

SUMMARY
The circuitry of the striatum is characterized by two organizational plans: the division into striosome and matrix compartments, thought to mediate evaluation and action, and the direct and indirect pathways, thought to promote or suppress behavior. The developmental origins of these organizations and their developmental relationships are unknown, leaving a conceptual gap in understanding the cortico-basal ganglia system. Through genetic fate mapping, we demonstrate that striosome-matrix compartmentalization arises from a lineage program embedded in lateral ganglionic eminence radial glial progenitors mediating neurogenesis through two distinct types of intermediate progenitors (IPs). The early phase of this program produces striosomal spiny projection neurons (SPNs) through fate-restricted apical IPs (aIPSs) with limited capacity; the late phase produces matrix SPNs through fate-restricted basal IPs (bIPMs) with expanded capacity. Notably, direct and indirect pathway SPNs arise within both aIPS and bIPM pools, suggesting that striosome-matrix architecture is the fundamental organizational plan of basal ganglia circuitry.
eTOC BLURB
In this article, Kelly et al. describe a lineage program embedded in lateral ganglionic eminence progenitors responsible for striosome-matrix compartmentalization in striatum.
INTRODUCTION
The striatum, the gateway of the basal ganglia, receives massive inputs from all areas of the cerebral cortex and the thalamus, and gives rise to the output circuits of the basal ganglia by way of multi-synaptic pathways leading to the brainstem and thalamus. Neural activity in the striatum is influenced by dopaminergic afferents from the substantia nigra, serotonergic afferents from raphe nuclei, cholinergic inputs from the brainstem, and other neuromodulatory circuits. The striatum thus constitutes a key substrate through which diverse functional regions of the neocortex and thalamus modulate not only downstream motor programs for the initiation of voluntary behaviors, but also neural systems underpinning emotion, motivation, evaluation and learning (Graybiel, 2008; Hikosaka et al., 2014).
The majority of neurons in the striatum are output spiny projection neurons (SPNs) that follow two fundamental organizational schemes. First, the striatum has a striking compartmental organization by which labyrinthine neurochemically specialized zones called striosomes lie embedded within the much larger matrix compartment (Graybiel and Ragsdale, 1978). This striosome-matrix architecture contributes to the functional input and output connectivity of the striatum. The SPNs of striosomes receive preferential input from particular limbic regions and have privileged output to dopaminergic neurons of the substantia nigra and indirectly to the lateral habenula, thus engaging crucial dopamine-and serotonin-related neuromodulatory systems (Crittenden and Graybiel, 2011). By contrast, SPNs of the matrix compartment receive inputs from sensorimotor and associative cortical regions and in turn project to the main pallidonigral output nuclei of the basal ganglia that target downstream premotor regions and thalamocortical circuits (Flaherty and Graybiel, 1994; Graybiel, 2008; Hikosaka et al., 2014). Whereas striosomal circuits have been implicated in evaluative functions related to value-based decision-making and reinforcement-based behaviors, the matrix appears to be involved in the translation of cortical action plans and strategies to basal ganglia circuits involved in action execution (Amemori et al., 2011; Friedman et al., 2015). The developmental basis of this striosome and matrix segregation is not well understood.
Superimposed upon striosome-matrix compartmentalization is a split between the projections of SPNs to the basal ganglia output nuclei: the pallidum and the substantial nigra pars reticulata (SNr) (Gerfen, 1992; Kim and Hikosaka, 2015). SPNs projecting to the internal pallidum and SNr constitute the so-called direct pathway SPNs (dSPNs), considered to release basal ganglia-mediated inhibition or inhibition-excitation sequences and to promote action. By contrast, the SPNs projecting to the external pallidum give rise to the indirect pathway (iSPNs), conventionally considered to inhibit actions, potentially those with unwanted characteristics (Freeze et al., 2013). The direct-indirect pathway dichotomy has been formulated as a center-surround system in which wanted movements are promoted by the direct pathway and competing, interfering motor programs are inhibited (Mink, 1996) and fresh evidence suggests different roles in aversive versus rewarding actions (Kravitz et al., 2012). With its large size and output organization, the matrix compartment is the main source of the direct and indirect pathways, but striosomes as well as matrix contain both dSPNs and iSPNs, at least as categorized by the expression of dopamine D1 (in dSPNs) and D2 (in iSPNs) receptors (Crittenden and Graybiel, 2011; Fujiyama et al., 2011).
Despite strong evidence for the coexistence of the striosome and matrix compartments and the direct and indirect pathways, little is known about the relationship between these two fundamental axes of striatal organization. A clue to the importance of developmental mechanisms underlying the compartmental divisions first came from findings that SPNs making up striosomes and matrix have different birthdates, with striosomal SPNs generated earlier than those of matrix (Graybiel and Hickey, 1982; Newman et al., 2015; van der Kooy and Fishell, 1987). Here we link this evidence to neural progenitors and lineage mechanisms known to be crucial in the striatal developmental program (Rubenstein and Campbell, 2013). The neuroepithelium of the lateral ganglionic eminence (LGE) of the embryonic ventral telencephalon consists of diverse types of progenitors that emerge and differentiate across several phases of striatal neurogenesis (Turrero García and Harwell, 2017). These include regenerative radial glial progenitors (RGs), which give rise to apical intermediate progenitors (aIPs) such as short neural progenitors (SNPs) and subapical progenitors (SAPs), as well as basal intermediate progenitors (bIPs) (Pilz et al., 2013). Although certain lineage relationships and neurogenic capacity of some of these progenitor types have been characterized (e.g., Pilz et al., 2013), their developmental trajectory during LGE neurogenesis and especially their link to different SPN types and striatal circuit organization remain unknown. Tapping into the molecular mechanisms expressed in different progenitor types, we performed systematic genetic fate mapping to resolve different progenitor types and trace their developmental trajectories from lineage progression to the distinct SPN types that they give rise to.
Here, we demonstrate that striosomal and matrix SPNs are sequentially generated from a RG lineage program through sequential allocation of distinct types of IPs with different amplification capacities. Whereas the early phase of this program produces striosomal SPNs through fate-restricted Ascl1+ aIPSs with limited neurogenic capacity, the late phase amplifies matrix SPN production through the deployment of fate-restricted Ascl1+/Dlx1+ bIPMs with expanded neurogenic capacity. During the final phase, Dlx1+ bIPs specifically generate SPNs of the annular compartment adjoining striosomes. Notably, a similarly clear temporal and progenitor type split was not observed for the genesis of D1- and D2-receptor bearing direct and indirect pathway SPNs. We found that each of these pathways is derived in parallel from both aIPSs and bIPMs throughout the course of RG lineage progression, with a bias toward D1 early and D2 late in post-mitotic stages of differentiation. These findings suggest that a primary LGE lineage program gives rise to striosome-matrix compartmentalization, superimposed upon which a secondary and different mechanism gives rise to direct and indirect pathway SPNs within each compartment. Thus the major components of striatal architecture are rooted in a radial glia lineage program at the inception of striatal development, which differentiates the striosome and matrix compartments through sequential phases of lineage progression and distinct intermediate progenitor types. These findings establish a lineage framework for exploring the assembly of cortico-basal ganglia circuitry and should provide new clues to the differential vulnerabilities of the striosome-matrix compartments and direct-indirect pathways in human disease states.
STAR METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Z. Josh Huang (huangj@cshl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
The following mouse strains were used for fate-mapping experiments studying striatal projection neurons: Ascl1-CreER (Kim et al., 2011), Dlx1-CreER (Taniguchi et al., 2011), Nkx2.1-ires-FLP-O (He et al., 2016), Ai14 (Madisen et al., 2010), Ai65 (Madisen et al., 2015), and Tis21–2A-CreER (unpublished). A gene-targeting vector for Tis21–2A-CreER was generated using a PCR-based cloning approach (Taniguchi et al., 2011) to insert a 2A-CreER construct immediately after the STOP codon of the Tis21 gene. The targeting vector was linearized and transfected into a 129SVj/B6 F1 hybrid ES cell line (V6.5, Open Biosystems). G418-resistant ES clones were first screened by PCR and then confirmed by Southern blotting using probes against the 5’ and 3’ homology arms of the targeted site. The loxP-Neo-loxP cassette in the founder line was removed by breeding with CMV-Cre transgenic mice (JAX 006054) before Tis21-CreER mice were used in experiments. Breeder mice used in fate mapping experiments were backcrossed 5 generations to Swiss-Webster or CD-1 background for genetic homogeneity and due to these backgrounds having reduced susceptibility to side effects from tamoxifen dosing in these strains. Tis21–2A-CreER mouse line was deposited to Jackson Laboratory for maintenance and sharing for research purposes.
Ascl1-CreER, Dlx1-CreER, or Tis21-CreER mice were bred separately to Ai14 reporter mice to label SPNs in fate-mapping experiments. Mouse brain tissue was harvested for histology either during embryonic development or on P28 according to the inducible fate-mapping protocols described below. Similarly, to perform intersectional fate mapping of interneurons Ascl1-CreER mice were bred with double-transgenic Nkx2.1-ires-FLP-O;Ai65 mice. In such experiments, the Ai65 reporter labeled cells with tdTomato only when both the lox-Stop-lox and Frt-STOP-Frt cassettes were excised by a Cre and Flp driver within a single cell. Datasets for all fate-mapping experiments were assembled from groups of a minimum of two litters for each experiment, after processing multiple male and female brains processed from each. For all experiments, no relevant morphological or cellular composition differences were observed between male and female brains.
All mouse colonies at Cold Spring Harbor Laboratory (CSHL) were maintained in accordance with husbandry protocols approved by the IACUC (Institutional Animal Care and Use Committee reference number 16-13-09-8). Mice were housed by gender in groups of 2–4 with access to food and water ad libitum and 12-hour light-dark cycle, except when placed in a breeder pair. Ascl1-CreER and Dlx1-CreER mice were also sent to the Massachusetts Institute of Technology (MIT), re-derived and maintained under a standard light/dark cycle with free access to food and water in MIT housing facilities. All experimental procedures were approved by the Committee on Animal Care at MIT and were performed in accordance with the U.S. National Research Council Guide for the Care and Use of Laboratory Animals.
METHOD DETAILS
Inducible Genetic Fate Mapping
Genetic fate-mapping experiments were carried out in parallel at CSHL and MIT. Inducible fate-mapping experiments were conducted by crossing either Dlx1-CreER or Ascl1-CreER mice to the Cre-dependent Ai14 reporter. Intersectional inducible fate-mapping experiments were conducted by crossing Nkx2.1-ires-FLP-O;Ai65 double-transgenic mice to a CreER driver line (backcrossed 6 generations to Swiss-Webster background). Upon observing a vaginal plug, with noon of that day counted as E0.5, tamoxifen was administered by gavage to the pregnant female at a desired embryonic time point (Taniguchi et al., 2011). Adult progeny mice with appropriate genotypes were anesthetized with 2.5% avertin before transcardial perfusion with 4% paraformaldehyde (PFA) on P28. Brains were post-fixed in 4% PFA for 12 hours. In 8-hour pulse-chase experiments, embryos were lightly fixed by immersion in 4% PFA starting 8 hours after TM induction and frozen for cryosectioning after overnight cryoprotection in 30% sucrose. Later stage embryos (E14–E18) were perfused with 4% PFA using a 30-gauge needle and syringe before being post-fixing brains for 6 hours and embedding in 1.8% agarose for vibratome sectioning (Taniguchi et al., 2011). Embryos appearing underdeveloped for expected age or exhibiting signs of poor circulation we excluded from pulse chase studies.
Tamoxifen Induction and BrdU Birth Dating
To reduce toxicity to pregnant females and enable sparse cellular labeling, pregnant female mice were submitted to gavage with low to medium dose tamoxifen (dissolved in corn oil by gentle agitation for 24 hours), using an 18-gauge 50 mm gavage tool. Doses were in the range of 0.15 mg/30 g body weight (BW) up to 5 mg/30 g BW (5 mg/Kg BW–165 mg/Kg BW), with most experiments performed using doses below 1.5 mg/30 g BW. For BrdU and EdU birth dating, pulses of BrdU or EdU were intraperitoneally injected into pregnant mice at a dose of 5 mg/100 g BW (2.5 mg/100 g BW for EdU), spaced 4, 24, or 72 hours after TM induction on E10, also previously described by Taniguchi et al. (2011).
