

SUMMARY
Regulatory T cells (Tregs) are critical for maintaining immune homeostasis, but their presence in tumor tissues impairs anti-tumor immunity and portends poor prognoses in cancer patients. Here, we reveal a mechanism to selectively target and reprogram the function of tumor-infiltrating Tregs (TI-Tregs) by exploiting their dependency on the histone H3K27 methyltransferase enhancer of zeste homolog 2 (EZH2) in tumors. Disruption of EZH2 activity in Tregs, either pharmacologically or genetically, drove the acquisition of pro-inflammatory functions in TI-Tregs, remodeling the tumor microenvironment and enhancing the recruitment and function of CD8+ and CD4+ effector T cells that eliminate tumors. Moreover, abolishing EZH2 function in Tregs was mechanistically distinct from, more potent than, and less toxic than a generalized Treg depletion approach. This study reveals a strategy to target Tregs in cancer that mitigates autoimmunity by reprogramming their function in tumors to enhance anti-cancer immunity.
In Brief
EZH2 plays an intrinsic role in neoplastic cells as an oncogene, prompting the development of EZH2 inhibitors for cancer therapy. Wang et al. show that disrupting EZH2 function also has immunomodulatory activities and, when blocked in Tregs, promotes potent cancer immunity.
Graphical Abstract

INTRODUCTION
Regulatory T cells (Tregs) are an immunosuppressive subset of CD4+ T cells that are essential for maintaining immune tolerance and preventing autoimmune disease. Defects in the Treg master regulatory transcription factor FOXP3, or Treg depletion, leads to rapid lymphoproliferation and the onset of multi-organ autoimmunity in both humans and mice (Sakaguchi et al., 2008). While critical for controlling inappropriate immune responses to self, Tregs have been found at extremely high frequencies in nearly all cancers (Curiel et al., 2004; Saito et al., 2016). It is hypothesized that cancers have co-opted this natural mechanism of immune tolerance to blunt anti-tumor immune responses because the presence of Tregs in tumor tissues is commonly associated with poorer prognoses (Curiel et al., 2004; Liu et al., 2016a; Saito et al.,2016;Schreiber et al., 2011). Therefore, targeting Tregs may provide a powerful means to unleash more potent immune responses against cancer.
Generalized depletion of Tregs in murine cancer models by treatment with antibodies against the high-affinity interleukin-2 (IL-2) receptor (CD25) or genetic ablation approaches have been shown to slow the progression or even lead to the rejection of several types of cancer (Bos et al., 2013; Klages et al., 2010; Shimizu et al., 1999; Teng et al., 2010a, 2010b). However, these strategies must be limited in duration because the generalized inactivation of Tregs incites severe systemic autoimmune toxicities (Joshi et al., 2015; Liu et al., 2016b). For these strategies to be most effective, methods to selectively target intratu-moral Tregs are needed that preserve Tregs at other locations in the body to prevent autoimmune reactions. Preferential ablation of intratumoral Tregs has been achieved in some instances, such as with depleting anti-CTLA-4 or anti-CCR4 antibody treatments (Selby et al., 2013; Simpson et al., 2013; Sugiyama et al., 2013), which has led to strong anti-tumor responses with reduced autoimmune toxicities. This supports the hypothesis that directly targeting the function of Tregs in tumor tissues is most efficacious. Alternatively, investigations have shown that the immunosuppressive phenotype of Tregs is vulnerable, and in the context of inflammatory environments, Tregs are reprogrammed to become pathogenic T cells with effector functions (Bailey-Bucktrout et al., 2013; Oldenhove et al., 2009; Zhou et al., 2009). In the setting of cancer, blocking the engagement of ligands with several critical receptors on Tregs, such as CD25, glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR), or neuropilin-1 (Nrp-1), has demonstrated that the immunosuppressive properties of Tregs can be replaced by pro-inflammatory activities that beneficially augment immune responses to cancers (Nakagawa et al., 2016; Overa-cre-Delgoffe et al., 2017; Rech et al., 2012; Schaer et al., 2013). Targeting the functional plasticity of immune cells represents a powerful new mechanism to promote immune responses to cancer because it can both subvert immune tolerance, by removing immunosuppressive cells from tumors, and directly boost anti-tumor immunity, by converting the Treg niche from immunosuppressive to immunostimulatory (DuPage and Bluestone, 2016).
The development of targeted small molecule anti-cancer agents designed to directly affect critical pathways in tumor cells has brought about new opportunities for targeting intracellular pathways that control immune plasticity. By determining how these agents impinge on immune cells or other accessory cells of the tumor microenvironment, it may be possible to repurpose these drugs to simultaneously alter key immune cell populations to complement immunotherapeutic treatments for cancer. Small molecule inhibitors of enhancer of zeste homolog 2 (EZH2) are being evaluated in clinical trials as direct anti-cancer agents, but their potential to disrupt regulatory immune cells to promote tumor immunity remains unexplored (Kim and Roberts, 2016; Tiffen et al., 2016). EZH2, a histone H3K27 methyltransferase of the polycomb repressor complex 2 (PRC2) that controls chromatin condensation, is induced upon Treg activation, functioning as an epigenetic switch necessary to maintain Treg stability and function in tissues (Arvey et al., 2014; DuPage et al., 2015). Previous studies have shown that the disruption of EZH2 in Tregs selectively altered the stability of FOXP3 expression in activated Tregs and led to the acquisition of pro-inflammatory properties in these cells (DuPage et al., 2015; Sarmento et al., 2017; Zhang et al., 2014). Here, we investigated the importance of EZH2 in regulating immune function within the tumor microenvironment and shaping anti-tumor immunity.
RESULTS
T Cell-Dependent Anti-cancer Activity of Pharmacological EZH2 Inhibitors
To investigate whether EZH2 inhibition in immune cells modulates cancer progression, we administered an EZH2 inhibitor (CPI-1205, Constellation Pharmaceuticals [Vaswani et al., 2016]) to mice transplanted with a murine colorectal tumor (MC38). The MC38 tumor model was chosen because EZH2 did not exhibit intrinsic oncogenic properties, as treatment of cells in vitro did not affect cell proliferation despite a clear reduction in the levels of H3K27me3 in cells (Figures S1A-S1C). Furthermore, treatment of mice with an EZH2 inhibitor did not slow MC38 tumor growth when transplanted into T cell-deficient Rag-1−/− mice. In fact, tumor growth was accelerated in this immunodeficient setting (Figure 1A). In sharp contrast, treatment of tumor-bearing immunocompetent mice with the EZH2 inhibitor led to dramatic inhibition of tumor progression, indicating a tumor cell extrinsic mechanism for EZH2 inhibition that involved adaptive immune cells (Figure 1B). Indeed, mice receiving an EZH2 inhibitor had enhanced proportions of CD8+ T cells in tumors, implicating an augmented effector T cell response for improving tumor control (Figure 1C). Recent studies have shown that the blockade of EZH2 function within tumor cells increased their expression of chemokines that enhanced the anti-tumor immune response (Nagarsheth et al., 2016; Peng et al., 2015; Zingg et al., 2017), but EZH2 inhibition of MC38 cells grown in vitro did not increase the expression or secretion of CXCL9 or CXCL10 (Figures S1D and S1E). Importantly, the contribution of EZH2 activity within tumor-infiltrating Tregs (TI-Tregs) has not been explored. Therefore, we assessed Tregs from tumor-bearing mice treated with the EZH2 inhibitor. Tregs retained normal levels of EZH2 protein expression, but they had significantly reduced levels of H3K27me3 resulting from the inhibition of the enzymatic activity of EZH2 in Tregs (Figure 1D). While EZH2 inhibition did not reduce the frequency of Tregs in tumors, the ratio of CD8+ T cells to Tregs was increased in tumors (Figures 1E and 1F). Most significant, pharmacologic blockade of EZH2 led to reduced levels of FOXP3 protein in Tregs (Figure 1G), which is in line with the results of studies in which Ezh2 was genetically disrupted in Tregs (DuPage et al., 2015). These data reveal a critical role for T cells in mediating the anti-cancer effects of EZH2 inhibitor treatment and implicate Tregs as a potential target of EZH2 inhibition.
