

Abstract
Genetic redirection of T lymphocytes with chimeric antigen receptors (CARs) has soared from treating cancers preclinically to FDA approval for hematologic malignancies and commercial-grade production scale in under 30 years. To date, solid tumors are less susceptible to CAR therapies and instead have been treated more successfully with immune checkpoint blockade or tumor-infiltrating lymphocyte therapy. Here, we discuss the current challenges in treating solid tumors with CAR T cells, and the obstacles within the host and tumor microenvironment hindering their efficacy. We present a novel three-pronged approach for enhancing the efficacy of CAR T cells whereby a single infusion product can synergize the power of an optimal CAR construct, a highly potent T cell subset, and rejuvenate the endogenous immune response to conquer therapeutically-resistant solid tumors.
Keywords: chimeric antigen receptor, T cell, solid tumor, adoptive cell transfer, checkpoint
Introduction
Healing is a matter of time, but it is sometimes also a matter of opportunity—Hippocrates
The interactions between antigen-presenting cells and T cells enable high fidelity host protection against foreign pathogens and malignant cells. T cells have unparalleled ability to not only respond to these antigens but also to formulate memory, permitting a rapid and robust response upon future challenge against the same antigen. In terms of cancer, this potentially means long-term protection against recurrence of tumor cells expressing those antigens. Tumors can express antigens that are rapidly recognized by T cells, where mutations of self-antigens or germline cancer antigens differ sufficiently from normal antigens, or those that are less robustly detected, such as overexpressed self-antigens or differentiation antigens expressed by tissue from which the tumor originates (1). As a result, tumors that are more similar to normal cells, or those with highly immune-suppressive qualities, escape surveillance, permitting their outgrowth and potential to cause great harm. Many technological advances have created opportunities for cancer immunotherapists to bolster the power of T cells against cancer through reeducation and intelligent design to overcome the evasive barriers established by solid tumors. Perhaps immunotherapy represents one such opportunity posited by Hippocrates—a chance for intervention that could heal cancer patients much more effectively than time itself.
Adoptive cell transfer (ACT) comprises one of two arms of immunotherapy and involves ex vivo enrichment of tumor-specific cells, expansion to large numbers, and reinfusion into the patient to specifically target and kill cancer cells. ACT is conducted via two approaches: (1) naturally arising T cells that infiltrate the tumor—called tumor-infiltrating lymphocytes (TILs)—can be expanded ex vivo from the malignant site or (2) non-therapeutic endogenous lymphocytes obtained from the peripheral blood can be rendered tumor specific via genetic redirection with a T-cell receptor (TCR) or chimeric antigen receptor (CAR). The second arm of immunotherapy includes immune checkpoint blockade (ICB), where enhancing priming or rejuvenating exhausted T cells can render a functional, albeit often transient, antitumor state. This review will focus on CAR T cell therapies and how future CARs may work synergistically with other immunotherapies to drive long-lasting cures in patients.
The CAR combines a single chain variable fragment (scFv) ectodomain that can target an antigen of choice with an endodomain comprised of the CD3ζ TCR signal and additional costimulatory domain. Its first use by Kuwana et al. and Gross et al. in the late 1980s revealed that redirection of a T cell with this receptor could induce antigen recognition without the major histocompatibility complex (2, 3). CAR-redirected T cell therapies have been successful in hematologic malignancies but are less effective in treating the majority of patients with solid tumors to date. For solid tumors, immunotherapy based in TIL generation or ICB has been more successful. Conceivably, harnessing a CAR therapy with mechanisms of success from TIL and ICB therapies is a logical approach to overcome the obstacles preventing their effective regression of solid tumors. This review will discuss the current status of CAR therapies for solid tumors and outline a three-pronged approach to enhance these therapies against treatment-resistant cancers based on lessons learned with adoptive immunotherapy.
Destinations of Car T Cell Immunotherapy
The ability to harness an immune response against cancer through ACT or ICB has reinvigorated cancer therapies by improving outcomes in patient populations previously resistant to conventional treatment. Genetic redirection of T cells with specificity against a chosen antigen provides theoretical opportunity to invoke long-term immunity, but with varied results based on type of tumors targeted (4, 5). Herein, we will review recent triumphs of CAR T cells against B cell hematologic malignancies, and the challenges currently preventing similar efficacy in treatment of aggressive solid tumors.
Success in Hematologic Malignancies
Since 2010, numerous clinical trials have demonstrated the ability of CAR T cells directed against CD19 to promote clinical responses in acute lymphoblastic leukemia (ALL) (6–10), diffuse large B cell lymphoma (DLBCL) (11–13), chronic lymphocytic leukemia (CLL) (14, 15), and other B-cell non-Hodgkin lymphomas (16, 17) with remissions of up to 90% in some of these cases. Because CD19 is expressed ubiquitously in the B cell lineage, targeting CD19 ablates this cell compartment in patients, though sparing of some plasma cells with long-term humoral immunity is possible (18). Fortunately, B cell aplasia can be treated with immunoglobulins to prevent infections, making this a serious but manageable on-target/off-tumor toxicity (19). As a result of excellent responses in patients refractory to standard of care therapies, two constructs of CD19-CAR T cells have been granted FDA approval. Tisagenlecleucel (KYMRIAH, Novartis), with the 4-1BB/CD3ζ costimulatory domain, was approved in August 2017 for B-ALL (20) and in May 2018 for DLBCL, and axicabtagene ciloleucel (YESCARTA, Kite Pharmaceuticals), with the CD28/CD3ζ costimulatory domain, was approved for DLBCL in October 2017. Administration of these CAR T cell therapies requires specialized training under the FDA Risk Evaluation and Mitigation Strategies to manage adverse events such as cytokine release syndrome or neurotoxicity. These approvals render CAR T cells the first FDA approved personalized gene therapy and establish a major milestone in the field of cancer immunotherapy.
Unfortunately, the dramatic responses reported in patients with B cell malignancies have not yet been consistently reproduced with analogous therapies for individuals with solid tumors. However, it is important to appreciate that CAR T cell development for patients with solid tumors is still in early stages. The historical progress, current status, and major obstacles facing success of these therapies in conquering solid tumors are discussed below.
Clinical Challenges in Solid Tumors
While the results of CAR T cells in B cell malignancies are encouraging, treatment of solid tumors with similar approaches has yielded less favorable results. Similar to therapies for hematologic malignancies, the difficulty in initial design begins with constructing the CAR against an antigen expressed in the tumor—but not in normal tissue—to bolster efficacy while reducing off-tumor toxicity (21). Thus far, clinical trials with CAR T cells in solid tumors have demonstrated severe toxicities since the targeted antigens are often not completely foreign to the host, and even low expression in distant tissues can instigate devastating effects in the presence of a potent T cell therapy (22, 23). Several examples of off-tumor responses in clinical trials are as follows: in renal cell carcinoma, targeting carbonic anhydrase IX (CAIX) led to liver toxicity in 4/8 patients in 2/3 cohorts due to basal expression of CAIX on bile duct epithelium even with low doses of CAR T cells (24, 25). CAR T cells engineered against ERBB2 given in a high dose to a patient with metastatic colorectal cancer caused multi-organ failure with acute pulmonary toxicity due to antigen expression on lung epithelium (26). This resulted in death of the patient within 5 days post-transfer of the cellular product (26). Similarly, a trial for gastrointestinal tumors with CEACAM5-CAR T cells was closed due to poor efficacy and persistence of cells, in addition to toxicity from expression of the targeted antigen on lung epithelium (27). Careful consideration of target antigens is therefore warranted so that a balance between safety and efficacy can be maintained for patients.
Some antigens specific to tumors have been identified that result in more limited off-tumor effects, but many of these targets for CAR T cells have mediated poor clinical efficacy in patients. Several studies using HER2-based CAR in sarcoma (28), mesothelin-specific CAR in mesothelioma and pancreatic cancer (29–31), carcinoembryonic antigen for colorectal cancer (32), EGFRvIII in glioblastoma (33), and α-folate receptor in ovarian carcinoma (34) have shown safer toxicity profiles but yield no better treatment outcomes than stable disease in most cases. Furthermore, similar to CD19+ B cell malignancies (9, 35), solid tumors treated with therapeutic CAR T cells can undergo antigen escape due to selection pressure favoring tumor cells lacking the targeted antigen. High frequency of EGFRvIII loss in glioblastoma patients, though indicating the CAR T cells are potent against their target, highlights the importance of heterogeneity in antigen targeting for future solid tumor CAR treatments to be successful (36). Despite these challenges, there has been recent success with CAR T cell therapy in glioblastoma. Localized delivery of CAR T cells engineered against IL-13Rα for recurrent glioblastoma resulted in an objective response lasting 7.5 months in one patient with several intracranial and spinal tumors (37, 38). Obtaining responses in such aggressive, end-stage cancers emphasizes the vast potential for CAR T cell therapies and the importance of their future development.
Theoretically, even if the perfect antigen for a solid tumor could be identified and targeted, CAR T cell therapies for solid tumors face further obstacles including poor trafficking to the tumor site (39), as well as limited persistence and proliferation within the host (27, 34, 40–42). Moreover, CAR T cells can be functionally suppressed within the hostile tumor microenvironment (43). These collective hurdles set solid tumor CAR-based therapies apart from liquid tumors (21, 44). The question puzzling the medical community today is how—or if—we can overcome these significant barriers and cure solid tumors with a CAR T cell therapy approach. Reflecting upon these challenges, we hypothesize that the ultimate CAR therapy for solid tumors may be established via a three-pronged approach, as illustrated in Figure 1. The most therapeutic strategy should (1) encompass specificity through the CAR construct, (2) select for a T cell subset with enhanced persistence, trafficking, and long-lived memory responses, and (3) synergize with the endogenous host response to neoantigens. We will review our field’s progress on encompassing these three axes thus far and present our blueprint for what may be necessary to combat solid tumors with next-generation CAR-based approaches.
Figure 1.