Immunohistochemistry and Microscopy
Fixed adult brains were serially sectioned at 55 µm using a vibratome and collected into well plates. Sections were blocked in 10% normal goat serum and 0.1% triton-X100 in 1XPBS, then incubated in primary antibodies (diluted in blocking solution) at 4°C overnight: anti-RFP (rabbit polyclonal antibody, Rockland Pharmaceuticals), anti-MOR1 (rabbit polyclonal, immunostar), anti-Calbindin 28K (rabbit polyclonal, Millipore), anti-CDG1 (rabbit polyclonal, Crittenden et al., 2009), anti-Tyrosine Hydroxylase (rabbit polyclonal, Millipore), anti-Ki67 (rabbit polyclonal, Vector), anti-Ascl1/Mash1 (mouse monoclonal, BD biosciences), anti-BrdU (rat monoclonal Accurate Chemical), anti-TuJ1 (mouse monoclonal, Covance), anti-Parvalbumin (mouse monoclonal, Sigma), anti-Somatostatin (rabbit polyclonal, Millipore), anti-ChAT (rabbit polyclonal, Santa Cruz biotechnology), anti-CTIP2 (mouse monoclonal, Abcam), anti-Nkx2.1 (rabbit polyclonal, Santa Cruz biotechnology), anti-Sox2 (human-derived mouse/rat/human monoclonal, R&D Systems). Sections were then rinsed and incubated with appropriate Alexa Fluor dye-conjugated IgG secondary antibodies (1:800 goat anti-rabbit/mouse/rat, Life Technologies) and mounted in Fluoromount-G (SouthernBiotech). Sections were counterstained with NeuroTrace Nissl Stain (Molecular Probes) or DAPI. For BrdU immunostaining, sections were denatured in HCl (2 N) at 37°C for 45 min, then neutralized in sodium borohydrate buffer (0.1 M pH 8.5) twice for 10 min at room temperature before the normal staining protocol.
At MIT, 40-µm thick coronal frozen sections through the striatum were cut with a sliding microtome and were stored in a solution of 0.1M NaKPO4 and 0.4% Na azide until use. For immunofluorescence immunohistochemistry, sections were rinsed in 3×2 min in 0.01M phosphate buffered saline (PBS) containing 0.2% Triton-X-1000 (Tx) and were blocked in tyramide signal amplification (TSA) blocking reagent (Perkin Elmer) for 20 min. The sections were then incubated with primary antibodies in TSA blocking reagent in PBS-Tx for 24 hours at 4°C with gentle shaking: anti-RFP (rabbit/mouse polyclonal/monoclonal, Rockland Pharmaceuticals), anti-MOR1 (rabbit polyclonal, Immunostar/Abcam), anti-CDG1 (rabbit polyclonal, Graybiel laboratory), anti-parvalbumin (mouse monoclonal, Millipore), anti-somatostatin (rabbit polyclonal, Millpore), and anti-ChAT (rabbit polyclonal, Millipore). After 24-hour incubation, sections were rinsed 3×2 min in PBS-Tx and incubated for 2 hours in secondary antibody solution with the appropriate Alexa Fluor dye-conjugated IgG secondary antibodies (1:300 goat anti-rabbit/mouse, ThermoFisher Scientific). Following incubation, sections were rinsed 3×2 min in 0.1M PBS, mounted onto subbed glass slides, and were coverslipped using ProLong Antifade Reagent containing DAPI (ThermoFisher Scientific).
Double Fluorescent in situ Hybridization
To obtain fresh frozen tissue for in situ hybridization, several Dlx1-CreER:Ai14 and Ascl1-CreER:Ai14 mice were selected following different time points of tamoxifen induction. Fresh tissue was harvested, rinsed in 0.9% saline, and submerged in Tissue-Tek O.C.T embedding media (VWR) in a cryomold container. This mold was quickly placed in 2-methyl butane (Sigma), which then was placed in liquid nitrogen until the OCT block was frozen solid. The resulting frozen tissue was stored in −80°C freezer before cryosectioning. Two-probe fluorescent in situ hybridization was performed on fresh frozen 12-µm thick cryosections of P28 mouse brains using the QuantiGene ViewRNA ISH assay from Affymetrix. Briefly, tissue was treated with formaldehyde fixation, protease digestion, target and label probe incubation, and buffer washing according to the QuantiGene Affymetrix protocol. Tissue was then co-labeled with fluorescent Nissl and imaged using a DeltaVision epifluorescent microscope (details below).
Imaging and Serial Reconstruction of Whole Brains
Fate-mapped neurons were imaged from serially mounted sections on a Zeiss LSM 780 confocal microscope (CSHL St. Giles Advanced Microscopy Center) and Zeiss LSM 510 confocal microscope with ZEN software (Ragon Institute of MGH, MIT and Harvard), including all featured images with immunostaining. Volumetric whole brain imaging of native fluorescent signal was completed with a 5-µm Z-step using previously described methods for serial two-photon tomography (Kim et al., 2015, Ragan et al., 2012). Briefly, brains were embedded in oxidized agarose and placed in a motorized stage in a TissueCye 1000 (Tissuevision). Whole brains were then imaged as 12 (X) x 16 (Y) tiles with 1 µm x 1 µm resolution for 270 Z sections (50-µm interval) or 1500 Z sections (5-µm interval). Image datasets were stitched and reassembled by custom built software and viewed as stacks or 3D volumes. Image processing was completed using ImageJ/FIJI software with all alterations applied only to entire images. Three-dimensional image stacks of striatal cells from in situ data were acquired at room temperature with a DeltaVision restoration microscope system consisting of an Olympus IX71 inverted microscope equipped with Olympus 20X/0.75 U Apo 340 dry objectives and a Photometrics coolsnap HQ2 camera configured at 0.1122 µm/pixel. Filters used were 430/24 ex/470/24 em and 645/30 ex/705/72 em. Z-stack images of cells of cells were subjected to deconvolution by using Deltavision softworx 6.5.1 software (Applied Precision). The maximum projection or the middle optical section of the de-convolved sections was exported to TIFF format and then processed by FIJI/ImageJ for the final images.
QUANTIFICATION AND STATISTICAL ANALYSIS
Manual scoring and counting of SPN cell types in striatal compartments was completed using Neurolucida 11 software either directly from slides on an Olympus Bx51 microscope or from serially collected Z-stack tile scan images (confocal or epifluorescent). Specifically, striosomes were identified positively as groups of clustered cells with thick spinous dendrites co-localizing to MOR1-positive zones and negatively by the absence of both Calbindin 28K (CB) and CDG1 co-localization, which are specifically expressed in the matrix compartment of the striatum. Unless otherwise indicated, each quantified sample consists of cell counts from 6 representative rostral-caudal coronal sections of striatum per hemisphere, with multiple (2–10) brains from at least two litters sampled in all cases. Multiple individuals from both MIT and CSHL scored image datasets independently before results were compared and pooled to ensure validity of findings. N represents number of animals, sections, or cells as indicated in all figure legends, error bars represent standard error of the mean (SEM) unless otherwise indicated. Cell densities in striosome and matrix compartments were calculated in two in areas bounded by drawing contours around the MOR1+ striosomes or CDG1+ matrix and counting the number of RFP+ cells within each compartment, averaging the cell density from replicate sections as done for all other cell counts. Comparisons of striosome content in rostral (bregma 1.54–0.5 mm) versus caudal (bregma 0.45–0.82 mm) sections through the striatum at E12.5 TM (Ascl1-CreER) and E13.5 TM (Dlx1-CreER) were quantified in counts in serial sections (on average 42 sections of 55-µm thickness). Rostral/caudal levels were defined according to the 4th edition of the Mouse Brain in Stereotaxic Coordinates atlas (Paxinos and Franklin, 2001). Finally, to quantify the co-localization of BrdU/EdU with RFP+ cells, the ratio of BrdU+RFP+ and EdU+RFP+ cells in striosomes or matrisomes to total RFP+ cells was determined for a minimum of three different brains of two different litters in all cases. Unpaired or paired Student’s t-tests were used to analyze data with two groups (see each figure legend for details), and one-way ANOVA and Tukey’s honest difference significance tests were used for unbiased comparisons of multiple time-points within a series. The threshold for significance was reached in statistical tests in which a p value was < 0.05 and the number of animals or cells for each graph were listed in figure legends and graphs where appropriate. Results were displayed graphically in all figures and statistics completed using Graphpad Prism software.
DATA AND SOFTWARE AVAILABILITY
No original software or data (sequences structures, biological macromolecules, or microarray) requiring repository were generated in this study.
RESULTS
Early LGE Radial Glial Cells Sequentially Give Rise to Both Apical and Basal Intermediate Progenitors and to Both Striosomal and Matrix SPNs
The formation of the LGE around embryonic day (E) 9.5 along the subpallium is followed shortly by the onset of neurogenesis, when proliferative neuroepithelial cells (NEs) begin to transform into neurogenic radial glial progenitors (RGs) (Sousa and Fishell, 2010). Although all striatal SPNs are generated from LGE RGs, recent studies have identified multiple types of progenitors with distinct molecular and morphological characteristics that are derived from RGs at different embryonic times (Pilz et al., 2013; Turrero García and Harwell, 2017). These include apical progenitors (aIPs, e.g. SNPs and SAPs) and also basal progenitors (bIPs). How these different progenitor types relate to distinct striatal SPN types remains largely unknown. We used several inducible CreER drivers to perform comprehensive genetic fate mapping of multiple LGE progenitor types.
Among the markers expressed in telencephalic progenitors (i.e., pallium and subpallium), the anti-proliferative protein TIS21 is unique in its specific expression in all progenitors undergoing neurogenic divisions, including subsets of RGs, aIPs, and bIPs (Attardo et al., 2008). To fate map different types of neurogenic progenitors, especially self-renewing RGs in the process of neurogenesis, we generated a knock-in Tis21-CreER driver and demonstrated that tamoxifen (TM) -induced activation of the Ai14 reporter reliably and efficiently captured neuron-producing progenitors (Figures 1 and S1). The Tis21-CreER driver thus provided a unique and powerful fate-mapping tool for LGE neurogenic RGs starting from the very onset of neurogenesis. In this context, transiently neurogenic IPs were distinguished from self-renewing RGs using multiple criteria, including high expression of Ascl1 and morphology of basal processes (Harwell et al, 2015). This enabled tracing of the RG lineage progression through sets of embryonic IPs to their SPN progenies in the postnatal striatum (Figures 1A and1B). At the onset of neurogenesis at ~E10, 8-hour pulse-chase of Tis21+ progenitors labeled both RGs and aIPs (Figures 1C, 1F and 1G). We could not strictly distinguish between SNPs and SAPs, as these aIP subtypes are defined by their cell division patterns best captured by live imaging experiments (Pilz et al., 2013). Importantly, 48 hours after E10 TM induction (i.e., at E12), dozens of RFP+ RGs and aIPs remained in the ventricular zone (VZ), indicating that fate-mapped RGs self-renewed and continued to generate aIPs (Tables 1 and S1). Sparse labeling allowed identification of elongated cell clusters comprised of RGs with multiple closely-associated IPs and newly postmitotic neurons. These clusters became increasingly spatially isolated over time and likely represented clonal descendants from an initial neurogenic RG (Figures 1D, 1H, 1I and S1D). Even at 96 hours after E10 TM induction (i.e., at E14), dozens of RFP+ RGs still remained in VZ of each brain sampled, indicating persistent self-renewal (Figures 1E, 1J-1L, S1E and S1F; Tables 1 and S1). Particularly large cell clones were frequent, which consisted of a “founder RG”, multiple Ascl1 + IPs, and larger number of postmitotic neurons likely migrating along the RG radial fiber (Figures 1L and S1F; Table S2, Supplemental Movie 1). Whereas E10-labeled RGs gave rise to aIPs contacting the lateral ventricle from E10- E12 (Figures 1D, 1H and 1I), they mainly led to the generation of bIPs in the subventricular zone (SVZ) by E14 (Figures 1E and1J-1L). All Ascl1+RFP+ contacted the lateral ventricle at 8 hours following E10 TM induction, distinguishing this population as aIPs and not bIPs.
Figure 1. A Radial Glial Lineage in LGE Generates Both Striosomal and Matrix SPNs, See also Figure S1 and Tables S1 and S2.
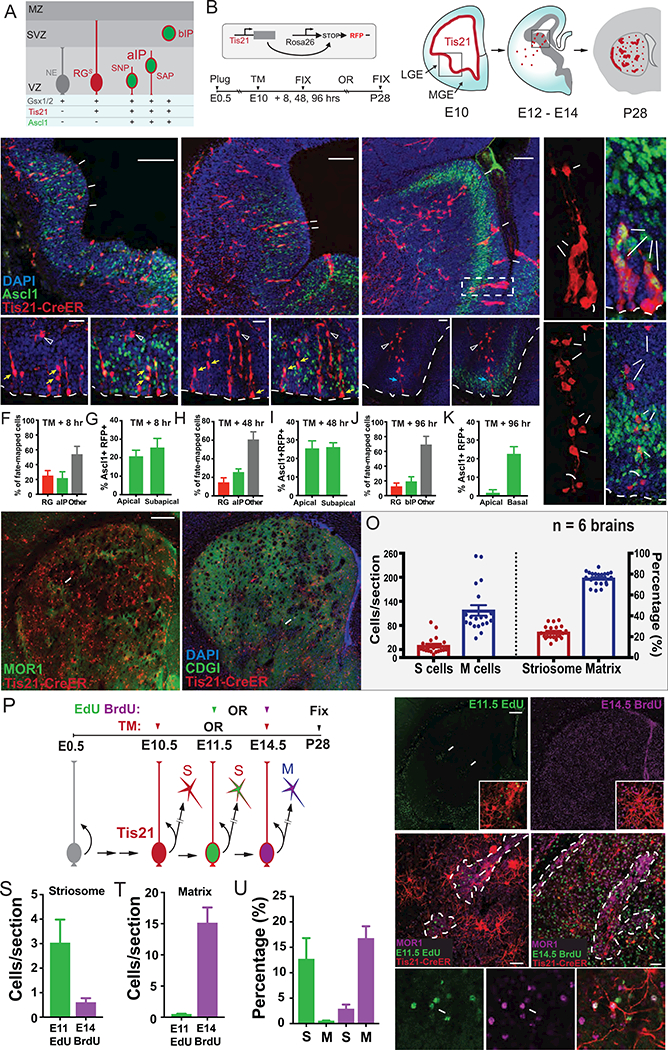
(A) Schematic of LGE progenitor types, including proliferating neuroepithelial cells (NE), self-renewing and neurogenic radial glial cells (RGS), apical intermediate progenitors (aIPs), and basal intermediate progenitors (bIPs). aIPs include short neural precursors (SNP) and sub-apical progenitors (SAP). These progenitor types are marked by Gsx1/2, Tis21, Ascl1 expression. Tis21 marks neurogenic RGs, aIPs and bIPs. VZ: ventricular zone. SVZ: subventricular zone. MZ: mantle zone.