Figure 1. EZH2 Inhibition Disrupts Tumor Progression via a T Cell-Dependent Mechanism.

(A and B) Growth curve of MC38 tumors in untreated or EZH2 inhibitor-treated Rag-1−/− mice (A) or C57BL/6 mice (B) beginning 1 day after tumor inoculation throughout the experiment. Inset numbers represent the number of mice that rejected tumors of total implanted.
(C) Percentage of CD8+ T cells of viable CD45+ immune cells in MC38 tumors in (B) by flow cytometric analysis.
(D) Mean fluorescent intensity (MFI) of antibody staining for H3K27me3 and EZH2 in Tregs from untreated or EZH2 inhibitor-treated mice from (B).
(E and F) Percentage of FOXP3+ Treg cells of CD4+ T cells (E) and ratio of CD8+ T cell to FOXP3+ Tregs (F) in tumors from (B).
(G) MFI of antibody staining for FOXP3 in Tregs from untreated or EZH2 inhibitor-treated mice from (B).
Data represent means ± SEMs; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from two-way ANOVA with Sidak’s multiple comparisons or by unpaired t tests. See also Figure S1.
Increased EZH2 Activity in Tregs Infiltrating Murine and Human Cancers
To determine whether TI-Tregs are sensitive to EZH2 inhibition because of increased expression of the protein, we compared the level of EZH2 expression and activity in T cells from different tissues of MC38 tumor-bearing mice. EZH2 protein and its histone modification (H3K27me3) were increased in Tregs compared to effector CD4+Foxp3− T cells (Teff) in tumors, but not when comparing the T cells from tumor-draining lymph nodes (dLNs) (Figures 2A and S1F). In addition, Tregs exhibited a clear induction of EZH2 and H3K27me3 levels specifically within tumor tissues as compared to other non-lymphoid tissue (Figure 2B).
Figure 2. Elevated Expression of EZH2 in Tumor-Infiltrating Tregs.
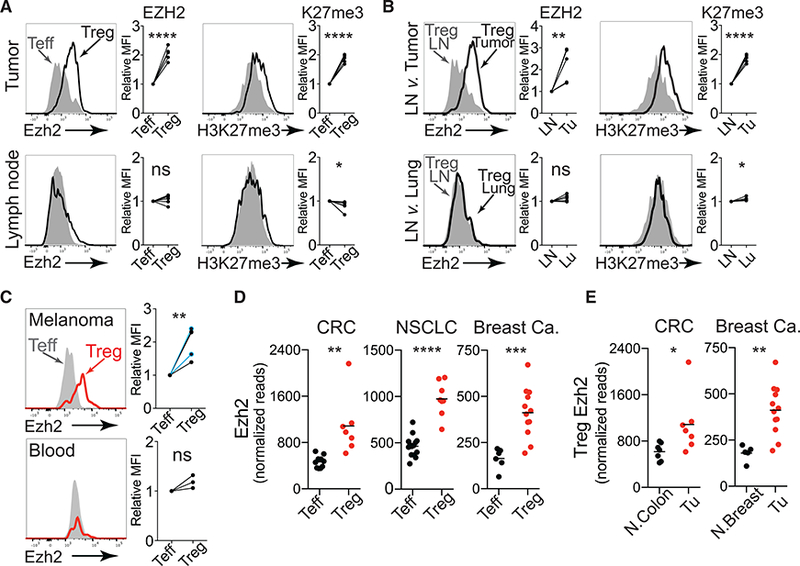
(A) Flow cytometric staining for EZH2 and H3K27me3 levels in Teff (filled histogram) and Treg (solid black line) cells isolated from mouse MC38 tumors (top) and tumor draining lymph nodes (bottom). Quantification of data from two independent experiments with two to three mice per group is shown with Treg MFI relative to Teff MFI.
(B) Flow cytometric staining for EZH2 and H3K27me3 levels in Tregs isolated from draining lymph node (filled histograms) versus Tregs from MC38 tumors (top) or lung (bottom) (black lines). Data quantified as in (A).
(C) Flow cytometric staining for EZH2 levels in Teff (filled histogram) versus Treg (solid red line) cells isolated from human melanoma and blood. Relative expression of EZH2 in Treg compared to Teff in all melanomas examined (n = 5, shown below). Two patients were treated with pembroluzimab (blue). Data quantified as in (A).
(D) Normalized read counts of Ezh2 from RNA sequencing of Teff and Treg cells isolated from primary human colorectal carcinoma (CRC), non-small-cell lung cancer (NSCLC), and breast cancer.
(E) Normalized read counts of Ezh2 from RNA sequencing of Treg cells isolated from tumors of patients with CRC and patients with breast cancer versus adjacent normal tissue.
Data represent means ± SEMs; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from unpaired t tests. See also Figure S1.
Examination of human Tregs infiltrating melanomas revealed a similar pattern of EZH2 induction in TI-Tregs. Within patient tumors, Tregs (CD4+FOXP3+) expressed higher levels of EZH2 protein compared to Teff (CD4+Foxp3−CD25−) despite equivalent activation states (both CD44+), but increased levels of EZH2 were not observed in Tregs from the peripheral blood (Figures 2C and S1G). Gene expression analysis of T cells from human colorectal cancer (CRC), non-small cell lung cancer (NSCLC), and breast cancers (De Simone et al., 2016; Plitas et al., 2016) also revealed an induction of EZH2 transcript levels in TI-Tregs compared to TI-Teff or in TI-Tregs compared to normal tissue-resident Tregs (Figures 2D and 2E). Taken together, these data support a hypothesis whereby TI-Tregs have enhanced EZH2 activity that when disrupted may impair their capacity to inhibit T cell responses to cancer.
Potent Anti-cancer Immunity in Mice with Targeted Disruption of Ezh2 in Tregs
To dissect the role of EZH2 activity specifically in Tregs, we transplanted tumors into mice genetically engineered to delete Ezh2 constitutively in Tregs (Foxp3-GFP-Cre;Ezh2fl/fl, abbreviated FP3;Ezh2fl/fl). Compared to littermate controls (FP3;Ezh2fl/+ or Ezh2fl/[fl/+]), FP3;Ezh2fl/fl mice completely rejected colon carcinomas (MC38) and prostate carcinomas (TRAMPC2) after an early tumor growth phase (Figures 3A, 3B, and S2A). In the context of a poorly immunogenic model of melanoma (B16F10), tumor growth was reduced significantly (Figure 3C). While in this setting tumors were not rejected, a prior vaccination regimen with irradiated granulocyte macrophage-colony-stimulating factor (GM-CSF)-expressing B16F10 cells (B16-GVAX) led to the rejection of melanomas in FP3;Ezh2fl/fl but not FP3;Ezh2fl/+ mice (Figure 3C). Similarly, treatment of wild-type mice with a combination of an EZH2 inhibitor and the B16-GVAX vaccination led to the rejection of B16F10 tumors (Figure S2B), supporting a hypothesis that EZH2 inhibition can synergize with cancer immu-notherapeutic modalities.
Figure 3. EZH2 Function in TI-Tregs Is Required to Block Anti-tumor T Cell Responses.

