

Abstract
Among the major challenges in quantitative genetics and personalized medicine is to understand how gene × gene interactions (G × G: epistasis) and gene × environment interactions (G × E) underlie phenotypic variation. Here, we use the intimate relationship between mitochondria and oxygen availability to dissect the roles of nuclear DNA (nDNA) variation, mitochondrial DNA (mtDNA) variation, hypoxia, and their interactions on gene expression in Drosophila melanogaster. Mitochondria provide an important evolutionary and medical context for understanding G × G and G × E given their central role in integrating cellular signals. We hypothesized that hypoxia would alter mitonuclear communication and gene expression patterns. We show that first order nDNA, mtDNA, and hypoxia effects vary between the sexes, along with mitonuclear epistasis and G × G × E effects. Females were generally more sensitive to genetic and environmental perturbation. While dozens to hundreds of genes are altered by hypoxia in individual genotypes, we found very little overlap among mitonuclear genotypes for genes that were significantly differentially expressed as a consequence of hypoxia; excluding the gene hairy. Oxidative phosphorylation genes were among the most influenced by hypoxia and mtDNA, and exposure to hypoxia increased the signature of mtDNA effects, suggesting retrograde signaling between mtDNA and nDNA. We identified nDNA-encoded genes in the electron transport chain (succinate dehydrogenase) that exhibit female-specific mtDNA effects. Our findings have important implications for personalized medicine, the sex-specific nature of mitonuclear communication, and gene × gene coevolution under variable or changing environments.
Keywords: mtDNA, epistasis, hypoxia, G ×, G ×, E, Drosophila, transcriptome, mitonuclear
Introduction
Understanding how an individual’s genes contribute towards their phenotypes is a fundamental tenet of quantitative genetics and evolutionary biology. Among the complications of complex phenotypes (and multifactorial diseases) are genes interacting with each other (gene × gene interactions: G × G) (Mackay and Moore 2014), and with their environment (gene × environment: G × E) (Hunter 2005), making first order genetic effects partly ephemeral, context-specific, phenomena. In a medical context, the success of personalized medicine and pharmacogenomics industries will rely on an acute understanding of how genes interact with their genetic and physical environments. It has been postulated that a significant proportion of “missing heritability” in complex diseases can be attributed to genetic interactions (Eichler et al. 2010; Zuk et al. 2012). Adding to this complexity is the reality that phenotypic variation also incorporates higher order simultaneous interactions between multiple genes and their environment (G × G × E) (Weinreich et al. 2013; Zhu et al. 2014; Mossman, Biancani et al. 2016).
Given the inherent complexities of fitness-related phenotypes, the underlying architecture of genetic and environmental interactions can be difficult to define clearly due to context-specificity of phenotypic expression. To help identify how gene interactions are sensitive to environment, we have developed a Drosophila melanogaster mitonuclear introgression model that specifically manipulates haplotype-associated mutations in the mitochondrial genome (mtDNA) on alternative nuclear genetic backgrounds (nDNA) (Montooth et al. 2010; Meiklejohn et al. 2013; Zhu et al. 2014; Mossman, Biancani et al. 2016; Mossman, Tross et al. 2016). The main motivation of this approach is to test how disrupted genome–genome communication can influence mitochondrial function and organismal fitness. Mitochondria are an attractive focus of study because they mediate numerous functions in the cell including ATP production, homeostasis, redox signaling, the innate immune response, and apoptosis (West et al.2011; Friedman and Nunnari 2014). Crucially, mitochondrial proteins are jointly encoded by mtDNA (13 protein coding genes) and nDNA (>1,000 protein coding genes) and protein products from both genomes are required to be produced and assembled in a regulated way for efficient mitochondrial function (Rand 2001; Smeitink et al. 2001).
Mutations in both mtDNA (Wallace 1999; Taylor and Turnbull 2005) and nuclear genes (Koopman et al. 2012) can influence mitochondrial health and maladies associated with mitochondria are termed “mitochondrial diseases”. Mitochondria are critical hubs for cell physiological processes and have been implicated in a wide spectrum of disorders in humans (DiMauro and Schon 2003; Lin and Beal 2006). We have previously used combinations of distinct mtDNA and nDNA strains of fruit flies to simulate the phenotypic variation generated by the pairing of novel mitonuclear genotypes (Montooth et al. 2010; Meiklejohn et al. 2013; Villa-Cuesta et al. 2014; Holmbeck et al. 2015). These studies have uncovered significant main effects of mtDNA and nDNA genetic variation as well as mitonuclear epistasis (G × G) (Mossman, Tross et al. 2016), some of which can be modified by environment (Hoekstra et al. 2013; Zhu et al. 2014; Mossman, Biancani et al. 2016). In this study, we extend the model to incorporate a physiologically relevant environmental interaction effect: hypoxia.
Mitochondria are sensitive to environmental stressors (e.g., oxygen availability) (Guzy et al. 2005; Guzy and Schumacker 2006) and are the primary sites of oxygen consumption and free radical production in the cell (Bunn and Poyton 1996). Hypoxia—a deficiency in oxygen—is associated with a number of disease phenotypes including cancer (Harris 2002; Denko 2008; Wallace 2012; Vyas et al. 2016) [see also “the Warburg effect” (Vander Heiden et al. 2009)], renal injury (Eckardt et al. 2005), ischemic stroke (Sims and Muyderman 2010), altitude sickness (Grocott et al. 2009; Murray 2016), along with long term complications of preterm birth in children (Askie et al. 2003). Oxygen is therefore a physiologically important environmental cue that we hypothesized would interact with mitonuclear genotypes.
Drosophila are highly adapted to hypoxic conditions and tolerate low O2 availability (Haddad 2006). Hypoxia exposure during any stage of development can have wide ranging effects on behavior (Farzin et al. 2014; Callier et al. 2015) and gross morphology of Drosophila including overall size (Harrison and Haddad 2011; Heinrich et al. 2011), and flies exposed to hypoxia have generally smaller wing cells and lower wing cell numbers (Peck and Maddrell 2005). At the gene expression level, a number of studies have investigated transcripts that are differentially expressed (DE) by hypoxia and have revealed functionally diverse gene sets among these genes (Liu et al. 2006; Romero et al. 2007). Among these studies, it has been shown that different degrees and durations of hypoxia (0.5% O2 and 5% O2, 1 h or 6 h) change the expression of different gene sets and with varying magnitude (Liu et al. 2006).
Here, we tested the hypothesis that hypoxia interacts with mitonuclear genotypes. We used a factorial mtDNA × nDNA × O2 design to test for first order effects of each factor, along with their genetic (G × G) and environmental (G × G, G × G × E) interactions. We quantified the effects of G × G × E using transcription profiles obtained by RNA-seq.
Results
nDNA, mtDNA, and Hypoxia Influence Adult Survival and Transcript Profiles in D.melanogaster
Using mitonuclear genotypes that have demonstrated significant variation in fitness (Montooth et al. 2010) and development time (Meiklejohn et al. 2013), we tested for variation in tolerance of hypoxia and found significant mitonuclear effects on adult survival (supplementary fig. S1, Supplementary Material online). This motivated the current study for transcriptional variation underlying the whole organism response of exposure to low oxygen. We first tested whether first order effects of nuclear DNA variation (nDNA), mtDNA variation (mtDNA) and hypoxia (hypoxia) were associated with changes in transcript expression. The nuclear genomes used in this study (Oregon R and Austria W132), hereon termed OreR and AutW132, encompass wide variation in nucleotide divergence across genes in these genomes, and this variation was reflected in a large number of genes that were differentially expressed (DE). We estimated that the nucleotide divergence between AutW132 and OreR nuclear backgrounds was at least 48,449 SNPs with a quality score ≥20 and CLR > 90 across the sequenced transcript. This is an underestimate of the total transcript coverage and does not include non-coding sequence variants, which were not sequenced. In total we assayed the expression of 14,095 transcripts in all RNAseq libraries and of these, 6,803 transcripts were DE by nDNA in females, and 5,918 were DE by nDNA in males at FDR < 0.1 (fig. 1A and B).
Fig. 1.