(B) Scheme of genetic fate mapping using Tis21-CreER;Ai14 mice. Upon TM induction, Tis21+ neurogenic progenitors are marked throughout the VZ (E10). Newborn neurons subsequently migrate to the striatum; self-renewing RGs remain in the VZ (E12-E14). Fate-mapped SPNs populate both the striosome (red patches) and matrix (red dots) compartments (P28).
(C) 8-hour (hr) pulse-chase analysis of Tis21+ progenitors (RFP, arrows in upper panel) (boxed region in B) by TM induction at E10.5 with immunostaining for Ascl1 and DAPI. Bottom panels depict RGs (Ascl1-, arrowhead) with radial processes, aIPs (putative SNP/SAPs) with soma located away from the lateral ventricle (Ascl1+, yellow arrow), and postmitotic neurons (open arrowhead).
(D) 48-hr pulse-chase of Tis21+ progenitors (arrows) from E10.5. Bottom panels show that self-renewing RGs (Ascl1−, arrowhead) remain in VZ at E12.5 with aIPs (including a putative Ascl1+ SNP; yellow arrows), and postmitotic neurons (open arrowhead).
(E) 96-hr pulse-chase analysis of Tis21+ progenitors (arrows) from E10.5, showing isolated clusters of progenitors and newborn neurons in the LGE at E14. Bottom panels show a RG (arrowhead) with a long radial fiber and associated Ascl1+ bIP in the SVZ (blue arrows) and postmitotic neurons reaching the developing striatum (open arrowhead).
(F, H and J) Percentage of fate-mapped RGs (Ascl1-) versus aIPs (Ascl1+) at 8 (F), 48 (H) and 96 (J) hr following E10.5 TM. “Other” represents cells that could not be readily discerned as RG or aIP. At 96 hr, Ascl1+ cells exclusively localized to SVZ and were scored as bIPs.
(G, I and K) Distribution of Ascl1+RFP+ progenitors within the LGE germinal zone at 8 (G), 48 (I) and 96 (K) hr following E10.5 TM. Apical progenitors, SAPs, and basal progenitors were scored.
(L) Two examples of E10.5 fate-mapped proliferative clusters shown at E14.5 consisting of a putative RG with a radial process running through intermingled Ascl1+ bIPs and migrating postmitotic neurons Top cluster correlates with the dashed box in (E). See also Figure S1.
(M and N) SPNs fate-mapped from E10.5 Tis21+ RGs distribute broadly to both the striosome (arrow; co-labeling with MOR1 in M) and matrix (co-labeled with CDGI in N).
(O) Number (left Y axis) and percentage (right Y axis) of fate-mapped SPNs localized to striosomes and matrix at P28 following TM induction of Tis21-CreER;Ai14 mice at E10.5. Dots represent individual 50 µm sections selected at 6 representative rostral caudal levels of 6 brains. Error bars = SEM.
(P) Scheme of combined fate-mapping and birth dating in Tis21-CreER;Ai14 mice using sequential injections of EdU (E11.5), and BrdU (E14.5) after E10.5 TM induction. Brains were harvested at P28 to identify co-labeling of RFP with EdU or BrdU.
(Q) Distribution of EdU+ cells in P28 striatum following E11 injection; arrows mark clusters of SPNs in striosomes. Inset shows fate-mapped neurons co-labeled by EdU.
(R) Distribution of BrdU+ cells in P28 striatum following E14 injection, showing broad distribution throughout striatum. Inset shows fate-mapped SPNs co-labeled with BrdU injection (magenta).
(S and T) Quantification of fate-mapped cells per section that incorporated EdU or BrdU within striosomes (S) and matrix (T), respectively. The vast majority of E11 EdU+ fate-mapped SPNs adopt S cell fate, while the vast majority of E14.5 BrdU+ fate-mapped SPNs adopt M cell fate (n = 3 brains from 2 litters). Scale bars: 50 µm
(U) Percentage of fate-mapped S versus M cells that co-incorporated E11.5 EdU and E14.5 BrdU, respectively.
(V) E10.5 fate-mapped and E11 born (EdU+) SPNs distribute to MOR1+ striosomes (outlined).
(W) E10.5 fate-mapped and E14 born SPNs (BrdU+) distribute to matrix (arrows) outside of MOR1+ striosomes (outlined).
(X) E10.5 fate-mapped cell (right) with co-labeling from both E11 EdU (left) and E14 BrdU (middle) injections.
Scale bars: 100 µm (top), 20 µm (bottom) in C, D, E; 300 µm in M, Q; 50 µm in V.
Table 1. Average Number of Progenitors Per Brain in Tis21-CreER;Ai14 Mice.
Scored as RG, aIP, bIP, and the total number RFP+ cells within the ventricular and subventricular zone of the LGE at 8, 48, or 96 hr following E10 TM induction.
| Average number of fate-mapped progenitors per brain* | ||||
|---|---|---|---|---|
| Radial Glia (Ascl1-) | Apical (Ascl1+) | Basal (Ascl1+) | Total RFP+ cells per LGE | |
| 8 hour pulse chase | 106 | 79 | 12 | 339 |
| 48 hour pulse chase | 35 | 28 | 31 | 149 |
| 96 hours pulse chase | 26 | 2 | 42 | 226 |
Rounded to the nearest single cell
To determine the identity of SPNs derived from early Tis21+ RGs, we assayed the striatum on postnatal day (P) 28 (Figures 1M–1O). Both striosomal (S) and matrix (M) SPNs were generated, roughly in a 1:4 ratio (Figure 1O). We then combined genetic fate mapping of RGs at E10 and consecutive birth dating by EdU at E11 and BrdU at E14. We found that RFP+ S cells were predominantly double-labeled by EdU at E11, whereas RFP+ M cells were predominantly double-labeled by BrdU at E14 (Figures 1P–1W). Additionally, rare examples of EdU+BrdU+RFP+ triple-labeled cells were observed in the matrix, suggesting that they derived from a single progenitor that was actively dividing both on E11 (S cell production period) and E14 (M cell production period; Figure 1X). These results suggest that along the lineage progression, the same early Tis21+ RG population first generates S cells and then M cells, likely via production of several types of IPs.
A Set of Early Apical Intermediate Progenitors Are Fate-Restricted to Generate Striosomal SPNs
The proneural protein ASCL1 is restricted more to differentiated neurogenic progenitors than to self-renewing RGs, and it is important in coordinating the balance between neural progenitor proliferation and cell cycle exit toward neurogenesis (Guillemot and Hassan, 2017). ASCL1 expression exhibits two distinct dynamic patterns: an oscillatory pattern of relatively low ASCL1 levels correlating with cell-cycle progression and proliferation, versus sustained expression at higher levels driving neurogenic cell division and neuronal differentiation (Imayoshi et al., 2013; Imayoshi and Kageyama, 2014). Thus the Ascl1-CreER driver is an effective tool for selectively labeling progenitors biased towards adopting a neurogenic path (Sudarov et al., 2011; Figures 2A–2D). Throughout the E10 LGE, we detected both low and high levels of ASCL1 immunoreactivity in cells within the VZ, suggesting intermingling of cells in various proliferative (oscillatory expression) and neurogenic (sustained expression) states, as supported by their expression patterns of progenitor or postmitotic cell markers (Figures 2G, 2H and S2A-SD). We used relatively low-dose TM induction to achieve sparse labeling and to target progenitors with high-level Ascl1 expression, which we verified by co-labeling with anti-ASCL1 antibody (Figure 2H). A brief 8-hour pulse-chase labeling with TM induction in E10.5 Ascl1-CreER;Ai14 embryos reliably labeled aIPs with end-feet at the ventricle surface, with or without basal radial fibers (Figure 2D). At 18 and 24 hours following induction at E10.5, few or no RFP+ progenitors remained in the VZ, in sharp contrast to Tis21+ RGs (Figure 1D), indicating that E10 Ascl1+ progenitors are aIPs, rather than RGs endowed with extensive capacity for self-renewal. It is possible that a lower level of Ascl1 might have been expressed in RGs as well as in aIPs, not captured by our low-dose TM induction. Newly postmitotic RFP+ cells expressed typical SPN markers such as CTIP2 as they reached the striatum, but not Nkx2.1, a marker of GABAergic interneurons derived from the medial ganglionic eminence (MGE), suggesting that all fate-mapped cells were SPNs (Figures S2G-S2J).
Figure 2. Early Ascl1+ aIPs Have Limited Proliferation and Are Fate-Restricted to Produce Striosomal SPNs, Figure S2 and S4.
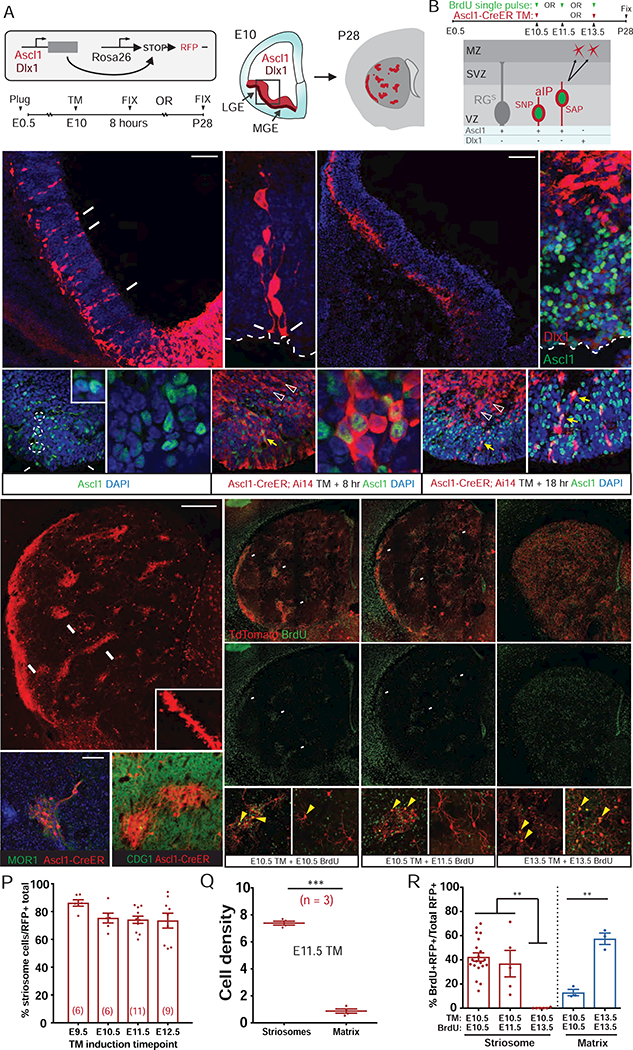
(A) Schematic of E10.5 short- and long-pulse fate mapping by TM induction in Ascl1-CreER;Ai14 or Dlx1-CreER;Ai14 mice. Ascl1 (red) and Dlx1 expression (darker red) in a LGE coronal section. Striosomes in the striatum (red patches) are depicted in a schematic P28 coronal brain section.
(B) Scheme of fate mapping aIPs (SNP, SAP) with E10.5 TM induction followed by BrdU birth dating at E10.5, E11.5, or E13.5 in Ascl1-CreER;Ai14 mice. The distribution of BrdU+ and RFP+ cells was assayed in P28 striatum. Differential expression of ASCL1 and DLX1 in RG, aIPs and postmitotic neurons are depicted.
(C) 48-hr pulse-chase of Ascl1+ progenitors (arrows) labeled by E10.5 TM, arrows indicate aIP with end-feet on the lateral ventricle. Image was taken from the boxed region in (A).
(D) E10.5 aIPs extending end-feet (arrow) to the ventricle surface (dashed line) with short (putative SAP) or no (putative SNP) vertical radial fibers.