(A-C) Growth curves of multiple syngeneic tumor models—MC38 (A), TRAMPC2 (B), and B16F10 (C) — in mice with constitutive deletion of Ezh2 in Tregs (FP3;Ezh2fl,fl) compared with littermate controls (FP3;Ezh2fl/+, Ezh2fl,[fl,+]). B16GVAX was administered 2 days before B16 tumor inoculation.
(D) Growth curves of MC38 tumors in mice with constitutive deletion of Jmjd3 in Tregs (FP3;Jmjd3fl/fl) compared with littermate controls (FP3;Jmjd3fl/l,+).
Inset numbers represent the number of mice that rejected tumors of total implanted pooled from all of the experiments. Data represent means ± SEMs; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from two-way ANOVA with Sidak’s multiple comparisons. See also Figure S2.
To more broadly investigate the capacity of H3K27me3 levels in Tregs to alter anti-cancer immune responses, we examined the growth of MC38 tumors in mice with a Treg-specific deficiency for the H3K27 demethylase JMJD3, which opposes EZH2 function by removing methylation marks from H3K27 (FP3;Jmjd3fl/fl versus FP3;Jmjd3fl/+ littermates). In these mice, loss of JMJD3 in Tregs leads to increased levels of H3K27me3, and consequently FP3;Jmjd3fl/fl mice exhibited a more rapid outgrowth of MC38 tumors (Figure 2D). These results support the hypothesis that control of the level of H3K27me3 by EZH2 in Tregs is crucial for their capacity to suppress anti-tumor immune responses.
Ezh2 Disruption in Tregs Enhances the Number and Function of Tumor-Infiltrating T Cells
To understand how Ezh2 deficiency in Tregs enhanced the immune response against tumors, we analyzed the composition of tumor-infiltrating lymphocytes by immunohistochemistry and flow cytometry. This revealed that FP3;Ezh2fl/fl mice harbored significantly increased numbers and proportions of CD8+ and eCD4+ (CD4+Foxp3−) T cells within their tumors, similar to what we observed in the tumors of mice treated with an EZH2 inhibitor (Figures 4A–4D, S2C, and S2D). We also investigated the functionality of tumor-infiltrating T cells by restimulation in vitro and staining for intracellular cytokine production by flow cytometry. In FP3;Ezh2fl/fl mice, the polyfunctionality of tumor-infiltrating T cells was markedly enhanced, with CD8+ and eCD4+ T cells capable of co-producing multiple cytokines, including interferon-γ (IFN-γ), TNF-a, and IL-2 (Figures 4E–4G). Both CD8+ and CD4+ T cells were required for tumor clearance because depletion of either population permitted the outgrowth of MC38 tumors in FP3;Ezh2fl/fl mice (Figure S2E). These data reveal that selectively disrupting EZH2 activity in Tregs is sufficient to enhance the number and function of tumor-infiltrating effector T cells to improve tumor control.
Figure 4. Enhanced Number and Function of Tumor-Infiltrating T Cells with EZH2 Disruption in Tregs.

(A and B) Multiplexed immunohistochemical staining for CD8 (teal), CD4 (yellow), and Foxp3 (dark brown nuclear stain) in MC38 tumors from control (A) or FP3;Ezh2fl/fl mice (B). Scale bars, 200 μm and 50 μm (inset).
(C and D) Percentage of CD8+ (C) or effector CD4+ T cells (eCD4+: CD4+Foxp3−) (D) of viable CD45+ immune cells in tumor draining lymph node (dLN) or tumor by flow cytometric analysis.
(E) Representative flow cytometry of IFN-γ and TNF-α produced in stimulated CD8+ T cells from tumors in control or FP3;Ezh2fl/fl mice and quantification of results from one of two experiments.
(F) Representative flow cytometry of IFN-γ and IL-2 produced in stimulated eCD4+ T cells from tumors in control or FP3;Ezh2fl/fl mice and quantification of results from one of two experiments.
(G) Pie chart depicting the fraction of CD8+ and eCD4+ T cells that produce multiple cytokines from control or FP3;Ezh2fl/fl mice in (E) and (F).
Asterisks indicating significance determined by unpaired t tests between groups are *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data represent means ± SEMs pooled from two or more experiments; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from unpaired t tests. See also Figure S2.
Foxp3 Instability and Altered Function of Ezh2-Deficient Tregs within the Tumor Microenvironment
Pharmacological inhibition of EZH2 led to a reduction of FOXP3 protein levels in Tregs (Figure 1G). Therefore, we assessed the stability of Foxp3 expression in Tregs after genetic deletion of Ezh2 by incorporating a Cre-activatable lineage reporter (R26lsl-Rfp) into FP3;Ezh2fl/fl mice to irreversibly mark Foxp3 expressing cells and trace Tregs that subsequently lose Foxp3 expression, defined as GFP−red fluorescent protein (RFP)+ cells, termed exFoxp3 cells (Zhou et al., 2009). As with pharmacologic inhibition of EZH2, stable Foxp3 expression was markedly reduced with genetic deletion of Ezh2, with a greater proportion of Tregs losing Foxp3 and becoming exFoxp3 cells (Figure 5A). In addition, loss of Foxp3 was most prevalent in tumor tissues as opposed to the lymphoid organs (Figure 5B). Thus, the instability for Foxp3 in the absence of Ezh2 lowered the number of tumor-infiltrating FOXP3+ Tregs and dramatically shifted the balance in favor of CD8+ T cells over Tregs in tumors (Figures 5C–5E).
Figure 5. Foxp3 Instability and Altered Function of Ezh2-Deficient Tregs within the Tumor Microenvironment.
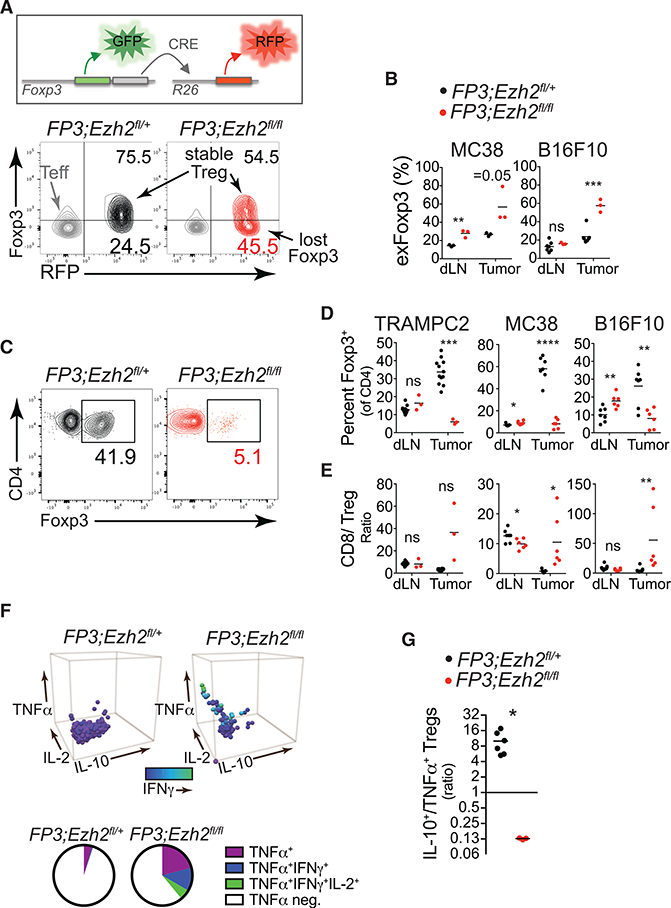
(A) Foxp3 lineage tracing by flow cytometric analysis of the frequency of Tregs that lost Foxp3 expression (exFoxp3 cells = GFP_RFP+) compared to stable Tregs (GFP+RFP+) in an MC38 tumor from control or FP3;Ezh2fl/fl mice. Effector CD4+Foxp3– T cells (Teff; CD4+Foxp3_RFP_) are shown in gray for reference.
