLAG-tagged UL37 or UL37C819S were purified as previously described (He et al., 2015; Zhang et al., 2016b). Briefly, HEK293T cells were transiently transfected with a plasmid containing UL37 or UL37C819S, harvested and resuspended in lysis buffer (20 mM Tris [pH 7.4], 150 mM NaCl, 10% (vol/vol) glycerol, 0.5% Triton X-100, and 0.5 mM DTT) supplemented with a protease inhibitor cocktail (Roche), and lysates were precipitated with 20 µL of FLAG M2-conjugated agarose (Sigma). After extensive washing with lysis buffer, precipitated proteins were eluted with FLAG peptide (100 µg/ml), analyzed by silver staining and used for in vitro deamidation assays.
For His-tagged human cGAS purification, pET28a-hcGAS construct was kindly provided by Dr. Fanxiu Zhu (Florida State University). cGAS or its mutants were expressed in E. coli BL21-DE3 cells by induction with 0.5 mM IPTG at 18°C overnight. Cells were collected and sonicated in lysis buffer (20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 5% glycerol, 10 mM imidazole, 1% Triton X-100, 0.2 mM PMSF). The supernatant was collected and incubated with Ni-NTA beads. After washing with buffer (20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 5% glycerol, 20 mM imidazole), bound protein was eluted with elution buffer (20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 300 mM imidazole). The proteins were further purified by HiTrap Heparin affinity purification (GE Healthcare). The purified protein was concentrated with Amicon Ultra Centrifugal Filters (15 mL; 30 kDa) and used for in vitro enzymatic assay.
Two-dimensional Gel Electrophoresis
Whole cell lysates or in vitro deamidation samples were resolved in 150 µl rehydration buffer (8 M urea, 2% CHAPS, 0.5% IPG buffer, and 0.002% bromophenol blue), and lysates were centrifuged at 20,000 g for 10 min. Centrifuged whole cell lysates were loaded to IEF strips with a program comprising 20 V, 10 hr; 100 V, 1 hr; 500 V, 1 hr; 1,000 V, 1 hr; 2,000 V, 1 hr; 4,000 V,1 hr; and 8,000 V, 4 hr. Then, strips were incubated with SDS equilibration buffer (50mM Tris-HCl [pH 8.8], 6M urea, 30% glycerol, 2%SDS, 0.001% bromophenol blue) containing 10 mg/ml DTT for 15 min and SDS equilibration buffer containing 2-iodoacetamide for 15 min. Strips were washed with SDS-PAGE buffer, resolved by SDS-PAGE, and analyzed by immunoblotting.
In Vitro Deamidation Assay
The in vitro deamidation assay was performed as previously described (He et al., 2015; Zhao et al., 2016b). Briefly, 5 µg of purified MBP-mcGAS or MBP-mcGAS-DDEE mutant on amylose resin was incubated with 0.5 µg of purified FLAG-UL37 or FLAG-UL37C819S at 30 °C fo r 45 min in deamidation buffer (50 mM Tris-HCl [pH 7.5], 100mM NaCl, and 5 mM MgCl2). The reaction was stopped by adding rehydration buffer. cGAS was eluted and analyzed by two-dimensional gel electrophoresis and immunoblotting.
cGAMP Reporter Assay
THP-1 monocytes were transfected with HT-DNA (2 µg/ml) or infected with HSV-1 virus (MOI=5) for 6 h. Cell extracts were prepared by heating at 95 °C for 5 mi n to denature most proteins, and precipitated proteins were removed by centrifugation. The cGAMP-containing supernatant was diluted and delivered to digitonin-permeabilized (2.5 µg/ml for 30 min) THP1-Lucia cells at 37 °C for 30 m in. Cells were cultured for another 20 h before Lucia reporter activity was measured according to the manufacturer’s instruction (InvivoGen). Pure cGAMP was diluted and used to establish a standard for the assay.