(E and F) 8 hr pulse-chase of Dlx1+ cells labeled by E10.5 TM induction in Dlx1-CreER;Ai14 embryo (E) No progenitors in VZ were co-labeled with Ascl1 (F); instead, postmitotic neurons in MZ were prominently labeled. See also Figure S2.
(G) Ascl1+ cells are distributed in a salt and pepper pattern throughout the E10 VZ, with neighboring progenitors exhibiting high and low levels suggestive of oscillatory expression (inset, white circles), including pairs bordering the lateral ventricle (arrows) and in the VZ but not in the MZ. Scale bars: 20 µm (left), 10 µm (right). See also Figure S2.
(H) 8-hr pulse-chase in Ascl1-CreER;Ai14 embryo at E10.5 selectively captures cells with high-level ASCL1 in LGE. Many RFP+ progenitors in the VZ are neurogenic (yellow arrows), while postmitotic neurons (RFP+, arrowheads) cease Ascl1 expression and migrate to the MZ.
(I) 18-hr pulse-chase shows that most Ascl1+ aIPs completed their neurogenic cycles in less than 18 hr, with their postmitotic progenies moving out of the VZ. Only a very small number of Ascl1+ RFP+ aIPs remain in VZ (arrows).
(J and K) SPNs fate-mapped from E10.5 Ascl1+ aIPs are destined for striosomes (arrow), which are MOR1+ (J) and CDGI− (K), in the P28 striatum (dendritic spines in inset).
(M and N) SPNs fate-mapped from E10.5 Ascl1+ aIPs birth-dated by BrdU injection at E10.5 (M) or E11.5 (N), showing BrdU/RFP co-labeling of striosomal SPNs (arrows, top), BrdU only (middle), and high magnification of co-labeled S cells (bottom left) versus RFP+/BrdU− M cells (bottom right).
(O) SPNs fate-mapped and birth dated from E13.5 Ascl1+ progenitors, showing BrdU/RFP co-labeling (top), BrdU only (middle), and high magnification of co-labeled RFP+/BrdU+ M cells and S cells.
(P) Percentage of fate-mapped SPNs localized in striosomes following TM induction at indicated embryonic days. Dots represent individual mice with their numbers in parentheses. Error bars, SEM.
(Q) Average cell density within the striosome versus the matrix compartment (# of cells/percent compartment area) following E11.5 TM induction. ***p < 0.0001.
(R) Quantification of BrdU birth dating in fate-mapped striosome and matrix population, showing the rate of BrdU incorporation at E10.5, E11.5 or E13.5 in S cells (red bars, labeled by E10.5 TM), relative to incorporation of BrdU in M cells at E10.5 or E13.5 (blue bars). Error bars, SEM; **p < 0.001; ***p < 0.0001. Note the absence of S cell production by E13.5.
Scale bars: 50 µm in C, E; 20 µm (left), 10 µm (right) in G, I; 300 µm in J, top of M; 100 µm in bottom of M, K.
We then examined the types of mature SPNs in P28 striatum derived from E10.5-labeled Ascl1+ aIPs (Figures 2J–2N). Strikingly, early Ascl1+ aIPs gave rise almost exclusively to SPNs located within striosomes. These neurons exhibited spiny dendrites and clustered into relatively discrete, widely distributed zones that were positive for the striosome marker mu opioid receptor 1 (MOR1) and negative for the matrix marker CalDAG-GEFI (CDGI) (Figures 2J–2L; Supplemental Movies 2 and 3) (Kawasaki et al., 1998). Both confocal microscopy (Figures S5A and S5C) and serial two-photon tomography (STP; Figure S7; Supplemental Movies 2-4) demonstrated cell morphology and showed that these early aIP-derived SPNs (herein designated as S cells) projected to the dopaminergic domain of the substantia nigra pars compacta (SNc), a defining feature of striosomal SPNs (Crittenden et al 2016; Gerfen, 1984). The small fraction of SPNs located in matrix might correspond to the cells described as “exo-patch SPNs” (Smith et al., 2016), long seen in most strisomal preparations (Graybiel and Hickey, 1982). These are likely cells that have not fully completed migration to striosomes, but are S cell in type, as the axons of labeled SPNs formed dense projections to SNc, characteristic of S cells. Throughout the period from E9.5 to E12.5, these Ascl1+ aIPs almost exclusively generated S cells and not M cells (Figures 2P, 2Q, 5 and S4B).
Figure 5. The Annular Sub-Compartment Is Generated through Intermediate Progenitors at the End of the RG Lineage, See also Figures S8.
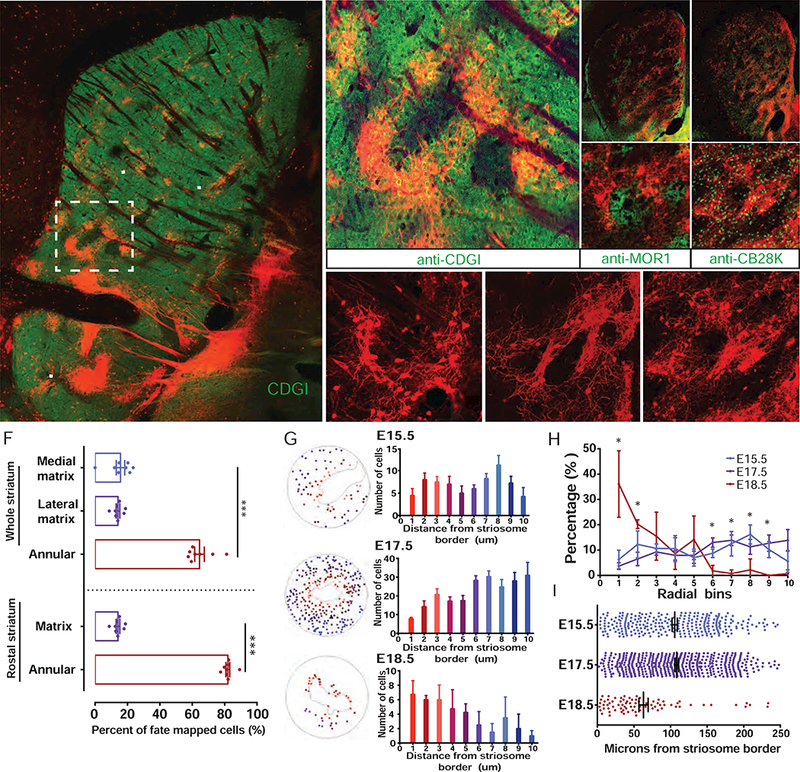
(A) Sagittal section of P28 Dlx1-CreER;Ai14 mouse brain following E18.5 TM induction, showing fate-mapped peri-striosomal M cells (arrows) bordering striosomes devoid of CDGI expression.
(B-D) Rings of peri-striosomal M cells express the matrix markers CDGI (B, rectangle in A) and calbindin D28K (CB28K, D), and adhere tightly to the borders of MOR1+ striosomes (C). S: striosomes.
(E) Both the neurites and somata of fate-mapped peri-striosomal M cells form dense lattice-like structures (arrowheads) at the interface between striosomes (S) and matrix (M).
(F) Fate-mapped SPNs throughout striatum following E18.5 TM induction in Dlx1-CreER;Ai14 mice. Peri-striosomal M cells represent 60–80% of labeled neurons, especially enriched in rostral regions, in addition to a dense band of matrix bordering the lateral ventricle in the medial striatum and sparsely distributed matrix cells throughout the lateral striatum. Error bars, SEM, ***p < 0.0001.
(G) Distribution of fate-mapped matrix neurons following E15.5, E16.5 and E18.5 TM induction in Dlx1-CreER;Ai14 mice, during the onset, peak or closing phase of matrix production. Cells were grouped into ten 20-µm concentric bins moving radially out from the nearest perpendicular point of a traced MOR1+ striosome (red bars closer to and blue bars farther from striosome surface), and the distribution was plotted as a bar graph. n = 4 striosomes from 2 mice per time point.
(H) The proportion of total fate-mapped cells in each 20-µm radial bin following E15.5, E17.5 or E18.5 TM inductions was plotted for all regions of interest, showing a significant enrichment in cells closer than 40 µm to the striosome border in the E18.5 cohort and a significant enrichment in cells between 120 and 180 µm from striosome borders for both the E15.5 and E17.5 cohorts. n = 4 striosomes from 2 mice per time point. *p < 0.05.
(I) Distribution of cells based on distance from striosome borders following E15.5 (purple), E17.5 (blue) or E18.5 (red) TM inductions. Dots represent individual cells, and mean and SEM are indicated in black for each group.
Scale bars: 300 µm in A, C (top pane); 100 µm in B, C (bottom panel), and E.
To further verify that S cells were indeed born from Ascl1+ aIPs within this time-window, we combined TM induction at E10.5 followed with BrdU birth dating of their neuronal progeny at several time points in different litters, including at 4, 24, and 48 hours after TM induction (Figure 2R). These experiments demonstrated that the peak of S cell production from E10.5 aIPs was between E10.5 and E11.5, followed by a sharp decline before E13.5 (Figure 2R). These findings suggest that the early cohorts of Ascl1+ aIPs were fate-restricted to produce striosomal SPNs from E10 to ~E12, and confirmed that early Ascl1+ aIPs do not linger in the VZ for >24 hours.
As the homeobox gene Dlx1/2 is also implicated in LGE neurogenesis and striatal development (Anderson et al., 1997), we performed similar fate mapping using the Dlx1-CreER driver (Taniguchi et al., 2011). Surprisingly, we found that during the early phase of LGE neurogenesis (E10.5), Dlx1 was not expressed in progenitors in the VZ, but instead was almost exclusively confined to postmitotic neurons in the mantle zone (MZ; Figures 2E and2F). This result was substantiated by co-labeling with BIII-tubulin1, which marked all RFP+ cells; and Ki67, which was excluded from all RFP+ cells (Figure S2E and S2F). Fate mapping of these early-born (E9.5-E12.5) Dlx1+ neurons to the mature brain showed that they were almost exclusively S cells, similar to those deriving from Ascl1+ aIPs during the same embryonic time (Figure 4). This result suggests that Dlx1 likely acts down-stream of Ascl1 during this early phase, possibly in regulating the migration and maturation of postmitotic SPNs.
Figure 4. Comprehensive Time-Course of IP-Mediated Neurogenesis Reveals a Rapid Transition from Striosome to Matrix SPN Production, See also Figures S4 – S7.
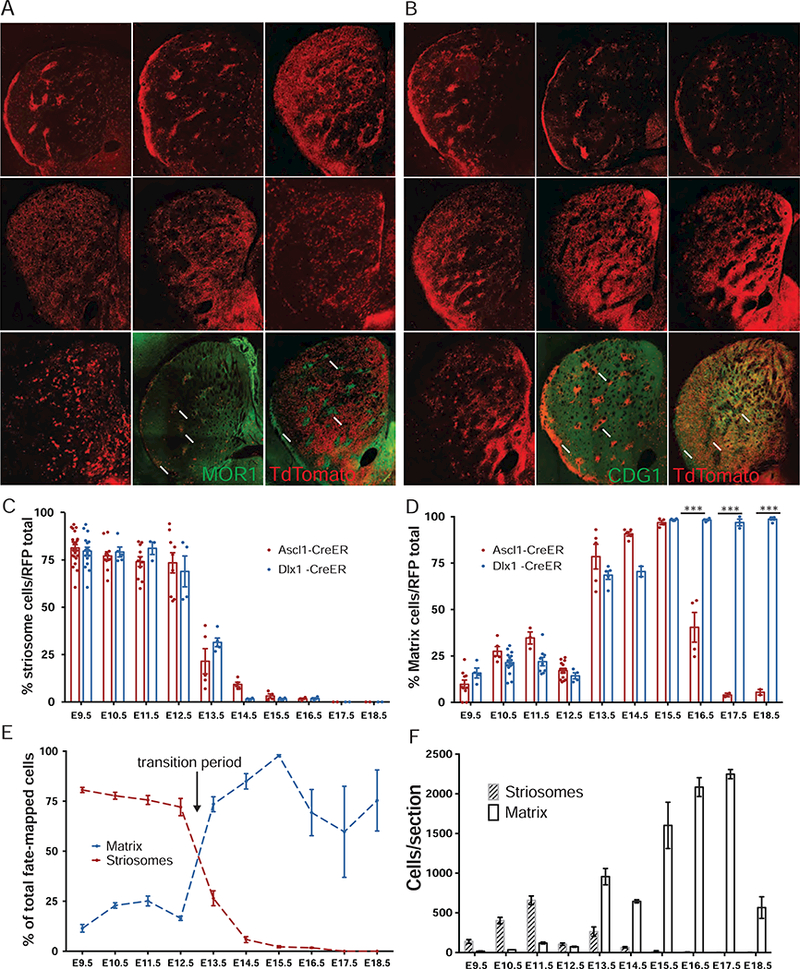
(A) Representative striatum panels of TM time points in Ascl1-CreER;Ai14 mice, including MOR1 co-labeling at peak striosome (E10.5) versus peak matrix (E14.5) cell birth times. Scale bars: 300 µm.