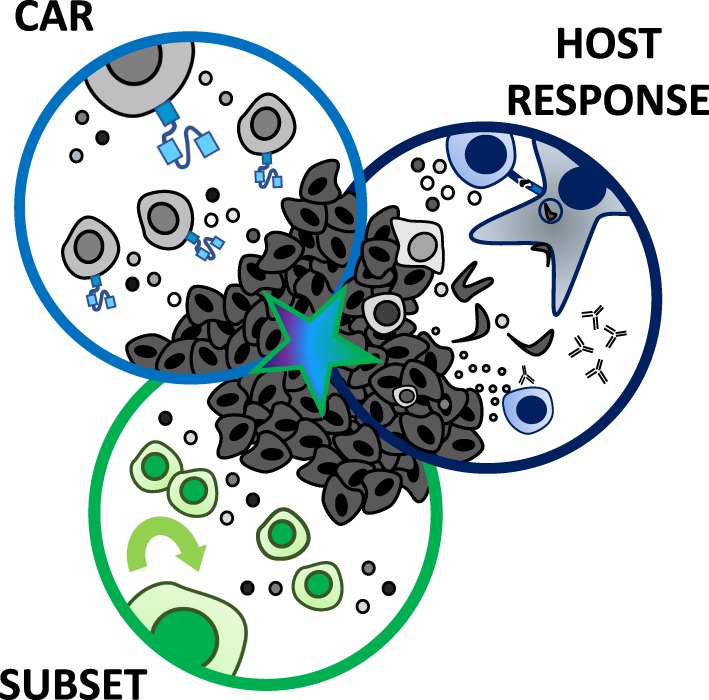
Three-pronged approach to improve chimeric antigen receptor (CAR) T cell therapies in solid tumors. A multi-faceted attack on solid tumors resistant to standard CAR T cell therapies may best augment their efficacy in clinical trials. The ultimate CAR T cell therapy should encompass three axes: (1) a CAR with high fidelity targeting of more than one tumor antigen and trafficking capacity, (2) selection of a T cell subset with potent self-renewal and migratory capacity for long-term persistence and immunity, and (3) ability to harness and rejuvenate the host response to tumor neoantigens. A single arm (CAR, subset, or host response) has not been sufficient for long-term responses against aggressive solid tumors to date.
Evolution of the Car Design
The first three generations of CAR construct design have evolved to incorporate two activating signals (TCR-signaling domains and costimulation) to enhance functionality of therapies in vivo and have been reviewed previously (45–47). Briefly, the first-generation CAR, pioneered by several groups in the late 1980s (2, 3, 48–50), consisted of only the scFv region and CD3ζ intracellular domain. These cells demonstrated poor efficacy and expansion in response to antigen, especially if given without exogenous IL-2 (51). The second-generation CAR includes an additional costimulatory domain while the third generation (Figure 2A) uses two costimulatory domains in tandem (52); both have greatly enhanced efficacy over the first generation. CD28 was incorporated first (53), followed by ICOS (54), OX40 (CD134) (54, 55), and 4-1BB (CD137) (54, 56, 57). While the optimal costimulatory signals are under debate and may depend on the T cell subset itself, 4-1BB signaling has been shown to improve persistence (15, 58) and enhance metabolic fitness and memory potential of CAR T cells over CD28 (59), and the combination of 4-1BB and ICOS appears promising preclinically (60).
Figure 2.

“Fourth-generation” chimeric antigen receptor (CAR) constructs incorporate novel mechanisms to enhance targeted antitumor efficacy. (A) The third-generation CAR incorporates the extracellular scFv with intracellular CD3ζ signaling and two tandem costimulatory domains. (B) CAR T cells with additional chemokine receptors have improved trafficking to tumors. (C,G) T cells secreting additional cytokines or engineered with cytokine signaling domains have enhanced activation and can modulate surrounding microenvironment. (D) Armored CARs redirect suppressive signals from the tumor to activating signals to resist exhaustion. (E) Suicide genes and (F) bispecificity mitigate off-tumor toxicity through the ability to deplete transferred cells or enhance specific targeting to tumors, respectively. (H) Switchable CAR targeting via adaptor molecules provides versatile opportunity to control CAR activation, specificity, and longevity after transfer of cells. Abbreviations: Ag, antigen; PNE, peptide neo-epitopes.
Due to a lack of clinically successful CAR therapies in patients with solid tumors, numerous groups have been inspired to design “fourth-generation” CAR constructs incorporating novel mechanisms to improve antitumor activity. These approaches include enhancing migration and efficacy of the engineered cell, as well as the ability to resist immunosuppression and off-tumor toxicity, illustrated in Figure 2 and discussed directly below.
Enhancing Migration
Tumors that express fewer chemokines often evade host surveillance via impairing effector T cell recruitment and infiltration into the tumor (61). Several different chemotherapeutics have been shown to induce CXCR3-ligand and CCL5, which enhance CD8+ T cell recruitment and reduce tumor growth (62). One chemokine in particular, MCP-1/CCL2, has been correlated with enhanced CCR2-expressing T cell trafficking when secreted by tumors such as gliomas, neuroblastoma, renal cell carcinoma, and mesothelioma (63). For CAR T cells, ex vivo activation protocols can alter expression of chemokine receptors, where those such as CCR2 are frequently downregulated (64). Two groups have shown that forced expression of CCR2 on CAR T cells (Figure 2B) targeting either GD2 in neuroblastoma (65) or mesothelin for malignant pleural mesothelioma (64) enhances T cell infiltration and augments antitumor activity of the transferred cells. In melanoma, poor T cell infiltration within tumor has been correlated with high tumor IL-8/CXCL8 expression (61); therefore, future CAR T cells engineered to express CXCR1 or CXCR2 may also be more efficient at targeting melanoma. As various solid tumors express unique combinations of chemokines, further understanding of these chemokine profiles could aid in the design of novel CAR T cells that can traffic more robustly to the particular cancer they are intended to destroy.
Augmenting Efficacy
As solid tumors have proven to be formidable foes, CAR T cells fortified with enhanced properties of cytokine secretion or cytokine signaling domains have several unique advantages to overcome limitations of the tumor microenvironment, as depicted in Figures 2C,G. If the T cell produces a cytokine related to cytotoxic effector programming upon ligation of the CAR, autocrine signaling can activate and support the antitumor activity, persistence, and survival of the transferred cells. In addition, tumor-targeting CAR T cells can deliver cytokines to modulate the cancer microenvironment in an advantageous manner to either activate host effectors or hinder host suppressors to bolster memory T cells in the patient long term.
These cytokine-producing “TRUCKs” (T cells Redirected for Universal Cytokine Killing) have shown efficacy when delivering IL-12, IL-15, IL-18, or IL-21 to the tumor microenvironment (Figure 2C) (66). Of particular clinical importance, IL-12-producing CARs were reported to be therapeutic against lymphoma even without preparative lymphodepletion (67), and significantly enhanced efficacy of MUC-16ecto CAR against a preclinical model of ovarian carcinoma (68). IL-12-producing CD8+ T cells modulated suppressive host myeloid cells within the tumor microenvironment, and as a result improved therapeutic efficacy (69). In a clinical trial for metastatic melanoma patients, autologous TIL engineered to secrete IL-12 yielded objective responses in lower doses compared with unmodified TILs and without systemic administration of IL-2; however, many of these responding patients developed severe liver toxicities and hemodynamic instability (70). Moving forward, it will be critical to deliver localized and inducible IL-12 production within the tumor microenvironment via TILs or CAR T cells to more specifically direct its potency while minimizing risk of unacceptable toxicity. IL-15 production similarly improved survival and proliferation of CAR T cells specific for CD19 in leukemia/lymphoma (71) and IL-13Rα2 in glioblastoma (72), as did membrane-bound IL-15 for CD19+ leukemia without significant toxicity (73). Recently, IL-18-producing CAR T cells have been developed. Administration of IL-18 has been shown to augment immunity in solid tumors via activating natural killer (NK) cells (74) and is known to induce IFN-γ production from Th1 cells in the presence of IL-12 (75, 76). In CD19+ tumors, IL-18-producing TRUCKS improved engraftment and long-term survival of hosts bearing established tumors (77). Importantly, in mouse models of pancreatic carcinoma and metastatic lung adenocarcinoma—classically highly resistant to treatment—Chmielewski and Abken established that IL-18 secretion and autocrine signaling can induce a T-BetHigh FoxO1low signature in the CAR T cells and augment tumor infiltration of NKG2D+ NK cells, while reducing the frequency of regulatory T cells (Tregs) and suppressive macrophages in the tumor microenvironment (78). While improved proliferation and cytokine production within the host are important to antitumor efficacy, the possibility of cytokine-induced dysregulation of CAR expansion or toxicity highlights the need for a form of safety switch or suicide gene within the CAR (71).
IL-21 is a homeostatic cytokine that has shown promise in preclinical TIL and CAR studies, and may be a desirable future candidate to bolster responses in adoptive transfer clinical trials. Programming CD8+ tumor specific lymphocytes ex vivo with IL-21 was reported to reduce the activation/exhaustion phenotype of terminally differentiated cells observed after long-term expansion with IL-2 (79). While ex vivo cytolytic function of CD8+ T cells upon antigenic stimulation was reduced with IL-21 priming, the in vivo melanoma regression was greatly enhanced long term compared with CD8+ T cells primed with IL-2 or IL-15. Systemic administration of IL-21 also enhanced efficacy of tumor-specific CD8+ T cells against melanoma in a preclinical model (80). IL-21 fosters generation of antitumor T cells expressing Tcf7, L-selectin, and Lef1 in the Wnt/β-catenin pathway, inducing a signature of stem-like properties that may support long-lived memory of transferred CAR T cells clinically (79). IL-21 programming of human CAR T cells ex vivo was also shown to improve efficacy against CD19+ tumors in vivo (81). Furthermore, in a direct comparison, CAR T cells producing IL-21 were superior to IL-15- or IL-2-producing CARs against CD19+ malignancies (82). Therefore, maintenance of memory characteristics in vivo through inducible IL-21 expression in CAR T cells, theoretically also supporting memory of endogenous tumor-specific T cells, may greatly improve the longevity of future CAR therapies for long-lasting curative responses.
Another related application of this concept has been described where the CAR construct encodes a costimulatory domain as well as a cytokine signaling domain for IL-2Rβ (Figure 2G) (83). Therefore, the CAR T cell does not produce the cytokine, but the pathway downstream of the desired signal is activated upon engagement of the scFv fragment with antigen. Unfortunately, this approach is restricted to augmenting the CAR T cell’s efficacy and not the endogenous host response. However, with cytokines like IL-2, which signal to both effector and Tregs, this approach can restrict signal activation to the effector arm of the antitumor response. Collectively, manipulating cytokine production or cytokine signaling has opened new possibilities for generating CARs with desirable traits to bolster their efficacy against tumors and improve immunity of other infiltrating immune cells.
