

Abstract
Ivacaftor is a drug used to treat cystic fibrosis (CF) patients carrying specific gating CFTR mutations. Interpatient variability in the lung response has been shown to be partly explained by rs7512462 in the Solute Carrier Family 26 Member 9 (SLC26A9) gene. In an independent and larger cohort, we aimed to evaluate whether SLC26A9 variants contribute to the variability of the lung phenotype and if they influence the lung response to ivacaftor. We genotyped the French CF Gene Modifier Study cohort (n = 4,840) to investigate whether SLC26A9 variants were involved in the lung phenotype heterogeneity. Their influence in the response to ivacaftor was tested in the 30 treated patients who met the inclusion criteria: older than 6 years of age, percent-predicted forced expiratory volume measured in 1 s (FEV1pp) in the 3 months before treatment initiation ranging between 40 and 90%. Response to treatment was determined by the change in FEV1pp from baseline, averaged in 15–75 days, and the 1st-year post-treatment. We observed that SLC26A9 variants were not associated with lung function variability in untreated patients and that gain of lung function in patients treated with ivacaftor was similar to clinical trials. We confirmed that rs7512462 was associated with variability in ivacaftor-lung response, with a significant reduction in lung function improvement for patients with the C allele. Other SLC26A9 SNPs also contributed to the ivacaftor-response. Interindividual variability in lung response to ivacaftor is associated with SLC26A9 variants in French CF patients. Pharmacogenomics and personalized medicine will soon be part of CF patient care.
Keywords: cystic fibrosis, lung, gene modifier, SLC26A9, ivacaftor, pharmacogenetic, individualized medicine
Introduction
Cystic fibrosis (CF) is the most common, severe, autosomal recessive genetic disease in Caucasians. It is caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR), a chloride channel expressed in epithelial cells throughout the body (Riordan et al., 1989). The disease affects several organs such as the lungs, pancreas, intestine, and liver. Over 2,000 variations in the CFTR gene have been described, including 312 CF-causing variant [The Clinical and Functional TRanslation of CFTR (CFTR2)1], which are usually classified into six classes, according to their resulting effect on the protein (Corvol et al., 2016). The most common mutation (70% of alleles) is p.Phe508del (F508del), which prevents normal CFTR expression at the apical surface of epithelia. CFTR genotype strongly influences pancreatic function, which is either deficient (PI for pancreatic insufficiency), or normal (PS for pancreatic sufficiency). It is recognized that in the major part of cases, patients carrying two PI-associated severe CFTR mutations have a classical form of CF, whereas others have a milder form of disease associated with PS (Corvol et al., 2016).
Until recently, treatment of CF was only symptomatic. However, in recent years, considerable efforts have led to the development of therapies that target the CFTR protein. Since 2012, patients carrying the CFTR gating mutation p.Gly551Asp (G551D) and who are older than 6 years can be treated with ivacaftor, a molecule called a potentiator, which targets CFTR directly to increase the probability of the channel being open (Van Goor et al., 2009). Significant clinical benefits of ivacaftor, such as gain of lung function and reduced number of pulmonary exacerbations, were initially observed in patients older than 12 years and carrying at least one G551D CFTR mutation (Ramsey et al., 2011). Subsequently, ivacaftor was approved for other CFTR-gating mutations: p.Gly1244Glu (G1244E), p.Gly1349Asp (G1349D), p.Gly178Arg (G178R), p.Gly551Ser (G551S), p.Ser1251Asn (S1251N), p.Ser1255Pro (S1255P), p.Ser549Asn (S549N), and p.Ser549Arg (S549R) (De Boeck et al., 2014) and younger patients (Davies et al., 2013). Now, ivacaftor is approved for patients with CF older than 2 years carrying at least one of these gating mutations (Davies et al., 2016).
The Solute Carrier Family 26 Member 9 gene, SLC26A9, was recently shown to modulate the airway response to CFTR-directed therapeutics. In particular, in CF patients carrying at least one CFTR-G551D mutation, the single nucleotide polymorphism (SNP), rs7512462, in the SLC26A9 gene was shown to explain 28% of the response variability to ivacaftor (Strug et al., 2016). In that study, rs7512462 was also associated with the lung function variability of patients carrying a CFTR-gating mutation. Moreover, SLC26A9 variants have been previously shown in genome wide association studies (GWAS) to contribute to the phenotype variability of meconium ileus (Sun et al., 2012) (rs7512462, rs4077468, rs4077469, rs7419153, rs12047830, rs12741299) and CF-related diabetes (CFRD, rs4077468, rs4077469, rs1874361) (Blackman et al., 2013).
In the current study, we examine the French cohort (n = 4,840) of the French CF Gene Modifier Study to investigate whether SLC26A9 variants firstly contribute to the variability of the lung phenotype, and secondly influence the response to ivacaftor.
Materials and Methods
Study Subjects and Lung Phenotype
Patients with CF treated in 38 out of the 47 French CF centres between January 2004 and January 2017 were enrolled in the French CF Modifier Gene Study. As of January 1, 2017, 4,840 patients with CF had been included (corresponding to ∼75% of all French patients with CF) (Vaincre la Mucoviscidose and Ined, 2017). The study was approved by the French ethical committee (CPP n°2004/15), and the information collection was approved by the Commission Nationale de L’informatique et des Libertés (n°04.404). Informed consent in writing was obtained from each patient and/or guardian.
Measurements of the forced expiratory volume measured in 1 s (FEV1) were either expressed as percent-predicted values (FEV1pp) using the Global Lung Function Initiative (GLI) equations (Quanjer et al., 2012) or transformed to the Kulich Normalized Mortality Adjusted CF-specific lung phenotype (SaKnorm Z-value) (Kulich et al., 2005; Taylor et al., 2011). This quantitative phenotype allows indeed direct comparison of lung phenotypes between patients with CF and accounts for differential survival.