(B) Quantification of exFoxp3 cells in dLN and tumorsfrom MC38 and B16F10 tumor-bearing mice.
(C) Flow cytometric analysis of the percentage of FOXP3+ Treg cells of CD4+ T cells in an MC38 tumor from control or FP3;Ezh2fl/fl mice.
(D and E) Quantification of the percentages of FOXP3+ Treg cells of CD4+ T cells (D) or the ratio of CD8+ T cells to FOXP3+ Tregs (E) in dLN and tumor from control or FP3;Ezh2fl/fl mice.
(F) Intracellular cytokine staining for IL-10, TNF-a, IL-2, and IFN-g in FOXP3+ Treg cells isolated from MC38 tumors of FP3;Ezh2fl/+ and FP3;Ezh2fl/fl mice. IFN-g staining is indicated by shading. Pie chart depicts the fraction of Tregs producing multiple cytokines shown below. Representative data from three mice per group and two experiments.
(G) Ratio of IL-10+ Tregs to TNF-a+ Tregs isolated from MC38 tumors in control versus FP3;Ezh2fl/fl mice.
Data represent means ± SEMs pooled from two or more experiments; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from unpaired t tests. See also Figure S3.
Ezh2-deficient TI-Tregs that retained Foxp3 expression also exhibited a pronounced change in the profile of cytokines that they produced. While wild-type TI-Tregs were characterized by the production of IL-10 and minimal production of TNF-α, Ezh2-deficient Tregs exhibited an inverse phenotype, with increased potential to produce the pro-inflammatory cytokines TNF-α, IFN-γ, and IL-2 and reduced production of immunosuppressive IL-10 (Figures 5F, 5G, and S3A). Therefore, Ezh2-deficiency in Tregs alters TI-Tregs not only by destabilizing Foxp3 expression, thereby reducing the frequency of suppressive Tregs in tumors, but also by altering the function of FOXP3+ Tregs, converting them from immunosuppressive to pro-inflammatory cytokine producers specifically in tumors.
To investigate whether Ezh2 deficiency during activation was sufficient to drive the changes in the profile of cytokines produced, we performed gene expression analysis of wild-type Tregs compared to Ezh2-deficient Tregs that were first sorted naive (CD62L+) and then activated by T cell receptor and CD28 stimulation in vitro for 4 days. This analysis revealed that early after activation, Ezh2-deficient Tregs were already enriched in gene sets associated with TNF-α signaling and inflammatory responses by gene set enrichment analysis (Figure S3B). In addition, while the expression of key immunosuppressive genes in Tregs Il10 and Tgfb1 was reduced, expression of pro-inflammatory genes (e.g., Il2) exhibited increased expression upon activation without EZH2 function (Figure S3C). However, many other pro-inflammatory cytokines trended toward enhanced expression in Ezh2-deficient Tregs but were not changed significantly. Therefore, longer periods of sustained activation within tumors or additional factors within the tumor microenvironment must be required to stably enforce changes in pro-inflammatory cytokine production. Collectively, these results indicate that disrupting EZH2 activity in Tregs enhances anti-tumor immune responses by selectively depleting intratumoral Tregs, altering their functionality, or a combination of both.
Temporal Disruption of EZH2 Function in Tumor-Bearing Mice Enhances Anti-cancer Immunity while Minimizing Systemic Autoimmunity
In FP3;Ezh2fl/fl mice, Ezh2 is deleted in all Tregs during the initial establishment of Foxp3 expression in the thymus. This is not applicable to therapeutic strategies in which targeting EZH2 activity in Tregs would be limited to patients after tumors are detected. Therefore, we generated mice to temporally delete Ezh2 in tumor-bearing mice using a Cre-estrogen receptor fusion protein driven by Foxp3 (FP3-ER;Ezh2f,/A) (Rubtsov et al., 2010). Tamoxifen (tam) administration for 14 days efficiently induced CreER activity, with 70%−90% of the in vivo-identified Tregs having deleted Ezh2 based on RFP reporter expression (R26lsl-rfp) and a reduced level of H3K27me3 in RFP+ cells (Figure S4). Acute disruption of Ezh2 in Tregs by tam treatment in FP3-ER;Ezh2fl/Δ mice led to significant tumor protection in the TRAMPC2 and MC38 tumor models when performed at tumor initiation or in established tumors (Figures 6A and S5A-S5C). More important, in this model of prolonged tam treatment to delete Ezh2, no symptoms of autoimmunity were observed (not shown). This was significant, because our previous studies showed that complete disruption of Ezh2 in Tregs leads to the development of autoimmune symptoms in all of the tissues examined (DuPage et al., 2015). Furthermore, when mice with Ezh2-deficient Tregs were challenged to maintain tolerance in adult mice (after permanent removal of wild-type Tregs), animals succumbed to autoimmune toxicities within 3–4 weeks. However, in that setting, Ezh2-deficient Tregs deleted Ezh2 from the time of initial expression of Foxp3, whereas here, in this model of tam-induced deletion of Ezh2, Tregs delete Ezh2 after their development and differentiation in the presence of EZH2. Therefore, these experiments more accurately recapitulate the consequences of pharmacological inhibition of EZH2 therapeutically in patients and indicate that in the setting of blocking EZH2 function late, during Treg effector activity, autoimmune toxicities can be prevented.
Figure 6. Temporal Disruption of EZH2 Function in Tregs Promotes Anti-tumor Immunity.
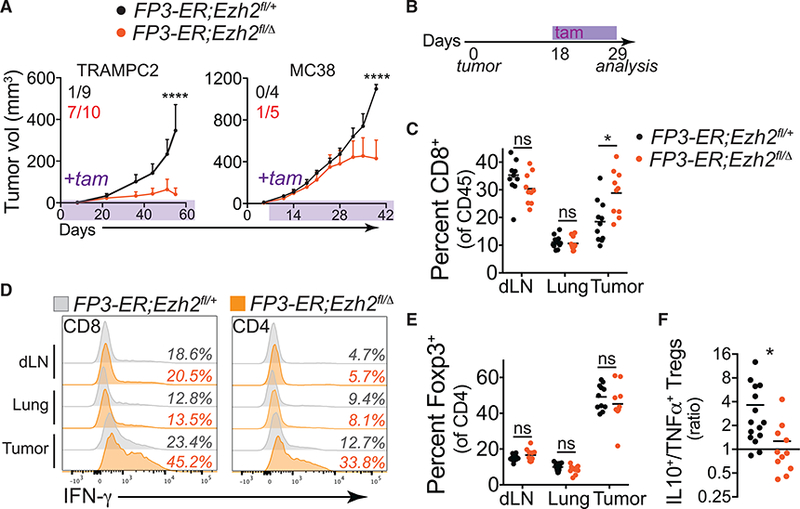
(A) TRAMPC2 and MC38 tumor growth in FP3-ER;Ezh2fl/+ or FP3-ER;Ezh2fl/Δ mice treated with tam (timing indicated by shading). Inset numbers represent the number of mice that rejected tumors of total implanted.
(B) Timeline for short-term tam administration (11 days) to FP3-ER MC38 tumor-bearing animals in (C), (D), and (E).
(C) Percentage of CD8+ T cells of CD45+ immune cells in draining lymph node (dLN), lung, and MC38 tumor tissues.
(D) Representative intracellular cytokine staining for IFN-γ in CD8+ and effector CD4+ T cells in control (gray histograms) or FP3-ER;Ezh2fl/Δ mice (orange histograms). Quantification of data in Figures S5D and S5E.