Evading Immunosuppression: Turning Lemons into Lemonade
Even when CAR T cells successfully invade the tumor, they face a microenvironment rich in suppressor cytokines, such as TGF-β and IL-4, and inhibitory molecules including PD-L1 that poise the cancer to escape immunity. To make these limitations advantageous, tumor immunologists are now redirecting TCR or CAR-specific T cells with additional domains that either (1) limit suppressive signaling or (2) convert suppressive signals into activating signals, thus “armoring” T cells against the suppressive elements of the tumor (Figure 2D). The earliest studies using this approach were with Epstein–Barr virus-specific T cells engineered with a dominant negative mutation of the TGF-β receptor, which allowed tumor-specific T cells to resist suppression by the tumor-derived TGF-β (84). Likewise, PSMA-specific CAR T cells for prostate cancer engineered with a dominant negative TGF-β receptor demonstrated enhanced proliferation post-transfer and are now being used in clinical trials (NCT03089203, Table 1) (85). New studies with CAR T cells have used a chimeric cytokine receptor that binds IL-4, a cytokine that suppresses immunity, via an ectodomain but transmits a therapeutic IL-7 signal via the endodomain. When IL-4 binds the receptor, instead of the anti-inflammatory STAT6 translocation, the IL-7 pathway phosphorylates STAT5 and polarizes the cell toward an inflammatory Th1 response (86). Similarly, a PD-1/CD28 chimeric switch receptor has been designed to convert an exhaustive stimulus into a costimulatory signal; this construct was shown to enhance cytokine production and in vivo efficacy in the presence of PD-L1+ prostate cancer cells compared with CAR-only transduced cells (87). Two clinical trials are ongoing in China with the use of chimeric switch receptors and are described in Table 1. These advances in T cell engineering may now enable reversal of mechanisms driving CAR T cell failure in solid tumors.
Table 1.
Clinical trials of fourth-generation chimeric antigen receptor (CAR) T cells in solid tumors.
| 4th Generation CAR T cells in solid tumors | ||
|---|---|---|
| ClinicalTrials.gov identification | Trial description | Location(s) |
| “Armored” CAR | ||
| NCT03089203 | CAR T cells targeting PSMA for castration-resistant prostate cancer with dominant negative TGF-β receptor | University of Pennsylvania |
| NCT02937844 | Pilot study of autologous chimeric switch receptor modified T cells in recurrent glioblastoma multiforme | Sanbo Brain Hospital Capital Medical University, Beijing, China |
| NCT02930967 | Chimeric switch receptor with PD-L1+ recurrent or metastatic malignant tumors | China Meitan General Hospital |
| Suicide genes | ||
| NCT00730613 | CAR T against IL-13Ra2 in glioblastoma with Hy/TK suicide switch | City of Hope Medical Center |
| NCT02992210 | 4SCAR-GD2 targeting CAR with iCaspase9 domain in refractory solid tumors | Shenzhen Geno-Immune Medical Institute |
| NCT02414269 | Malignant pleural disease treated with Meso-CAR T cells, modified with iCasp9/M28ζ | Memorial Sloan Kettering Cancer Center |
| NCT01822652 | GD-2-CAR T (28-Ox40ζ) and iCaspase9 Suicide safety switch for Neuroblastoma | Baylor College of Medicine |
| NCT03185468 | 4SCAR-GS2 with iCaspase9 domain in advanced/metastatic urothelial carcinoma | Shenzhen Geno-Immune Medical Institute |
| Antibody-producing CAR T cells | ||
| NCT03179007 | CTLA-4/PD-1 antibody expressing MUC-1 CAR T for MUC1+ advanced solid tumors | Shanghai Cell Therapy Research Institute |
| NCT03182803 | CTLA-4/PD-1 antibody expressing mesothelin-CAR T for Meso+ advanced solid tumors | Shanghai Cell Therapy Research Institute |
| NCT03182816 | CTLA-4/PD-1 antibody expressing EGFR-CAR T for EGFR+ advanced solid tumors | Shanghai Cell Therapy Research Institute |
| NCT02862028 | PD-1 antibody expressing CAR T cells for EGFR family member positive advanced solid tumor (liver, lung, stomach) | Shanghai International Medical Center, Shanghai, China |
| NCT02873390 | PD-1 antibody expressing CAR T cells for EGFR family member positive advanced solid tumor | Ningbo Cancer Hospital, Zhejiang, China |
| NCT03030001 | PD-1 antibody expressing mesothelin-specific CAR T cells for meso+ malignant tumors (recurrent or refractory) | Ningbo Cancer Hospital, Zhejiang, China |
| NCT03170141 | 4SCAR-IgT against EGFRvIII on glioblastoma multiforme, producing PD-1 and PD-L1 antibodies | Shenzhen Geno-Immune Medical Institute |
Mitigating Off-Tumor Toxicity
Finally, CAR T cell depletion in patients experiencing uncontrolled toxicity and engineering approaches to enhance specificity to solid tumor antigens are two methods to reduce severe toxicities previously discussed. Suicide genes to deplete CAR T cells, incorporation of epitopes for antibody neutralization, and logic gate control of CAR T cell function have been described. The first examples of suicide genes involved use of HSV-thymidine kinase, which converts ganciclovir into a toxic metabolite (88). However, the problem with this approach is that the response is slow (89) (several days) and the viral proteins themselves may be immunogenic leading to rejection of the cells (90). In recent development, the inducible-caspase 9 system armors the CAR with a homodimer iCasp9 domain that dimerizes upon administration of a small molecule (Figure 2E) (89). Dimerization leads to cleavage of caspase 3 and apoptosis of the CAR T cells. Several clinical trials are now incorporating such safety switches into their CAR programs, which are outlined in Table 1. In addition, incorporating epitopes like RQR8/CD20 into the CAR construct provides a target for their depletion with antibodies such as rituximab (91). This approach depletes the majority of CAR T cells within a few hours (91). As rituximab is widely used clinically, this is a non-toxic and relatively inexpensive method for rapid deletion of CAR T cells in case of severe toxicity. Though protective against severe toxicities, the iCasp9 and antibody-directed depletion approaches do not differentiate cells causing off-tumor side effects from cells with therapeutic efficacy, which could result in loss of any clinical benefit against tumors.
To improve the discriminatory nature of strategies used to reduce toxicity, design of CAR T cells equipped with tetracycline-inducible systems or AND/NOT Boolean logic gates permit enhanced control over effector responses and improved sensing of tumor targets. Sakemura and colleagues established a Tet-on inducible system for CD19+ malignancies, where administration of a tetracycline turns on CAR expression—useful for a period of heavy tumor burden—while withdrawal of the drug ceases CAR expression but permits survival of the cell—important for periods of off-tumor toxicity (92, 93). Boolean logic gates aim to prevent toxicity while maintaining efficacy, rather than irreversibly deleting CAR T cells that are toxic against both tumor and host. First, AND gates require a combination of antigens for full T cell activation. In prostate cancer, Kloss and colleagues demonstrated that high affinity CAR and chimeric costimulatory receptors targeting two antigens, such as PSMA and PSCA, leads to eradication of cells bearing either target (94). However, with low affinity receptors, activation of one receptor was not sufficient for full T cell activation, making the presence of both antigens necessary for activation (94). Wendell Lim and colleagues have pioneered the use of syn-Notch receptors in CAR T cells where engagement of a tissue-specific antigen by a surface receptor induces transcription of a CAR against a tumor-specific antigen (Figure 2F) (95–97). These approaches allow increased sensitization to tumor cells and reduced toxicity against healthy tissues bearing only one of the targeted antigens. Alternatively, NOT gates employ receptors that prevent T cell activation. For example, the iCAR developed by Fedorov et al. has two receptors with opposite functions: first, a receptor for an off-target antigen such as one found on healthy tissue signals the inhibitory cascade downstream of CTLA-4 or PD-1, while a second tumor-specific receptor signals CD3ζ and costimulation for T cell activation (98). Therefore, CAR T cells can be designed to discriminate between on- and off-tumor targets without compromising survival of the transferred T cells. With these novel CAR T cell designs, toxicities can be managed without loss of antitumor function, though indication of each approach may vary depending upon the type of tumor and immunogenicity of the antigens targeted.
While combinatorial or logical sensing may enhance specificity of CAR T cells to tumor targets in the future, the search for antigens specific for tumors remains an important ongoing approach. Self-antigens are frequently modified through processes such as glycosylation as they undergo mutagenesis and cells experience malignant transformation (99). CAR T cells targeting glycosylated self-antigens in the tumor are potent against several solid tumor types and minimally toxic to the host due to the specificity of glycosylation sites for the tumor (100). Overall, a better understanding of how self-antigens are modified in tumors may represent a simpler approach to achieve high potency and low toxicity clinically.
Remote-Controlled CARs
Very recently, CAR T cells active only in the presence of a soluble, inert adaptor molecule have been brought to life in preclinical systems (Figure 2H). Early studies incorporated CAR T cells engineered for specificity against FITC (101) or PNEs (102), which are linked to antibodies specific for antigens on tumor. Recently, “SUPRA” (split, universal, and programmable) CAR T cells were developed where a “zipCAR” domain links an intracellular costimulatory domain and an extracellular leucine zipper (103). This zipper can be targeted with a complementary zipper fused to an scFv region to render the SUPRA CAR T cell tumor specific. These approaches would be particularly useful for generating universal CAR T cells for various tumors; adaptor molecules could be designed for tumor specificity and would provide options for altering specificity post-adoptive transfer, key for situations of selection pressure and antigen escape. The feasibility and speed of developing a new adaptor with specificity for tumors is likely to be much greater than generating a new, personalized CAR T cell product.
As collectively revealed in Figure 2, the scientific community’s response to challenges in treating solid tumors has been robust and impressive. Indeed, many opportunities now exist for design of future clinical trials incorporating more specialized CAR constructs. However, since persistence of T cells and a long-lasting memory response are ideal for a successful therapy, it is likely that the quality of the lymphocyte itself is as important for building a better CAR to target the antigen. Consequently, we will next discuss what is known about the optimal properties of a T cell for adoptive transfer and future implications of their clinical use in patients with solid tumors.
Beyond the Car: Pursuit of the Optimal T Cell
Ex vivo manipulation of T cells provides a unique opportunity to select the most highly therapeutic cells before transfer, including generation of CD8+ lymphocytes with a distinct memory lineage or polarized CD4+ helper T cell subsets. Despite the advantages of precisely defining the most effective infusion product composition through cell sorting, most clinical trials of CAR T cells to date infuse bulk products in efforts to transfer large numbers of cells (104). One recent clinical trial at Fred Hutchinson used this selective approach by infusing a 1:1 ratio of CD4+ and CD8+ cells with a central memory (CM) signature to treat patients with B-ALL; however, only 16 of the 30 patients had enough CD8+ TCM cells in the peripheral blood to meet their minimum criteria to enrich this population (105). As technologies advance to permit more efficient T cell purification, so will the feasibility of selecting the optimal T cells for achieving long-term cures in patients. By enriching cell subsets with greater potency, reliance on large doses of T cells may become obsolete. Highlighted below are current novel ways in which investigators are generating T cell subsets with enhanced properties for ACT.