Details on the 4,840 CF patients are reported in Table 1 and in the Flowchart (Figure 1). Only patients with severe CFTR mutations were considered (pancreatic sufficient patients excluded). Among those, 119 carried at least one gating mutation for which ivacaftor therapy has been approved in Europe (i.e., G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R), 81 were prescribed ivacaftor. Finally, 60 patients on ivacaftor had lung function measurements available before and after treatment initiation. To assess the association of SLC26A9 with lung function response to ivacaftor, we included the 30 patients older than 6 years of age and with FEV1pp in the 3 months before treatment initiation ranging between 40 and 90%; their CFTR genotypes are depicted in Supplementary Table S1. The response to treatment was determined by the change in FEV1pp from baseline, averaged in the 15–75 days after treatment, as well as that averaged over the 1st-year post-treatment, as used in an earlier study (Table 1) (Strug et al., 2016). Besides, these two timelines were chosen to evaluate: (1) an “early” response (15–75 days), as it takes several days for ivacaftor to reach a maximal response; and (2) a “long-term” response (1 year), computed by averaging the FEV1pp over the 1st year of ivacaftor treatment, illustrating the overall response of the patients.
Table 1.
Baseline characteristics of the patients.
| Patient characteristics |
CFTR variants |
||
|---|---|---|---|
| F508del/ F508del |
Gating$/ other |
G551D/ other |
|
| Pancreatic Insufficient (n = 4,045) | n = 2,143 | n = 119 | n = 72 |
| Sex (male/female) | 1,125/1018 | 64/55 | 42/30 |
| European origin, n (%) | 2,061 (96) | 105 (88) | 67 (93) |
| Treated by ivacaftor (n = 81) | |||
| FEV1pp (baseline) $ | n = 60 | n = 40 | |
| >90% | 17 | 16 | |
| 40–90% | 30 | 16 | |
| <40% | 13 | 8 | |
| FEV1pp change analysis (n = 30) | n = 30 | n = 16 | |
| Age, mean (SD) | 20.0 (14.0) | 17.6 (12.4) | |
| Sex (male/female) | 18/12 | 10/6 | |
$Ivacaftor-approved CFTR gating mutations: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R; ∗∗FEV1pp (forced expiratory volume in 1 s percent-predicted) value in the 3 months before treatment. Data are means (SD) or numbers (%) unless otherwise indicated. CFTR: Cystic Fibrosis Transmembrane Conductance Regulator.
FIGURE 1.
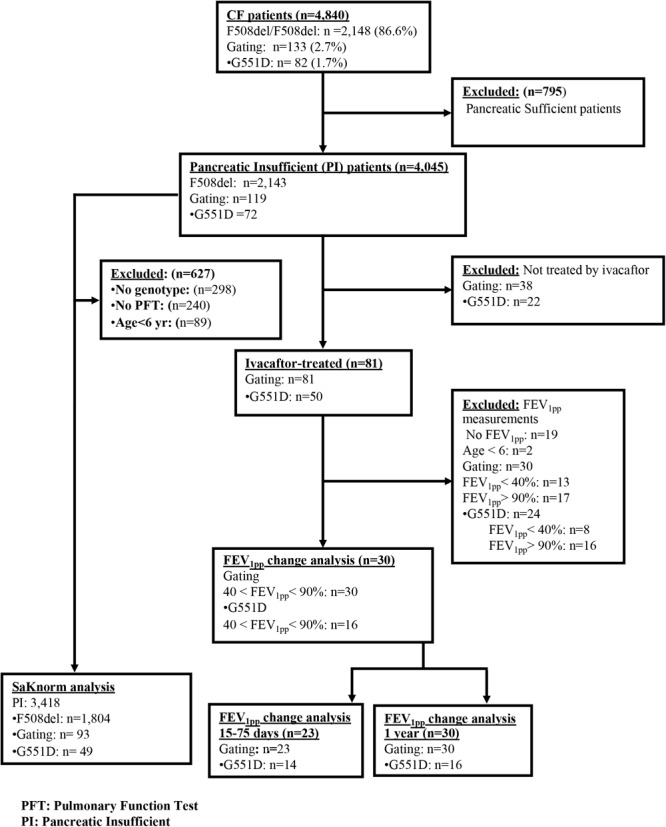
Flowchart of the study.
Genotyping
The genotyping of SLC26A9 SNPs (rs7512462, rs4077468, rs7419153, rs12047830, rs4077469, rs12741299, and rs1874361) was carried out using Kompetitive Allele Specific PCR (KASP) genotyping chemistry (LGC, Teddington, United Kingdom).
Statistical Analysis
Lung function was analyzed as FEV1pp (GLI) (Quanjer et al., 2012) or CF-specific quantile-Z value (SaKnorm Z-value) (Kulich et al., 2005; Taylor et al., 2011). For each patient, FEV1pp measurements in the 15-75 days following treatment initiation were averaged to determine the early response. We used the trapezoidal rule to compute average FEV1pp over the 1st year to account for irregular measurements. The change in FEV1pp from baseline was then analyzed by linear regression, adjusting for baseline measurement. We used additive coding to estimate the effect of SNPs in SLC26A9. Reference alleles were taken from annotations of the human genome2. Among SNPs in SLC26A9, rs7512462 had previously shown association with treatment response and, therefore, was analyzed independently from the other SNPs. We also analyzed five other SNPs in SLC26A9, adjusting the P-values for multiplicity in this situation.
Conformance of the allele frequencies with the Hardy–Weinberg equilibrium (HWE) was tested using a Fisher’s exact test. As shown in Supplementary Table S2, the population did not deviate significantly from the HWE indicating no issue with the genotyping method or population stratification.
We reconstructed haplotypes using the EM algorithm with all patients (n = 4,045) keeping loci in their physical order on chromosome 1 (using haplo.stats package in the R software) (Supplementary Table S3) (Lake et al., 2003). We analyzed the association of haplotypes with FEV1pp using additive haplotype coding (see Supplementary Material).
A P-value of less than 5% was interpreted as evidence of a statistically significant difference or association. Multiple comparisons utilized the Bonferroni correction. All association analyses were carried out using the software, R (version 3.4.03).