In Vitro cGAMP Activity Assay
One µM of cGAS or mutant cGAS was mixed with 100 µM ATP, 100 µM GTP and 10 µCi [α-P32]-ATP in reaction buffer (20 mM Tris-Cl [pH 7.5], 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol [DTT]). After 2 h of incubation at 37 °C, 2 µl of reaction solution was spotted onto PEI-Cellulose thin layer chromatography plate (Sigma). Reaction products were resolved with running buffer (1 M (NH4)2SO4, 1.5 M KH2PO4, pH 3.8). TLC plates were dried and scanned with a phosphoimager (Fuji).
Stable Cell Line Generation
Lentivirus production was carried out in 293T cells as described previously (He et al., 2015; Zhang et al., 2016b). THP-1 or L929 cells were infected with lentivirus containing UL37 or UL37 C819S mutant. L929 cGAS KO cells were infected with lentivirus containing cGAS of different species (wild-type or mutants). After 36 h, THP-1 cells were selected with puromycin at 1 µg/ml and L929 cells or L929 CGAS KO cells were selected with puromycin at 5 µg/ml for 2 days. Cells were maintained in culture medium containing puromycin.
Deamidation Site Analysis by LC/MS/MS
Purified mcGAS(141–507) was used for deamidation by purified FLAG-UL37 in vitro. Samples were resolved by SDS-PAGE and mcGAS bands were excised and subjected to LC/MS/MS analysis. The analysis of samples was carried out using a Thermo Scientific Q Exactive hybrid Quadrupole-Orbitrap Mass Spectrometer and a Thermo Dionex UltiMate 3000 RSLCnano System. Peptide mixtures from each sample were loaded onto a peptide trap cartridge at a flow rate of 5 µL/min. Trapped peptides were eluted onto a reversed-phase PicoFrit column (New Objective, Woburn, MA) using a linear gradient of acetonitrile (3–36%) in 0.1% formic acid. The elution duration was 120 min at a flow rate of 0.3 µl/min. Eluted peptides from the PicoFrit column were ionized and sprayed into the mass spectrometer, using a Nanospray Flex Ion Source ES071 (Thermo) under the following settings: spray voltage, 2.0 kV, Capillary temperature, 250°C. Other settings were empirical ly determined. Raw data files were searched against mouse protein sequence database obtained from NCBI website using the Proteome Discoverer 1.4 software (Thermo, San Jose, CA) based on the SEQUEST algorithm. Carbamidomethylation (+57.021 Da) of cysteines was fixed modification, and Oxidation and Deamidation Q/N-deamidated (+0.98402 Da) were set as dynamic modifications. The minimum peptide length was specified to be five amino acids. The precursor mass tolerance was set to 15 ppm, whereas fragment mass tolerance was set to 0.05 Da. The maximum false peptide discovery rate was specified as 0.01. The resulting Proteome Discoverer Report contains all assembled proteins with peptides sequences and matched spectrum counts and peak area.
Mouse infections
Age- and gender-matched wild-type (C57BL/6J), cGAS KO or STING KO mice were infected with 5 × 107 PFU of HSV-1 UL37WT or HSV-1 UL37C819S virus via intraperitoneal injection for viral pathogenesis analysis. Mouse survival was monitored daily for 4 weeks. Sera were collected at 8 hours post-infection to measure cytokine production by ELISA.
Five mice from each group were sacrificed at 3 days post-infection (dpi). Homogenates from brains of infected cGAS KO and STING KO mice were centrifuged at 12,000 rpm for 10 min at 4 °C a nd supernatants were collected and used to measure viral titer by plaque assay. Brains of infected wild-type mice were homogenized and genomic DNA was extracted and HSV-1 genome copy number was determined by real-time PCR. No mice were excluded from all the quantifications and no randomization method was used to divide the mice. The experiments were carried out non-blinded.