(E) Percentage of FOXP3+ Tregs of CD4+ T cells.
(F) Ratio of IL-10+ FOXP3+ Tregs to TNF-α+ FOXP3+ Tregs isolated from MC38 tumors in tam-treated FP3-ER;Ezh2fl/+ versus FP3-ER;Ezh2fl/Δ mice.
Data represent means ± SEMs from two or more experiments; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from two-way ANOVA with Sidak’s multiple comparisons or by unpaired t tests. See also Figures S4 and S5.
To elucidate the mechanisms driving improved tumor immunity, we assessed the frequency and function of T cells shortly after deleting Ezh2 in Tregs of mice with established tumors (11 days of tam treatment, Figure 6B). Acute deletion of Ezh2 in Tregs increased the percentage of CD8+ T cells in tumors but not in tumor-dLNs or lung tissues (Figure 6C). In addition, the capacity of both CD8+ and CD4+ T cells to produce IFN-g was enhanced selectively in both CD8+ and eCD4+ T cells infiltrating tumors (Figures 6D, S5D, and S5E). While the effector T cell response phenocopied the response observed with constitutive Ezh2 deletion in Tregs, this occurred without a quantitative reduction in the frequency of TI-Tregs (Figure 6E). However, TI-Tregs temporally deleted for Ezh2 exhibited similar changes in the profile of cytokines produced in the constitutive Ezh2-deficient Tregs, producing more TNF-a and less IL-10 (Figures 6F and S5F). This supports the hypothesis that disruption of EZH2 function in Tregs acts predominantly by altering the function of tumor-infiltrating FOXP3+ Tregs to promote tumor immunity. It also indicates that the enhanced anti-tumor immune response in FP3;Ezh2fl/fl mice is not the result of deleting Ezh2 during thymic development of Tregs nor of FP3;Ezh2fl/fl mice being preconditioned to reject tumors. Finally, because temporal disruption of Ezh2 using the FP3-ER system restricts the deletion of Ezh2 to established Tregs (Rubtsov et al., 2010), it is clear that the effects of EZH2 disruption are mediated by alterations of bona fide Tregs rather than T cells that transiently induce Foxp3 expression.
The Presence of Ezh2-Deficient Tregs Potentiates Anticancer Immunity
Ezh2 deficiency in TI-Tregs alters their function, but in this setting, we also observed reductions in the frequency of intratumoral Tregs. To determine whether the potency of Ezh2 deletion was partially the result of selectively eliminating Tregs from tumors, we assessed the effect of Treg depletion directly, using mice with a Foxp3-driven diphtheria toxin receptor (FP3-DTR) (Kim et al., 2007). Administration of diphtheria toxin (7 days on, 7 days off) to FP3-DTR mice, but not tam to FP3-ER;Ezh2fl/Δ expansion of CD8+ and eCD4+ T cells in lung tissue and dLNs from tumor-bearing mice (Figure 7D). In contrast, the temporal deletion of Ezh2 in Tregs did not generate any apparent autoimmunity and only increased the number of CD8+ and eCD4+ T cells specifically infiltrating tumors (Figures 7C and 7D). Most strikingly, short-term depletion of Tregs at tumor initiation did not confer complete protective immunity because no tumors were rejected (Figure 7E). Longer-term depletion of Tregs beyond 7–10 days in the FP3-DTR model was not possible because it led to fulminant and lethal autoimmune pathologies. These results have two implications. First, it would appear that like many immunotherapies, global Treg depletion leads to autoimmune toxicities that will limit its use therapeutically (June et al., 2017). Thus, because acute disruption of EZH2 in Tregs preserved Treg functionality in non-tumor tissues and did not generate systemic autoimmune toxicity, it would be a more desirable treatment. Second, temporal deletion of Ezh2 in Tregs can confer more robust tumor protection than short-term pan Treg depletion. This result suggests that blocking EZH2 function in Tregs is distinct from Treg depletion when mediating enhanced anti-tumor immunity.
Figure 7. The Presence of Ezh2-Deficient Tregs Are Required to Promote Tumor Rejection.

(A) Timeline for diphtheria toxin (DT) administration to FP3-DTR mice or tam administration to FP3-ER mice bearing MC38 tumors in (B), (C), and (D).
(B) Percentage of FOXP3+ Tregs of CD4+ T cells in lymph nodes.
(C) Representative pictures of mice temporally disrupted for EZH2 in Tregs (left) or depleted of Tregs (right).
(D) Absolute number of CD8+ T cells (top) and effector CD4+ T cells (eCD4+: CD4+Foxp3—, bottom) in tumor, lung, and draining lymph node (dLN) of indicated mice.
(E) TRAMPC2 and MC38 tumor growth in FP3-DTR micewith orwithout DT treatment (time ofadministration indicated by shading). Inset numbers represent the number of mice that rejected tumors of total implanted.
(F) Scheme (top) and representative flow cytometry plot (bottom) illustrating chimeric female model fortransiently eliminating wild-typeTregs in the presence of Ezh2-deficient Tregs.
(G) MC38 tumor growth in indicated chimeric female mice treated with diphtheria toxin from tumor inoculation throughout.
(H) MC38 tumor growth in indicated chimeric female mice or FP3-DTR/FP3-DTR mice treated short term with diphtheria toxin from days—2 to 4 after tumor inoculation. Inset numbers represent the number of mice that rejected tumors of total inoculated in one of two experiments(see Figure S6 for cumulative results). p values indicate comparison between red (Ezh2fl/fl) and blue (FP3-DTR) lines, but comparison of red to black (Ezh2fl/+) lines at these three time points also was significant. No significant difference at any time point observed between black and blue lines.
Data represent means ± SEMs from two or more experiments; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from two-way ANOVA with Sidak’s multiple comparisons or by unpaired t tests. See also Figure S6.
To test directly whether the presence of Ezh2-deficient Tregs directly contributes to tumor protection, we bred female mice that harbored an FP3-DTR or an FP3-Cre knockin allele on each X chromosome (Rubtsov et al., 2010). Because of X inactivation, these female mice are naturally chimeric for two populations of Tregs: wild-type DTR+ Tregs that can be eliminated by DT treatment and Cre+ Tregs deficient for Ezh2 that are insensitive to DT-mediated depletion (Figure 7F). Using this system, we confirmed that the continued depletion of wild-type Tregs in chimeric females harboring residual Ezh2-deficient Tregs phe-nocopied our results with FP3;Ezh2fl/fl mice (Figure 7G). Chimeric female mice not treated with DT did not exhibit tumor protection in this setting, likely because the wild-type Tregs made up the majority of Tregs-infiltrating tumors (not shown). Next, we directly compared mice in which all Tregs were transiently depleted to chimeric littermates, wherein more than half of their Tregs were transiently depleted but in the context of a remaining pool of Ezh2-deficient (or Ezh2-sufficient) Tregs. Mice homozygous for FP3-DTR alleles, in which all Tregs were transiently depleted following DT administration, never rejected their tumors (Figure 7H). However, in chimeric females in which identical conditions were used to transiently deplete wild-type Tregs but Ezh2-defi-cient Tregs remained, there was robust anti-tumor immunity, with the majority of tumors being rejected (Figures 7H and S6). These results were striking, because they definitively indicate that the presence of Ezh2-deficient Tregs are necessary to drive tumor protection in the context of Treg depletion, and that disruption of EZH2 function in Tregs is more potent than Treg depletion alone. These results are in line with the hypothesis that Ezh2-deficient Tregs acquire a pro-inflammatory functionality that supports stronger tumor immunity.