(B) Representative striatum panels of TM induction time points in Dlx1-CreER;Ai14 mice, including CDGI co-labeling at peak striosome (E10.5) versus matrix (E17.5) cell birth times.
(C) Normalized histogram of striosomal SPN production from Ascl1+ (red) and Dlx1+ (blue) progenitors from E9.5 to E18.5. Error bars, SEM.
(D) Normalized histogram of matrix SPN production from Ascl1+ and Dlx1+ progenitors from E9.5 to E18.5. ***p < 0.0001.
(E) Total relative production of striosomes and matrix from Dlx1+ and Ascl1+ progenitors pooled from E9.5 to E18.5.
(F) Absolute numbers (cells/section) of striosomal or matrix SPNs produced from Dlx1 and Ascl1 progenitors (pooled) from E9.5 to E18.5. (n = 6–15 sections in 3 mice from 2 litters).
A Set of Basal Intermediate Progenitors Are Fate-Restricted to Generate Matrix SPNs
The nearly exclusive fate restriction of early aIPs for S cell production prompted us to examine later phases of LGE neurogenesis from different types of progenitors. At E14.5, 8 hour pulse-chase in Tis21-CreER;Ai14 embryos continued to label VZ RGs with apical end-feet (Figures 3A and 3E), and large numbers of Ascl1+ bIPs were labeled in the SVZ (Figures 3A and3F). These were frequently engaged in symmetric cytokinesis (Figure 3G), suggestive of neurogenic cell division. Among the total RFP labeled cells, ~5% were putative RGs (located in VZ with end-feet and expression of the proliferation marker Sox2), ~70% were bIPs (Ascl1+ in SVZ with no end-feet); and the remaining ~25% of Ascl1− cells in SVZ likely represented other types of bIPs (e.g., Dlx1+) as well as new-born postmitotic neurons (Figures 3H and3I). In addition to RGs, 24 and 72-hour pulse-chase showed the generation of almost exclusively bIPs and not aIPs (Figures 3B–3D and S3A). In particular, 72-hour pulse-chase demonstrated that Tis21+ RGs continued multiple rounds of self-renewal during the late stage of embryonic development with ongoing generation of Ascl1+ progenitors in the SVZ (Figures 3C, 3D and S3B). These findings suggest that during the late phase of LGE neurogenesis (from E14.5 on), Tis21+ RGs persist to the end of neurogenesis (Figures S3E and S3F) and begin to predominantly generate a new population of bIPs, possibly in addition to production of a much smaller number of aIPs, as described below.
Figure 3. LGE RGs During Late Phase Lineage Progression Give Rise to Ascl1+/Dlx1+ bIPs Committed to the Generation of Matrix SPNs, See also Figures S3 – S5.
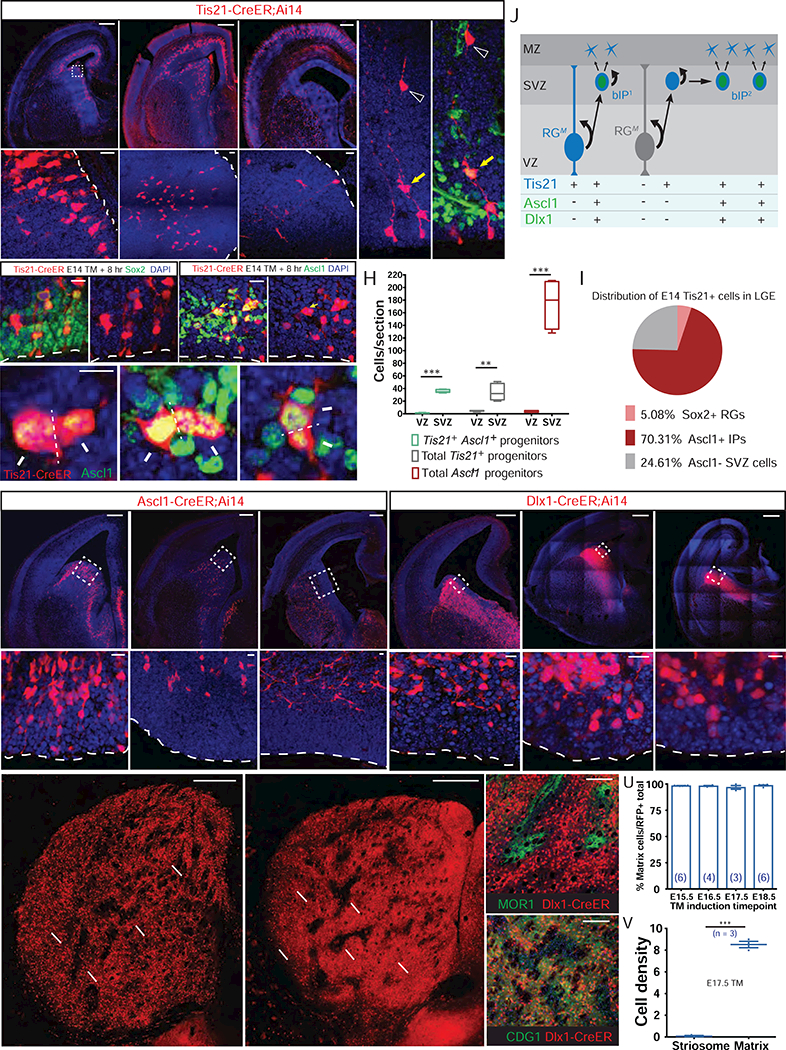
(A-C) Pulse-chase of E14.5 Tis21+ RGs and IPs at 8 (A), 24 (B), and 72 (C) hr in Tis21-CreER;Ai14 embryos. Although post-mitotic SPNs migrate into the striatum, self-renewing RGs (arrowheads) remain in the VZ for at least 72 hr (bottom panels).
(D) Example of a continually self-renewing RG in the VZ (arrowhead) 72 hr following fate-mapping from E14.5 Tis21+ RGs. Note the Ascl1+ bIP likely derived from the RG (yellow arrow) and the postmitotic progeny migrating to striatum (open arrowhead).
(E) 8-hr TM pulse-chase of Tis21+ RGs at E14.5. These RGs were Sox2+, Ascl1−, and extended end-feet to ventricle surface and radial processes (arrowheads). See also Figure S3.
(F and G) In the same sample as in (E), a large number of bIPs in the SVZ are Ascl1+ (F; yellow arrow), many of them in mitosis (G; dashed lines show plane of cytokinesis), generating putative post-mitotic progenies (arrows).
(H) Distribution of Ascl1+ IPs in VZ and SVZ following 8-hr pulse-chase in Tis21-CreER;Ai14 embryo, showing that the vast majority of Tis21+, Ascl1+ Tis21+ and Ascl1+ progenitors reside in SVZ at E14, in contrast to the distribution pattern of the same pulse-chase at E10.5 (compare with Figure 1C).
(I) Percentage of Sox2+ RGs, Ascl1+ IPs, and Ascl1- SVZ cells among the total population of fate-mapped Tis21+ progenitors at E14.
(J) A schematic showing that during the late phase of LGE neurogenesis RGs mostly generate bIPs, which may proliferate further and amplify before completing the neurogenic divisions that generate SPNs. Tis21 expression is generally activated in progenitors entering such a neurogenic phase, whether in RG or bIP.
(K-M) Pulse-chase of E14.5 Ascl1+ IPs at 8 (K), 24 (L), and 72 (M) hr in Ascl1-CreER;Ai14 embryos. Fate-mapped IPs were depleted from VZ by 24 hr after TM induction, and have differentiated into young neurons by 72 hr. Lower panels show magnified view of the boxed areas.
(N-P) 8-hr pulse-chase of Dlx1+ IPs at E13 (N), E15 (O) and E17 (P). Lower panels show that Dlx1+ IPs did not extend apical or basal processes at any of these stages and thus were exclusively bIPs. Dlx1 is also expressed in postmitotic neurons during this period.
(Q) SPNs fate-mapped from E14.5 Ascl1+ IPs localized to matrix (double arrows), but not striosomes (arrows).
(R-T) SPNs fate-mapped from E17.5 Dlx1+ IPs localized to matrix (double arrows) but not striosomes (arrows) (R); They surround MOR1+ striosomes (S) and were co-labeled by CDGI (T), with arrowheads indicating a void in the immediate region surrounding striosomes.
(U) Percentage of fate-mapped SPNs that localize to matrix following TM induction to map the output from Dlx1+ IPs at indicated embryonic times.
(V) Average cell density within the striosome versus the matrix compartment (# of cells/percent compartment area) following E17.5 TM induction in Dlx1+ IPs. ***p < 0.0001.
Scale bars: 300 µm in top of A, B, C, K, L, M, N, O, P; 20 µm in bottom of A-C, E, F, K-P; 5 µm in G; 300 µm in Q, R; 100 µm in S, T.
For a more detailed analysis of the neuronal output of Ascl1+ bIPs, we performed analogous embryonic fate mapping in E14.5 Ascl1-CreER;Ai14 mice. Eight-hour pulse-chase of Ascl1+ IPs labeled few aIPs and many bIPs that were identified as progenitors located in the SVZ without apical end-feet (Figure 3K). Some E14.5-labeled bIPs remained in the SVZ at 24 hours post-induction (Figures 3L S3C and S3D). As the cell-cycle length of LGE IPs at this time is only ~12 hours, this finding corroborated results from live imaging experiments (Pilz et al., 2013) and suggested that some bIPs may serve as transit-amplifying progenitors that proliferate in the SVZ before neurogenesis (Figure 3J). To identify the SPN types derived from late-phase Ascl1+ bIPs, we analyzed the P28 striatum following E14.5 induction of Ascl1-CreER;Ai14 mice. We detected only M cells (Figures 3Q, 3U and 3V). These results indicate that late-phase Ascl1+ bIPs are fate-restricted to generating M cells.
As Dlx1 may act downstream and/or in parallel to Ascl1 in the LGE (Long et al., 2009, Wang et al. 2013), we also performed systematic fate mapping using the Dlx1-CreER;Ai14 mice throughout LGE neurogenesis (Figures 3N–3P and 5). Similar to the early-phase Dlx1+ cells’ positions in the MZ, late phase (E13.5 to 17.5), pulse-chase of Dlx1+ cells labeled post-mitotic neurons in the MZ, but did not label RGs and aIPs in the VZ (Figures 3N–3P and S3G-S3J). In striking contrast to the early-phase pulse-chase results, however, late-phase pulse-chase did prominently label bIPs with no apical end-feet throughout the SVZ (Figures 3N–3P). Thus, the recruitment of the Dlx1 transcription factor distinguished putative bIPs from aIPs. When assaying fate-mapped SPNs in the mature striatum, we found that late-phase Dlx1+ bIPs also specifically produced SPNs of the matrix compartment (Figures 3R–3V). These M cells formed an expansive and dense field of SPNs within which unlabeled striosomes were embedded (Figures 3R–3T). Over 98% of these IP progeny were co-labeled with the matrix marker CDGI, but not with the striosome marker MOR1. These SPNs projected primarily to the non-dopamine-containing SNr, but apparently including labeling of some ventrally extending dendrites arising from the SNc (Figures S4C, S5B, and S5D; Supplemental Movie 5; Crittenden et al., 2016). Together with evidence from analysis of Ascl1 and Dlx1 mutant mice (Yun et al., 2002), these results support the idea that during the late phase, Dlxl acts in coordination with Ascl1 in bIPs to promote M cell production, in addition to its role in postmitotic SPNs.
The Sequential Production of Striosomal and Matrix SPNs is Punctuated by a Sharp Transition Period
We further performed an extensive set of fate-mapping with Ascl1- and Dlx1-drivers in time-steps of 0.5–1.0 day from E9.5 to E18.5 (Figures 4 and S4). The results (Figure 4) indicated a surprisingly sharp shift in the generation of striosomal and matrix SPNs, likely driven by largely distinct S cell-producing aIPs (aIPS) and M cell-producing bIPs (bIPM) pools.
Thus, during an early phase (E9.5-E12.5), Ascl1 is expressed in aIPs and Dlx1 in postmitotic neurons, and TM induction of both drivers predominantly labeled S cells (Figures 4A and4C). This early period was followed by a transition phase in which there was a rapid decline of S cell production starting around E12.5. From E13.5 on, S cell production became negligible, whereas the labeling of M cells from both drivers was initiated at ~E13.5 and rose sharply within ~24 hours to peak levels. In the subsequent late phase, Ascl1 expression shifted from aIPs in the VZ to bIPs in the SVZ, whereas Dlx1 expression emerged specifically in SVZ bIPs in addition to postmitotic SPNs in the MZ. Between E13.5 and E18.5, the Ascl1 driver initially labeled progenitors producing M cells until ~E16.5, before a sharp decline (Figures 4A and4D) correlated with a switch to the generation of glial cells from the LGE (Figures S6B-S6D). By contrast, the Dlx1 driver persisted in marking progenitors in an active phase of M cell production until the end of neurogenesis at ~E18.5. During both the early and transition phases, the onset and progression of S cell production and the switch from S to M cell production occurred earlier in the Ascl1 driver than in the Dlx1 driver, suggesting that Dlx1 might act downstream of Ascl1 in a transcription cascade that regulates neurogenesis and SPN specification. The MZ expression of Dlx1 in the early and late phase neurogenesis is consistent with this notion (Figures 2E and S3G). The divergence of Ascl1 and Dlx1 cell progeny in the late phase, with Ascl1 driving the production of glia and Dlx1 driving production of M cells, suggests that Ascl1 and Dlx1 probably also act in parallel neurogenetic programs during cell type specification.