Cytokine Measurement
THP-1 cells were transfected with HT-DNA (2 µg/ml) or cGAMP (2 µg/ml) for 16 h or were infected with HSV-1 for 16 h. The supernatant was collected and used to determine cytokine concentration. Mice were infected with HSV-1 UL37WT or HSV-1 UL37C819S virus (5 × 107) and sera were collected at 8 hours post-infection for cytokine detection.
ELISA kit was used to determine the concentration of human interferon-β (PBL assay science), murine interferon-α (PBL assay science), interferon-β (PBL assay science), CCL5 (R&D systems) and IL-6 (BD Biosciences) according to the manufacturer’s instructions.
Next Generation Sequencing and Analysis
THP-1 cells were mock-infected or infected with HSV-1 UL37WT or UL37C819S (MOI=5) for 4 h. Total RNA was extracted from the infected cells with a RNeasy kit (QIAGEN). mRNA was isolated and library was constructed by using KAPA mRNA kit (KAPA biosystems) according to the manufacturer’s instruction. Next generation sequencing was performed at the USC molecular genomics core (NextSeq500). The global gene expression difference was analyzed with Partek Genomics Suite and Ingenuity Pathway Analysis (IPA) software. Global gene expression profiles rank ordered by relative fold-change values were analyzed using Gene Set Enrichment Analysis.
Evolutionary analysis
29 simian primate cGAS sequences were collected from GenBank for evolutionary analysis (human NM_138441, Pongo abelii XM_002817062, Erythrocebus patas KR062009,Colobus angolensis palliatus XM_011944436, Macaca nemestrina XM_011737409, Macaca mulatta XM_001109400, Cercopithecus wolfi KR062006, Saguinus fuscicollis KR062013, Papio anubis NM_001168940, Cebus capucinus imitator XM_017524347, Chlorocebus aethiops KR062008, Pan troglodytes XM_009451552, Lagothrix lagotricha KR062014, Callithrix jacchus XM_008994663, Chlorocebus sabaeus XM_008013489, Saimiri boliviensis boliviensis XM_003926328, Cercocebus atys XM_012067433, Nomascus leucogenys XM_012502825, Aotus nancymaae XM_021670299, Rhinopithecus roxellana XM_010372214, Gorilla gorilla KR062003, Hylobates lar KR062004, Colobus guereza KR062012, Macaca fascicularis XM_015449182, Lophocebus aterrimus KR062010, Pithecia pithecia KR062015, Cercopithecus ascanius KR062005, Allenopithecus nigroviridis KR062007, Rhinopithecus bieti XM_017871337). Sequences were aligned using the Muscle algorithm(Edgar, 2004).
Detection of recurrent positive selection was carried out using the codeml method in the PAML 4.3 software package(Yang, 2007). First, the cGAS alignment from the simian primate species listed above was fit to two different codon models (M8 and M8a). The M8a model is a neutral model of selection where all codons are constrained to evolve with a dN/dS value ≤ 1. M8, by contrast, allows for one group (“class”) of codons to evolve with dN/dS > 1. Using a likelihood ratio test, we found that the positive selection model (M8) was a significantly better fit to the data than the neutral model (M8a) (p<<0.01). The actual dN/dS calculated for each codon in model M8, and the posterior probability (PP) for each codon being correctly assigned to the dN/dS > 1 class, are illustrated in Figure 4B.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded during experiments and assessment. No data or subjects were excluded from all the quantifications
Data represent the mean of at least three independent experiments, and error bars denote SEM unless specified otherwise. Exact values for n and what n represents are listed in each figure legend. For Figures 2C–F, 3B–D, 3G, 3H, 5A, 5B, 5D–F, 6F, 7D, 7E, 7I, 7J, S3A, S4A–C, S4E, S4G, S4I, S6A–D, S6F, S8A, S8B, S8D, S8E and S9C–G, n represents the number of biological replicates per group. For Figure S1E–J, n=3 (n represents biological replicates per group) BMDM cells from 2–3 mice. For mouse infection experiments, Figures 1A–F and Figure S1A–D, n indicates individual mice. A two-tailed Student’s t test was used for statistical analysis except the mouse survival analysis. The mouse Kaplan-Meier survival curve was generated using GraphPad Prism 5, and the log-rank test applied to the mouse survival data was also performed in GraphPad 5. For all statistical analysis, *p<0.05, **p<0.01, ***p<0.005.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNAseq data of THP-1 mock infected or infected with HSV-1UL37WT or HSV-1UL37C819S: GSE114966
Supplementary Material
Highlights.