Results
SLC26A9 Gene Variants and Lung Function
In the absence of ivacaftor treatment, the effect of rs7512462 on lung function did not reach statistical significance for any CFTR genotype group. We considered all patients (n = 3,418), F508del homozygous patients (n = 1,804), carriers of at least one ivacaftor-approved gating mutation (n = 93), and carriers of one G551D allele (n = 49) (Figure 2 and Table 2). The results were similar with other SNPs in the SLC26A9 gene (i.e., rs1874361, rs12741299, rs4077468, rs4077469, rs12047830, rs7419153) (Table 3).
FIGURE 2.
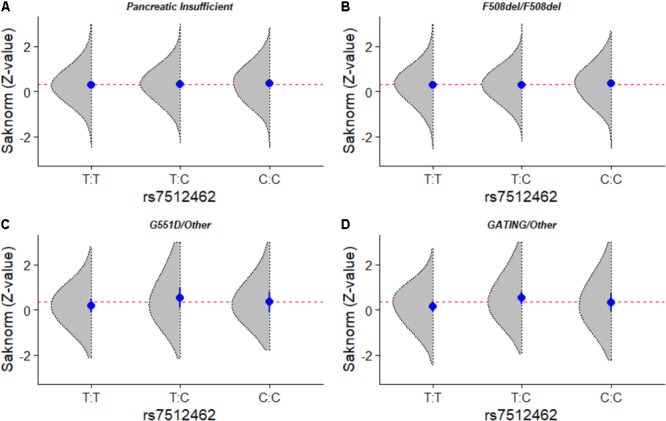
Lung phenotype distribution according to SLC26A9 rs7512462 variant in patients with cystic fibrosis. Smoothed histograms of SaKnorm Z-values by SLC26A9 rs7512462 genotypes (gray areas), genotype specific mean ± SE (dot and bars) and overall mean (dashed). (A) All pancreatic insufficient CF patients (n = 3,418), (B) CFTR F508del homozygous CF patients (n = 1,804), (C) CF patients with at least one G551D allele (n = 49) and (D) CF patients with at least one ivacaftor-approved gating mutation (n = 93).
Table 2.
Mean differences in lung function according to CFTR and SLC26A9 variants.
| All pancreatic insufficient | F508del/F508del | G551D/other | Gating$/other | |||||
|---|---|---|---|---|---|---|---|---|
| patients (n = 3,418) |
(n = 1,804) |
(n = 49) |
(n = 93) |
|||||
| SLC26A9 | Mean change | P-value | Mean change | P-value | Mean change | P-value | Mean change | P-value |
| variants$ | in CF SaKnorm∗ | in CF SaKnorm∗ | in CF SaKnorm∗ | in CF SaKnorm∗ | ||||
| rs7512462 | 0.03 ± 0.02 | 0.20 | 0.03 ± 0.03 | 0.29 | 0.14 ± 0.16 | 0.40 | 0.13 ± 0.12 | 0.27 |
| rs1874361 | –0.02 ± 0.02 | 0.30 | –0.00 ± 0.03 | 0.97 | –0.07 ± 0.16 | 0.66 | –0.17 ± 0.11 | 0.13 |
| rs12741299 | 0.01 ± 0.03 | 0.70 | –0.02 ± 0.04 | 0.64 | –0.06 ± 0.33 | 0.86 | 0.23 ± 0.23 | 0.32 |
| rs4077468 | 0.05 ± 0.02 | 0.01 | 0.05 ± 0.03 | 0.07 | 0.22 ± 0.18 | 0.22 | 0.14 ± 0.13 | 0.29 |
| rs4077469 | 0.05 ± 0.02 | 0.01 | 0.05 ± 0.03 | 0.08 | 0.22 ± 0.17 | 0.21 | 0.14 ± 0.13 | 0.26 |
| rs12047830 | 0.04 ± 0.02 | 0.03 | 0.03 ± 0.02 | 0.19 | 0.37 ± 0.18 | 0.05 | 0.27 ± 0.13 | 0.05 |
| rs7419153 | –0.02 ± 0.02 | 0.359 | –0.02 ± 0.03 | 0.47 | –0.35 ± 0.18 | 0.06 | –0.18 ± 0.14 | 0.20 |
$See description Supplementary Table S2; $Ivacaftor-approved CFTR gating mutations: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R; ∗change per minor allele (linear regression of SaKnorm Z-value with additive model). SaKnorm is normalized function of FEV1 (forced expiratory volume in 1 s) adjusted for age, sex, height, and cohort-specific survival.
Table 3.
Change in lung function evolution after 15–75 days and after 1 year on ivacaftor.
| n | Before∗∗ | After | Difference | 95% CI | P-value∗ | |
|---|---|---|---|---|---|---|
| Average FEV1pp measures within 15–75 days on ivacaftor | ||||||
| Gating$/other | 23 | 66.13 | 80.85 | 11.72 | 7.32–16.06 | <0.0001 |
| G551D/other | 14 | 70.66 | 85.05 | 14.39 | 7.89–20.66 | 0.00012 |
| Average FEV1pp measures in the 1st year on ivacaftor | ||||||
| Gating$/other | 30 | 69.07 | 78.90 | 9.83 | 4.91–14.77 | <0.0001 |
| G551D/other | 16 | 69.14 | 84.2 | 15.06 | 7.10–23.14 | <0.0001 |
$Ivacaftor-approved CFTR gating mutations: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R; ∗∗FEV1pp (forced expiratory volume in 1 s percent-predicted) value in the 3 months before treatment. ∗Paired Mann–Whitney–Wilcoxon test.