DISCUSSION
Immunosuppressive Tregs are recruited to nearly all cancers, where they can negatively affect anti-tumor immune responses. Methods are needed to selectively and safely target the Tregs that prevent anti-tumor immunity without instigating adverse autoimmune toxicities. In this study, we investigated whether targeting the histone H3K27 methyltransferase EZH2 in Tregs provides a method to selectively alter TI-Tregs. We found that Tregs infiltrating tumors exhibit increased EZH2 expression and activity. As a consequence, we hypothesized that TI-Tregs may be especially sensitive to EZH2 inhibition. We showed that EZH2 activity is critical to Treg-mediated suppression of anti-tumor immunity by genetic disruption of Ezh2 in all Tregs. In this setting, mice were protected from multiple cancers because of enhanced CD8+ and CD4+ T cell responses, and this was associated with a reduced frequency and altered function of Tregs (increased pro-inflammatory cytokine production) specifically within tumors. Temporal disruption of Ezh2 after tumor development, which more accurately mimics therapeutic targeting of EZH2, also led to significantly enhanced anti-tumor T cell immunity, but affected only the function and not the number of TI-Tregs. More important, pharmacologic inhibition of EZH2 also led to improved CD8+ T cell responses within tumors that impeded tumor progression and was associated with reduced FOXP3 expression in Tregs. Finally, by comparing Treg depletion in the absence or presence of Ezh2-deficient Tregs, we showed that the presence of Ezh2-deficient Tregs blocked tumor progression more potently than Treg depletion alone and without the devastating autoimmunity that ensued upon complete Treg depletion. Thus, inhibiting EZH2 activity selectively reprograms the function of TI-Tregs without systemically altering Treg function, leading to enhanced tumor immunity without autoimmune toxicity.
Targeting Tregs to enhance immune responses against cancer has been a long-standing goal. Support for the hypothesis was shown shortly after the initial identification of Tregs as CD25+ (IL-2Rα+) cells by anti-CD25 antibody-mediated ablation of Tregs (Shimizu et al., 1999). While attempts to generally ablate Tregs has shown remarkable efficacy in various preclinical tumor models (Bos et al., 2013; Simpson et al., 2013; Teng et al., 2010b), clinical efficacy in cancer trials has been discouraging (Liu et al., 2016a; Rech et al., 2012). This is partly because of a lack of targets that provide sufficient specificity for Tregs. Even the prototypical target—the IL-2/IL-2R pathway—is problematic because this pathway also is important for promoting effector T cell responses that can act against tumors (Curiel, 2007). Furthermore, while attempts to ablate Tregs in patients have yet to exhibit significant toxicity, the potential to drive dangerous systemic inflammation is a major concern (Joshi et al., 2015; Liu et al., 2016a). Therefore, methods to selectively deplete the Tregs suppressing anti-tumor immune responses are essential. It is important to note that when Tregs have been specifically depleted within tumors, it appears to be sufficient to liberate productive anti-tumor immune responses in preclinical models of cancer (Chen et al., 2007). Antibodies directed toward CTLA-4 in murine cancer models have been demonstrated to deplete predominantly intratumoral Tregs leading to improved anti-tumor immunity (Selby et al., 2013; Simpson et al., 2013; Sugiyama et al., 2013). However, in patients receiving clinically approved anti-CTLA-4 agents, this mechanism has not been consistently demonstrated (Liu et al., 2016a; Tanaka and Sakaguchi, 2017). The selectivity of anti-CTLA-4 for intratumoral Tregs has been attributed to the antibodies’ preferential binding of the most activated Tregs, which are known to have the greatest levels of surface-exposed CTLA-4.
In this study, we found that constitutive disruption of EZH2 in Tregs leads to dramatic reductions in intratumoral Treg frequencies without disrupting the frequency of Tregs in lymphoid and non-tumor tissues. This is likely because of the decreased stability of Foxp3 expression in highly activated Tregs that lack functional EZH2 (DuPage et al., 2015). EZH2 is induced upon activation via the T cell receptor and CD28; thus, intratumoral Tregs likely have a particularly strong dependence on EZH2 activity. It is noteworthy that by targeting other pathways that are induced in activated Tregs, a similar selectivity for disrupting intratumoral Treg stability and function has been observed (Liu et al., 2016a; Luo et al., 2016). Recently, blockade of the nuclear factor kB (NF-kB) pathway in Tregs was shown to selectively disrupt the activity of intratumoral Tregs and promote stronger anti-tumor immune responses (Grinberg-Bleyer et al., 2017). The NF-kB pathway directly regulates the activation of Ezh2 expression (Neo et al., 2014), potentially connecting our studies and providing strong support for a general methodology to target intratumoral Tregs based on their highly activated state in tumor tissues.
Short-term disruption of EZH2 activity in Tregs by pharmacological inhibition or temporal genetic deletion also led to enhanced anti-tumor CD8+ T cell responses. However, this occurred without significant reductions in the intratumoral frequency of Tregs, but it was associated with the rapid acquisition of an immunostimulatory cytokine profile in tumor-infiltrating FOXP3+ Tregs. Our study provides further support for a shifting paradigm for disabling Tregs in cancer by altering their functionality. This approach is superior to Treg depletion because, as we and others have demonstrated, Treg ablation drastically disrupts immune homeostasis and can lead to irreversible autoimmunity. By instead tapping into pathways that control the natural context-specific plasticity of Tregs, it is possible that we can reprogram the functionality of Tregs with great anatomical and context specificity that only narrowly disrupts immunoregulation without causing systemic autoimmune toxicity (DuPage and Bluestone, 2016). In the context of cancer, mechanisms to induce Foxp3 instability and alter Treg function have been demonstrated by targeting the critical Treg receptors, such as CD25, GITR, and Nrp-1 (Nakagawa et al., 2016; Overacre-Delgoffe et al., 2017; Rech et al., 2012; Schaer et al., 2013). Administration of blocking antibodies against these receptors or genetic disruption of the pathways activated downstream of them has been shown to destabilize Foxp3 expression and drive the production of IFN-γ in Tregs isolated from tumor tissues. Treg reprogramming toward pro-inflammatory activities has been shown to be critical for the efficacy of anti-tumor immune responses and boosting immunotherapy in preclinical models (Sharma et al., 2010). However, few studies have demonstrated Treg reprogramming with small-molecule drugs that target the downstream pathways that these receptors engage. Epigenetic regulators that control chromatin states, such as EZH2, may be ideal targets for reprogramming Treg function (Liu et al., 2013). Targeting EZH2 is significant because inhibitors of EZH2 are being evaluated in clinical trials as direct anti-cancer agents in which EZH2 is predicted to have tumor-promoting oncogenic activity, especially in cancers harboring gain-of-function mutations in EZH2 such as melanomas, diffuse large-cell B cell lymphomas, and follicular lymphomas (Kim and Roberts, 2016 ). Moreover, EZH2 has been shown to regulate chemokine production within cancer cells, shaping their immune microenvironment and making it more permissive to cancer progression (Nagarsheth et al., 2016; Peng et al., 2015). Among the myriad of pleiotropic effects induced by EZH2 inhibition that have been described, we have revealed here an additional activity of EZH2 in activated TI-Tregs that, if disrupted, synergizes with cancer immunotherapeutic strategies.
Finally, we identified EZH2 because of its CD28-dependent induction. Therefore, EZH2 regulation is likely to be critical for immune checkpoint blockade therapies that target the CD28 pathway such as anti-CTLA-4 and anti-programmed cell death protein-1 (PD-1)/programmed death ligand-1 (PD-L1) antibodies (Hui et al., 2017; Kamphorst et al., 2017; Leach et al., 1996). Checkpoint blockade antibodies, by enhancing CD28 signaling, may induce EZH2 in Tregs, thereby enhancing immunosuppression in tumors and resistance to checkpoint blockade immunotherapy. In this setting, the inhibition of EZH2 function may reverse this effect and strengthen the efficacy of these therapies. Furthermore, because preexisting immune infiltration into tumors is a strong indicator of responsiveness to checkpoint blockade, the capacity for EZH2 inhibition to increase the infiltration of CD8+ T cells in tumors also may augment the effectiveness of these therapies (Tumeh et al., 2014; Vonderheide et al.,2017). In conclusion, we demonstrate that the blockade of EZH2 function in Tregs can selectively break tolerance in the tumor microenvironment without unleashing systemic autoimmune toxicities, revealing that context-specific activities of epigenetic enzymes may be targeted for cancer immunotherapies.