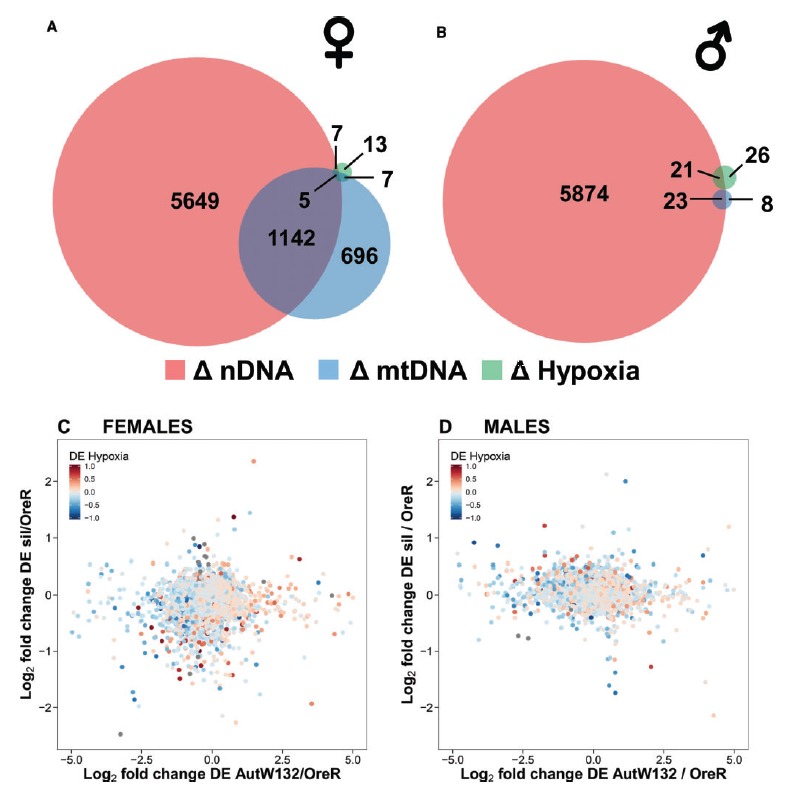
First order genetic and hypoxia effects in females and males. Scaled Venn diagrams describe the number of differentially expressed genes by nDNA (red), mtDNA (blue) and hypoxia (green) in females (A) and males (B) at FDR < 0.1. There was some overlap between the DE gene sets and some genes were sensitive to all three genetic and hypoxia perturbations. Females were generally more sensitive to nDNA and mtDNA variation. Biplots with a heat component (females: C and males: D) show the log2-fold change in expression for nDNA (AutW132/OreR) (abscissa), mtDNA (siI/OreR) (ordinal) and hypoxia [combined 30 min and 120 min of hypoxia/0 min (control)] (heat). The heat component is the hypoxia effect and the scale is from −1 to 1. Data in grey are outside of the −1 to 1 log2-fold range but still contribute to the mtDNA–nDNA correlation reported in the main text.
The effects of mtDNA substitution on gene expression were smaller in magnitude and differed between the sexes. It has been estimated there are at least 94 amino acid substitutions between OreR and siI haplotypes and approximately 418 synonymous SNPs between the D. melanogaster (OreR and Zim53) and siI haplotypes (Ballard 2000). There are therefore substantial SNP and amino acid differences between the mtDNA haplotypes in this study and we hypothesized these mutations would influence transcript expression. In females, a relatively large proportion of genes from both nuclear and mitochondrial genomes were DE by mtDNA (FDR < 0.1; n = 1,850), however the same analysis in males revealed a smaller impact of mtDNA substitution (FDR < 0.1; n = 31).
For the hypoxia analysis, we grouped the two hypoxia treatments (decreased atmospheric O2 from 21% to 6% for 30 min or 120 min; see “Materials and Methods” section) and treated these combined treatments as a main effect of hypoxia. In a later analysis, we consider this temporal variation in fast and slow responding genes. First order hypoxia was associated with DE in n = 32 transcripts in females (FDR < 0.1) and a similar number of transcripts in males (FDR < 0.1, n = 47) (fig. 1A and B). A list of hypoxia sensitive genes is given in supplementary table S1, Supplementary Material online.
Of the genes DE by nDNA variation, n = 3,434 were shared between females and males (FDR < 0.1). For mtDNA variation, n = 18 (FDR < 0.1) genes were DE in both females and males. For hypoxia induced genes, n = 7 (FDR < 0.1) were shared between females and males, indicating that some genes behaved consistently across sexes. A large proportion of the genes behaved in a sex-specific manner, suggesting a context (sex) specific expression as a result of nDNA, mtDNA and hypoxia variation. Moreover, there was a degree of overlap between the genes that were DE by the three forms of experimental manipulation (fig. 1A and B). In other words, genes that were DE by nuclear genetic variation were often the same genes that were DE by mtDNA and hypoxia. This effect was more evident in females who demonstrated a generally larger response to genetic and environmental variables [total number of genes in Venn diagrams; females (fig. 1A) = 7,519; males (fig. 1B) =5,952].
We next asked whether genes that were DE on one experimental axis were also DE on another without the requirement of a significance threshold. We found a general pattern that genes showed larger log2 fold changes as a consequence of nDNA variation than mtDNA variation (fig. 1C and D), consistent with multidimensional scaling (MDS) plots (supplementary figs. S2 and S3, Supplementary Material online). There were significant correlations between log2 fold change on the nDNA and mtDNA axes (females: r = 0.149, t = 15.35, df = 10316, P < 2.2e−16; males: r = −0.115, t = −11.83, df = 10316, P < 2.2e−16). It is important to note that the sign of the correlation is arbitrary and a consequence of the directions of the genetic contrasts. However, between the sexes, the direction of correlation did differ in spite of the genetic contrasts being the same (fig. 1C and D). Genes that were down-regulated by nDNA or mtDNA variation often demonstrated up-regulation by hypoxic environment and vice versa, suggesting transcript expression is sensitive to interactions between nDNA, mtDNA and hypoxia environments.
Mitonuclear Genotypes Have Distinct Expression Profiles
Having established that nDNA, mtDNA and hypoxia variation all contribute to transcript expression as first order effects, we next tested whether there were distinct expression profiles across the 12 treatments: 2 mtDNAs × 2 nDNAs × 3 hypoxia conditions. To conduct this analysis, we clustered genes by their expression profiles (using MBCluster.Seq; Si et al. 2014) and asked whether there were genes that showed correlated gene expression and whether these were distinguishable by genotype. By clustering genes into 70 clusters for each sex, we found gene clusters that responded to each form of experimental treatment (see supplementary figs. S4–S7). In general, the clustering approach revealed that transcripts in females adopt a genotype-specific signature of expression, whereas the majority of genes in males can be delineated only on the nuclear axis (fig. 2). Multidimensional scaling plots (supplementary figs. S2 and S3) confirmed distinct genotype-specific clusters in females yet no major mtDNA or hypoxia effect in males, consistent with the first order effect quantifications (see above). Figure 2 describes the transcript co-expression clusters (Si et al. 2014) and shows that for females, transcript expression patterns can be clearly separated by nDNA and mtDNA treatments (fig. 2A). In contrast, males did not show any appreciable signal of transcript expression in either mtDNA or hypoxia treatments (fig. 2B). Low signal of transcript expression due to hypoxia treatments in both sexes suggests there are either (i) weak hypoxia effects across all genotypes, or (ii) that higher order interactions exist between genes and environment, and these are dependent on sex. This proposes the question: how do individual genotypes respond differently to environmental cues?
Fig. 2.
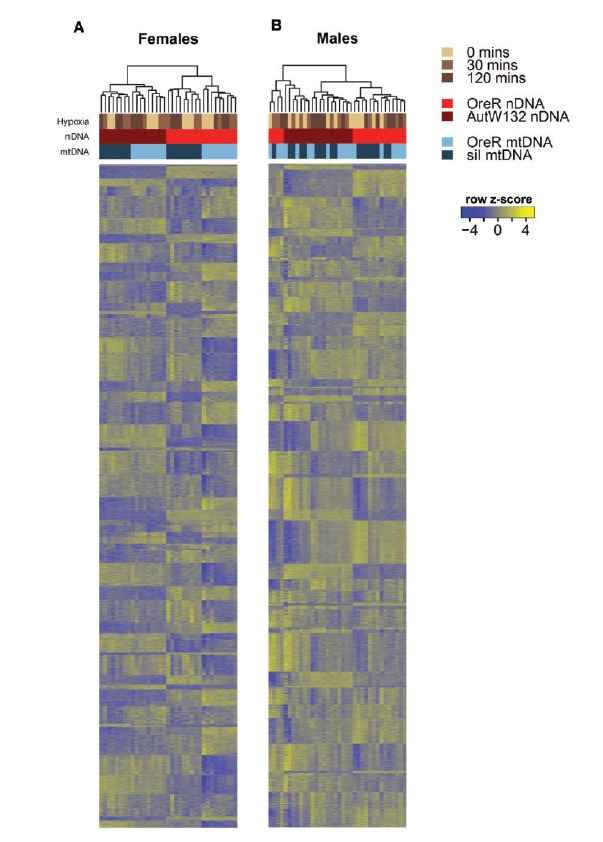
Gene and library clusters of the top 70% of genes by read count. Heat maps show the clustering of libraries in females (A) and males (B) using k = 70 clusters; calculated using MBCluster.Seq. The three forms of experimental variation are shown as colored bars at the top of each heat map. Browns show hypoxia treatment, reds are alternative nuclear backgrounds and blues are alternative mtDNA haplotypes. Column (library) dendrograms describe the clustering of libraries using the hierarchical clustering approach in the heatmap3 function (Zhao et al. 2014). Each row in the heatmap is a gene and these are ordered based on their cluster membership (1–70). The rows do not correspond with the same gene across sexes. The relative expression (row z-score) of each gene is scaled on a per gene basis. Relatively high expression is shown in yellow; relatively low expression is shown as blue. In females, mitonuclear genotypes are clearly differentiated by expression, whereas males show less defined genetic signal in the expression data.