CD8+ Memory Subsets
Debate exists about which memory CD8+ T cell subset is ideal for sustaining durable responses to cancer. Some investigators believe that effector CD8+ T cells that secrete heightened IFN-γ are more effective against tumors while others argue that less-differentiated or even naïve CD8+ T cells are the most ideal lymphocytes to foster long-lasting immunity (106, 107). Therefore, we review previous work defining the role of CD8+ T cell differentiation and memory in the context of adoptive T cell transfer therapy.
When activated with a cognate tumor antigen, CD8+ T cells differentiate into a short-lived effector phase poised with cytotoxicity against their target. The exact mechanism of this differentiation pathway remains under dispute and two differentiation models have been proposed. One model suggests that naïve cells differentiate directly into the effector phase, followed by de-differentiation into long-lived memory cells. New evidence supporting this model is highlighted by the ability of an individual cell to lose and regain expression of L-selectin without cell division (108). A second model, known as the linear differentiation model, suggests that naïve cells are programmed into TSCM [stem-cell memory (SCM)], TCM [central memory (CM)], and TEM [effector memory (EM)] subsets with varied capability of responding to antigenic rechallenge, terminating with differentiation into effector cells (Figure 3A) (109, 110). Recent epigenetic findings add to this body of literature supporting the linear model of T cell differentiation by showing that after priming, the histone methyltransferase Suv39h1 silences memory genes to direct CD8+ T cells into the effector phenotype (111). Without Suv39h1, the memory subsets are preserved after activation while generation of effector subsets is impaired, suggesting that memory phenotypes are enriched before effector phenotypes (111). For a patient in complete remission from CLL after CD19-CAR T therapy, CAR integration into the tumor suppressor gene, TET2, resulted in robust clonal proliferation of CAR T cells with a predominantly CM phenotype (112). While this particular integration site was not by design, this clinical example highlights the intricacy of memory differentiation and the implications of driving the T cell toward a particular memory phenotype on patient outcomes. This suggests that epigenetic or genetic manipulation of T cells ex vivo could be a novel approach to control memory differentiation of cells and generate a more therapeutic product before transferring cells into patients.
Figure 3.

Antitumor efficacy of memory CD8+ T cell subsets diminishes with differentiation. (A) Once activated with cognate antigen, CD8+ T cells progressively differentiate from stem-cell memory (SCM), with the highest capacity of self-renewing properties, to central memory (CM), effector memory (EM), and finally to terminal effector (EFF) phenotypes. (B) Antitumor immunity of TSCM cells is enhanced due to establishing long-term memory responses to tumor antigens and heightened ability to persist. As cells become more differentiated through the TCM, TEM, and TEFF stages, they lose capacity for self-renewal and become exhausted, resulting in poor antitumor immunity.
The antitumor efficacy of adoptively transferred memory subsets has been shown to progressively worsen as cells expand logarithmically and often approach the TEFF phase (Figure 3B) (106, 113, 114). By contrast, TSCM cells, characterized by CD45RO−CD45RA+CD95+CD62L+ expression (107), were most potent in a direct comparison of human meso-CAR-engineered memory subsets due to their enhanced proliferative capacity and survival (110). In addition, TSCM cells have the ability to self-renew across several cell divisions when reactivated (110). Following the path of differentiation, tumor specific TCM cells are traditionally more effective for long-term regression of established solid tumors than TEM (114), while all memory subsets are superior to TEFFs (113).
As a result of finding that less-differentiated memory cells are superior in regressing tumors in ACT models, there is now a growing clinical interest in the ability to expand T cells to large numbers for ACT, while concomitantly inhibiting phenotypic differentiation to foster more stem-like features and enhanced potency against tumors. One approach to accomplish this objective includes targeting downstream of the IL-2 pathway during ex vivo expansion through inhibiting subunits of GSK-3β (115), AKT (116), and PI3K (Figure 4) (117). GSK-3β inhibition was shown to bolster Wnt/β-catenin signaling and maintain the TCM phenotype with ex vivo expansion, thus improving efficacy of infused antitumor CD8+ T cells (115). Inhibition of AKT (AKT inhibitor VIII) (116) or the p110δ subunit of PI3K (Idelalisib/CAL-101) (117) ex vivo were also two strong approaches to enrich the frequency of TCM cells in infusion products and improve ACT with CAR-engineered cells for leukemia and mesothelioma models, respectively. However, when directly compared in a transgenic model of melanoma, CAL-101 improved persistence of CD8+KLRG1loCD62Lhi cells in the peripheral blood and significantly enhanced tumor regression compared with AKTi (117). CAL-101 has also been shown to improve antitumor efficacy of Th17 cells by enhancing the proportion of TCM cells and reducing Tregs in culture (118). These reports reveal that adding small molecules to cultures can propagate T cells with a stem-like memory signature. This approach presents a simple and translatable way to improve both the quality and longevity of antitumor responses.
Figure 4.

Two approaches for generating less-differentiated T cells after ex vivo expansion for adoptive cell transfer. (A) Naïve T cells sorted from peripheral blood can be activated and transduced with chimeric antigen receptors (CARs) for antigen specificity. Adding pharmacologic inhibitors of AKT, GSK-3β, PI3K, or mTOR to the T cell culture helps retain cells in a less-differentiated state as they expand. This approach can enrich TSCM and TCM phenotypes in CAR T cells from naïve populations before adoptive transfer to enhance long-term immunity. (B) Differentiated T cells can be reprogrammed with stem-like qualities using iPSC technology. In brief, bulk T cells are isolated from the blood, programmed into iPSCs, and transduced with a CAR before lymphoid differentiation into naïve T cells. The most efficient approaches for lymphoid differentiation into naïve phenotypes are still under development.
An alternate approach to generating more naïve-like T cells for ACT involves genetic reprogramming of induced pluripotent stem cells (Figure 4) (iPSCs). Theoretically, reprogramming T cells in this manner poses the opportunity to de-differentiate terminally exhausted tumor- or neoantigen-specific T cells, such as found in a TIL culture, into “younger” more memory-like cells, while retaining their rearranged TCR (119). Early reports on this concept showed the feasibility of generating iPSCs from peripheral blood mononuclear cells via induction of Oct4, Sox2, Klf4, c-Myc, and Lin28 (120–122). To move this approach into CAR T cell therapies, Sadelain and colleagues engineered peripheral T lymphocyte-derived iPSC cells to express a CD19-CAR, and subsequently differentiated them back into the lymphoid lineage (123). However, upon phenotypic analysis, they were genetically more closely related to innate γδ T cells and functionally demonstrated weaker antitumor efficacy compared with the desired αβ T cells normally used in ACT (123). This process can also take up to 2 months to generate these CAR T cells, making the time investment on par with or even greater than expanding TIL ex vivo (123). Other attempts to program iPSCs down the lymphoid pathway in vitro have resulted in abnormal T cell development due to the absence of thymic selection or have generated T cells with effector-like phenotypes (123–125). To generate a potent response against tumors, the αβ+ TCR indicative of a more natural T cell is required.
In response to this need, the Restifo lab devised a new approach for generating tumor-specific T cells from iPSCs in vitro with a phenotype closer to endogenous, thymic-derived T cells (126). Their 3D thymic culture system generated tumor specific CD8αβ+ naïve-like T cells that regressed melanoma and prolonged survival comparably with bona-fide naïve T cells obtained from the pmel-1 transgenic mouse spleen (126). This new approach is exciting as it may permit generation of a more robust supply of CAR-engineered naïve-like T cells to mediate long-term cures in patients whose peripheral T cells were previously dysfunctional. Moving forward, inhibition of memory differentiation pathways in ex vivo culture and further developments in the feasibility of genetically reprogramming iPSCs will support generation of memory-like CD8+ subsets with enhanced antitumor properties, thereby improving patient outcomes.
CD4+ T Cell Subsets
While ACT with CD8+ T cells has been more thoroughly studied, the impact of CD4+ T helper cells on tumor immunity has recently emerged both preclinically and clinically (127, 128). This body of work indicates that CD4+ T lymphocytes may play a key role in enhancing cancer immunotherapy. Since CD4+ T cells classically support CD8+ T cell activation and proliferation through cytokine secretion, an infusion product containing only CD8+ cytotoxic T cells, as is used frequently in the clinic, may show poor persistence simply due to flawed design. Recently, adoptive transfer of a CD4+ dominant T cell product resulted in tumor regression in a patient with metastatic cholangiocarcinoma (127) and a complete durable remission in a patient with metastatic breast cancer (128). These cases hint that CD4+ lymphocytes may be a powerful subset that should not be selected against. Could it be possible that contrary to accepted dogmas, CD4+ T cells may be able to lyse tumor cells themselves without reliance on CD8+ T cells? The quality of tumor immunity may ultimately depend upon the CD4+ subset transferred, and whether these subsets require CD8+ T cells to exert antitumor effects is unclear and will be discussed further below. Herein, we will examine the role of CD4+ T cells in tumor immunity (Figure 5), new discoveries of potent subsets within the CD4+ lineage, and clinical implications of engineering human CD4+ T cell subsets with CAR-specificity to extend treatment outcomes.
Figure 5.

Antitumor immunity of CD4+ T cells is dependent upon the subset to which they are polarized. Regulatory T cells (Tregs) (top left) and Th2 cells (top right) are classically tumor promoting. Tregs downregulate effector T cell responses via secretion of suppressive cytokines or engagement of inhibitory checkpoint molecules like CTLA-4 or TIGIT. Th2 cells secrete suppressive cytokines that hinder a Th1-mediated antitumor response. Conversely, transfer of Th1 cells (bottom left) and Th17 cells (bottom right) enhance antitumor responses. Th1 cells produce IFN-γ and enhance CD8+ cell-mediated immunity. Th17 cells produce proinflammatory cytokines that have controversially been implicated in carcinogenesis; however, adoptive transfer of Th17 cells has shown robust immunity in several solid tumors. Transferred Th17 cells have stem-like self-renewal capabilities and enhanced persistence long term over Th1 cells.