The HSV-1 UL37 tegument protein antagonizes the cGAS-STING pathway in mice.
UL37 deamidates a critical Asp on cGAS of humans and mice but not many non-human primates
cGAS deamidation abolishes cGAMP synthesis and downstream innate immune activation
Variation of the cGAS UL37 deamidation site defines HSV-1 species-specific permissiveness
Acknowledgments
We thank Drs. Fanxiu Zhu (Florida State University), Zhijian James Chen (UT southwestern medical center), Dustin Hancks (UT southwestern medical center) and Nels Elde (University of Utah) for cells and plasmids. We thank Dr. Yibu Chen (USC Norris Medical Library Bioinformatics Service) for assisting with sequencing data analysis (the bioinformatics software and computing resources used in the analysis are funded by the USC Office of Research and the Norris Medical Library). We thank Drs. Jon Hao (Poochon Scientific), Steve Gygi and Ross Tomaino (Taplin Mass Spectrometry) for protein deamidation analysis. This work is supported by grants from NIH [(R35DE027556, R01DE026003, R01CA221521, R21AI134105 (PF) and P01CA180779 (JUJ), R01GM093086 (SLS), and F30AI112277 (ACS)], Joint Research Fund for Oversea Chinese, Hong Kong and Macao Scholars (81528011, PF), and National Key Research and Development Program of China (2016YFD0500300, 2016YFC1200200, ZX). SLS is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease. Core services were provided by Norris Cancer Center grant P30CA014089-34.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.Zhan and P.F. conceived the study. J.Zhan, J.Zhao, S.X., J.L., S.H., Y.Z., L.X., N.X., T.L., K.L., Z.X., C.H., and P.F. performed the experiments. G.J.S., L.C., and J.J. contributed valuable reagents and helped with data analysis. A.C.S and S.L.S. performed evolutionary analysis. J. Zhang and P.F. prepared the manuscript. All authors read and approved the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, Fredericks AC, Tripathi S, Zhu T, Pintado-Silva J, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nature microbiology. 2017;2:17037. doi: 10.1038/nmicrobiol.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012;8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, Hopfner KP, Gack MU. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-Imediated immunity. Nat Immunol. 2018;19:53–62. doi: 10.1038/s41590-017-0005-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Yao Q, Li S, Ding X, Lu Q, Mao H, Liu L, Zheng N, Chen S, Shao F. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science. 2010;329:1215–1218. doi: 10.1126/science.1193844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yu H, Zheng X, Peng R, Wang Q, Zhou Y, Wang R, Wang J, Qu B, Shen N, et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog. 2017;13:e1006156. doi: 10.1371/journal.ppat.1006156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol. 2012;12:687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC. Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet. 2015;11:e1005203. doi: 10.1371/journal.pgen.1005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Zhao J, Song S, He X, Minassian A, Zhou Y, Zhang J, Brulois K, Wang Y, Cabo J, et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol Cell. 2015;58:134–146. doi: 10.1016/j.molcel.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, Yin L, Shu HB. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity. 2016;45:555–569. doi: 10.1016/j.immuni.2016.08.014. [DOI] [PubMed] [Google Scholar]