Genotype-Specific Hypoxia Response
Our clustering approach suggested females demonstrate greater sensitivity to all three forms of experimental treatment (nDNA, mtDNA, and hypoxia). We conducted DE analyses across all genotypes and characterized the main effects of nDNA (fig. 3A and I) and mtDNA treatment (fig. 3B and J), but estimated transcript response between the control hypoxia treatment (0 min at 6% O2) and both 30 min of hypoxia (fig. 3C and K), and 120 min of hypoxia (fig. 3D and L). With all genotypes grouped together, the responses to hypoxia in females were roughly equal at both timepoint comparisons (fig. 3C and D), suggesting the hypoxic response is more immediate and sustained. In contrast, males showed a slower response to hypoxia, and a larger number of genes responded in the 0–120 min contrast (fig. 3L) than in the 0–30 min comparison (fig. 3K).
Fig. 3.
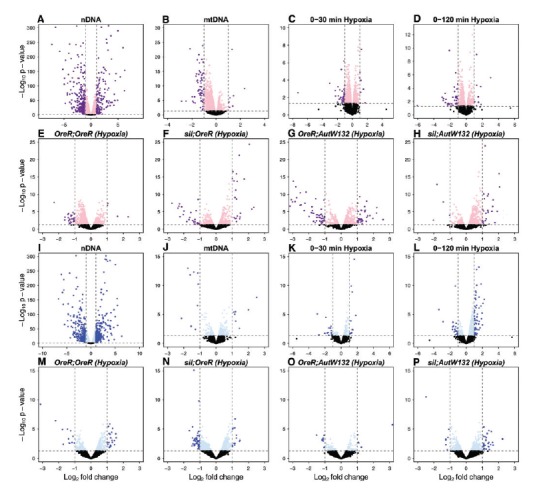
First order DE and genotype signatures of hypoxia. Volcano plots show female (A–H) and male (I–P) differential expression responses to nDNA (A, I), mtDNA (B, J), initial hypoxia (C, K), and longer hypoxic exposure (0–120 min) (D, L). Individual (mtDNA;nDNA) genotype responses to combined hypoxic treatments are shown for OreR;OreR (E, M), siI;OreR (F, N), OreR;AutW132 (G, O) and siI;AutW132 (H, P). Log2-fold changes are on the abscissa and −log10P-values are shown in the ordinal. Horizontal dashed grey lines highlight P-value =0.05. Vertical dashed grey lines highlight ± 1 on a log2 fold scale (2 fold up- or down-regulated). Dark purple and dark blue data highlight genes with ± 1 log2 fold change and P < 0.05, in females and males, respectively.
We then focused on individual mitonuclear genotypes to test the hypothesis that genotypes respond in distinct ways to hypoxia, using edgeR (Robinson et al. 2010). The four mitonuclear genotypes (noted as mtDNA;nDNA: OreR;OreR, siI;OreR, OreR;AutW132, siI;AutW132) showed diverse patterns of gene expression in response to overall “hypoxia” (normoxia vs. combined hypoxic treatments). In general, the response in OreR;OreR in females (fig. 3E) and males (fig. 3M) were small compared with siI;OreR (females: fig. 3F, males: fig. 3N). In particular, siI;OreR males demonstrated the most sensitive response to initial hypoxia (fig. 3N) with a large number of transcripts down-regulating expression. Females with a AutW132 nDNA were more sensitive than females with a OreR nDNA to initial hypoxia whereas the male response was similar across nuclear backgrounds but exaggerated in the siI mtDNA background. The differences in sensitivity to hypoxia dependent on nDNA and mtDNA backgrounds suggest a wide range of mitonuclear interactions are mediating the hypoxia response.
Are Hypoxia-Response Genes Consistent across Genotypes?
Given that mitonuclear genotypes respond with different sensitivities to hypoxia we next asked whether there were consistent hypoxia-response genes in each genotype and whether these were known hypoxia genes in the literature. In this analysis, we grouped the hypoxia treatments and the contrast was between 0 min (control) and combined 30 min and 120 min exposures. Figure 4(A–H) shows interaction plots that describe the norms of reaction across time points for each individual genotype in both sexes (females: A–D; males: E–H). The red lines show the reaction norms of genes that were consistently up-regulated across all genotypes in females. Lines in black represent all up-regulated genes across all intersections including private, genotype-specific response genes, e.g., all genes included in figure 4I for females, and all genes in figure 4J for males. The intersections between these genes are shown in Venn diagrams in figure 4I and J. Eight genes were consistently upregulated with FDR < 0.1 in females (fig. 4I) and zero genes (FDR < 0.1) were consistently upregulated in males (fig. 4J). The consistently upregulated genes with FDR < 0.1 are described in table 1. Of those differentially expressed genes between genotypes (both up- and down-regulated), a number were also associated with first order mtDNA variation at FDR < 0.1. In females, 5/8 (62.5%) were both hypoxia-responsive and mtDNA responsive. Zero genes were both hypoxia responsive and mtDNA responsive in males. This analysis reveals a novel interaction between sex, mtDNA and hypoxia whereby females show a simultaneous response to both mtDNA and hypoxia variation for a subset of genes.
Fig. 4.
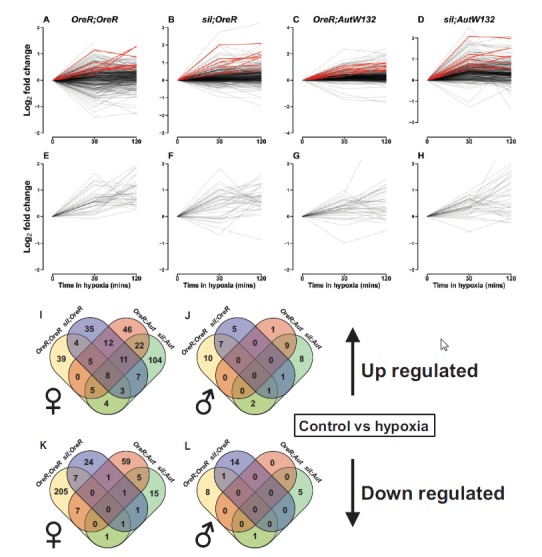
Genotypes show both conserved (intersected) and private gene expression responses to combined hypoxia treatments. (A–H) show the norms of reaction for each genotype for all upregulated genes across all genotypes, measured as the log2 fold change relative to t = 0 min in hypoxia. Females (A–D) and male (E–H) norms of reaction are shown. Red lines in A–D show the eight consistently upregulated genes across all four genotypes in females (FDR < 0.1). Black lines show the remaining genes that are differentially upregulated in at least one genotype. Genes that are upregulated in at least one genotype are often downregulated in another. Venn intersections in (I–L) show the numbers of genes that are intersected and private to individual genotypes. Female up- (I) and down-regulated (K) genes are those genes DE by combined hypoxic treatments relative to the control (0 min in hypoxia). Equivalent male plots are shown in (J) and (L). A large majority of genes were private to individual genotypes in both up- and down-regulated genes (larger numbers on margins of Venn diagrams).
Table 1.
Consistently Upregulated Hypoxia Genes across All Mitonuclear Genotypes in Females
| Females | |
|---|---|
| Gene ID | Flybase ID |
| Pepck | CG10924 | FBgn0003067 |
| CG12105 | FBgn0035241 |
| CG14757 | FBgn0033274 |
| CG31324 | FBgn0051324 |
| CG3739 | FBgn0038702 |
| CG5724 | FBgn0038082 |
| flightin | FBgn0005633 |
| black | FBgn0000153 |
Note.—There were eight consistently upregulated genes in females and zero genes in males. The intersections are described in figures 4I for females. Gene symbols and Flybase IDs are shown. Gene IDs separated by a (|) overlap.
In both sexes, the majority of genes were not consistent in their response to hypoxia. In other words, genes that were upregulated in one genotype were often down regulated in another genotype (black genes in fig. 4A–H). Interestingly, there were zero genes consistently down regulated across all genotypes and very few included in multiple genotype intersections. Instead, those downregulated genes were almost always private to a single genotype (fig. 4K and L) with the exception of genes shared within a nuclear background. Of the genes that responded to hypoxia, there were mixed norms of reaction wherein some genes initially increased then decreased their expression. In contrast, other genes initially increased then maintained high levels of expression.