Cytokine and costimulatory cues can polarize naïve CD4+ T cells into distinct subsets, such as Th1, Th2, Th17, Th9, Th22, T follicular helper, and Treg. The presence of various cytokines needed during activation by antigen-presenting cells to generate these various subsets is reviewed elsewhere (129–131). In the context of tumor immunity, CD4+ T helper cells aid activation of CD8+ cytotoxic lymphocytes (132, 133), but can also eradicate tumors in the absence of CD8+ T cells (134, 135). The relative antitumor immunity of Th1, Th2, Th17, and Tregs has been thoroughly studied, and emerging reports on the potency of human CD4+CD26high T cells that possess improved migration, persistence, and multi-functionality underscores the rationale for translating adoptive transfer of CD4+ T cells clinically (135).
The historical understanding of T helper subsets originated with a hypothesis of two opposing helper subsets, termed Th1 and Th2, with distinct functions in promoting cell-mediated or humoral immunity, respectively (136). While both Th1 and Th2 have demonstrated some degree of antitumor efficacy in vivo, Th1 cells were shown to induce a CD8+ CTL memory response against antigen rechallenge while Th2 cells did not (137). The mechanism of Th1-mediated immunity relies on their production of IFN-γ, which can augment CD8+ T cell infiltration and macrophage production of nitric oxide to induce tumor cell apoptosis (138). In a recent clinical trial, CD4+ Th1 cells specific for ERBB2IP were successful in regressing a patient’s metastatic cholangiocarcinoma (127). Conversely, Th2 cells, as producers of IL-4, have largely been regarded to promote tumor growth because they inhibit the Th1 polarization program and produce suppressive IL-10 (139). Other reports reveal that Th2 cells stimulate tumor necrosis through inhibition of angiogenesis (140). Recently, in a prophylactic myeloma model, adoptive transfer of Th2 cells induced a strong type II inflammatory response at the tumor site and prevented tumor growth via M2-macrophages producing arginase (141). However, these cells were transferred into a host deficient in IFN-γ, which may itself support persistence of Th2 cells, so translational relevance of their efficacy is debatable. Also, dissent over the role for Th2 cells in ACT is furthered since arginase activity has previously been correlated with tumor progression (142). This body of work underscores a need to further understand the role of Th2 cells in antitumor immunity.
Th17 cells, characterized by high IL-17 production, play a contested role in tumor immunity but have been shown highly potent in several preclinical ACT models. Th17 cells are phenotypically polarized by the cytokines IL-6, TGF-β, and IL-1β via induction of STAT3 and RORγT and are maintained long term by IL-21 and IL-23 (143–146). ICOS costimulation fosters differentiation and expansion of Th17 cells (147) as well as the function of IL-17-producing CD8+ T cells (148). Incorporation of an ICOS costimulatory domain in CAR T cells augments persistence of co-adoptively transferred CD8+ T cells in a humanized model of mesothelioma (60). In several different cancer models, transfer of Th17-polarized cells enhanced survival and tumor regression superiorly to Th1 or unpolarized CD4+ cells (147, 149, 150). In addition, when expanded ex vivo long term, Th17 cells retain their antitumor efficacy while Th1 cells lose tumor control (151). Phenotypically, Th17 cells express more stem-like markers (CCR7, Lef1, TCF7) and fewer exhaustion markers (PD-1, KLRG-1, Tim3) compared with their Th1 counterparts, possibly contributing to longevity (150, 151). Important to the field of CAR therapies, Th17-polarized human meso-CAR T cells exhibit enhanced immunity against mesothelioma versus Th1-polarized cells after both short and long expansion (151). Also, in patients with CLL treated with CD19-CAR T cells, complete responders had CAR T cells with a transcriptomic profile of STAT3/IL-6 signaling, generating a type-17 signature with higher production of IL-17 and IL-22 compared with non-responders (152). Thus, it is truly possible that isolating human PBMCs and polarizing cells to a Th17 phenotype during CAR transduction and activation may generate a therapy with enhanced persistence and thus a long-lived response in patients with solid tumors refractory to treatment with standard bulk CAR T cell preparations.
Despite such preclinical evidence of antitumor potency, adoptive transfer of engineered cells polarized to Th17 phenotype has not been translated yet clinically. The numerous cytokines required to polarize may generate a T cell with enhanced stem-like properties and persistence but also represent a major hurdle halting ease of translation. Our lab has recently described a novel method for isolating potent CD4+ T cells via surface expression of CD26, an ectoenzyme with costimulatory properties (130, 135). In work pioneered by Nelson and Bailey, CD4+ T cells expressing high levels of CD26 are polyfunctional, secreting up to five cytokines simultaneously including IL-17 and IFN-γ, and have robust migratory capacity. CD4+CD26high meso-CAR T cells are highly potent against difficult to treat mesothelioma and pancreatic tumors, and have superior persistence compared with other subsets expressing intermediate or low levels of CD26 (135). Clinical translation of CAR-engineered CD26high cells could support superior trafficking, long-term persistence and cytotoxicity at baseline, which could be further enhanced with fourth-generation CARs; thus, these cells are a strong candidate for overcoming major barriers to successful solid tumor CAR therapies.
T cell memory, persistence, and therapeutic efficacy are tightly related to metabolic state, and within an unfavorable environment such as a solid tumor, their ability to use nutrients for energetic needs may mean the difference between life and death of the cell. Just as different memory or helper subsets have varied capacity to kill solid tumors, T cells armed with a superior metabolic state are more equipped to exert their effector functions and generate long-lasting memory responses against tumor antigens. Therefore, we present metabolic manipulation of antitumor T cells as another approach to generating potent therapies below.
Fine-Tuning Metabolic Fitness
How T cells use energy to survive in the tumor microenvironment has recently gained the interest of cancer immunotherapists. Manipulation of T cell bioenergetics to elicit immunity to solid tumors has shown great promise recently in the preclinical setting. At the fundamental level, it is now clear that lymphocytes engage specific metabolic pathways to best support their functions, intricately regulated by nutrient demand and availability (153). Resting T cells favor energy production through the TCA cycle and fatty acid oxidation (154). Once activated, however, both CD4+ and CD8+ T cells become quickly poised to exert an effector response, and thus upregulate biosynthetic pathways and rely on aerobic glycolysis, where glucose is rapidly consumed and shuttled through glycolysis to lactate to support their proliferation and effector functions (154, 155). Conversely, memory and Treg cells operate using mitochondrial metabolism and fatty acid β-oxidation in a similar manner as naïve T cells (156). Induction of anabolic, glycolytic pathways may augment proliferation and the inflammatory nature of T cells but correlates with poorer persistence in vivo, which in adoptive transfer therapy directly associates with a less effective antitumor response (157, 158). As memory-like T cells are most effective in mediating long-term responses to solid tumors, new data implicates that modulation of their metabolism to favor catabolic pathways may generate a lymphocyte population with enhanced antitumor functions in vivo.
Yet, complete denial of anabolic pathways is not a quality of successful T cell therapies. In fact, blunting the anabolic pathway in T cells prevents their capacity to lyse targeted antigens. For example, in models of autoimmunity, genetic deletion of Glut1 prevented effector T cells from causing pathology in inflammatory bowel disease (159). Inhibition of fatty acid synthesis in T cells limited Th17-induced autoimmunity and promoted a Treg signature, notoriously implicated in promoting tolerance to tumors (160). Pharmacologic inhibition of glycolytic enzyme GAPDH with dimethyl fumarate prevented acquisition of effector function in Th17 cells and skewed their polarization ex vivo toward a Treg phenotype, reducing autoimmune pathology in models of experimental autoimmune encephalitis (161). Therefore, direct inhibition of glycolysis in T cells is likely to be deleterious for cancer therapies. Augmentation of fatty acid oxidation in CD8+ T cells by treating mice with metformin, on the other hand, promoted memory T cell formation and enhanced immunity to tumor challenge post vaccination (162). Fostering a balance between memory-like metabolism and intrinsic support of glycolysis in CAR T cells may be important for maintaining T cell function and fate within the metabolically restricted tumor microenvironment when the supply of glucose and oxygen is limited (163).
Interestingly, several groups have demonstrated that metabolic manipulation of T cells in vitro can benefit antitumor efficacy of transferred cells in vivo. Overexpression of glycolytic enzyme phosphoglycerate-mutase 1 limited persistence of transferred CD8+ T cells, while inhibition of glycolysis with 2-deoxyglucose augmented stem memory characteristics like Tcf7 and Lef1 expression, and significantly enhanced survival of tumor-bearing hosts (164). Inhibition of AKT signaling, discussed previously as a method for reducing T cell differentiation ex vivo, was also shown to decrease glycolytic function and enhance mitochondrial spare respiratory capacity in CD8+ T cells (165). Moreover, when these AKTi-treated T cells were transferred into mice, they persisted superiorly to untreated cells (165). Similarly, inhibition of the inositol triphosphate receptor, an important second messenger for calcium release from intracellular storage, in CD4+ T cells ex vivo prevented glycolytic initiation due to altered calcium flux, fostered a CM phenotype, and augmented their therapeutic efficacy against established melanoma tumors (166). Interestingly, the integrity of the mitochondria in T cells also profoundly impacts their capacity to mount durable immunity to tumors. For example, Pearce and colleagues showed that mitochondrial morphology is tightly related to T cell metabolism; fused mitochondria, described as tubular and closely associated, were characteristic of memory T cells. Conversely, effector T cells were composed of “fissed” or distinct mitochondria dispersed throughout the cytoplasm (167). Forced mitochondrial fusion and inhibition of fission of T cells via pharmaceutical approaches ex vivo using M1 and Mdivi-1, respectively, promoted a superior antitumor response once transferred in vivo (167).
Emerging data reveal that programming and polarization of CD4+ T cells also critically determines metabolic commitments and modulates their antitumor properties. Recently, the Mehrotra lab reported that ex vivo polarized Th1/Th17 hybrid cells upregulate glutaminolysis and rely on oxidative phosphorylation compared with glycolytic-Th1 cells, ultimately supporting their superior antitumor capacity over traditional Th1 or Th17 cells (168). Homeostatic gamma chain cytokines have also been shown to alter the metabolic fate of antitumor T cells. For example, priming T cells with IL-15 (169) or IL-21 (170), previously discussed as a potential method for enhancing their stemness, redirects metabolism away from glycolysis in favor of fatty acid β-oxidation. This bioenergetic signature directly correlates with T cells possessing longer-lived memory responses to tumors and foreign antigen. Thus, it seems that holding back acquisition of full effector glycolytic capacity in CAR T cells ex vivo before infusion could greatly enhance persistence of cells in patients, augmenting therapeutic outcomes in solid tumors.