Change in FEV1 With Ivacaftor
In total, 30 pancreatic insufficient CF patients had at least one ivacaftor-approved gating mutation, baseline FEV1pp between 40 and 90%, and post-treatment data (see Flowchart, Figure 1). FEV1pp measurements within 15–75 days post-treatment and in the 1st year on ivacaftor were available for 23 and 30 patients, respectively, among whom 14 and 16, respectively, were carriers of at least one G551D allele (see Flowchart, Figure 1). Overall, patients showed an improvement in FEV1pp after 15–75 days and in the 1st year on ivacaftor. For individuals with ivacaftor-approved gating mutations, the baseline-adjusted change in FEV1pp was +11.72% (95% CI: 7.32–16.06) at 15–75 days and +9.83% (95% CI: 4.91–14.77) in the 1st year of treatment (P < 0.0001, Table 3 and Figure 3). In patients with at least one G551D allele, the improvement in FEV1pp was +14.39% (95% CI: 7.89–20.66) at 15–75 days and +15.06% (95% CI: 7.1–23.14) over the 1st year of treatment (P < 0.0001, Table 3 and Figure 3).
FIGURE 3.

FEV1pp change from baseline in patients carrying at least one ivacaftor-approved gating mutation (light blue) or one G551D variant (dark blue) within the first 15–75 days and over the 1st year of ivacaftor.
SLC26A9 Variants and Ivacaftor Treatment Response
In patients carrying at least one G551D allele, the response to treatment changed with the SLC26A9 rs7512462 genotype, with less change in FEV1pp (-7.7% over 15–75 days and -7.8% over 1 year of treatment) for each C allele (P = 0.0007 and 0.006, respectively; Table 4 and Figure 4). Other SLC26A9 variants showed similar associations with the changes in FEV1pp. In particular, the following SNPs demonstrated significant association after adjustment for multiplicity: with reduced FEV1pp over 15–75 days: rs4077468, -7.9 FEV1pp (Padj = 0.0007); and rs4077469, -9.3 FEV1pp (Padj = 0.0035); or with increased FEV1pp over 15–75 days: rs7419153, +9.8 FEV1pp (Padj = 0.0049) (Table 4).
Table 4.
Change in FEV1pp within 15–75 days and over 1 year on ivacaftor treatment according to SLC26A9 variants in patients carrying at least one G551D mutation.
| Period of evaluation | 15–75 days after ivacaftor treatment start |
Over the 1st year of ivacaftor treatment |
||||
|---|---|---|---|---|---|---|
| SLC26A9 variants$ | G551D/other (n = 14) |
G551D/other (n = 16) |
||||
| SNP | FEV1pp change∗ | P-value | Adjusted P-value∗∗ | FEV1pp change∗ | P-value | Adjusted P-value∗∗ |
| rs7512462 | –7.7 ± 1.7 | 0.0007 | 0.0049 | –7.8 ± 2.4 | 0.0063 | 0.0441 |
| rs1874361 | 4.4 ± 1.5 | 0.0107 | 0.0749 | 3.1 ± 3.0 | 0.3129 | 1.0000 |
| rs12741299 | 2.9 ± 6.0 | 0.6336 | 1.0000 | 2.1 ± 5.9 | 0.7331 | 1.0000 |
| rs4077468 | –7.9 ± 1.3 | 0.0001 | 0.0007 | –8.4 ± 1.7 | 0.0003 | 0.0027 |
| rs4077469 | –9.3 ± 1.9 | 0.0005 | 0.0035 | –10.5 ± 2.3 | 0.0006 | 0.0042 |
| rs12047830 | –10.9 ± 2.7 | 0.0020 | 0.014 | –7.5 ± 3.5 | 0.0518 | 0.3626 |
| rs7419153 | 9.8 ± 2.1 | 0.0007 | 0.0049 | 8.1 ± 2.6 | 0.0089 | 0.0623 |
$See description Supplementary Table S2; ∗linear regression of FEV1pp (forced expiratory volume in 1 s percent-predicted) change with additive model adjusted on baseline; ∗∗Bonferroni adjustment.
FIGURE 4.
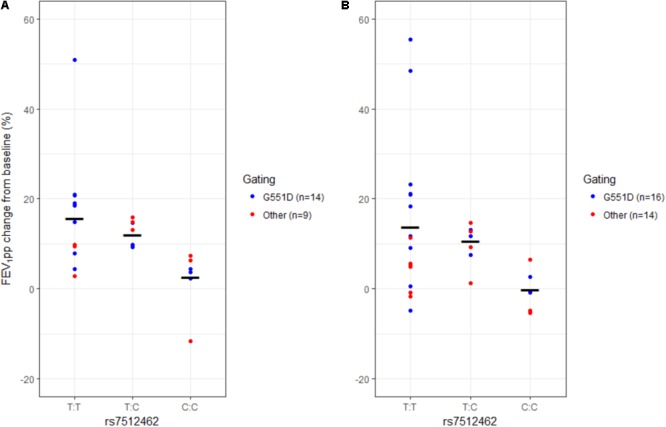
FEV1pp change from baseline in patients with ivacaftor treatment according to rs7512462 SLC26A9 genotype. Individual change in patients with at least one G551D mutation (blue) and with other gating mutations (red) and model fitted mean change (horizontal bar) within the first 15–75 days of treatment (A) and over the 1st year of treatment (B).
The results for individuals with gating mutations were similar. Indeed, the response to treatment also changed with the SLC26A9 rs7512462 genotype, with less change in FEV1pp (-5.9% over 15–75 days and -5.2% over 1 year of treatment) for each C allele (P = 0.0031 and 0.0042, respectively; Table 5 and Figure 4). Moreover, other SLC26A9 variants also showed similar associations with, in particular, significant association after adjustment for multiplicity for the following SNPs with reduced FEV1pp over 15–75 days: rs4077468, -7.0 FEV1pp (Padj = 0.0231); and rs4077469, -7.8 FEV1pp (Padj = 0.0490) (Table 5).
Table 5.