- Hurst LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486. doi: 10.1016/s0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Fitzgerald K, Kurt-Jones E, Finberg R, Knipe DM. Herpesvirus tegument protein activates NF-kappaB signaling through the TRAF6 adaptor protein. Proc Natl Acad Sci U S A. 2008;105:11335–11339. doi: 10.1073/pnas.0801617105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JS, Giotis ES, Moncorge O, Frise R, Mistry B, James J, Morisson M, Iqbal M, Vignal A, Skinner MA, et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature. 2016;529:101–104. doi: 10.1038/nature16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou DI, Kim ET, Meyerson NR, Pancholi NJ, Mohni KN, Enard D, Petrov DA, Weller SK, Weitzman MD, Sawyer SL. An Intrinsically Disordered Region of the DNA Repair Protein Nbs1 Is a Species-Specific Barrier to Herpes Simplex Virus 1 in Primates. Cell Host Microbe. 2016;20:178–188. doi: 10.1016/j.chom.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Damania B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe. 2016;19:150–158. doi: 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Meyerson NR, Zhou L, Guo YR, Zhao C, Tao YJ, Krug RM, Sawyer SL. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe. 2017;22:627–638. e627. doi: 10.1016/j.chom.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian A, Zhang J, He S, Zhao J, Zandi E, Saito T, Liang C, Feng P. An Internally Translated MAVS Variant Exposes Its Amino-terminal TRAF-Binding Motifs to Deregulate Interferon Induction. PLoS Pathog. 2015;11:e1005060. doi: 10.1371/journal.ppat.1005060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MR, Loo YM, Horner SM, Gale M, Jr, Malik HS. Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol. 2012;10:e1001282. doi: 10.1371/journal.pbio.1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada T, Kim M, Mimuro H, Suzuki M, Ogawa M, Oyama A, Ashida H, Kobayashi T, Koyama T, Nagai S, et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature. 2012;483:623–626. doi: 10.1038/nature10894. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387:725–729. doi: 10.1038/42735. [DOI] [PubMed] [Google Scholar]
- Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, Jung JU. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015;13:440–449. doi: 10.1016/j.celrep.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P. Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J Biol Chem. 2002;277:48664–48676. doi: 10.1074/jbc.M207029200. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe. 2015;18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, Fan Z. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol. 2016;17:369–378. doi: 10.1038/ni.3356. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, Pich A, Brinkmann MM, Schulz TF. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci U S A. 2016a;113:E1034–1043. doi: 10.1073/pnas.1516812113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Feng H, Zhao J, Feldman ER, Chen SY, Yuan W, Huang C, Akbari O, Tibbetts SA, Feng P. IkappaB Kinase epsilon Is an NFATc1 Kinase that Inhibits T Cell Immune Response. Cell Rep. 2016b;16:405–418. doi: 10.1016/j.celrep.2016.05.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, He S, Wang Y, Brulois K, Lan K, Jung JU, Feng P. Herpesviral G protein-coupled receptors activate NFAT to induce tumor formation via inhibiting the SERCA calcium ATPase. PLoS Pathog. 2015a;11:e1004768. doi: 10.1371/journal.ppat.1004768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhu L, Lu X, Feldman ER, Keyes LR, Wang Y, Fan H, Feng H, Xia Z, Sun J, et al. Recombinant Murine Gamma Herpesvirus 68 Carrying KSHV G Protein-Coupled Receptor Induces Angiogenic Lesions in Mice. PLoS Pathog. 2015b;11:e1005001. doi: 10.1371/journal.ppat.1005001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Chen ZJ. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014;6:421–430. doi: 10.1016/j.celrep.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Li J, Xu S, Feng P. Emerging Roles of Protein Deamidation in Innate Immune Signaling. Journal of virology. 2016a;90:4262–4268. doi: 10.1128/JVI.01980-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zeng Y, Xu S, Chen J, Shen G, Yu C, Knipe D, Yuan W, Peng J, Xu W, et al. A Viral Deamidase Targets the Helicase Domain of RIG-I to Block RNA-Induced Activation. Cell Host Microbe. 2016b;20:770–784. doi: 10.1016/j.chom.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.