Impact of Host Immunity
It is possible that targeting solid tumors via a single or combination of several known surface antigens, even with the most persistent or metabolically fit T cell subset, will not be sufficient to evoke cures in patients with heterogenous hard-to-treat solid tumors. Thus far, TIL therapies and ICB have shown greater responses in treating these types of tumors, likely through their ability to induce or bolster an endogenous response of exhausted cells against a highly personalized repertoire of neoantigens existing within the tumor (171–173). TIL therapies in melanoma have shown response rates of up to 50% in contrast to previously FDA approved therapies such as interleukin-2 with response rates near 15% at best (174), and melanoma patients with the highest neoantigen load have the best progression-free survival (175). PD-1 blockade success in clinical trials has led to FDA approval for solid tumors such as metastatic melanoma, advanced NSCLC, recurrent or metastatic SCC of head and neck, refractory classical Hodgkin lymphoma, urothelial carcinoma (176), and as second line in MMR/microsatellite instability-high tumors as of May 2017.
As TIL therapies and checkpoint blockade have generated robust results in patients in several solid tumors, it is likely that incorporating the mechanisms of TIL/ICB into CAR constructs may improve their efficacy. Activated CAR T cells within the tumor microenvironment do express high concentrations of exhaustion markers such as PD-1, Tim-3, Lag3, and 2B4 (177). PD-1 expression also contributes to reduced efficacy of transferred cells regardless of tumor specificity (177). Strategies to improve efficacy of PD-1 expressing, exhausted CAR T cells or to rejuvenate host tumor-specific exhausted T cells along with CAR therapy are threefold: (1) genetic removal of PD-1 from CAR T cells, (2) combination PD-1 blockade with CAR infusion, or (3) CAR T cell production of PD-1 blockade within the host. These strategies and our recommendations for designing next-generation CAR therapies with highest efficacy are discussed below.
The first evidence of reducing PD-1 signaling from a CAR T cell was shown through a PD-1 dominant negative receptor, where engagement of PD-1/PD-L1 would not generate a signal (178). The dominant negative receptor enhanced the functionality of CAR T cells and survival of mice treated with meso-CAR against mesothelioma compared with control CAR with the ability to signal PD-1 (178). Recent advances in genome editing using CRISPR/Cas9 technology have permitted removal of PD-1 entirely from T cells, and in two solid tumor models (prostate and glioma) have shown benefits of this intervention for tumor regression (179, 180). While important for efficacy of transferred cells, and likely to be incorporated into more T cell therapies in the near future, removal of PD-1 would not benefit endogenous exhausted cells specific for potentially unknown antigens. In addition, genetic deficiency of PD-1 has been shown to induce terminally exhausted cytotoxic CD8+ T cells; without PD-1, T cells have robust cytokine production and proliferation upon early exposure to antigen, but contract more rapidly and have compromised long-term survival compared with T cells with normal PD-1 expression (181). Thus, genetic removal of PD-1 may not benefit CAR T cell survival long term.
Theoretically, CAR-mediated destruction of tumor cells could also lead to generation of new antigen-specific lymphocytes via epitope spreading (Figure 6) (182). These newly activated cells are susceptible to suppression within the tumor similar to CAR T cells. To overcome this limitation, PD-1 blockade could be given in combination with or could be encoded by CAR T cells to both support the transferred cells and the endogenous tumor-specific lymphocytes. Preclinically, combination therapies in solid tumors have demonstrated enhanced proliferation, function, and antitumor efficacy of HER2-CAR T cells in breast cancer and sarcoma (183). At the time of writing, clinical trials with such combinations are heavily skewed toward blood cancers (NCT02926833, NCT02706495, and NCT03287817 in DLBCL, NCT03310619 in B cell NHL, and NCT02650999 in DLBCL, follicular lymphoma, and mantle cell lymphoma; ClinicalTrials.gov identifiers). Preliminary results in these hematologic malignancies suggest that PD-1 blockade may enhance CAR T cell persistence and could improve objective responses in patients (184, 185). Thus, there is rationale for combining these approaches to improve persistence of CAR T cells and generate more robust responses.
Figure 6.

CAR-mediated tumor destruction can synergize with host immunity through epitope spreading. (A,B) Chimeric antigen receptor (CAR)-mediated tumor cell lysis induces inflammation, and release of tumor antigens. (C) DAMPs from dying cells recruit APCs to tumor site, which take up and process the released antigens for presentation. (D) APCs present newly processed tumor antigens to naïve T cells in lymph nodes. Activated T cells migrate to the tumor site. (E) Tumor-specific lymphocytes synergize with CAR T cells to eradicate difficult to treat solid tumors.
This principle could be streamlined even further if the CAR T cells produced monoclonal antibodies that inhibit checkpoint molecules themselves. Preclinical evidence in lung and ovarian tumors shows that CAR T cells producing PD-1 blocking antibodies are more therapeutic than control CAR T cells against the same target (186). Importantly, CAR T cell production of PD-1 antibody was more effective than systemic administration of the antibody, which could be related to localized, high dose delivery (186). Similar results were found in a renal cell carcinoma model where production of antibodies to PD-L1 enhanced CAR T cell function, though the results were less dramatic (187). Since both of these studies were conducted in NSG mice, the efficacy of ICB-producing CAR T cells may be even more striking in a host with an intact immune system. These preclinical results were rapidly translated to several clinical trials in China for variety of solid tumors, described in Table 1.
The Ultimate Car T Cell Therapy
Chimeric antigen receptor T cell therapies exemplify an incredible opportunity—one like Hippocrates described—to take control of healing patients through empowering and redesigning a patient’s own T cells to destroy tumor cells. As depicted in Figure 1, for a CAR T cell therapy to be more successful in solid tumors, the design should encompass three axes. Illustrated in Figure 7, we posit that this therapy would incorporate bispecificity through the CAR construct, generate enhanced potency via engineering a superior T cell subset, and revitalize the host immune response through cytokine and checkpoint antibody secretion. First, to enhance specificity, syn-Notch inducible CAR expression upon engagement of a tissue-specific antigen could improve sensitivity of the CAR to target the tissue and reduce off-tumor effects. Secondly, to enhance persistence, trafficking, and self-renewal properties, a CAR-engineered TSCM CD8+ T cell expanded with pharmacologic inhibitors or generated from iPSCs, or either a Th17 cell or a CD4+CD26high T cell could overcome these limitations of poor quality T cell infusion products. Use of a multipotent T cell may permit adoptive transfer of fewer cells, thereby streamlining and reducing the cost and time investment to generate T cell products for infusion. A lower dose of T cells could reduce risk for severe toxicities and cytokine storms; however, engineering such a potent cell could alternatively be more toxic to patients when infused. Therefore, safety switches or Boolean logic gates should be incorporated to prevent life-threatening adverse events. Finally, taking advantage of the host’s response to personalized neoantigens, PD-1 antibody-producing CAR T cells that also produce cytokines like IL-12, IL-15, IL-18, or IL-21 locally in the tumor after engaging a tumor-specific antigen would counteract the highly suppressive environment and synergize the power of the endogenous immune response with the genetically redirected CAR T cell response.
Figure 7.

The trifecta of successful chimeric antigen receptor (CAR) T cell therapies in solid tumors. The ultimate CAR T cell therapy has tumor specificity, potent migratory capacity and persistence, and improves the host immune response. (A) Bispecificity through syn-Notch technology augments targeting to tumor/tumor-specific tissue. (B) Engineering a T cell with enhanced persistence and migratory capacity—such as a Th17 or CD4+CD26high cell—or with self-renewing properties—such as a CD8+ TSCM cell—will enhance long-term memory responses to prevent tumor recurrence. (C) Secretion of PD-1 blockade and cytokines such as IL-12, IL-15, IL-18, or IL-21 locally could overcome the suppressive tumor microenvironment, reinvigorate the exhausted host immune response to other tumor antigens, and synergize with CAR-specific T cells to destroy large heterogenous solid tumors.
Though the combination of these approaches is theoretically appealing, a T cell incorporating the several mechanisms proposed has not yet been engineered. Such a construct may prove difficult to generate without interruption of normal gene function. Use of CRISPR/Cas9 could direct incorporation to a specific location in the genome to enhance efficacy, such as the TRAC locus (188). Generating a universal CAR that is not MHC restricted, where infusion products could be mass-produced versus individually developed for each patient, could also make this CAR design feasible. The ease of developing a T cell as proposed is likely to improve over time as academic and industrial facilities expand and commercial-grade production becomes streamlined through automation and improved quality control (189). While it may make for a complex construct, harnessing capabilities of genetic redirection, optimal T cell subsets, and augmented crosstalk to other infiltrating immune cells may be one attainable approach to eradicate heterogenous and therapeutically resistant solid tumors.
Conclusion
Adoptive cell transfer with CAR-redirected T cells is a potentially curative approach for patients with previously treatment-resistant tumors. CAR T cells have proven their potency against hematologic cancers evidenced by their recent FDA approvals for B-ALL and DLBCL. For solid tumors, these therapies remain in early development but may require a new approach to enhance their efficacy. Herein, we have presented a combinatorial approach to augment the ability of CAR T cells to overcome challenges they face within the tumor microenvironment. We posit that a future CAR T cell armored with (1) a superior targeting system specific to the tumor and tumor tissue, (2) engineering of a highly potent, persistent, and self-renewing T cell subset, and (3) rejuvenation of the endogenous host response through CAR T cell production of monoclonal antibodies against immune checkpoint molecules will bolster the immune attack on the solid tumor to best reduce toxicity and support a long-lived memory response against targeted antigens and personalized neoantigens. Elegant findings from investigators worldwide will continue moving forward the solid tumor CAR T cell approach to generate cures for patients with previously therapeutically resistant cancers.
Author Contributions
HK and CP conceptualized, wrote, and edited the manuscript. AS, CD, MW, and SM provided feedback and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Arman Aksoy and Pinar Aksoy for their helpful comments and feedback on this manuscript. We also thank cancer immunotherapists worldwide for improving the lives of cancer patients and inspiring this work.
Footnotes
Funding. This work was supported by NIH Training grant T32 GM08716 to HK, NIH Training grant T32 AI132164-01 to CD, CP grants R01 CA175061 and R01 CA208514, KL2 South Carolina Clinical & Translational Research grant UL1 TR000062, ACS-IRG grant 016623-004, and MUSC Start-up funds to CP, R21 CA198646 to SM, and 5 PO1 CA154778-05 to SM, CP, and M. Nishimura.