These results support the existence of two distinct and sequential types of LGE IPs that generate the SPNs of the two major neurochemical compartments of the striatum. An early set of aIPS in the VZ generate striosomes with limited neurogenic capacity, whereas a late-activated set of bIPM generate matrix SPNs with expanded neurogenic capacity. Quantification of the total number of fate-mapped S and M cells across the embryonic neurogenic period indicated that the proportion of S and M cells produced was, respectively, approximately 20% and 80% (Figure 4F), matching the approximate ratio of total S and M cells estimated for the mature striatum (Johnston et al., 1990).
Given that both Ascl1 and Dlx1 are expressed not only in the LGE but also in the MGE, which generates both cortical and striatal interneurons (e.g., Marin et al., 2000). To address the degree to which MGE-derived GABAergic interneurons might be intermixed with fate-mapped SPNs derived from the LGE, we devised an intersectional strategy specifically to fate map Ascl1+ or Dlx1+ progenitors within the MGE (Figure S6). We combined Ascl1-CreER or Dlx1-CreER drivers with a constitutively active Nkx2.1-Flp driver (which targets all Nkx2.1 positive MGE progenitors; He et al., 2016) and the intersectional Ai65 reporter (See Star Methods) by crossing the corresponding mouse lines. This approach allowed us to restrict fate mapping to Ascl1+ and Dlx1+ progenitors in the MGE and preoptic area, yielding only GABAergic interneurons following TM induction at any given time point (Figure S6). Consistent with previous reports, the overwhelming majority of MGE-derived GABAergic interneurons were born during late embryonic days (~90% from E16.5-E18.5), with very few originating during the period of striosome neurogenesis (Figures S6B-S6D). Parvalbumin-positive and somatostatin-positive interneurons were scarce within fate-mapped striosomes, and were located in the matrix compartment when labeled (Figure S7). Cholinergic interneurons were not part of Ascl1 and Dlx1 fate-mapped cohorts regardless of induction time, whether from MGE only (intersection) or MGE/LGE (Ai14) (Figure S7).
The Temporal and Spatial Sequence of SPN Settlement in the Striatum Include a Final Cohort That Constitutes Peri-Striosomal Rings
Consistent with previous studies ( Newman et al., 2015, Hagimoto et al., 2017), we observed strong spatial gradients in the settling of SPNs along both the lateral-medial and caudal-rostral axes according to the time of Ascl1 and Dlx1expression (Figures 4 and S8; Supplemental Movie 6). Specifically, the SPNs of the matrix compartment is further divided into mosaics of cell-clusters (“matrisomes”), which become obvious in tracing experiments demonstrating the input-output organization of the matrix (Flaherty and Graybiel, 1994). Here we found a striking non-uniformity in the temporal production of matrix in our fate-maps: a ring-like void of labeled SPNs around the immediate border of striosomes was present in E15-E17 cohorts (Figure 3S), gaps that were subsequently filled in by the last-born E18 cohort of SPNs distributing specifically in the peri-striosomal space (Figure 5). These E18-derived SPNs adhered tightly to the borders of striosomes (Figures 5A–5E), and were reminiscent of the peri-striosomal rings (“annular compartment”) demarcated at maturity by immunochemical markers and interneuron distributions observed in primates and humans (Faull et al., 1989; Graybiel and Ragsdale, 1980).
We found that peri-striosomal cells were born almost exclusively during the terminal rounds of neurogenesis at E18.5 (Figure 5). These cells accounted for ~65% of total E18.5 fate-mapped SPNs in whole striatum at P28. They were more enriched in the rostral striatum (~80% of fate-mapped SPNs), reflecting the fact that there were also a significant number of caudal matrix SPNs still being generated at this late time, which mainly populated the medial matrisomes bordering the lateral ventricle (Figures 5C–5F). We also noted similar ring-like structures surrounded MOR1-positive patchy zones in the ventral striatum and nucleus accumbens (Figure 5A). It is thought that such peri-striosomal regions may functionally link striosome and matrix compartments (Banghart et al., 2015; Miura et al., 2007). Overall, we conclude that M cells generated from E15.5-E18.5 were deployed in a distal to proximal sequence in relation to the striosomes (Figures 5G–5I). As the RFP-labeled axons of the late-born peri-striosomal M cells innervated both pallidal segments and the SNr, the typical pattern of M cell axons, it is highly likely that at least part of the peri-striosomal cells correspond to the annular compartment (Faull et al., 1989).
Direct and Indirect Pathway SPNs in the Striosomes and Matrix Originate Independently within Both aIPS and bIPM
How the neurogenic programs producing striosome-matrix architecture relate to the generation of the direct and indirect pathway SPNs has long been a major question about basal ganglia organization. Direct and indirect pathway SPNs are distinguished from one another by multiple molecular markers, including their differential expression of D1 or D2 dopamine receptor (Gerfen et al., 1990). These markers occur in both striosomes and matrix, but in sharp contrast to the ~1:5 ratio of spatial compartmentalization of striosomal and matrix SPNs, dSPNs and iSPNs are intermixed in both compartments (Flaherty and Graybiel, 1994).
To determine whether S-cell and M-cell fates relate to the acquisition of direct and indirect pathway fates, we distinguished the dSPNs and iSPNs in our genetic fate mapping of the striosome and matrix compartments by relying on the tight correlation between dSPN with D1 receptor expression and iSPN with D2 receptor expression. We identified these SPNs by fluorescent mRNA double in situ hybridization with D1 and tdTomato probes or with D2 and tdTomato probes performed on brain sections in Ascl1-CreER;Ai14 and Dlx1-CreER;Ai14 mice induced at specific embryonic times. This allowed simultaneous identification of striosome and matrix compartments based on their D1 and D2 expression levels (Figure 6).
Figure 6. Integration of Direct and Indirect Pathway SPNs within the Striosome-Matrix System.
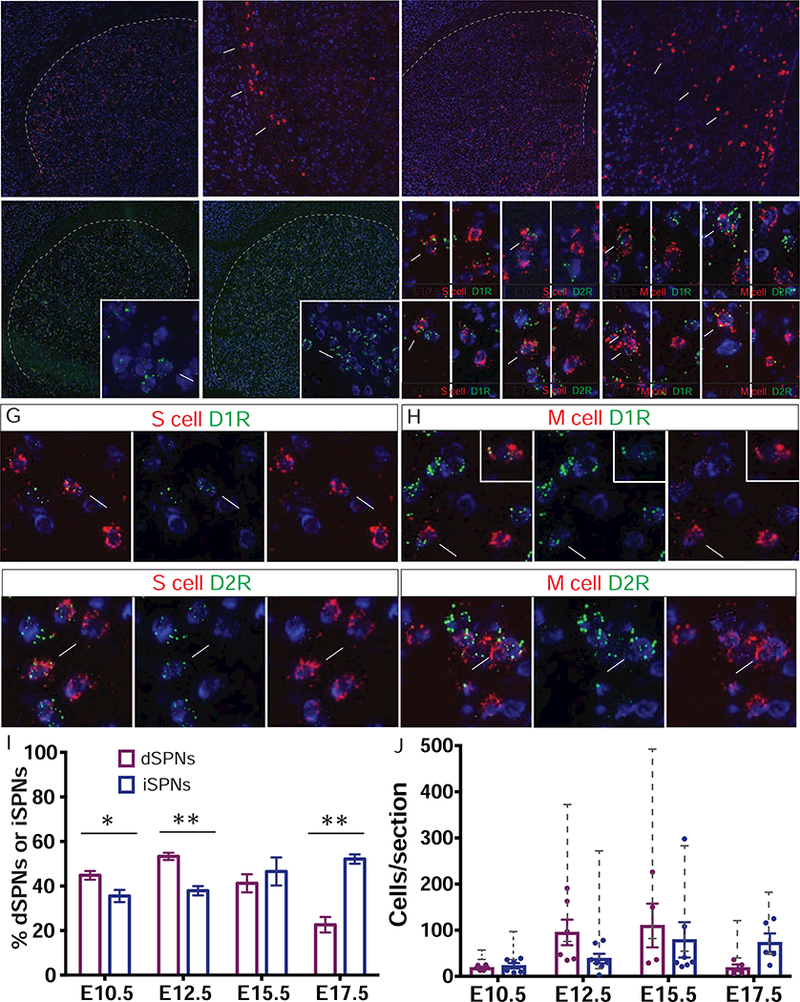
(A) Fluorescent in situ hybridization (FISH) signal from a branched-chain DNA (bDNA) probe targeting the tdTomato mRNA (red) in P28 SPNs following E10.5 TM in Ascl1-CreER;Ai14 mice matches the signature pattern (arrows) of striosomal cells along the lateral border of striatum. Blue: Nissl. Scale bars: 300 µm in left panel, 100 µm in right panel.
(B) tdTomato mRNA in P28 SPNs following E17.5 TM in Dlx1-CreER;Ai14 mice matches the matrix pattern (arrows) in medial striatum.
(C) FISH signal of dopamine D1 receptor mRNA (D1R, green) is distributed throughout about 50% of Nissl positive cells in striatum (inset: arrows: positive cell, arrowhead: negative cell). Scale bars: 300 µm, 10 µm in inset.
(D) Dopamine D2 receptor mRNA (D2R, green) is distributed throughout about 50% of Nissl positive cells in striatum (inset: Arrows: positive cell, arrowhead: negative cell)
(E) Representative panels of double FISH marking either D1 or D2 expressing cells in striosomes born at E10.5 (top) or E12.5 (bottom). Arrows: co-labeled, arrowheads: tdTomato-only. Scale bar: 10 µm.
(F) Representative panels of double FISH marking either D1 or D2 expressing cells in matrix born at E15.5 (top) or E17.5 (bottom). Arrows: co-labeled, arrowheads: tdTomato-only.
(G and H) Differential expression of the D1R or D2R in fate-mapped striosomal (G) and matrix (H) SPNs. Arrows: co-labeled, arrowheads: tdTomato-only. Scale bar: 10 µm.
(I) Proportions of dSPNs and iSPNs within fate-mapped striosome cohorts following E10.5 or E12.5 TM induction and within fate-mapped matrix cohorts following E15.5 or E17.5 TM induction. Error bar: SEM; *p < 0.05, **p < 0.001.
(J) Number of dSPNs and iSPNs within fate-mapped striosome and matrix cohorts (dots represent individual sections, bars project mean values, error bar: SEM). Grey brackets display the range of the number of total RFP+ cells per section detected by in situ probe in samples pooled from both high and low TM dose litters.
The spatial pattern of tdTomato mRNA-expressing S cell clusters was readily distinguishable from the M cell distributions. By contrast, D1 and D2 mRNA signals were evenly distributed and intermixed throughout the striatum (Figures 6C and 6D). Among S cells labeled by TM induction of Ascl1-CreER;Ai14 mice at E10.5 and E12.5, approximately equal ratios of D1 and D2 cells were detected, with some enrichment of D1 cells, especially in Dlx1-CreER cohorts (Figure 6I). Among M cells labeled by TM induction of Ascl1-CreER;Ai14 mice at E15.5, we found similar numbers of D1 and D2 cells (Figure 6I). Both D1 and D2 M cells were labeled by TM induction of Dlx1-CreER;Ai14 mice at E17.5, with a significant enrichment of D2 cells (Figure 6I). Thus, at least at the population level, a neurogenic program analogous to the one that generates the striosome and matrix compartments, punctuated by an abrupt transition during embryonic development, does not specify the generation of dSPNs and iSPNs. These results suggest that the specification of a direct-indirect pathway fate appears to be mainly independent of striosome-matrix fate and manifests itself within both the aIPS and bIPM progenitor pools, with a somewhat skewed distribution early and late toward a D1-striosome/D2-matrix bias.