In a separate analysis, we combined all significant hypoxia-associated genes (all genes in fig. 4I and K—females; all genes in fig. 4J and L—males) for each sex and intersected these with a list of 36 known hypoxic genes (downloaded from Flybase with the category “response to hypoxia”), including genes that are known to interact with sima (HIF1α). We identified four genes in the female dataset [phurba tashi (phu), Major Facilitator Superfamily Transporter 14 (MFS14), pasang lhamu (plh), hairy (h)] and two genes in the male dataset with known hypoxic response (MFS14, h). Of these, there were two core genes intersected between females and males. These were: Major Facilitator Superfamily Transporter 14 and hairy. With more relaxed criteria, we intersected all genes that were significant in at least one genotype from each sex and found 31/651 genes that overlapped between the sexes. In the relaxed intersection, we identified zero additional genes that were differentially expressed in at least one genotype in both sexes. This analysis allowed genes to either increase or decrease in expression and not all genotypes were fully intersected for a given gene. The gene lists are therefore different from those reported for fully intersected upregulated genes in table 1.
Genotype-Specific, but Not Always Significant Overall
So far, we have shown that overall transcript responses to hypoxia are largely genotype-specific. To test whether canonical hypoxia-response genes were identified in statistical models across all genotypes combined we conducted DE analyses with the complete dataset. Of the known hypoxia-response genes, we found one gene in females that was hypoxia-responsive as a first order term (fig. 5A), and two genes in males (fig. 5B). These were not always the same genes that were identified when genotypes were tested individually (red gene IDs in fig. 5, and see above). This result underpins the complexity of identifying gene targets for gene therapy, since genes do not have the same response in all genotypes and only a small number of genes respond consistently across all genotypes. Using models with interaction terms fitted, we found a number of the hypoxia-response genes also demonstrated significant higher order interaction effects of G × G, although these did not remain after strict P-value adjustment (FDR < 0.1; fig. 5). Taken together, these results demonstrate context-specific response to hypoxia and that not all canonical hypoxia genes respond in the same way to hypoxia treatment.
Fig. 5.
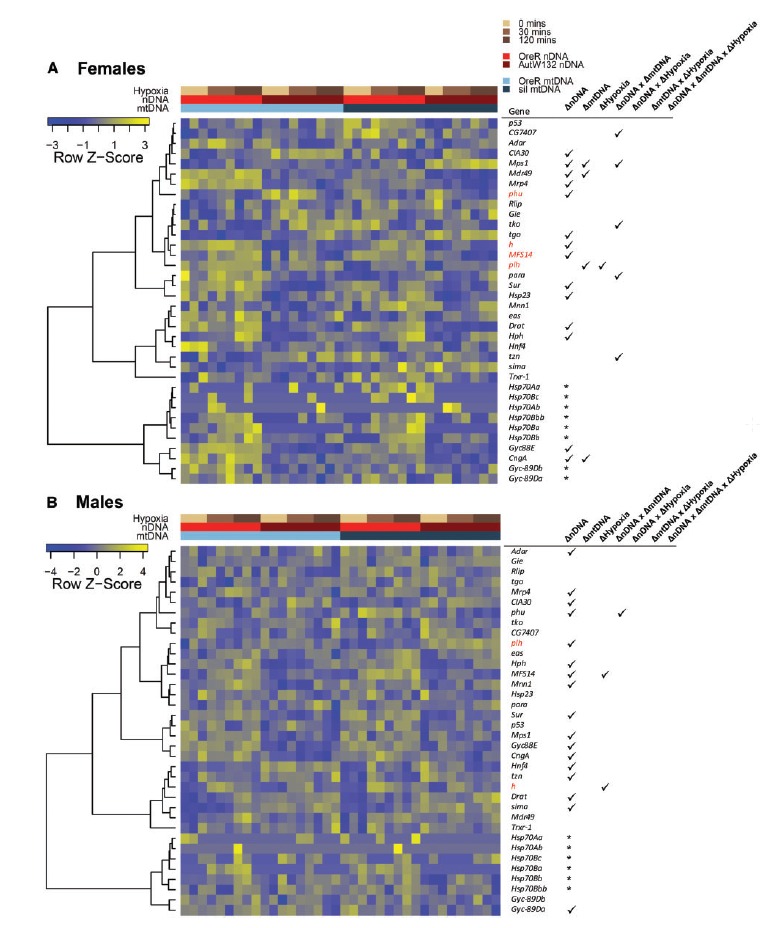
Known hypoxia genes show first order genetic and environment effects, along with significant interaction effects on expression. Heatmaps show relative gene expression patterns of known hypoxia-response genes in females (A) and males (B). Heatmaps are clustered by gene expression patterns using the hierarchical clustering as implemented in the heatmap3 function (Zhao et al. 2014). Dendrograms of genes describe the gene clusters. Libraries (columns) are sorted by mitonuclear genotype and time in hypoxia. Colored bars above the heatmaps describe the experimental treatment of individual RNA-seq libraries. Tables to the right of heatmaps show the gene ID and ticks highlight if that gene was significantly DE (FDR < 0.1) by a genetic or environmental effect, and any significant interaction effect. Genes in red were significantly differentially expressed (up- or down-regulated) by hypoxia in at least one genotype. The hypoxia term in the table describes whether there was an overall significant hypoxia effect with all genotypes combined in the analysis. An asterisk means that gene was not included in the combined genotype model due to insufficient read counts.
Higher Order Genetic Effects on Transcript Expression
We identified large numbers of genes that were sensitive to interactions between nDNA × mtDNA (FDR < 0.1 females: n = 1,865, males: n = 72; fig. 6A), yet only small numbers for the remaining interaction terms: mtDNA × Hypoxia (FDR < 0.1 females: n = 1, males: n = 0; fig. 6B), nDNA × Hypoxia (FDR < 0.1 females: n = 1, males: n = 0; fig. 6C), and nDNA × mtDNA × hypoxia (FDR < 0.1 females: n = 2, males: n = 0; fig. 6D). Female and male datasets were analyzed separately and all libraries were included in the analyses. Generally, the numbers of transcripts that were DE by the interaction effects were larger in females than males and there was little overlap of genes between the sexes (fig. 6). Higher order interactions are by definition context-dependent and our results further suggest that higher order genetic and environmental effects are sex specific. Of the genes that were DE by interaction terms, we conducted GO enrichment analyses. The results of significant GO process, GO function and GO component terms for all higher order interactions are shown in supplementary tables S2–S8, Supplementary Material online. The G × G gene enrichments were most significantly associated with “‘peptide biosynthetic process” in females, and “humoral immune response” in males.
Fig. 6.
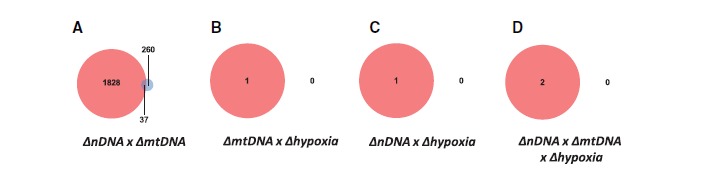
Higher order genetic and hypoxia interactions across sexes. Venn diagrams describe the numbers of genes that were differentially expressed by a higher order interaction (FDR < 0.1). Red circles are female DE genes; blue are male. The numbers of genes that intersected between the sexes are also shown. nDNA × mtDNA significant interactions are shown in (A). mtDNA × hypoxia effects are shown in (B). There were no overlapping genes between the sexes for any of the remaining higher order interactions (B, C, D) with a strict FDR < 0.1 cut-off. nDNA × hypoxia effects (C) and nDNA × mtDNA × hypoxia (D) are shown.
Hypoxia Sensitive Genetic Effects
Clearly, transcript expression is a dynamic process that is sensitive to genetic and environmental interactions. To test how DE genes changed over the three experimental timepoints, we treated each timepoint as a separate experiment event and tested for statistically significant first order genetic effects of nDNA and mtDNA, along with their nDNA × mtDNA interaction. In females, the numbers of significant (FDR < 0.1) nuclear genes showed a monotonic increase with increasing time in hypoxia (fig. 7A). The opposite effect was observed in males, whose numbers declined monotonically with time in hypoxia (fig. 7G). For genes DE by mtDNA, there was an initial increase in numbers, followed by a subsequent drop. Qualitatively similar effects were observed in females (fig. 7B) and males (fig. 7H). In both females and males, the number of significant nDNA × mtDNA genes decreased with time in hypoxia. This effect was monotonic in females (fig. 7C) and males (fig. 7I).
Fig. 7.