References
- 1.Vigneron N. Human tumor antigens and cancer immunotherapy. Biomed Res Int (2015) 2015:948501. 10.1155/2015/948501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuwana T, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun (1987) 149(3):960–8. 10.1016/0006-291X(87)90502-X [DOI] [PubMed] [Google Scholar]
- 3.Gross G, Waks T, Eshar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A (1989) 86:10024–8. 10.1073/pnas.86.24.10024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359:1361–5. 10.1126/science.aar6711 [DOI] [PubMed] [Google Scholar]
- 5.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer (2016) 16(9):566–81. 10.1038/nrc.2016.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. 10.1056/NEJMoa1215134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude S, Frey N, Shaw P, Aplenc R, Barrett D, Bunin N, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. 10.1126/scitranslmed.3005930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6(224):224ra25. 10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33(6):540–9. 10.1200/JCO.2014.56.2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory b-cell lymphomas. N Engl J Med (2017) 377(26):2545–54. 10.1056/NEJMoa1708566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. 10.1056/NEJMoa1707447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. 10.1056/NEJMoa1103849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. 10.1126/scitranslmed.aac5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116(20):4099–102. 10.1182/blood-2010-04-281931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119(12):2709–20. 10.1182/blood-2011-10-384388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood (2016) 128(3):360–70. 10.1182/blood-2016-01-694356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doan A, Pulsipher MA. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr Blood Cancer (2018) 65(4):e26914. 10.1002/pbc.26914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasad V. Tisagenlecleucel — the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol (2017) 15:11. 10.1038/nrclinonc.2017.156 [DOI] [PubMed] [Google Scholar]
- 21.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis (2018) 9(3):282. 10.1038/s41419-018-0278-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics (2016) 3:16011. 10.1038/mto.2016.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med (2012) 14(6):405–15. 10.1002/jgm.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24(13):e20–2. 10.1200/JCO.2006.05.9964 [DOI] [PubMed] [Google Scholar]
- 25.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21(4):904–12. 10.1038/mt.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother (2017) 66(11):1425–36. 10.1007/s00262-017-2034-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. 10.1200/JCO.2014.58.0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beatty GL, O’Hara MH, Nelson AM, McGarvey M, Torigian DA, Lacey SF, et al. Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. J Clin Oncol (2015) 33(15_suppl):3007. 10.1200/jco.2015.33.15_suppl.3007 [DOI] [Google Scholar]
- 30.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res (2014) 2(2):112–20. 10.1158/2326-6066.CIR-13-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res (2013) 1(1):26–31. 10.1158/2326-6066.CIR-13-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res (2015) 21(14):3149–59. 10.1158/1078-0432.CCR-14-1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Rourke DM, Nasrallah M, Morrissette JJ, Melenhorst JJ, Lacey SF, Mansfield K, et al. Pilot study of T cells redirected to EGFRvIII with a chimeric antigen receptor in patients with EGFRvIII+ glioblastoma. J Clin Oncol (2016) 34(15_suppl):2067. 10.1200/JCO.2016.34.15_suppl.2067 [DOI] [Google Scholar]
- 34.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20 Pt 1):6106–15. 10.1158/1078-0432.CCR-06-1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J (2016) 14:357–62. 10.1016/j.csbj.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Rourke D, Nasrallak M, Desai A, Melenhorst J, Mansfield K, Morrissette J, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9:eaaa0984. 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375(26):2561–9. 10.1056/NEJMoa1610497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. 10.1158/1078-0432.CCR-15-0428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14(11):1264–70. 10.1038/nm.1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer (2017) 5:22. 10.1186/s40425-017-0222-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther (2016) 24(6):1135–49. 10.1038/mt.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamers CHJ, Gratama JW, Pouw NMC, Langeveld SCL, Krimpen BAV, Kraan J, et al. Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: implications for loss of transgene expression. Hum Gene Ther (2005) 16(12):1452–62. 10.1089/hum.2005.16.1452 [DOI] [PubMed] [Google Scholar]
- 43.Scarfò I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer (2017) 5:28. 10.1186/s40425-017-0230-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunol Cell Biol (2017) 95:356–63. 10.1038/icb.2016.128 [DOI] [PubMed] [Google Scholar]
- 45.Dotti G, Gottschalk S, Savoldo B, Brenner M. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev (2014) 257:107–26. 10.1111/imr.12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov (2013) 3(4):388–98. 10.1158/2159-8290.CD-12-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson MH, Paulos CM. Novel immunotherapies for hematologic malignancies. Immunol Rev (2015) 263(1):90–105. 10.1111/imr.12245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eshhar Z, Waks T, Gross G, Schindler D. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A (1993) 90:720–4. 10.1073/pnas.90.2.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Becker MLB, Near R, Mudgett-Hunter M, Margolies MN, Kubo RT, Kaye J, et al. Expression of a hybrid immunoglobulin-T cell receptor protein in transgenic mice. Cell (1989) 58(5):911–21. 10.1016/0092-8674(89)90943-4 [DOI] [PubMed] [Google Scholar]
- 50.Goverman J, Gomez SM, Segesman KD, Hunkapiller T, Laug WE, Hood L. Chimeric immunoglobulin-T cell receptor proteins form functional receptors: Implications for T cell receptor complex formation and activation. Cell (1990) 60(6):929–39. 10.1016/0092-8674(90)90341-B [DOI] [PubMed] [Google Scholar]
- 51.Brocker T. Chimeric Fv-zeta or Fv-ε receptors are not sufficient to induce activation of cytokine production in peripheral T cells. Blood (2000) 96(5):1999–2001. [PubMed] [Google Scholar]
- 52.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther (2007) 18(8):712–25. 10.1089/hum.2007.028 [DOI] [PubMed] [Google Scholar]
- 53.Finney H, Lawson A, Bebbington C, Weir A. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol (1998) 161:2791–7. [PubMed] [Google Scholar]
- 54.Finney HM, Akbar AN, Lawson ADG. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR chain. J Immunol (2003) 172(1):104–13. 10.4049/jimmunol.172.1.104 [DOI] [PubMed] [Google Scholar]
- 55.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther (2005) 12(5):933–41. 10.1016/j.ymthe.2005.04.016 [DOI] [PubMed] [Google Scholar]
- 56.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther (2009) 17(8):1453–64. 10.1038/mt.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A (2009) 106(9):3360–5. 10.1073/pnas.0813101106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res (2011) 71(13):4617–27. 10.1158/0008-5472.CAN-11-0422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44(2):380–90. 10.1016/j.immuni.2016.01.021 [DOI] [PubMed] [Google Scholar]
- 60.Guedan S, Posey AD, Jr, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight (2018) 3(1):96976. 10.1172/jci.insight.96976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. 10.1158/0008-5472.CAN-08-2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res (2011) 71(22):6997–7009. 10.1158/0008-5472.CAN-11-1466 [DOI] [PubMed] [Google Scholar]
- 63.Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, et al. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol (2007) 179(5):3332–41. 10.4049/jimmunol.179.5.3332 [DOI] [PubMed] [Google Scholar]
- 64.Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res (2011) 17(14):4719–30. 10.1158/1078-0432.CCR-11-0351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (2010) 33(8):780–8. 10.1097/CJI.0b013e3181ee6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor–redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother (2012) 61(8):1269–77. 10.1007/s00262-012-1202-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (2002) 298(5594):850–4. 10.1126/science.1076514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology (2015) 4(3):e994446. 10.4161/2162402X.2014.994446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest (2011) 121(12):4746–57. 10.1172/JCI58814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res (2015) 21(10):2278–88. 10.1158/1078-0432.CCR-14-2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24(6):1160–70. 10.1038/leu.2010.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krenciute G, Prinzing BL, Yi Z, Wu M-F, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res (2017) 5(7):571–81. 10.1158/2326-6066.CIR-16-0376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A (2016) 113(48):E7788–97. 10.1073/pnas.1610544113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Osaki T, Peron J, Cai Q, Okamura H, Robbina P, Kurimoto M, et al. IFN-γ-inducing factor/IL-18 administration mediates IFN-γ and IL-12-independent antitumor effects. J Immunol (1998) 160:1742–9. [PubMed] [Google Scholar]
- 75.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev (2001) 12:53–72. 10.1016/S1359-6101(00)00015-0 [DOI] [PubMed] [Google Scholar]
- 76.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. 10.3389/fimmu.2013.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse car T cells secreting IL-18. Cell Rep (2017) 20(13):3025–33. 10.1016/j.celrep.2017.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep (2017) 21(11):3205–19. 10.1016/j.celrep.2017.11.063 [DOI] [PubMed] [Google Scholar]
- 79.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood (2008) 111(11):5326–33. 10.1182/blood-2007-09-113050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med (2005) 201(1):139–48. 10.1084/jem.20041057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res (2011) 71(10):3516–27. 10.1158/0008-5472.CAN-10-3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood (2010) 115(17):3508–19. 10.1182/blood-2009-09-241398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med (2018) 24(3):352–9. 10.1038/nm.4478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-[beta] receptor. J Immunother (2008) 31(5):500–5. 10.1097/CJI.0b013e318177092b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther (2018) 26:1855–66. 10.1016/j.ymthe.2018.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther (2014) 22(6):1211–20. 10.1038/mt.2014.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res (2016) 76(6):1578–90. 10.1158/0008-5472.CAN-15-2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant (2010) 16(9):1245–56. 10.1016/j.bbmt.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol (2014) 5:235. 10.3389/fphar.2014.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berger C, Flowers M, Warren E, Riddell S. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood (2006) 107(6):2294–302. 10.1182/blood-2005-08-3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood (2014) 124(8):1277. 10.1182/blood-2014-01-545020 [DOI] [PubMed] [Google Scholar]
- 92.Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, et al. A tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res (2016) 4(8):658–68. 10.1158/2326-6066.CIR-16-0043 [DOI] [PubMed] [Google Scholar]
- 93.June CH. Remote controlled CARs: towards a safer therapy for leukemia. Cancer Immunol Res (2016) 4(8):643. 10.1158/2326-6066.CIR-16-0132 [DOI] [PubMed] [Google Scholar]
- 94.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol (2013) 31(1):71–5. 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roybal Kole T, Rupp Levi J, Morsut L, Walker Whitney J, McNally Krista A, Park Jason S, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell (2016) 164(4):770–9. 10.1016/j.cell.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell (2016) 167(2):419–32.e16. 10.1016/j.cell.2016.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell (2016) 164(4):780–91. 10.1016/j.cell.2016.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med (2013) 5(215):215ra172. 10.1126/scitranslmed.3006597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell (2006) 126(5):855–67. 10.1016/j.cell.2006.08.019 [DOI] [PubMed] [Google Scholar]