EXPERIMENTAL PROCEDURES
Mice
All of the mice used were bred onto a C57BL/6 background for a minimum of five generations. All of the mouse experiments (or cells from mice ofgiven genotypes) used comparisons between littermates or age-matched control mice. Foxp3-GFP-hCre transgenic, Foxp3YFP-cre knockin, and Foxp3EGFP-cre-ERT2 mice express Cre recombinase in FOXP3+ cells using distinct technologies (Rubtsov et al., 2008, 2010; Zhou et al., 2008). Ezh2fl mice harbor loxP sites flanking exons 16–19 encoding the SET domain (Su et al., 2003). For tam treatments, mice were treated by oral gavage with a dose of 160 mg/kg suspended in corn oil two times per week. For diphtheria toxin treatments, Foxp3DTR mice were treated as indicated withadose of 0.04 mg/kg upto three times per week every other day (Kimet al., 2007). For tumor studies, syngeneicC57BL/6 mice were inoculatedwith 1 × 105MC38 cells engineered to express luciferase and GFP, 1 × 106 TRAMPC2 cells (all in male mice), or 1 × 104 B16F10 cells expressing GFP in 100 μL of PBS in the flank. Vaccination studies with B16-GVAX (GM-CSF expressing B16cells) were performed with 1 × 106 irradiated cells (50 Gy) injected subcutaneously in the flank 2 days before inoculating 1 × 105B16F10 cells. For Ezh2 inhibitor studies, mice were treated twice daily by oral gavage with 300 mg/kg CPI-1205 made in a vehicle of 0.5% methyl cellulose and 0.2% Tween 80. The treatment regimen began 1 day after tumor inoculation and continued throughout the course of the experiment. Tumor measurements were performed by a single operator in three dimensions using calipers two or more times per week. All of the experiments were conducted according to the Institutional Animal Care and Use Committee guidelines of the University of California, San Francisco.
Murine Lymphocyte Isolation
Resected tumors and lung tissues were minced to 1 mm3 fragments and digested in RPMI media supplemented with 4-(2-hydroxyethyl)-1-piperazi-neethanesulfonic acid (HEPES), 20 mg/mL DNase I (Roche), and 125 U/mL collagenase D (Roche) using a gentleMACS tissue dissociator (Miltenyi) for ~45 min at 37°C. Cells from lymphoid organs were prepared by mechanical disruption between frosted slides. All of the suspensions were passaged over 40 μm filters before cell staining or in vitro stimulation. Cytokine staining was performed with 3 × 106 cells after 210 min of in vitro stimulation in Opti-MEM media with Brefeldin A (eBioscience), phorbol 12-myristate 13-acetate (PMA) (Sigma), and ionomycin (Sigma). Fixation and permeabilization of cells were conducted for intracellular staining using BD Cytofix/Cytoperm (BD Biosciences) for cytokines or the eBioscience Foxp3 fixation/permeabilization kit (ThermoFisher).
Human Lymphocyte Isolation
For human melanomas, freshly isolated samples were minced and digested overnight with a buffer consisting of collagenase type 4 (4188; Worthington Biochemical), DNase (SDN25–1G; Sigma-Aldrich), 10% fetal bovine serum (FBS), 1% HEPES, and 1% penicillin in RPMI medium. Single-cell suspensions were double filtered, centrifuged, and counted. Biopsies of melanoma tumors were performed after patients provided informed consent under the University of California, San Francisco (UCSF) Committee on Human Research Protocol 138510.
FlowCytometry
Surface marker stains for murine samples were carried out with BV605-conjugated anti-mouse CD4 (RM4–5, BioLegend), allophycocyanin (APC)-Cy7-conjugated anti-mouse CD8α (53–6.7, BD Biosciences), phycoerythrin (PE)-Cy7-conjugated anti-mouse PD1 (RMP1–30, BioLegend), BUV395-con-jugated anti-mouse CD45 (30-F11, BD Biosciences), and LIVE/DEAD Fixable Blue Dead Cell Stain (Invitrogen). Intracellular staining of cells was accomplished with fluorescein isothiocyanate (FITC)-conjugated antimouse Foxp3 (FJK-16 s, eBioscience), PerCP-Cy5.5-conjugated antimouse TNF-α (MP6-XT22, BioLegend), eFluor450-conjugated anti-mouse IFN-γ (XMG1.2, eBioscience), APC-conjugated anti-mouse IL-10 (JES5– 16E3, eBioscience), and BV711-conjugated anti-mouse IL-2 (JES6–5H4, BioLegend). Antibodies against H3K27me3 (C36B11, Cell Signaling Technology) and total H3 (D1H2, Cell Signaling Technology) were used after 1.6% formalin fixation and methanol (50%) permeabilization. For human samples, all of the antibodies used for staining were from eBioscience unless otherwise stated: anti-CD3 (UCHT1), anti-hCD8 (RPA-T8), anti-hCD45 (HI30), anti-CD4 (SK3), anti-FOXP3 (PCH101), anti-hCTLA-4 (14D3), anti-PD-1 (EH12.2H7, BioLegend), anti-EZH2 (D2C9, Cell Signaling Technology), and LIVE/DEAD Fixable Aqua Dead Cell Stain (Life Technologies, Thermo Fisher Scientific). Flow cytometry was performed on an LSR II or LSRFortessa (BD Biosciences), and datasets were analyzed using FlowJo software (Tree Star).
Histology
Tissues fixed in 10% formalin for 24 hr, preserved in 70% ethanol, and embedded in paraffin were cut and sections stained with H&E. Chromogenic multiplex immunohistochemistry was performed with anti-CD4 (EPR19514, Abcam) at 1:1,000 dilution, anti-CD8α (4SM15, eBioscience) at 1:50 dilution, and anti-FoxP3 (FJK-16 s, eBioscience) at 1:50 dilution.
Statistical Methods
p valueswere obtained from unpaired two-tailed Student’sttests for all of the statistical comparisons between two groups, and data were displayed as means ± SEMs. For tumor growth curves, two-way ANOVA was used with Sidak’s multiple comparisons test performed at each time point. p values are denoted in the figures by *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
Highlights.
EZH2 expression is elevated in tumor-infiltrating (TI) Tregs
Pharmacological inhibition of EZH2 destabilizes FOXP3 expression and slows tumor growth
Genetic disruption of Ezh2 function in Tregs leads to robust anti-tumor immunity
Blockade of EZH2 in Tregs reprograms TI-Tregs to gain pro-inflammatory activity
ACKNOWLEDGMENTS
We thank Alexander Marson, Mariela Moreno Ayala, and David Raulet for reviewing this manuscript; Vinh Nguyen, Michael Lee, and the UCSF singlecell analysis core facility; Jennifer Bolen, Scott Vandenberg, and the UCSF immunohistochemistry core facility; David Erle, Andrea Barczak, Rebecca Barbeau, Walter Eckalbar, and the UCSF Sandler Center Functional Genomics Core; and Constellation Pharmaceuticals for providing the EZH2 inhibitor. Support for this research came from NIH grants R01 AI046643 and UM1 AI-12–059 and Associazione Italiana per la Ricerca sul Cancro (AIRC) grant IG2013-ID14596. This research also was supported by the Parker Institute for Cancer Immunotherapy. The investigators M.D., L.F., and J.A.B. are members of the Parker Institute for Cancer Immunotherapy, which supported the UCSF Cancer Immunotherapy Program. D.W. was supported by the Frank A. Campini Foundation and NIH/National Cancer Institute (T32 5T32CA128583–09). M.D. also was supported by the Helen Hay Whitney Foundation. J.A.B. is the A.W. and Mary Margaret Clausen Distinguished Professor in Metabolism and Endocrinology.