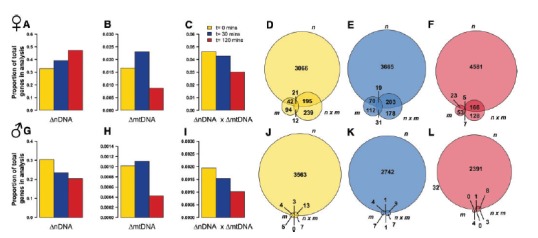
Genetic effects and interactions are modified by hypoxia. Barplots in (A, B, C) show the proportion of genes in females that were differentially expressed by nDNA, mtDNA, and nDNA x mtDNA, respectively, at different hypoxia treatments: 0 min (yellow), 30 min (blue), and 120 min (red) (FDR < 0.1). Equivalent barplots for males are shown in (G, H, and I). Under 30 min of hypoxia, the signature of mtDNA variation increased in proportion in both sexes. The proportion of nDNA × mtDNA decreased with increasing time in hypoxia in both sexes. In females, the number of DE nDNA genes increased with time in hypoxia, whereas the proportion in males decreased. Venn diagrams in (D, E, and F) show the intersections between first order nDNA, mtDNA, and higher order nDNA × mtDNA interactions for females at hypoxia treatments: 0 min (yellow), 30 min (blue), and 120 min (red). The equivalent Venn diagrams for males are shown in (J, K, and L).
Electron Transport Genes
Mitonuclear introgression provides an experimental method to disrupt mtDNA–nDNA crosstalk and influence gene expression in gene pathways that rely on the interaction between nDNA and mtDNA protein partners. There is an intimate relationship between nuclear-encoded and mtDNA-encoded protein subunits in four out of five oxidative phosphorylation (OXPHOS) complexes. Complex II of the ETC, however, does not contain any proteins encoded by mtDNA genes and instead this complex is solely encoded by nDNA. Complex II genes are therefore a special case of genes to examine a functional connection to mtDNA variation since they are in the same electron transport chain, yet do not directly contain genetic variants associated with mtDNA haplotypes. We tested the hypothesis that respiratory chain genes encoded by mtDNA demonstrate differential expression under hypoxia. We further tested the prediction that succinate dehydrogenase (complex II) genes would show a signature of mtDNA variation, since complex II is closely coupled with other proteins encoded by both nDNA and mtDNA genomes.
Figure 8 describes the responses of mitochondrial respiratory chain genes to hypoxia in individual genotypes in females (fig. 8A–D) and males (fig. 8E–H). There were qualitative and quantitative differences between the mitonuclear genotypes and between the sexes. The norms of reaction in red (female) and blue (male) represent the log2 fold change in mtDNA-encoded gene expression over time in hypoxia, relative to the control (time = 0 min in hypoxia) baseline. Respiratory chain genes were among the most responsive to hypoxia and there was a general enrichment for electron transport chain and oxidative-reduction processes in gene ontology (GO) enrichment analyses (supplementary table S4, Supplementary Material online). Interestingly, there was no uniform response to hypoxia and different mitonuclear genotypes showed unique signatures of hypoxia response in their respiratory chain genes. For example, OreR;OreR females show a strong mtDNA-encoded gene response and these genes were mainly up-regulated during hypoxia. In contrast, OreR;AutW132 males show the opposite effect for mtDNA-encoded genes and all mtDNA-encoded genes were down-regulated during hypoxia. In both these genotypes, the nuclear-encoded components of the respiratory chain (black lines) showed a dampened response to hypoxia.
Fig. 8.
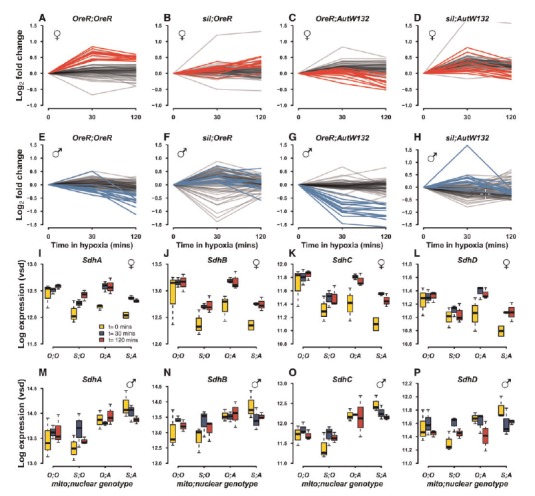
Oxidative phosphorylation genes encoded by mtDNA and nDNA are among the most differentially expressed by hypoxia. Norms of reaction for log2 fold change relative to hypoxia timepoint = 0 are shown for individual genotypes in females (A–D) and males (E–H). Red lines in (A–D) and blue lines in (E–H) show the mtDNA-encoded genes in females and males, respectively. Black lines show the remaining nDNA-encoded genes that comprise the Complexes I–IV and ATP synthetase. Boxplots in (I–L: females) and (M–P: males) show the expression of succinate dehydrogenase genes (SdhA, SdhB, SdhC, and SdhD) of complex II of the electron transport chain.
Sex-Specific Succinate Dehydrogenase Expression
We next tested whether mtDNA influenced the expression of succinate dehydrogenase (complex II) genes: SdhA, SdhB, SdhC, SdhD, in both sexes. Three of the four genes (SdhA, SdhC, and SdhD) were differentially expressed by mtDNA in females across all timepoints, and there was a significant up-regulated response to hypoxia in the female genotypes with the AutW132 nuclear genome (FDR < 0.1) (fig. 8I–L). The effect of hypoxia was not significant in the OregonR nuclear background in females. In contrast, males showed neither a significant mtDNA effect nor a significant hypoxia effect in any of the four succinate dehydrogenase genes (FDR > 0.1) (fig. 8 M–P). This result reveals there are sex-specific mtDNA effects in complex II, and that hypoxia does not directly influence complex II gene expression in males, at least for these mitonuclear genotypes. Nuclear background does, however, affect sensitivity to hypoxia in females. Interestingly, all succinate dehydrogenase genes were significantly DE by nDNA as first order effects in males, but none were significantly DE by nDNA in females.
RT-qPCR Validation
We measured the expression of ten nuclear markers by qPCR, to validate that RNA-seq was capturing repeatable fold changes as a consequence of hypoxia. The correlation between RNA-seq log2 fold changes and qPCR log2 fold changes was as follows: r = 0.76, t = 7.18, df = 38, P = 1.387e−08: (see supplementary fig. S8, Supplementary Material online). Full details and methods are described in the Supplementary Material online.
Discussion
We found that the gene expression profiles of adult Drosophila were altered by gene interactions between nDNA and mtDNA and were modified by hypoxia, and that 30 min in a hypoxic environment increased the number of genes associated with mtDNA variation in both sexes. Overall, we demonstrate there is extensive G × G × E in this panel of D. melanogaster genotypes, and that G × G × E also affect the sexes in different ways (there was only a small overlap between significant G × G × E genes). Importantly, we show that, with the exception of two individual genes (e.g., hairy, Major Facilitator Superfamily Transporter 14), the shared hypoxia response across genotypes is limited and the majority of genes that responded to hypoxia were private to individual genotypes. We discuss these results in the context of first order effects, mitonuclear interactions (epistasis) and the implications for understanding the architecture of complex phenotypes.
First-Order nDNA Effects
Nuclear genetic variation in our study was associated with a large number of DE genes, which was predicted since there are many SNP variants between OreR and AutW132 nDNAs. In a large mapping DGRP reference panel (Mackay et al. 2012), there is remarkable sequence polymorphism between strains and an estimated single genetic marker every 33 bp on average (Massouras et al. 2012). The nuclear backgrounds in the present study are differentiated by an estimated >48,449 SNPs in the transcriptome alone, and this estimate does not include genome rearrangements. In both sexes, we found a large number of genes were DE by nDNA when averaging across all other factors (mtDNA, and hypoxia). Our estimates of nDNA DE genes across the sexes (females n = 6,803; males n = 5,918) are similar to those of Gibson et al. (2004), who identified that at least 5,820 genes were differentially expressed between genotypes in D. melanogaster. Gibson et al. found 5,040 genes that were DE in females, and 4,098 in males. The ratio of female:male DE genes was approximately 1.23, and we found the same qualitative pattern and a similar quantitative ratio of 1.15. It should be noted that some of the nDNA encoded differences in gene expression are likely due to both cis- and trans-mediated factors, but our experimental design using homozygous nuclear backgrounds does not allow distinction between these effects.
The numbers of DE genes were similar in females and males for nDNA variation, however, the numbers of mtDNA-associated DE genes were generally higher in females suggesting a greater sensitivity to disrupted mtDNA-nDNA crosstalk. Variation in nDNA showed consistent enrichment for the GO function category—serine endopeptidase activity—in both sexes. Serine catabolism has been linked with tumor growth and mitochondrial redox balance during severe hypoxia (Ye et al. 2014), thus integrating both mitochondrial function and oxygen availability; two main objectives of the present study. The current mitonuclear-hypoxia experiment was not conducted in severe hypoxia (0.5% O2), but instead, at physiological hypoxia (6% O2). In spite of this difference, we were able to identify known pathways associated with hypoxia in human cells, demonstrating the value of the Drosophila model organism as a research tool for human disease genetics.