Change in FEV1pp within 15–75 days and over 1 year on ivacaftor treatment according to SLC26A9 variants in patients carrying at least one ivacaftor-approved CFTR gating mutation.
| Period of evaluation | 15–75 days after ivacaftor treatment start |
Over the 1st year of ivacaftor treatment |
||||
|---|---|---|---|---|---|---|
| SLC26A9 variants$ | Gating$/other (n = 23) |
Gating$/other (n = 30) |
||||
| SNP | FEV1pp change∗ | P-value | Adjusted P-value∗∗ | FEV1pp change∗ | P-value∗ | Adjusted P-value∗∗ |
| rs7512462 | –5.9 ± 1.7 | 0.0031 | 0.0217 | –5.2 ± 1.8 | 0.0042 | 0.0294 |
| rs1874361 | 3.7 ± 1.3 | 0.0120 | 0.0840 | 3.9 ± 1.9 | 0.0479 | 0.3346 |
| rs12741299 | –2.8 ± 4.8 | 0.5656 | 1.0000 | –2.7 ± 4.2 | 0.5270 | 1.0000 |
| rs4077468 | –7.0 ± 2.1 | 0.0033 | 0.0231 | –7.3 ± 2.2 | 0.0030 | 0.0210 |
| rs4077469 | –7.8 ± 2.6 | 0.0070 | 0.0490 | –8.2 ± 2.6 | 0.0040 | 0.0280 |
| rs12047830 | –6.3 ± 2.4 | 0.0138 | 0.0966 | –6.6 ± 2.6 | 0.0161 | 0.1127 |
| rs7419153 | 6.3 ± 2.6 | 0.0233 | 0.1631 | 7.9 ± 2.5 | 0.0038 | 0.0266 |
$See description Supplementary Table S2; ∗linear regression of FEV1pp (forced expiratory volume in one second percent-predicted) change with additive model adjusted on baseline; ∗∗Bonferroni adjustment; $Ivacaftor-approved CFTR gating mutations: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R.
In these patients, the most frequent haplotypes (7 SNPs in physical order, Supplementary Table S2) were CCCGTAG (35%) and TACACGA (18%) (Supplementary Table S3). In agreement with the direction of association in the SNP analysis, FEV1pp in carriers of at least one CCCGTAG haplotype increased on average by 17 ± 12% over 15–75 days and by 17 ± 17% over 1 year of treatment, while the FEV1pp of the carriers of at least one TACACGA increased by 6 ± 5% over 15–75 days and 4 ± 5% over 1 year of treatment. Overall, this analysis did not provide significant evidence for a heterogeneity in FEV1pp change with haplotypes (P = 0.2 over 15–75 days, P = 0.08 over 1 year), but was underpowered given the large diversity of haplotypes in the sample (Supplementary Table S3).
Discussion
We have shown that the response to ivacaftor measured as lung function modulation varied between individuals and is associated with SLC26A9 variants, as previously described (Strug et al., 2016). SLC26A9 is a key candidate in CF as it has been shown to play a pleiotropic role across CF phenotypes, associated with meconium ileus (Sun et al., 2012; Li et al., 2014), immunoreactive trypsinogen at birth (Soave et al., 2014; Miller et al., 2015), pancreatic damage (Li et al., 2014), and CFRD (Blackman et al., 2013; Soave et al., 2014). With the development of new curative treatments, such as CFTR-targeted therapies, pharmacogenomics will become a major step toward functional personalized medicine (Corvol et al., 2016).
SLC26A9 Gene Modulates Ivacaftor Lung Response
We observed that SLC26A9 variants were associated with the variability in lung responses to ivacaftor measured as FEV1pp change over 1 year with treatment. Indeed, we have shown that, although the response varied between individuals, FEV1pp improved after 15–75 days to 1 year on ivacaftor, in agreement with several clinical trials that led to the approbation of this drug (Ramsey et al., 2011; Davies et al., 2013, 2016; De Boeck et al., 2014; McKone et al., 2014). Further, we observed, as previously shown by Strug et al. (2016), that SLC26A9 variants modulate this drug response. Focusing on SLC26A9 rs7512462 in the patients carrying at least one G551D CFTR mutation, we observed that the CC genotype was associated with a decrease in FEV1pp of -7.7%. Surprisingly, this effect was inverse to that observed in the pilot study of Strug et al. (2016) who showed an increase in FEV1pp of approximately 8.5% for each additional C allele at rs7512462. Since rs7512462 is not presumed to be functional (Strug et al., 2016), it must be linked to the causal variants so that population-specific differences could explain this difference in direction. In fact, the North-American CF Gene Modifier consortium found significant population admixture in the North-American CF patients, with a large portion of patients reporting African–Caucasian, Mexican–Caucasian, and Indian–Caucasian ancestries (Li et al., 2011), whereby French patients are predominantly of Caucasian origin. Besides, it has been found that the pharmacogenetic response to drugs varied across ethnic groups, which might play a role in the differences observed here between French and Canadian cohorts (Corvol and Burchard, 2008). Nevertheless, Strug et al. (2016) observed in CF patients carrying at least one CFTR-G551D mutation, that the rs7512462 SNP in the SLC26A9 gene explained 28% of the response variability to ivacaftor, a result similar to ours (22%) (Strug et al., 2016). We observed similar results when evaluating both G551D carriers and patients carrying other ivacaftor-approved gating mutations. There remains, however, a large part of interindividual variation besides the SNP status.
SLC26A9 Gene Is Not a Modifier of Lung Function in CF Patients
In this study, SLC26A9 variants were not associated with variation in lung function of French patients with CF, regardless of their CFTR genotype (i.e., two copies of the F508del mutation and/or at least one gating mutation, the most frequent being G551D). These results are in agreement with previous, large international GWAS studies (Wright et al., 2011; Corvol et al., 2015). The latest and largest study, a meta-analysis of 6,365 French and North-American CF patients, identified five regions outside the SLC26A9 locus that displayed significant association with variation in lung disease: the locus of the mucin genes MUC4 and MUC20, of the solute carrier genes SLC9A3 and SLC6A14, and of the HLA Class II region (Corvol et al., 2015). No association was observed between lung function and the SLC26A9 gene. In the pilot study of Strug et al. (2016) SLC26A9 rs7512462 also was not associated with lung function variation in CF patients who were homozygous for the F508del mutation. However, an association was observed for patients carrying at least one G551D variant, for whom the number of C alleles was positively associated with improved lung function (Strug et al., 2016). Another independent study of a smaller Brazilian CF cohort with various CFTR mutations also did not show an association of this variant with FEV1 heterogeneity (Pereira et al., 2017). We tested other SLC26A9 variants that had been previously shown in GWAS to contribute to the variability of other phenotypes, such as meconium ileus (rs4077468, rs4077469, rs7419153, rs12047830, rs12741299) (Sun et al., 2012) and CFRD (rs4077468, rs4077469, rs1874361) (Blackman et al., 2013). However, we found that none of these variants contributed to the lung function heterogeneity in our CF cohort.