DISCUSSION
In mammals, action selection and evaluation are crucially regulated by basal ganglia circuitry, through which the cerebral cortex and thalamus can modulate wide range of volitional and habitual behaviors. To support such sophisticated operations, the circuit architecture of the striatum is subdivided into striosome and matrix compartments, each further containing direct and indirect pathway subdivisions of SPNs. Here, we have discovered that the basic plan for assembling the striatal circuit architecture has its root in a developmental program embedded in the LGE RGs and their lineage progression, which generate distinct fate-restricted IPs to specify SPNs into striosome and matrix types upon which an independent and secondary mechanism divides SPNs into direct and indirect pathways. These findings raise the possibility that the deployment of different progenitor types may reflect an evolutionarily conserved mechanism to assemble striosomal and matrix circuits that mediate distinct behavioral categories involving action selection and evaluation, whereas intermixed direct and indirect pathway SPNs within the two compartments could represent a mechanism for flexible configuration of functional sub-circuits based for outcome evaluation and motor execution (i.e., experience). This view further raises the intriguing and more general possibility that, as in invertebrates in which cell lineage and developmental genetic program shape circuits and behavior (e.g., Harris et al., 2015), a developmental ground plan in mammalian telencephalic neural progenitors may direct the assembly of core forebrain circuits underlying high-order behaviors.
Lineage Dependent and Independent Production of Different Types of SPNs
Specific transcription factors, such as Gsx1/2, Ascl1, and Dlx1/2, as well as Notch signaling, mediate cell autonomous and non-autonomous regulation of neurogenesis in the LGE and control ordered production of striatal neurons (Mason, 2005; Yun et al., 2002). It has been suggested that the early LGE contains Gsx1/2+ neuroepithelial (NE) cells that give rise to multiple progenitor states characterized by Ascl1 and Dlx expression (Martín-Ibáñez et al., 2012; Yun et al., 2002). Ascl1+/Dlx1/2- and Ascl1+/Dlx1/2+ progenitors are inferred to emerge in a sequence (Martín-Ibáñez et al., 2012) and to interact through Notch-mediated lateral inhibition to coordinate proliferation and neurogenesis (Mason, 2005). The orderly production of early- and late-born SPNs is also regulated in part through downstream transcription factors (e.g., Ebf1, Isl1, Sp9) (Zhang et al., 2016; Merchan-Sala., 2017). Here, by combining cellular resolution genetic fate mapping and neuronal birth dating, we identify distinct progenitor types and trace their lineage progression to their neuronal progenies and SPN types. Based on our findings, we propose a model of the progenitor and lineage mechanisms underlying the production of SPN types and their influence on striatal architecture (Figure 7).
Figure 7. A Model on the Progenitor and Lineage Basis of SPNs that Constitute the Striosome and Matrix Compartments, and Direct and Indirect Pathways.
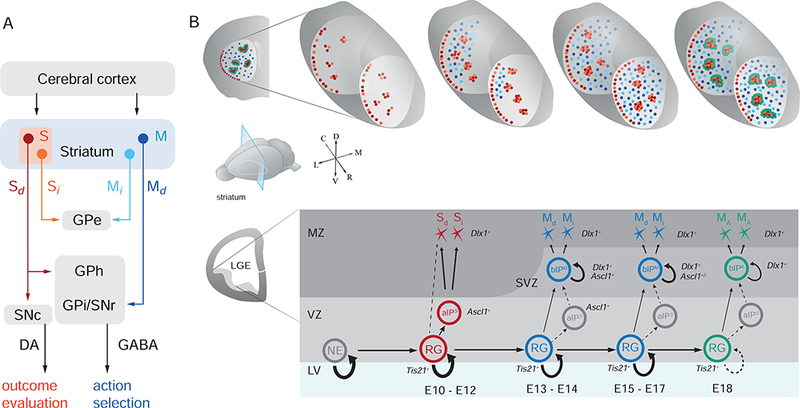
(A) schematic of the organization of striatal compartments and pathways that mediate different behaviors (GPe: globus pallidus externus, GPi: globus pallidus internus, GPh: globus pallidus habenula, Sd: striosome direct pathway, Si: striosome indirect pathway, Md: matrix direct pathway, Mi: marix indirect pathway).
(B) A model on the progenitor and lineage mechanisms underlying the production of SPN types and striatal architecture. See Discussion text for detailed description. MA: matrix annular compartment.
According to this model, a multi-potent radial glial lineage sequentially allocates distinct IP types to build striosome-matrix compartmentalization, and the division of direct and indirect pathways occurs within each IP type secondary to the compartmental divide. Specifically, the onset of neurogenesis in the early LGE (~E10) is marked by Tis21 expression in RGs, which self-renew over multiple rounds to generate several types of IPs and all types of SPNs. In the early phase of this lineage, RGs may generate neurons directly and/or through aIPs with dynamic ASCL1 expression patterns. Early aIPs (herein termed aIPS cells) with sustained high ASCL1 levels are committed to produce striosomal SPNs with likely no more than one round of amplification, thus limiting their neurogenic capacity. These aIPSs produce dSPNs and iSPNs in relatively similar ratios through striosomal genesis, but with an early bias toward D1-expressing SPNs. During an abrupt transition period starting ~E12.5, RGs shift to generating bIPs that are fate-restricted to produce M cells (i.e., bIPM). Dlx1 is activated downstream and/or in parallel to Ascl1 in bIPM. A possible depletion of aIPSs may result in the sharp decline of S cell production, whereas the rise in the production of bIPMs promotes M cell genesis. In the next phase (E14.5-E17.5), Ascl1+ bIPMs first promote M cell production and then switch to generating glial cells. By contrast, Dlx1+ bIPMs persist in driving M cell production throughout the late phase of LGE neurogenesis. Many bIPMs may undergo one or more rounds of transit-amplifying divisions before the final neurogenic division. Such divisions likely contribute to the significant numerical amplification of matrix relative to the striosome compartment, while RGs also continue to proliferate and generate new bIPs. We cannot distinguish whether a single bIPM generates either Md or Mi, or both. M cells populate the striatum in a caudal-to-rostral and lateral-to-medial sequence by birth time. At the final stage (~E18.5), Dlx1+ IPs generate SPNs destined to form peri-striosomal ring structures, of which some subset contributes to the annular compartment. Throughout bIPM-mediated neurogenesis, as during the aIPS-mediated period that precedes it, a similar ratio of matrix dSPNs and iSPNs are generated, with a bias toward iSPNs at the end of bIPM neurogenesis.
Our fate-mapping method cannot distinguish direct and indirect neurogenesis from Tis21+ RGs. Additionally, the mechanism underlying the shift of RG state from aIP S to bIPM production is unknown and might involve RG intrinsic (e.g., tracking cell division number) or extrinsic (e.g., signaling from increasing number of S cells) processes. Transcriptomic analysis of early and late RGs may uncover the molecular basis of this shift.
The Relation of Striosome-Matrix to Direct-Indirect Pathway SPN Origins
A notable finding is that dSPNs and iSPNs appear to derive from a different developmental process than that of the striosome and matrix compartments. Both dSPNs and iSPNs derive from both aIPS and bIPM pools, in similar or biased ratios, depending on the embryonic day of neurogenesis. How could such a pathway division be set up within the developmental process giving rise to striosomes and matrix? One possibility is that when S or M cells are first specified, they are neutral with respect to the D1-D2 distinction, so that these phenotypes are acquired in response to environmental factors or cell-cell interactions. An alternative possibility is that the D1 and D2 fates emerge together with the fundamental distinction into striosome or matrix identities. It is formally possible that, within the aIPS and bIPM pools, subsets of IPs are further fate-restricted to generate direct versus indirect pathway S cells, and direct versus indirect pathway M cells. A stochastic mechanism (Boije et al., 2014) might be well suited to generate the characteristic ratio of the two pathway SPNs, as stochastic decisions can produce a mosaic of fates within a population of cells, and they can be integrated into developmental programs and directed to yield robust and reproducible outcomes (Boije et al., 2014. Such a stochastic mechanism for D1 vs. D2 fate could manifest itself either at the step of IP generation from RGs or at the final neurogenic division of individual IPs.
The Significance of Distinct IPs and Their Differential Amplification for the Production of Striosomal and Matrix SPNs
The striosome and matrix compartments differ substantially in size and maintain an approximately 1:4 ratio that is conserved across mammalian species. Our finding that distinct IP types with different neurogenic capacities are fate restricted for S versus M cell production provide a plausible mechanism for this. First, a larger number of bIPsM are generated from RGs during a longer late phase and possibly through late-phase aIPs that further amplify bIPsM numbers. Second, individual bIPsM undergo more rounds of transit amplification compared to early aIPsS. We suggest that aIPS-mediated generation of SPNs in the early phase forms an evolutionarily conserved basic template for evaluative circuits. On the other hand, bIPM-mediated amplification dramatically increases the production of matrix SPNs and could underlie the development of the much larger matrix compartment. This may provide the basis for flexibility in building a large repertoire of basal ganglia circuits contributing to action selection and motor learning in parallel with the expansion of the neocortex.
As the striatal compartmental and pathway organizations are known to harbor different disease vulnerabilities (e.g. striosomes in Huntington’s disease, substance use, or mood disorders; D2 SPNs in Parkinson’s disease), these findings may have significance for understanding the developmental and circuitry etiology of such conditions (Hedreen and Folstein, 1995; Tippett et al., 2007).
Supplementary Material
STP of striatonigral projection from striosomes, Related to Figure 4.
Whole hemisphere STP of fate-mapped matrisomes, Related to Figure 4.
Whole hemisphere STP of transition period fate mapping, Related to Figure 4.
HIGHLIGHTS
Striatal compartment architecture arises from lineage progression of LGE radial glia
Striosomal SPNs largely derive from fate restricted apical intermediate progenitors
Matrix SPNs arise from later-emerging fate restricted basal intermediate progenitors
Both direct and indirect pathway SPNs arise from each intermediate progenitor pool
ACKNOWLEDGMENTS
We thank S. Kim for help with mouse engineering, N. El-Amine for help with in situ sample imaging, G. Fishell, S. Shi, M. Stepheson-Jones, and Y. Kubota for comments on the manuscript. This work was supported by a grant from the Simons Foundation to Z.J.H. and A.M.G., by CSHL Robertson Neuroscience Fund and a shared instrument grant from the NIH (1S10OD021759–01) to Z.J.H, by support from the CHDI Foundation (A-5552), the Nancy Lurie Marks Family Foundation, and the Saks Kavanaugh Foundation to A.M.G. An NRSA F30 Medical Scientist Predoctoral 5F30MH102002–03 supported S.M.K., and a Postdoctoral Fellowship from the William N. & Bernice E. Bumpus Foundation supported L.G.G.
Footnotes
AUTHOR CONTRIBUTIONS
Z.J.H. and A.M.G. conceived and organized the study. S.M.K. and Z.J.H. designed the experiments. S.M.K., P.W. and M.H. generated Tis21–2A-CreER and Nkx2.1-ires-Flp knock-in mouse lines. S.M.K and R.R. conducted primary fate-mapping, embryonic pulse-chase and FISH experiments. J.H.L., L.G.G. and A.M.G. performed 0.5 d tamoxifen inductions and histology. S.M.K., Y.K. and P.O. performed 2-photon imaging. Z.J.H, A.M.G. and S.M.K. wrote the manuscript.