First-Order mtDNA Effects
We found mtDNA had a larger effect in females than males averaging across all other variables: (nDNA and hypoxia). This effect was not the consequence of averaging across hypoxia timepoints since results at individual timepoints revealed the same pattern; females always showed greater numbers of mtDNA DE genes. These results are dramatically different from a previous study of gene expression as a consequence of mtDNA variation. Using a common nDNA background and five genetically distinct mtDNAs, Innocenti et al. (2011) found 1,172 genes that were DE by mtDNA variation in males and only seven genes that were DE by mtDNA variation in females. Our estimates differ both qualitatively and quantitatively from those of Innocenti et al. We found a larger proportion of genes were differentially expressed by mtDNA in females (n = 1,850; FDR < 0.1) than in males (n = 31; FDR < 0.1). These differences could be in part due to analyses being performed on different gene expression platforms [Illumina RNAseq (current study) vs. Affymetrix Drosophila 2.0 microarray (Innocenti et al. 2011)]. However, we favor the hypothesis that the different mtDNA haplotypes, with different degrees of polymorphism, are likely to explain the majority of the differences between studies. The same mtDNA haplotype was not present across studies and a different number of haplotypes were tested between studies. We are likely to have greater sensitivity to detect DE genes using a single mtDNA haplotype contrast, than in an analysis with five haplotypes, however, this would not explain the opposing qualitative differences we observed between females and males. It remains possible that the effect observed by Innocenti et al. is enhanced in the w1118 nuclear background used in that study, and not evident in other nuclear backgrounds.
A simple mtDNA genotype-phenotype divergence model predicts that greater numbers of nucleotide substitutions would confer greater phenotypic effects. However, previous work in our laboratory has revealed that mtDNA sequence divergence is a poor correlate of phenotypic divergence in mitonuclear introgression lines (Montooth et al. 2010; Mossman, Biancani et al. 2016), but see (Camus et al. 2012). Instead, main effects of mtDNA are largely confined to individual “lines”, with polymorphisms associated with “species” divergence demonstrating no consistent directional effects on phenotypic expression.
The mtDNA haplotype contrast in the present study (siI vs. OreR) is more genetically divergent than a typical intraspecific contrast, and may help explain why mtDNA effects were both quantitatively and qualitatively different from other mtDNA studies on transcription (Innocenti et al. 2011). In the context of personalized medicine and mitochondrial replacement therapies, it is clear that mtDNA and nDNA effects are important for transcription responses to hypoxia, and future work should focus on medically relevant environmental interactions and genotypes and how these may affect therapy outcomes. Our assessment of mtDNA effects may overestimate the number of genes whose transcripts are modified, however we establish here that G × G × E is a phenomenon that has the potential to affect gene expression and should be subject to future study.
We did identify enrichment for electron transport chain GO process in females (FDR Q < 0.01), as we would expect by manipulating genetic variation in the electron transport chain. However, this result was restricted to females. Similar to Innocenti et al., we found “reproduction” GO process was enriched at a nominal P < 0.05 in males, and there were some examples of male specific genes (seminal proteins) that demonstrate mtDNA effects, although these GO categories did not remain using strict FDR < 0.1 transcripts. However, these significant mtDNA effects were only present under hypoxic conditions, and not at normoxia. The increase in mtDNA DE genes under 30 min (initial) hypoxia was a general genome-wide finding and likely reflects the hypoxic state exposing mtDNA variants to exert larger effects on genes that interact with mtDNA-encoded proteins.
First-Order Hypoxia Effects
Environment can also exert a large influence on gene expression, even within closely related populations (Gilad et al. 2008). Using our inbred mitonuclear lines, we found only a small number of genes that were sensitive to hypoxia as first order effects when averaging over all genotypes. However, we found very clear genotype-specific effects of hypoxia and very little overlap between genotypes for genes that are DE by hypoxia. Previous selection experiments in D. melanogaster have shown that approximately half of DE genes under hypoxia were downregulated (Zhou et al. 2007). We found a similar effect in females, and of the significant DE hypoxia genes, n = 21 were upregulated and n = 11 were down regulated (∼34%). In males, however, n = 44 were upregulated and n = 3 were down regulated (∼6.4%).
Sima is the Drosophila homolog of hypoxia-inducible factor 1α (HIF1α) (Wang et al. 1995; Nambu et al. 1996); the transcription factor that is central to the cellular responses to hypoxia, yet does not directly sense oxygen (Jaakkola et al. 2001; Schofield and Ratcliffe 2004). The HIF1-β subunit—Tango (tgo)—is known to be constitutively expressed, independently of O2 levels (Romero et al. 2007). Under normoxia, the alpha subunit of HIF is targeted for degradation by the von Hippel-Lindau ubiquitin ligase complex (Ivan et al. 2001) and this process is mediated by the oxygen sensing HIF prolyl hydrolase (Hph) (Bruick and McKnight 2001; Kaelin and Ratcliffe 2008; Lendahl et al. 2009; Rytkönen et al. 2011). We found the Hph gene was consistently upregulated during hypoxia in both females and males, suggesting components of the canonical and evolutionarily conserved HIF pathway show a repeatable response with hypoxia, although these trends did not survive correction for strict false discovery rates. We consistently identified the transcriptional suppressor, hairy (h), in our hypoxia-sensitive gene list. Hairy has been suggested as a metabolic switch that reduces the expression of tricarboxylic acid (TCA) genes and glycolytic enzymes (Zhou et al. 2008). In flies adapted to low O2 levels, hairy is upregulated relative to naïve flies and flies with mutations in hairy demonstrate significantly lower hypoxia tolerance (Zhou et al. 2008). We also observed consistent upregulation of hairy during hypoxia in flies naïve to hypoxia.
In spite of identifying known hypoxia response genes, the suites of other genes that are DE by hypoxia were largely genotype-specific, signifying that interactions between nDNA and mtDNA can alter the identity of hypoxia response genes. These genotype-specific signatures of hypoxia further suggest there are HIF-dependent and HIF-independent hypoxic responses, which have been previously described in D. melanogaster (Li et al. 2013) and C. elegans (Shen et al. 2005). In humans, alternative O2 sensing pathways involve the mammalian target of rapamycin (mTOR) kinase and signaling through the unfolded protein response (UPR) (Wouters and Koritzinsky 2008). While specific mechanisms of hypoxia induction of transcription were not the focus of this study, we found a number or mTOR interacting genes to be significantly modified by mtDNA (data not shown). Previous studies have substantiated a link between dietary intake and hypoxia (Vigne et al. 2009; Vigne and Frelin 2010), and disrupted mitonuclear communication with advanced aging (Gomes et al. 2013). Future studies should incorporate mtDNA variation as an important factor incorporating nutrient sensing, hypoxia, and ageing.
Under hypoxia, cells generally decrease OXPHOS and increase glycolytic activity for adenosine triphosphate (ATP) production (Lendahl et al. 2009). In cancer, tumor cells paradoxically show increased aerobic glycolysis, even in the presence of functional mitochondria in which OXPHOS would be the more ATP-efficient process (Gatenby and Gillies 2004; Vyas et al. 2016). Initial observations led Otto Warburg to postulate that the “irreversible injuring of respiration” was a common factor in cancer (Warburg 1956). Succinate dehydrogenase is a tumor suppressor found in complex II in mitochondria (Selak et al. 2005) and is suspected as being involved in the pseudohypoxic response that enhances glycolysis (King et al. 2006). Mutations in succinate dehydrogenase turn on the HIF response pathway in tumors (Baysal 2003; Pollard et al. 2003). In one study in D. melanogaster, sdhB mutants were hypersensitive to oxygen and showed structural defects in flight muscle mitochondria which influenced longevity (Walker et al. 2006). We found at least three of the four succinate dehydrogenase genes to upregulate in response to hypoxia and express mtDNA effects in females. In contrast in males, transcripts increased when initially low, and decreased when initially high, but there was no mtDNA effect. Succinate dehydrogenase is a shared protein complex between the TCA cycle, which occurs in the mitochondrial matrix, and OXPHOS, which occurs on the inner mitochondrial membrane. Our observation that females show signatures of mtDNA variation on Sdh gene expression suggests there is some form of retrograde signaling between products of joint mitonuclear encoding and the nucleus (Liu and Butow 2006; Wallace 2012; Chae et al. 2013), and this signaling pathway is sex-specific. A previous analysis in D. melanogaster has suggested retrograde signaling from mitochondria to the nucleus that causes a mtDNA-dependent change in succinate dehydrogenase enzyme activity (Villa-Cuesta et al. 2014). More generally, mitochondria-to-nucleus retrograde signaling has been shown in tumor progression and metastasis (Amuthan et al. 2001) and is hypothesized to involve cytosolic calcium signaling as a main intermediate that transduces the message of metabolic stress (Biswas et al. 1999; Wallace 2012). The retrograde signaling is likely to be coordinated by a number of transcription factors (Chae et al. 2013).