- 100.Posey AD, Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44(6):1444–54. 10.1016/j.immuni.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A (2016) 113(4):E450–8. 10.1073/pnas.1524193113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A (2016) 113(4):E459–68. 10.1073/pnas.1524155113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell (2018) 173(6):1426–38.e11. 10.1016/j.cell.2018.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Onc (2016) 13:273–90. 10.1038/nrclinonc.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126(6):2123–38. 10.1172/JCI85309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest (2016) 126(1):318–34. 10.1172/JCI81217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer (2012) 12(10):671–84. 10.1038/nrc3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature (2017) 552(7685):404–9. 10.1038/nature25144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity (2014) 41(1):116–26. 10.1016/j.immuni.2014.05.018 [DOI] [PubMed] [Google Scholar]
- 110.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. 10.1038/nm.2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall J, et al. The epigenetic control of stemness in CD8+ T cell fate commitment. Science (2018) 359:177–86. 10.1126/science.aah6499 [DOI] [PubMed] [Google Scholar]
- 112.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature (2018) 558:307–12. 10.1038/s41586-018-0178-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115(6):1616–26. 10.1172/JCI24480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A (2005) 102(27):9571–6. 10.1073/pnas.0503726102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8 + memory stem cells. Nat Med (2009) 15(7):808–13. 10.1038/nm.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight (2017) 2(23):95103. 10.1172/jci.insight.95103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. PI3Kδ inhibition enhances the antitumor fitness of adoptively transferred CD8+ T cells. Front Immunol (2017) 8:1221. 10.3389/fimmu.2017.01221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Majchrzak K, Nelson MH, Bowers JS, Bailey SR, Wyatt MM, Wrangle JM, et al. beta-Catenin and PI3Kdelta inhibition expands precursor Th17 cells with heightened stemness and antitumor activity. JCI Insight (2017) 2(8):e90547. 10.1172/jci.insight.90547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Loh YH, Hartung O, Li H, Guo C, Sahalie JM, Manos PD, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell (2010) 7(1):15–9. 10.1016/j.stem.2010.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 121.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell (2010) 7(5):618–30. 10.1016/j.stem.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Okita K, Yamakawa T, Matsumura Y, Sato Y, Amano N, Watanabe A, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells (2013) 31(3):458–66. 10.1002/stem.1293 [DOI] [PubMed] [Google Scholar]
- 123.Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol (2013) 31(10):928–33. 10.1038/nbt.2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell (2013) 12(1):114–26. 10.1016/j.stem.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 125.Themeli M, Riviere I, Sadelain M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell (2015) 16(4):357–66. 10.1016/j.stem.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vizcardo R, Klemen ND, Islam SMR, Gurusamy D, Tamaoki N, Yamada D, et al. Generation of tumor antigen-specific iPSC-derived thymic emigrants using a 3D thymic culture system. Cell Rep (2018) 22(12):3175–90. 10.1016/j.celrep.2018.02.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tran EH, Turcotte S, Gros A, Robbins P, Lu Y, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. 10.1126/science.1251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med (2018) 24(6):724–30. 10.1038/s41591-018-0040-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol (2018). 10.1038/s41423-018-0004-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol (2014) 5:276. 10.3389/fimmu.2014.00276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Majchrzak K, Nelson MH, Bailey SR, Bowers JS, Yu XZ, Rubinstein MP, et al. Exploiting IL-17-producing CD4+ and CD8+ T cells to improve cancer immunotherapy in the clinic. Cancer Immunol Immunother (2016) 65(3):247–59. 10.1007/s00262-016-1797-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gao F, Khammanivong V, Liu W, Leggatt G, Frazer I, Fernando J. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res (2002) 62:6438–41. [PubMed] [Google Scholar]
- 133.Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4 T helper cells and hindered by naturally occurring T regulatory cells. J Immunol (2005) 174(5):2591–601. 10.4049/jimmunol.174.5.2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med (2010) 207(3):651–67. 10.1084/jem.20091921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26highT cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat Commun (2017) 8(1):1961. 10.1038/s41467-017-01867-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mossman T, Cherwinski H, Bond M, Giedlin M, Coffman R. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136:2348–57. [PubMed] [Google Scholar]
- 137.Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med (1999) 190(5):617–28. 10.1084/jem.190.5.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med (1998) 188(12):2357–68. 10.1084/jem.188.12.2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dennis KL, Blatner NR, Gounari F, Khazaie K. Current status of IL-10 and regulatory T-cells in cancer. Curr Opin Oncol (2013) 25(6):637–45. 10.1097/CCO.0000000000000006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Volpert OV, Fong T, Koch AE, Peterson JD, Waltenbaugh C, Tepper RI, et al. Inhibition of angiogenesis by interleukin 4. J Exp Med (1998) 188(6):1039. 10.1084/jem.188.6.1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lorvik KB, Hammarstrom C, Fauskanger M, Haabeth OA, Zangani M, Haraldsen G, et al. Adoptive transfer of tumor-specific Th2 cells eradicates tumors by triggering an in situ inflammatory immune response. Cancer Res (2016) 76(23):6864–76. 10.1158/0008-5472.CAN-16-1219 [DOI] [PubMed] [Google Scholar]
- 142.Mills CD, Shearer J, Evans R, Caldwell MD. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol (1992) 149(8):2709. [PubMed] [Google Scholar]
- 143.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. 10.1038/ni1254 [DOI] [PubMed] [Google Scholar]
- 144.Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol (2008) 181(9):5948–55. 10.4049/jimmunol.181.9.5948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. 10.1016/j.immuni.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 146.Mailer RK, Joly AL, Liu S, Elias S, Tegner J, Andersson J. IL-1beta promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep (2015) 5:14674. 10.1038/srep14674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator (ICOS) is critical for the development of human Th17 cells. Sci Transl Med (2010) 2(55). 10.1126/scitranslmed.3000448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nelson MH, Kundimi S, Bowers JS, Rogers CE, Huff LW, Schwartz KM, et al. The inducible costimulator augments Tc17 cell responses to self and tumor tissue. J Immunol (2015) 194(4):1737–47. 10.4049/jimmunol.1401082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Muranski P, Boni A, Antony P, Cassard L, Irvine K, Kaiser AD, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood (2008) 112(2):362–72. 10.1182/blood-2007-11-120998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35(6):972–85. 10.1016/j.immuni.2011.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bowers JS, Nelson MH, Majchrzak K, Bailey SR, Rohrer B, Kaiser AD, et al. Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight (2017) 2(5):e90772. 10.1172/jci.insight.90772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. 10.1038/s41591-018-0010-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. 10.1084/jem.20151159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. 10.1146/annurev-immunol-032712-095956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Frauwirth K, Riley J, Harris M, Parry R, Rathmell J, Plas D, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. 10.1016/S1074-7613(02)00323-0 [DOI] [PubMed] [Google Scholar]
- 156.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. 10.4049/jimmunol.1003613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Verbist KC, Guy CS, Milasta S, Liedmann S, Kaminski MM, Wang R, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature (2016) 532(7599):389–93. 10.1038/nature17442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol (2016) 17(6):704–11. 10.1038/ni.3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. 10.1016/j.cmet.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. 10.1038/nm.3704 [DOI] [PubMed] [Google Scholar]
- 161.Kornberg M, Bhargava P, Kim P, Putluri V, Snowman A, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science (2018) 360:449–53. 10.1126/science.aan4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. 10.1038/nature08097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol (2017) 46:14–22. 10.1016/j.coi.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. 10.1172/JCI69589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res (2015) 75(2):296–305. 10.1158/0008-5472.CAN-14-2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thaxton JE, Wallace C, Riesenberg B, Zhang Y, Paulos CM, Beeson CC, et al. Modulation of endoplasmic reticulum stress controls CD4+ T-cell activation and antitumor function. Cancer Immunol Res (2017) 5(8):666–75. 10.1158/2326-6066.CIR-17-0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166(1):63–76. 10.1016/j.cell.2016.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD(+)axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab (2017) 27(1):85–100.e8. 10.1016/j.cmet.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. 10.1016/j.immuni.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Loschinski R, Bottcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget (2018) 9(17):13125–38. 10.18632/oncotarget.24442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. 10.1038/nature13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. 10.1038/nm.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439–42. 10.1200/JCO.2012.47.7521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. 10.1158/1078-0432.CCR-11-0116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun (2017) 8(1):1738. 10.1038/s41467-017-01460-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hsu FS, Su CH, Huang KH. A comprehensive review of US FDA-approved immune checkpoint inhibitors in urothelial carcinoma. J Immunol Res (2017) 2017:6940546. 10.1155/2017/6940546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res (2014) 20(16):4262–73. 10.1158/1078-0432.CCR-13-2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126(8):3130–44. 10.1172/JCI83092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res (2017) 23(9):2255–66. 10.1158/1078-0432.CCR-16-1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of PD-1 in CD133-specific CAR T cells. Hum Gene Ther (2018). 10.1089/hum.2017.234 [DOI] [PubMed] [Google Scholar]
- 181.Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med (2015) 212(7):1125–37. 10.1084/jem.20142237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Johnson LA, Sanchez-Perez L, Suryadevara CM, Sampson JH. Chimeric antigen receptor engineered T cells can eliminate brain tumors and initiate long-term protection against recurrence. Oncoimmunology (2014) 3(7):e944059. 10.4161/21624011.2014.944059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19(20):5636–46. 10.1158/1078-0432.CCR-13-0458 [DOI] [PubMed] [Google Scholar]
- 184.Maude SL, Hucks GE, Seif AE, Talekar MK, Teachey DT, Baniewicz D, et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J Clin Oncol (2017) 35(15_suppl):103. 10.1200/JCO.2017.35.15_suppl.103 [DOI] [Google Scholar]
- 185.Chong E, Melenhorst J, Lacey S, Ambrose D, Gonzalez V, Levine B, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood (2017) 129(8):1039–41. 10.1182/blood-2016-09-738245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res (2017) 23(22):6982–92. 10.1158/1078-0432.CCR-17-0867 [DOI] [PubMed] [Google Scholar]
- 187.Suarez E, Chang D, Sun J, Sui J, Freeman G, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7(23):34341–55. 10.18632/oncotarget.9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543(7643):113–7. 10.1038/nature21405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Levine B, June C. Assembly line immunotherapy. Nature (2013) 498:S17. 10.1038/498S17a [DOI] [PubMed] [Google Scholar]