DECLARATION OF INTERESTS STATEMENT
The authors have no personal financial interests, professional affiliations, advisory positions, board memberships, or patent holdings to disclose related to the subject matter or preparation of this publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alloway KD, Crist J, Mutic JJ, and Roy SA (1999). Corticostriatal projections from rat barrel cortex have an anisotropic organization that correlates with vibrissal whisking behavior. J. Neurosci 19, 10908–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemori K, Gibb LG, and Graybiel AM (2011). Shifting responsibly: the importance of striatal modularity to reinforcement learning in uncertain environments. Frontiers in Human Neuroscience 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, and Rubenstein JL (1997a). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, and Rubenstein JL (1997b). Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron 19, 27–37. [DOI] [PubMed] [Google Scholar]
- Attardo A, Calegari F, Haubensak W, Wilsch-Bräuninger M, and Huttner WB (2008). Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS ONE 3, e2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart MR, Neufeld SQ, Wong NC, and Sabatini BL (2015a). Enkephalin Disinhibits Mu Opioid Receptor-Rich Striatal Patches via Delta Opioid Receptors. Neuron 88, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boije H, MacDonald RB, and Harris WA (2014). Reconciling competence and transcriptional hierarchies with stochasticity in retinal lineages. Curr. Opin. Neurobiol 27, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR, and Cragg SJ (2017). The Striosome and Matrix Compartments of the Striatum: A Path through the Labyrinth from Neurochemistry toward Function. ACS Chem Neurosci 8, 235–242. [DOI] [PubMed] [Google Scholar]
- Brown KN, Chen S, Han Z, Lu C-H, Tan X, Zhang X-J, Ding L, Lopez-Cruz A, Saur D, Anderson SA, et al. (2011). Clonal production and organization of inhibitory interneurons in the neocortex. Science 334, 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LL, Feldman SM, Smith DM, Cavanaugh JR, Ackermann RF, and Graybiel AM (2002). Differential metabolic activity in the striosome and matrix compartments of the rat striatum during natural behaviors. J. Neurosci 22, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Cantuti-Castelvetri I, Saka E, Keller-McGandy CE, Hernandez LF, Kett LR, Young AB, Standaert DG, and Graybiel AM (2009). Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc Natl Acad Sci U A 106, 2892–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, and Graybiel AM (2011). Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR and Graybiel AM (2016). Disease-associated changes in the striosome and matrix compartments of the dorsal striatum Chapter for Handbook of Basal Ganglia Structure and Function, 2e, edited by Steiner H and seng K-YT [Google Scholar]
- Crittenden JR, Tillberg PW, Riad MH, Shima Y, Gerfen CR, Curry J, Housman DE, Nelson SB, Boyden ES, and Graybiel AM (2016). Striosome-dendron bouquets highlight a unique striatonigral circuit targeting dopamine-containing neurons. Proc. Natl. Acad. Sci. U.S.A 113, 11318– 11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblen F, and Graybiel AM (1995). Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J. Neurosci 15, 5999–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull RL, Dragunow M, and Villiger JW (1989). The distribution of neurotensin receptors and acetylcholinesterase in the human caudate nucleus: evidence for the existence of a third neurochemical compartment. Brain Res. 488, 381–386. [DOI] [PubMed] [Google Scholar]
- Flaherty AW, and Graybiel AM (1994). Input-output organization of the sensorimotor striatum in the squirrel monkey. J. Neurosci 14, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, and Huttner WB (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Gil-Sanz C, Martinez-Garay I, Espinosa A, Harkins-Perry SR, Ramos C, and Müller U (2012). Fate-restricted neural progenitors in the mammalian cerebral cortex. Science 337, 746–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze BS, Kravitz AV, Hammack N, Berke JD, and Kreitzer AC (2013). Control of basal ganglia output by direct and indirect pathway projection neurons. J. Neurosci 33, 18531–18539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Homma D, Gibb LG, Amemori K-I, Rubin SJ, Hood AS, Riad MH, and Graybiel AM (2015). A Corticostriatal Path Targeting Striosomes Controls Decision-Making under Conflict. Cell 161, 1320–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama F, Sohn J, Nakano T, Furuta T, Nakamura KC, Matsuda W, and Kaneko T (2011). Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron-tracing study using a viral vector. Eur. J. Neurosci 33, 668–677. [DOI] [PubMed] [Google Scholar]
- Gerfen CR (1984). The neostriatal mosaic: compartmentalization of corticostriatal input and striatonigral output systems. Nature 311, 461–464. [DOI] [PubMed] [Google Scholar]
- Gerfen CR (1992). The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu. Rev. Neurosci 15, 285–320. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, and Young WS (1988). Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res 460, 161–167. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, and Sibley DR (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432. [DOI] [PubMed] [Google Scholar]
- Giménez-Amaya JM, and Graybiel AM (1991). Modular organization of projection neurons in the matrix compartment of the primate striatum. J. Neurosci 11, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM (1984). Correspondence between the dopamine islands and striosomes of the mammalian striatum. Neuroscience 13, 1157–1187. [DOI] [PubMed] [Google Scholar]
- Graybiel AM (1997). The basal ganglia and cognitive pattern generators. Schizophr Bull 23, 459–469. [DOI] [PubMed] [Google Scholar]
- Graybiel AM (2008). Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci 31, 359–387. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, and Grafton ST (2015). The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol 7, a021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Hickey TL (1982). Chemospecificity of ontogenetic units in the striatum: demonstration by combining [3H]thymidine neuronography and histochemical staining. Proc. Natl. Acad. Sci. U.S.A 79, 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW (1978). Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc. Natl. Acad. Sci. U.S.A 75, 5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW (1980). Clumping of acetylcholinesterase activity in the developing striatum of the human fetus and young infant. Proc. Natl. Acad. Sci. U.S.A 77, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillner S, Hellgren J, Ménard A, Saitoh K, and Wikström MA (2005). Mechanisms for selection of basic motor programs--roles for the striatum and pallidum. Trends Neurosci. 28, 364–370. [DOI] [PubMed] [Google Scholar]
- Guillemot F, and Hassan BA (2017). Beyond proneural: emerging functions and regulations of proneural proteins. Curr. Opin. Neurobiol 42, 93–101. [DOI] [PubMed] [Google Scholar]
- Hagimoto K, Takami S, Murakami F, and Tanabe Y (2017). Distinct migratory behaviors of striosome and matrix cells underlying the mosaic formation in the developing striatum. J. Comp. Neurol 525, 794–817. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Flandin P, Yoshikawa K, Rubenstein JL, Alvarez-Buylla A, and Kriegstein AR (2013). Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat. Neurosci 16, 1576–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RM, Pfeiffer BD, Rubin GM, and Truman JW (2015). Neuron hemilineages provide the functional ground plan for the Drosophila ventral nervous system. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwell CC, Fuentealba LC, Gonzalez-Cerrillo A, Parker PRL, Gertz CC, Mazzola E, Garcia MT, Alvarez-Buylla A, Cepko CL, and Kriegstein AR (2015). Wide Dispersion and Diversity of Clonally Related Inhibitory Interneurons. Neuron 87, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, and Huttner WB (2004). Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc. Natl. Acad. Sci. U.S.A 101, 3196–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, et al. (2016). Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron 91, 1228–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, and Folstein SE (1995). Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol 54, 105–120. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Kim HF, Yasuda M, and Yamamoto S (2014). Basal ganglia circuits for reward value-guided behavior. Annu. Rev. Neurosci 37, 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houk JC, Davis JL, and Beiser DG (1994). Models of information processing in the basal ganglia (Cambridge, Mass: MIT Press; ). [Google Scholar]
- Iacopetti P, Michelini M, Stuckmann I, Oback B, Aaku-Saraste E, and Huttner WB (1999). Expression of the antiproliferative gene TIS21 at the onset of neurogenesis identifies single neuroepithelial cells that switch from proliferative to neuron-generating division. Proc. Natl. Acad. Sci. U.S.A 96, 4639–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, and Kageyama R (2014). bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 82, 9–23. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Isomura A, Harima Y, Kawaguchi K, Kori H, Miyachi H, Fujiwara T, Ishidate F, and Kageyama R (2013). Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342, 1203–1208. [DOI] [PubMed] [Google Scholar]
- Jiménez-Castellanos J, and Graybiel AM (1989). Compartmental origins of striatal efferent projections in the cat. Neuroscience 32, 297–321. [DOI] [PubMed] [Google Scholar]
- Johnston JG, Gerfen CR, Haber SN, and van der Kooy D (1990). Mechanisms of striatal pattern formation: conservation of mammalian compartmentalization. Brain Res. Dev. Brain Res 57, 93–102. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Toki S, Canales JJ, Harlan P, Blumenstiel JP, Chen EJ, Bany IA, Mochizuki N, Ashbacher A, et al. (1998). A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. U.S.A 95, 13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HF, and Hikosaka O (2015). Parallel basal ganglia circuits for voluntary and automatic behaviour to reach rewards. Brain 138, 1776–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Ables JL, Dickel LK, Eisch AJ, and Johnson JE (2011). Ascl1 (Mash1) defines cells with long-term neurogenic potential in subgranular and subventricular zones in adult mouse brain. PLoS ONE 6, e18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Venkataraju KU, Pradhan K, Mende C, Taranda J, Turaga SC, Arganda-Carreras I, Ng L, Hawrylycz MJ, Rockland KS, et al. (2015). Mapping social behavior-induced brain activation at cellular resolution in the mouse. Cell Rep. 10, 292–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kooy D, and Fishell G (1987). Neuronal birthdate underlies the development of striatal compartments. Brain Res. 401, 155–161. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci 15, 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, and Martínez-Cerdeño V (2006). Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci 7, 883–890. [DOI] [PubMed] [Google Scholar]
- Krushel LA, Johnston JG, Fishell G, Tibshirani R, and van der Kooy D (1993). Spatially localized neuronal cell lineages in the developing mammalian forebrain. Neuroscience 53, 1035–1047. [DOI] [PubMed] [Google Scholar]
- Long JE, Swan C, Liang WS, Cobos I, Potter GB, and Rubenstein JLR (2009). Dlx1&2 and Mash1 transcription factors control striatal patterning and differentiation through parallel and overlapping pathways. J. Comp. Neurol 512, 556–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, and Kriegstein AR (2011). Development and evolution of the human neocortex. Cell 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Anderson SA, and Rubenstein JL (2000). Origin and molecular specification of striatal interneurons. J. Neurosci 20, 6063–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Ibáñez R, Crespo E, Esgleas M, Urban N, Wang B, Waclaw R, Georgopoulos K, Martínez S, Campbell K, Vicario-Abejón C, et al. (2012). Helios transcription factor expression depends on Gsx2 and Dlx1&2 function in developing striatal matrix neurons. Stem Cells Dev. 21, 2239– 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Drechsel D, and Guillemot F (2012). Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason HA, Rakowiecki SM, Raftopoulou M, Nery S, Huang Y, Gridley T, and Fishell G (2005). Notch signaling coordinates the patterning of striatal compartments. Development 132, 4247– 4258. [DOI] [PubMed] [Google Scholar]
- Merchan-Sala P, Nardini D, Waclaw RR, and Campbell K (2017). Selective neuronal expression of the SoxE factor, Sox8, in direct pathway striatal projection neurons of the developing mouse brain. J. Comp. Neurol 525, 2805–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink JW (1996). The basal ganglia: focused selection and inhibition of competing motor programs. Prog. Neurobiol 50, 381–425. [DOI] [PubMed] [Google Scholar]
- Miura M, Saino-Saito S, Masuda M, Kobayashi K, and Aosaki T (2007). Compartment-specific modulation of GABAergic synaptic transmission by mu-opioid receptor in the mouse striatum with green fluorescent protein-expressing dopamine islands. J. Neurosci 27, 9721–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman H, Liu F-C, and Graybiel AM (2015). Dynamic ordering of early generated striatal cells destined to form the striosomal compartment of the striatum. J. Comp. Neurol 523, 943–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, and Kriegstein AR (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci 7, 136– 144. [DOI] [PubMed] [Google Scholar]
- Pilz G-A, Shitamukai A, Reillo I, Pacary E, Schwausch J, Stahl R, Ninkovic J, Snippert HJ, Clevers H, Godinho L, et al. (2013). Amplification of progenitors in the mammalian telencephalon includes a new radial glial cell type. Nat Commun 4, 2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragan T, Kadiri LR, Venkataraju KU, Bahlmann K, Sutin J, Taranda J, Arganda-Carreras I, Kim Y, Seung HS, and Osten P (2012). Serial two-photon tomography for automated ex vivo mouse brain imaging. Nat. Methods 9, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JLR, and Campbell K (2013). Chapter 24 - Neurogenesis in the Basal Ganglia In Patterning and Cell Type Specification in the Developing CNS and PNS, (Oxford: Academic Press; ), pp. 455–473. [Google Scholar]
- Smith JB, Klug JR, Ross DL, Howard CD, Hollon NG, Ko VI, Hoffman H, Callaway EM, Gerfen CR, and Jin X (2016). Genetic-Based Dissection Unveils the Inputs and Outputs of Striatal Patch and Matrix Compartments. Neuron 91, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa VH, and Fishell G (2010). Sonic hedgehog functions through dynamic changes in temporal competence in the developing forebrain. Curr. Opin. Genet. Dev 20, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, and Tseng KY (2010). Handbook of Basal Ganglia Structure and Function (Academic Press; ). [Google Scholar]
- Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, and Joyner AL (2011). Ascl1 genetics reveals insights into cerebellum local circuit assembly. J. Neurosci 31, 11055–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Kvitsani D, Fu Y, Lu J, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippett LJ, Waldvogel HJ, Thomas SJ, Hogg VM, van Roon-Mom W, Synek BJ, Graybiel AM, and Faull RLM (2007). Striosomes and mood dysfunction in Huntington’s disease. Brain J. Neurol 130, 206–221. [DOI] [PubMed] [Google Scholar]
- Turrero García M, and Harwell CC (2017). Radial glia in the ventral telencephalon. FEBS Lett. Wang B, Long JE, Flandin P, Pla R, Waclaw RR, Campbell K, and Rubenstein JLR (2013). Loss of Gsx1 and Gsx2 function rescues distinct phenotypes in Dlx1/2 mutants. J. Comp. Neurol 521, 1561–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, and Uchida N (2012). Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74, 858–873. [DOI] [PubMed] [Google Scholar]
- Yun K, Fischman S, Johnson J, Hrabe de Angelis M, Weinmaster G, and Rubenstein JLR (2002). Modulation of the notch signaling by Mash1 and Dlx1/2 regulates sequential specification and differentiation of progenitor cell types in the subcortical telencephalon. Development 129, 5029–5040. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhang Y, Wang C, Xu Z, Liang Q, An L, Li J, Liu Z, You Y, He M, et al. (2016). The Zinc Finger Transcription Factor Sp9 Is Required for the Development of Striatopallidal Projection Neurons. Cell Rep. 16, 1431–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
STP of striatonigral projection from striosomes, Related to Figure 4.
Whole hemisphere STP of fate-mapped matrisomes, Related to Figure 4.
Whole hemisphere STP of transition period fate mapping, Related to Figure 4.