Sex-Specific Effects
MtDNAs in males are not subject to direct selection due to maternal inheritance (Frank and Hurst 1996) and mitochondrial efficiency may be constrained in males and more exposed to selection for efficient function in females. Such sexually antagonistic effects and their impacts on organismal sex differences in phenotypes (e.g., ageing) have been speculated and may explain why females and males often vary phenotypically (Tower 2006; Tower and Arbeitman 2009). However, sex differences in mitochondrial function alone are not prima facie evidence in support of the Frank and Hurst Hypothesis (Frank and Hurst 1996).
These sex differences we observed could be a consequence of overall gender differences in mitochondrial phenotypes, including transcript expression of mtDNA genes (Mossman, Tross et al. 2016). In D. simulans, females contain a higher mtDNA: nDNA copy number ratio in thoraces than males (Ballard et al. 2007), although in humans, differences in mtDNA copy number variation do not correlate with changes in mtDNA gene expression variability (Wang et al. 2014). There is good evidence in rodent models that females and males can have different mitochondrial phenotypes (Justo et al. 2005; Colom et al. 2007; Valle et al. 2007). Sex differences in mouse mitochondrial content and function have also been implicated in the sex differences on susceptibility to amyotrophic lateral sclerosis (ALS), a disease with a distorted male-biased sex ratio among sufferers (Logroscino et al. 2010). Importantly, male mouse brains also demonstrate lower brain ETC activity, lower mitochondrial mass, and lower mitochondrial membrane potential (Renolleau et al. 2008), although different mouse strains can sometimes demonstrate no sex differences in mitochondrial bioenergetics (Sanz et al. 2007). Our sex-limited mtDNA effect in succinate dehydrogenase genes warrants further study and future work should aim to understand how sex differences in mtDNA transcript response are intimately related to hypoxia. Our findings pose three main questions: (i) why do genes consistently sensitive to hypoxia also show significant mtDNA effects, (ii) why are these effects dramatically sex-specific, and (iii) could these possibly linked phenomena be explained by mitochondrial physiology alone, which is known to differ between the sexes?
We found inconsistent responses of mtDNA genes to hypoxia across the two hypoxia timepoints and evidence of non-overlapping (sex-limited) mtDNA x hypoxia interactions. Surprisingly, mtDNA transcripts generally increased in some mitonuclear backgrounds (e.g., siI;AutW132), yet decreased in others (e.g., OreR;AutW132). Only one pairing of D. melanogaster mtDNAs with D. melanogaster nDNA showed the predicted down-regulation of OXPHOS genes response (after a switch to a hypoxic environment) across both sexes (OreR;AutW132). These results are consistent with a mitonuclear coadaptation hypothesis suggesting co-evolved mtDNA and nDNA molecules should demonstrate more robust phenotypes when compared with more distantly related mtDNA and nDNA pairings (see above). In spite of its theoretical appeal, the evidence for mtDNA “species” effects on phenotypes is sparse (Montooth et al. 2010; Mossman, Biancani et al. 2016), and we have found the majority of mitonuclear incompatibilities are unique to capricious mtDNA–nDNA pairings. These significant epistatic interactions are clearly sensitive to hypoxia too and we posit this is likely a reflection of the intimate relationship between oxygen, oxygen sensing and mitochondria.
Epistasis and Complex Mitonuclear Interactions with Hypoxia
To our knowledge, this is the first study to simultaneously assess nDNA, mtDNA, and hypoxia variation in Drosophila and integrate these axes of genetic and environmental variation. Previous studies in the copepod Tigriopus californicus have also incorporated mtDNA, nDNA variation (hybrids), and environmental stress to disentangle the effects of mtDNA, nDNA, and temperature (Willett and Burton 2003), or salinity effects on gene expression in oxidative phosphorylation genes (Ellison and Burton 2008), evidencing that incompatibilities between nDNA and mtDNA could contribute to hybrid breakdown (Ellison and Burton 2006). Studies of hypoxia in Drosophila are numerous [reviewed in Harrison and Haddad (2011) and Zhao and Haddad (2011)] and Drosophila has emerged as a useful organism to test systems biology phenomena relating to oxidative stress (Romero et al. 2007). Selection followed by resequencing experiments in Drosophila have also revealed genes that have likely adapted to hypoxia and that also show signals of positive selection in four high altitude human populations (Sherpas, Tibetans, Ethiopians, and Andeans) (Jha et al. 2015). We explicitly tested the effects of mtDNA mutations in the electron transport chain via direct introgression onto alternative nDNAs. Using this approach, we identified a number of genes whose expression is modified by, and conditional upon, the nuclear genome the mtDNA is paired with. These results suggest there are interactions between mutations within these genomes that are associated with differential transcript expression. In females the significant mitonuclear interaction genes were largely associated with translational processes, which are consistent with a disruption of a key role of mitochondria in the cell: protein synthesis (Fox 2012; Barreto and Burton 2013). In males, we found enrichment for serine-type endopeptidase activity involving lysozymes and these are integrated in the humoral immune response. Our finding is consistent with a previous study in Drosophila larval fat body, which revealed a genetic link between hypoxia and proteasome inactivation via the canonical sima (HIF1α) pathway (Lőw et al. 2013).
Previous studies using the Drosophila mitonuclear introgression model have found translational defects to be main phenotypes and these have been related to development time (Montooth et al. 2010; Mossman, Biancani et al. 2016) and bristle length (Montooth et al. 2010). There are similarities between deleterious mitonuclear phenotypes and Minute loci; known to affect ribosomal proteins and hence translational machinery in the cell, along with OXPHOS deficiencies (Farnsworth 1965). We found among the most enriched GO terms relating to nDNA × mtDNA × hypoxia co-variation was “translation”, and we hypothesize that mitonuclear variation partly disrupted cross talk between mtDNA and nDNA molecules and this was revealed in related DE transcripts. We have previously isolated and fine mapped a two-nucleotide mitonuclear epistasis for development time in Drosophila (Meiklejohn et al. 2013). In that case, one nucleotide in the mtDNA (tRNATyr) and one nucleotide in the nDNA (mitochondrial tyrosyl-tRNA synthetase) interact epistatically and cause a translation syndrome, mimicking a mitochondrial disease in humans. Other Drosophila models of mitochondrial disease, including the tko(25t) strain (Chen et al. 2012), have shown mitoribosomal protein gene mutations can confer OXPHOS deficiencies and translational defects, and these can be modified by putative mtDNA variation. Together, these results suggest Drosophila mitonuclear covariation can provide a useful model of mitochondrial and translational defects.
Mitonuclear introgression introduces mutations into a co-evolved mitonuclear genetic unit (Rand 2001; Rand et al. 2004) and can experimentally mimic the phenotypic consequences of mitochondrial replacement therapy in a genetically tractable organism. Recently, changes in mitochondrial content have been pinpointed as sources of ∼50% of the variability in nuclear protein levels (Guantes et al. 2015). Given that mtDNA can effect gene expression, and new therapies to ameliorate mitochondrial disease by mtDNA replacement are being developed, extra care must be made to ensure mitonuclear incompatibilities are avoided (Muir et al. 2016).
Concluding Remarks
The canonical response to hypoxia is a complex cascade of gene action, although the basic mechanism is well understood (Lendahl et al. 2009). There are complications when comparing hypoxic responses between studies, because often different durations and severities of hypoxia are experimentally conducted, with different gene pathways being targeted. Also, there is often variation between the cell stages or ages of the organism between studies. Very few studies have made the separation between sexes and pool the sexes prior to expression arrays (Zhou et al. 2007). We show that this approach restricts the possibility of detecting sex-specific DE genes and pathways. We found overall large differences in sensitivity between the sexes to hypoxia, along with nDNA and mtDNA variation. These results have wide implications for the effects of mitonuclear communication and how these can often exert genotype-specific signatures of hypoxia. Not only did we find small overlap of DE genes between genotypes, but we also identified that some canonical hypoxia response genes are also significantly influenced by G × G and G × E effects, and these were often sex-specific responses. A meta-analysis approach to identify human HIF-target genes revealed that cell type alone can have a large influence on the number of DE genes detected, e.g., monocytes showed 486 genes, whereas HeLa cells revealed 2,119, with little overlap between the gene sets (Benita et al. 2009). The hypoxic transcriptome response is cell type specific in human cancer cells too (Denko et al. 2003; Chi et al. 2006) and we show here that mitonuclear genotypes of the same organism, at the same age, in the same sex, can display significantly different transcriptome responses, averaging across cell types. While the core sets of hypoxia response genes can be identified, as we show here, the interactions between mtDNA and nDNA are likely to underpin a large number of privately expressed DE genes to each genotype, which can be further influenced by oxygen availability. While this study has focused on hypoxia, the implications for the genetic and environmental complexities underlying personalized genomic medicine are clear: each genotype can have a unique response to a genetic or environmental perturbation. Quantifying the extent of these genotype- and environment-specific responses of organisms is a critical first step in addressing the common and unique components of the causal connection between genotypes and phenotypes.