Mechanisms of SLC26A9 Variants to Modulate Ivacaftor Lung Response
SLC26A9 is a highly conserved protein predominantly expressed in the lung (Lohi et al., 2002). It functions as a chloride channel with minimal bicarbonate conductance (Loriol et al., 2008; Bertrand et al., 2009), and may constitute an attractive alternative ion channel strategy to compensate for the CFTR defect (Mall and Galietta, 2015). Indeed, in human bronchial epithelial cells, it has been shown that SLC26A9 contributes to constitutive and cAMP-dependant chloride secretion (Bertrand et al., 2009; Avella et al., 2011). Physical interaction between SLC26A9 and CFTR has been shown in several studies (Bertrand et al., 2009; Chang et al., 2009; Avella et al., 2011), even if the consequences of this interaction [reviewed in (El Khouri and Toure, 2014)] are still controversial. According to these studies, SLC26A9 interaction with CFTR enhances CFTR activity (Bertrand et al., 2009; Avella et al., 2011). Reciprocally, CFTR has been shown to modulate SLC26A9 function (Bertrand et al., 2009). However, there has also been evidence showing that CFTR inhibits the activity of SLC26A9 (Chang et al., 2009; Ousingsawat et al., 2012). More recently, SLC26A9 membrane expression and activity was shown to be decreased by CFTR-F508del in co-expression experiments using HEK cells. Interestingly, the correction of F508del CFTR by VX-809 (lumacaftor) was also shown to restore SLC26A9 activity (Bertrand et al., 2017). Finally, a previous functional analysis of eight non-synonymous coding SNPs (p.Y70N, p.T1247N, p.I384T, p.R575W, p.606L, p.V622L, p.V744M, and p.H748R) revealed several functional modifications, including increased or decreased channel activity and altered protein expression that could modify disease (Chen et al., 2012).
The exact mechanism explaining how SLC26A9 variants affect the ivacaftor responses that we observed in our patients is unknown (depicted in Figure 5) and requires future investigation. Based on previous reports, it is reasonable to hypothesize that the variants have an impact on SLC26A9-CFTR-G551D interactions, which could result in altered intracellular trafficking of CFTR-G551D and/or transporter activation.
FIGURE 5.
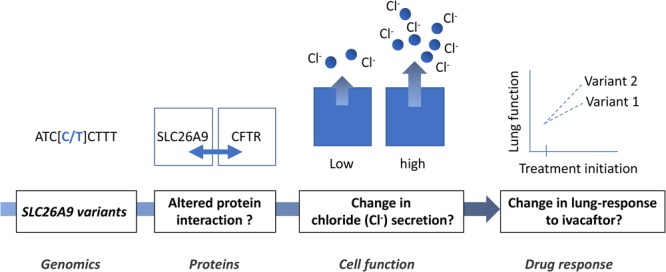
Proposed mechanism of the modulation of ivacaftor lung response in CF patients by SLC26A9 gene.
Our study has several limitations, mainly related to the small sample size of the cohort treated with ivacaftor, due to the rarity of CFTR gating mutations (∼4% of individuals with CF). Moreover, baseline FEV1 measurements at ivacaftor treatment initiation was missing for about one third of the patients (see flowchart), which reduced even more the size of the analyzed cohort. Nevertheless, this study highlights that the interindividual variability in the lung response to ivacaftor might genetically be associated with SLC26A9 variants in French CF patients and confirms the key pleiotropic role of this gene in CF. To confirm these results, it will be important to extend this study to patients with CF from different countries worldwide. The elucidation of the biological mechanisms beyond this variability is also necessary and will require functional genetic studies. In the exciting current era of curative treatment development in CF, pharmacogenomics will soon be an integral part of patient care, to modify treatment accordingly to provide the ultimate personalized medicine.