Footnotes
AUTHOR CONTRIBUTIONS
M.D. and J.A.B. designed the study. D.W., M.D., and J.Q. performed all of the experiments, with assistance from C.-C.P., A.Y., S.S., T.H., A.A., and F.V.G. Human melanoma experiments were performed by K.M. V.R. performed RNA sequencing analyses of human CRC and NSCLC samples. M.D.R., M.P., and L.F. provided conceptual advice and materials. M.D., D.W., and J.A.B. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA sequencing data reported in this paper is GEO: GSE114298.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.050.
REFERENCES
- Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, and Rudensky AY (2014). Inflammation-induced repression of chromatin bound bythetranscriptionfactorFoxp3 in regulatoryTcells. Nat. Immunol 15, 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, and Bluestone JA (2013). Self-antigen-driven activation induces instability of regulatoryT cellsduring an inflammatory autoimmune response. Immunity 39, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Plitas G, Rudra D, Lee SY, and Rudensky AY (2013). Transient regulatoryT cell ablation deters oncogene-driven breast cancerand enhances radiotherapy. J. Exp. Med 210, 2435–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Liu S, Park D, Kang Y, and Zheng G (2007). Depleting intratumoral CD4+CD25+ regulatory T cells via FasL protein transfer enhances the therapeutic efficacy of adoptive T cell transfer. Cancer Res. 67, 1291–1298. [DOI] [PubMed] [Google Scholar]
- Curiel TJ (2007). Tregs and rethinking cancer immunotherapy. J. Clin. Invest 117, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. (2004). Specific recruitment of regulatoryT cells in ovarian carcinomafosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949. [DOI] [PubMed] [Google Scholar]
- De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, et al. (2016).Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, and Bluestone JA (2016). Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol 16, 149–163. [DOI] [PubMed] [Google Scholar]
- DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, Zhang R, Turka L, Marson A, and Bluestone JA (2015). The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 42, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, Schmid RM, Klein U, Hayden MS, and Ghosh S (2017). NF-kB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 170, 1096–1108.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, Du-Page M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity 43, 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, Warshauer JT, and Bluestone JA (2017). Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med 23, 540–547. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. (2017). Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, and Roberts CWM (2016). Targeting EZH2 in cancer. Nat. Med 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MWL, Ngiow SF, Smyth MJ, Hamann A, Huehn J, and Sparwasser T (2010). Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 70, 7788–7799. [DOI] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, and Allison JP (1996). Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang L, Predina J, Han R, Beier UH, Wang L-CS, Kapoor V, Bhatti TR, Akimova T, Singhal S, et al. (2013). Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat. Med 19, 1173–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C,Workman CJ, and Vignali DAA (2016a). Targeting regulatoryT cells in tumors. FEBS J. 283, 2731–2748. [DOI] [PubMed] [Google Scholar]
- Liu J, Blake SJ, Harjunpaa H, Fairfax KA, Yong MCR, Allen S, Kohrt HE, Takeda K, Smyth MJ, and Teng MWL (2016b). Assessing immune-related adverse events of efficacious combination immunotherapies in preclinical models of cancer. Cancer Res. 76, 5288–5301. [DOI] [PubMed] [Google Scholar]
- Luo CT, Liao W, Dadi S, Toure A, and Li MO (2016). Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoim-munity. Nature 529, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L, et al. (2016). PRC2 epigenetically silences Th1-type cytokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 76, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, and Kim H-J (2016). Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc. Natl. Acad. Sci. USA 113,6248–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neo WH, Lim JF, Grumont R, Gerondakis S, and Su I-H (2014). c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J. Biol. Chem 289, 31693–31707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O’Brien S, Blank R, Lamb E, Natarajan S, et al. (2009). Decrease of Foxp3+Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. (2017). Interferon-g drives Treg fragility to promote anti-tumor immunity. Cell 769, 1130–1141.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, and Rudensky AY (2016). Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45, 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ, Colligon TA, Trosko JA, Leinbach LI, Pletcher CH, et al. (2012). CD25 blockade depletesand selectively reprogramsregulatoryT cellsin concert with immunotherapy in cancer patients. Sci. Transl. Med 4, 134ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY (2010). Stability of the regulatoryT cell lineage in vivo. Science 329, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, et al. (2016). Two FOXP3(+) CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med 22, 679–684. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Sarmento OF, Svingen PA, Xiong Y, Sun Z, Bamidele AO, Mathison AJ, Smyrk TC, Nair AA, Gonzalez MM, Sagstetter MR, et al. (2017). The role of the histone methyltransferase enhancer of zeste homolog 2 (EZH2) in the pathobiological mechanisms underlying inflammatory bowel disease (IBD). J. Biol. Chem 292, 706–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, Zhong H, Yang X, Houghton AN, Merghoub T, and Wolchok JD (2013). GITR pathway activation abrogatestumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol. Res 7, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, and Smyth MJ (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 337,1565–1570. [DOI] [PubMed] [Google Scholar]
- Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, and Korman AJ (2013). Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res 1, 32–42. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Hou D-Y, Baban B, Koni PA, He Y, Chandler PR, Blazar BR, Mellor AL, and Munn DH (2010). Reprogrammed foxp3(+) regulatoryT cells provide essential help to support cross-presentationand CD8(+) T cell priming in naive mice. Immunity 33, 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu J, Yamazaki S, and Sakaguchi S (1999). Induction oftumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol 163, 5211–5218. [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su I-H, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, and Tarakhovsky A (2003). Ezh2 controls B cell developmentthrough histone H3 methylation and Igh rearrangement. Nat. Immunol 4, 124–131. [DOI] [PubMed] [Google Scholar]
- Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, et al. (2013). Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 110, 17945–17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, and Sakaguchi S (2017). Regulatory T cells in cancer immunotherapy. Cell Res. 27, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MWL, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, and Smyth MJ (2010a). Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. 70, 7800–7809. [DOI] [PubMed] [Google Scholar]
- Teng MWL, Swann JB, von Scheidt B, Sharkey J, Zerafa N, McLaughlin N, Yamaguchi T, Sakaguchi S, Darcy PK, and Smyth MJ (2010b). Multiple antitumor mechanisms downstream of prophylactic regulatory T-cell depletion. Cancer Res. 70, 2665–2674. [DOI] [PubMed] [Google Scholar]
- Tiffen JC, Gallagher SJ, Tseng HY, Filipp FV, Fazekas de St. Groth B, and Hersey P (2016). EZH2 as a mediator of treatment resistance in melanoma. Pigment Cell Melanoma Res. 29, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, Good AC, et al. (2016). Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl) methyl)-2-methyl-1 -(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1 H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J. Med. Chem 59, 9928–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH, Domchek SM, and Clark AS (2017). Immunotherapy for breast cancer: what are we missing? Clin. Cancer Res 23, 2640–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, Zhang J-G, Zhan Y, Lew AM, Silke J, et al. (2014).The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 124, 737–749. [DOI] [PubMed] [Google Scholar]
- Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, and Bluestone JA (2008). Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med 205, 1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, and Bluestone JA (2009). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol 10, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, Sommer L, and Boyman O (2017). The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 20, 854–867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.