Author Contributions
HC, P-YB, and LG designed the study and wrote the manuscript. P-YB, JM, and I-HD performed the data analysis. LS critically revised the manuscript. HC and JM participated in patient recruitment, sample collection, and phenotyping.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the patients, care providers and clinic coordinators at CF Centres, as well as the Principal Investigators in the several contributing French CF Centres:
ABELY, M., American Memorial Hospital, Reims/BASSINET, L., Centre Hospitalier Intercommunal de Créteil, Créteil/ BELLEGUIC, C., Hôpital Pontchaillou, Rennes/BESSACI, K., American Memorial Hospital, Reims/BILLON, Y., Hôpital de Brabois, Vandoeuvre les Nancy/BRÉMONT, F., Hôpital des Enfants de Toulouse, Toulouse/BROUARD, J., Centre Hospitalier Universitaire de Caen, Caen/BUI, S., Hôpital Des Enfants Groupe Pellegrin, Bordeaux/CAMARA, B., Hôpital de la Tronche, Grenoble/CAMPBELL, K., Centre Hospitalier Universitaire de Caen, Caen/CHIRON, R., Hôpital Arnaud de Villeneuve, Montpellier/COIRIER-DUET, E., Hôpital André Mignot, Le Chesnay/DALPHIN, J. C., CNRS-UFC, UMR 6249 Chrono-environnement, Hôpital Jean Minjoz, Besançon/DALPHIN, M. L., Centre Hospitalier Universitaire de Besançon, Besançon/DAVID, V., Hôpital Mère-Enfant, Nantes/DENEUVILLE, E., Hôpital Sud Annexe Pédiatrique, Rennes/DERELLE, J., Hôpital d’Enfants, Vandoeuvre les Nancy/DOMBLIDES, P., Hôpital Haut Lévêque, Pessac/DOMINIQUE, S., Centre Hospitalier Universitaire Charles Nicolle, Rouen/DUBUS, J. C., Hôpital d’Enfants de la Timone, Marseille/DUFEU, N., Hôpital Nord, Marseille/DURIEU, I., UCBL1, Groupe Hospitalier Lyon Sud, Hospices Civils de Lyon, Pierre Bénite/DURY, S., Hôpital Maison Blanche, Reims/EPAUD, R., Centre Hospitalier Intercommunal de Créteil, Créteil/FANTON, A., Hôpital d’Enfants du Bocage, Dijon/FAYON, M., Hôpital Des Enfants Groupe Pellegrin, Bordeaux/FLEURENCE, E., Hôpital d’Enfants, Saint-Denis de la Réunion/FOUCAUD, P., Hôpital André Mignot, Le Chesnay/GINIES, J.L., Centre Hospitalier Universitaire d’Angers, Angers/GRENET, D., Hôpital Foch, Suresnes/HUBERT, D., Hôpital Cochin, Paris/HUET, F., Hôpital d’Enfants du Bocage, Dijon/KESSLER, R., Hôpital Civil, Strasbourg/LABBÉ, A., Centre Hospitalier Estaing, Clermont-Ferrand/LANGUEPIN, J., Centre Hospitalier, Limoges/LE BOURGEOIS, M., Necker Hôpital d’Enfants Malades, Paris/LE ROUX, P., Hôpital Jacques Monod, Montivilliers/LEROY, S., CHU de Nice, Nice/LLERENA, C., Hôpital de la Tronche, Grenoble/LOEUILLE, G. A., Centre Hospitalier de Dunkerque, Dunkerque/MARCHAND, S., Hôpital Clocheville, Tours/MARGUET, C., Centre Hospitalier Universitaire Charles Nicolle, Rouen/MELY, L., Hôpital Renée Sabran, Giens/MUNCK, A., Hôpital Robert Debré, Paris/MURRIS-ESPIN, M., Hôpital Larrey, Toulouse/NOVE JOSSERAND, R., Groupe Hospitalier Lyon Sud, Hospices Civils de Lyon, Pierre Bénite/PIN, I., INSERM U823 Université Joseph Fourier, Hôpital de la Tronche, Grenoble/PREVOTAT, A., Hôpital Calmette, Lille/RAMES, C., Hôpital Nord, Amiens/RAULT, G., Centre de Perharidy, Roscoff/REIX, P., Hôpital Femme Mère Enfant, Bron/REMUS, N., Centre Hospitalier Intercommunal de Créteil, Créteil/RENOUIL, M., Groupe Hospitalier Sud Réunion, Saint-Pierre de la Réunion/REYNAUD-GAUBERT, M., Hôpital Nord, Marseille/SERMET-GAUDELUS, I., Necker Hôpital d’Enfants Malades, Paris/STREMLER, N., Hôpital d’Enfants de la Timone, Marseille/STORNI, V., Centre Hospitalier Bretagne Atlantique, Vannes/TASSET, C., Hôpital d’Enfants, Saint-Pierre de la Réunion/URBAN, T., Centre Hospitalier Universitaire d’Angers, Angers/VIGNERON, P., Centre Hospitalier Bretagne Sud, Lorient/WALLAERT, B., Hôpital Calmette, Lille/WEISS, L., Hôpital de Hautepierre, Strasbourg/WIZLA, N., Hôpital Jeanne de Flandre, Lille.
Funding. This study was supported by Institut National de la Santé et de la Recherche Médicale, Assistance Publique-Hôpitaux de Paris, Sorbonne Université, Agence Nationale de la Recherche (R09186DS), DGS, Association Vaincre La Mucoviscidose, Association Agir Informer Contre la Mucoviscidose, GIS-Institut des Maladies Rares, Cystic Fibrosis foundation (CORVOL16G0).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00828/full#supplementary-material
References
- Avella M., Loriol C., Boulukos K., Borgese F., Ehrenfeld J. (2011). SLC26A9 stimulates CFTR expression and function in human bronchial cell lines. J. Cell. Physiol. 226 212–223. 10.1002/jcp.22328 [DOI] [PubMed] [Google Scholar]
- Bertrand C. A., Mitra S., Mishra S. K., Wang X., Zhao Y., Pilewski J. M., et al. (2017). The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell. Mol. Physiol. 312 L912–L925. 10.1152/ajplung.00178.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand C. A., Zhang R., Pilewski J. M., Frizzell R. A. (2009). SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J. Gen. Physiol. 133 421–438. 10.1085/jgp.200810097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman S. M., Commander C. W., Watson C., Arcara K. M., Strug L. J., Stonebraker J. R., et al. (2013). Genetic modifiers of cystic fibrosis-related diabetes. Diabetes Metab. Res. Rev. 62 3627–3635. 10.2337/db13-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M. H., Plata C., Sindic A., Ranatunga W. K., Chen A. P., Zandi-Nejad K., et al. (2009). Slc26a9 is inhibited by the R-region of the cystic fibrosis transmembrane conductance regulator via the STAS domain. J. Biol. Chem. 284 28306–28318. 10.1074/jbc.M109.001669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. P., Chang M. H., Romero M. F. (2012). Functional analysis of nonsynonymous single nucleotide polymorphisms in human SLC26A9. Hum. Mutat. 33 1275–1284. 10.1002/humu.22107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvol H., Blackman S. M., Boelle P. Y., Gallins P. J., Pace R. G., Stonebraker J. R., et al. (2015). Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 6:8382. 10.1038/ncomms9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvol H., Burchard E. G. (2008). Pharmacogenetic response to albuterol among asthmatics. Pharmacogenomics 9 505–510. 10.2217/14622416.9.5.505 [DOI] [PubMed] [Google Scholar]
- Corvol H., Thompson K. E., Tabary O., le Rouzic P., Guillot L. (2016). Translating the genetics of cystic fibrosis to personalized medicine. Transl. Res. 168 40–49. 10.1016/j.trsl.2015.04.008 [DOI] [PubMed] [Google Scholar]
- Davies J. C., Cunningham S., Harris W. T., Lapey A., Regelmann W. E., Sawicki G. S., et al. (2016). Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir. Med. 4 107–115. 10.1016/S2213-2600(15)00545-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. C., Wainwright C. E., Canny G. J., Chilvers M. A., Howenstine M. S., Munck A., et al. (2013). Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 187 1219–1225. 10.1164/rccm.201301-0153OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck K., Munck A., Walker S., Faro A., Hiatt P., Gilmartin G., et al. (2014). Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 13 674–680. 10.1016/j.jcf.2014.09.005 [DOI] [PubMed] [Google Scholar]
- El Khouri E., Toure A. (2014). Functional interaction of the cystic fibrosis transmembrane conductance regulator with members of the SLC26 family of anion transporters (SLC26A8 and SLC26A9): physiological and pathophysiological relevance. Int. J. Biochem. Cell Biol. 52 58–67. 10.1016/j.biocel.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Kulich M., Rosenfeld M., Campbell J., Kronmal R., Gibson R. L., Goss C. H., et al. (2005). Disease-specific reference equations for lung function in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 172 885–891. 10.1164/rccm.200410-1335OC [DOI] [PubMed] [Google Scholar]
- Lake S. L., Lyon H., Tantisira K., Silverman E. K., Weiss S. T., Laird N. M., et al. (2003). Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum. Hered. 55 56–65. 10.1159/000071811 [DOI] [PubMed] [Google Scholar]
- Li W., Soave D., Miller M. R., Keenan K., Lin F., Gong J., et al. (2014). Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum. Genet. 133 151–161. 10.1007/s00439-013-1363-7 [DOI] [PubMed] [Google Scholar]
- Li W., Sun L., Corey M., Zou F., Lee S., Cojocaru A. L., et al. (2011). Understanding the population structure of North American patients with cystic fibrosis. Clin. Genet. 79 136–146. 10.1111/j.1399-0004.2010.01502.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohi H., Kujala M., Makela S., Lehtonen E., Kestila M., Saarialho-Kere U., et al. (2002). Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J. Biol. Chem. 277 14246–14254. 10.1074/jbc.M111802200 [DOI] [PubMed] [Google Scholar]
- Loriol C., Dulong S., Avella M., Gabillat N., Boulukos K., Borgese F., et al. (2008). Characterization of SLC26A9, facilitation of Cl(-) transport by bicarbonate. Cell. Physiol. Biochem. 22 15–30. 10.1159/000149780 [DOI] [PubMed] [Google Scholar]
- Mall M. A., Galietta L. J. (2015). Targeting ion channels in cystic fibrosis. J. Cyst. Fibros. 14 561–570. 10.1016/j.jcf.2015.06.002 [DOI] [PubMed] [Google Scholar]
- McKone E. F., Borowitz D., Drevinek P., Griese M., Konstan M. W., Wainwright C., et al. (2014). Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2 902–910. 10.1016/S2213-2600(14)70218-8 [DOI] [PubMed] [Google Scholar]
- Miller M. R., Soave D., Li W., Gong J., Pace R. G., Boelle P. Y., et al. (2015). Variants in solute carrier SLC26A9 modify prenatal exocrine pancreatic damage in cystic fibrosis. J. Pediatr. 166 1152.e6–1157.e6. 10.1016/j.jpeds.2015.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousingsawat J., Schreiber R., Kunzelmann K. (2012). Differential contribution of SLC26A9 to Cl- conductance in polarized and non-polarized epithelial cells. J. Cell. Physiol. 227 2323–2329. 10.1002/jcp.22967 [DOI] [PubMed] [Google Scholar]
- Pereira S. V., Ribeiro J. D., Bertuzzo C. S., Marson F. A. L. (2017). Association of clinical severity of cystic fibrosis with variants in the SLC gene family (SLC6A14, SLC26A9, SLC11A1 and SLC9A3). Gene 629 117–126. 10.1016/j.gene.2017.07.068 [DOI] [PubMed] [Google Scholar]
- Quanjer P. H., Stanojevic S., Cole T. J., Baur X., Hall G. L., Culver B. H., et al. (2012). Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur. Respir. J. 40 1324–1343. 10.1183/09031936.00080312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey B. W., Davies J., McElvaney N. G., Tullis E., Bell S. C., Drevinek P., et al. (2011). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365 1663–1672. 10.1056/NEJMoa1105185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., et al. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245 1066–1073. 10.1126/science.2475911 [DOI] [PubMed] [Google Scholar]
- Soave D., Miller M. R., Keenan K., Li W., Gong J., Ip W., et al. (2014). Evidence for a causal relationship between early exocrine pancreatic disease and cystic fibrosis-related diabetes: a mendelian randomization study. Diabetes Metab. Res. Rev. 63 2114–2119. 10.2337/db13-1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strug L. J., Gonska T., He G., Keenan K., Ip W., Boelle P. Y., et al. (2016). Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet. 25 4590–4600. 10.1093/hmg/ddw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Rommens J. M., Corvol H., Li W., Li X., Chiang T. A., et al. (2012). Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat. Genet. 44 562–569. 10.1038/ng.2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C., Commander C. W., Collaco J. M., Strug L. J., Li W., Wright F. A., et al. (2011). A novel lung disease phenotype adjusted for mortality attrition for cystic fibrosis genetic modifier studies. Pediatr. Pulmonol. 46 857–869. 10.1002/ppul.21456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaincre la Mucoviscidose and Ined (2017). Registre Français de la Mucoviscidose - Bilan Des Données 2016. Paris: Vaincre la Mucoviscidose. [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Cao D., Neuberger T., et al. (2009). Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 106 18825–18830. 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. A., Strug L. J., Doshi V. K., Commander C. W., Blackman S. M., Sun L., et al. (2011). Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 43 539–546. 10.1038/ng.838 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.