Materials and methods
Mitonuclear Flies
We conducted our experiments on four mitonuclear genotype in a 2 × mtDNA haplotype × 2 × nDNA background matrix with four genotypes in total. MtDNA haplotypes were from: (i) OregonR D. melanogaster haplotype and (ii) siI D. simulans haplotype. These mtDNAs were introgressed with two nDNA types both from D. melanogaster; OregonR and AutW132. The four (mtDNA; nDNA) introgressed genotypes were: OregonR;OregonR, siI;OregonR, OregonR;AutW132, and siI;AutW132. The introgressions were performed via precise balancer chromosome replacement as described in (Montooth et al. 2010). Following balancer chromosome replacement, flies were repeatedly backcrossed to remove residual nuclear genetic variants that may have been retained during the chromosome replacement. Some fraction of SNPs associated with mitonuclear covariation may be due to underlying genetic differences at those loci and not necessarily a consequence of mitonuclear interactions. This is because (i) no Drosophila strain is truly isogenic >1 generation post-construction (due to mutation), and (ii) the lines were maintained for ∼100 generations prior to experimentation allowing opportunity for mutations to segregate within a nuclear background and between mitonuclear genotypes. In particular, nuclear SNPs that differ between strains carrying different mtDNA variants within the same nuclear background could associate with significantly differentially expressed genes. To test whether this was evident in our dataset, we merged .bam files from technical replicates of the same genetic background from the timepoint = 0 RNAseq libraries (4 in total/one per genotype). We then asked whether there were SNPs within the nuclear backgrounds that segregated between strains carrying different mtDNA variants, using the bcftools pair algorithm and a CLR cut-off > 90; in much the same way that we estimate between nuclear background differences (see below). We identified some SNPs in genes that were also significantly associated with mitonuclear variation. These were: 3/72 (4.17%) for male AutW132; 39/1865 for female AutW132 (2.09%); 1/72 male OregonR (1.39%); and 15/1865 (0.80%) for female OregonR. To test whether these genes were associated with the significant GO terms we identified in the mitonuclear gene lists, we conducted GO category analyses on those genes that were identified as containing SNPs. We did not find any GO functional categories, significant or otherwise, that overlapped with our identified gene sets that we report in the supplementary tables S2–S8, Supplementary Material online. We interpret this as good evidence that our identified genes that contain SNPs are not associated with the pathways we observed to be enriched in our mitonuclear gene sets.
The reason for introgressing these specific mtDNA haplotypes is because they contain a large number of synonymous SNP polymorphisms (418) and amino acid substitutions (94), therefore are likely to influence phenotypic variation. Using the same panel of genotypes, we have observed wide variation in whole organismal phenotypes (development time and egg-to-adult viability; Montooth et al. 2010) and introgression of OregonR and siI mtDNAs onto Drosophila Genetic Reference Panel (DGRP) nuclear backgrounds highlighted numerous mitonuclear epistases associated with development time and egg-to-adult survival (Mossman, Biancani et al. 2016). Prior to this study, all genotypes were treated with tetracycline to remove any source of Wolbachia infection. Wolbachia negative status was confirmed in all genotypes using a diagnostic Wolbachia-specific PCR with positive controls (Montooth et al. 2010).
Hypoxia Exposure
The hypoxia experiment was fully factorial and all genotypes were exposed to three hypoxia treatment conditions: (i) 0 min of hypoxia (control), (ii) 30 min of hypoxia (6% O2) (initial hypoxic response), and (iii) 120 min of hypoxia (6% O2). Adult Drosophila can tolerate acute hypoxia (0% O2) for a few hours without measurable tissue damage (Haddad et al. 1997) and selection experiments over several generations (>13) have successfully selected for flies that can tolerate ambient 5% O2 (Zhou et al. 2007). At 6% O2, the proportion of embryos surviving to adult stage is reduced to ∼10% (Zhou et al. 2007) and at 4% O2, no unselected adult flies are viable. The genotypes used in this study demonstrate variable survival under extreme hypoxia (1.5% O2) (see supplementary fig. S1, Supplementary Material online). Using this prior information along with previous studies, we selected 6% oxygen as a hypoxia environment that was likely to generate informative results, and which is in the range of “physiological hypoxia” (Koh and Powis 2012) that is non-lethal for short durations in adult Drosophila.
All experiments were performed at the same location in Providence, Rhode Island, at approximately sea level. Our hypoxia treatments were conducted by decreasing the level of O2 via nitrogen (N) gas injection into a BioSpherix animal chamber (Biospherix, Lacon, NY, USA) to establish an equilibrated 6% oxygen, creating a “physiological hypoxia” environment (sensu; Koh and Powis 2012). All experiments were performed at 25 °C. At sea level at 25 °C, the partial pressure of oxygen under normoxia (21% O2) is 20.33 kPa, while the partial pressure under our hypoxic condition was 5.81 kPa. Oxygen concentration was monitored and controlled using a BioSpherix ProOx Model 110 gas oxygen controller (BioSpherix, Lacona, NY, USA).
Prior to the experiment, flies were reared on standard laboratory food at 25 °C on a 12 h light: 12 h dark cycle, and density-controlled for one generation. After density control, eclosed flies were allowed to mate for 3 days on the standard laboratory food, and then separated by sex into vials with 50 individuals per vial. Flies were used in this experiment after two days of recovery from CO2 anesthesia. Hypoxia exposure commenced at 15:30 for both hypoxia treatments and the control. All flies were handled the same way across treatments to minimize any handling effects. After 0, 30, or 120 min of hypoxia, flies were transferred to a 1.5 mL micro centrifuge tube and immediately flash frozen in liquid nitrogen.
RNA Extraction
RNA extraction was performed as previously outlined (Mossman, Tross et al. 2016). Briefly, we extracted RNA from 30, 5-day-old healthy flies in each genotype x hypoxia × sex treatment. There were three replicates per genotype × hypoxia × sex condition, with the exception of one sample of siI;AutW132 females, which had two replicates at hypoxia timepoint = 0; resulting in a total of 71 RNA libraries. RNA extraction followed a modified RNA-Seq sample preparation protocol from the Gilad Laboratory (Marioni et al. 2008). Messenger RNA was first extracted, followed by RNA fragmentation, cDNA first strand synthesis, second strand synthesis, end repair, poly adenylation, adapter ligation and PCR enrichment. Throughout, RNA and DNA were quantified using the Qubit® Kits (RNA Broad Range, ds DNA Broad Range, and ds DNA High Sensitivity) with a Qubit® 1.0 Fluorometer. All Qubit® reagents were obtained from Molecular Probes™ (ThermoFisher Scientific). Following PCR enrichment, we size selected PCR products with size range = 334–500 bp using a Caliper Lab Chip XT (DNA 750 chip) (Caliper Life Sciences, Inc. Hopkington, MA, USA).
Gene Expression Assays
Gene expression was assayed in both sexes in all four genotypes at three hypoxia treatments (0 min, 30 min, and 120 min) using Illumina RNA-seq (HiSeq: Illumina, San Diego, USA) with a 50 bp, single end protocol. Samples were processed at Brown University’s Genomics Core Facility using an Illumina HiSeq2000 platform.
RNA-Seq Data Preprocessing
RNA-seq read quality was first assessed using the fastqc v0.11.5 program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, last accessed November 14, 2016). We then filtered the reads using a fastq quality filter (fastq_quality_filter) with –q 20 and –p 80 flags, as implemented in the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/commandline.html#fastq_quality_filter_usage, last accessed November 14, 2016). These values correspond to removing reads with 80% bases having a quality score lower than 20. Adapters were clipped from the reads using the fastx_clipper tool in the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/commandline.html#fastx_clipper_usage, last accessed November 14, 2016) and fastqc was repeated. We used Tophat v2.0.12 (Trapnell et al. 2012): https://ccb.jhu.edu/software/tophat/index.shtml, last accessed November 14, 2016) with Bowtie2 v2.2.3 (http://bowtie-bio.sourceforge.net/index.shtml, last accessed November 14, 2016) to map the reads to the dm3 reference genome using the dm3flybase.gtf annotation file obtained from the UCSC Genome Browser (https://genome.ucsc.edu/, last accessed November 14, 2016). BAM files generated by Tophat were converted to .SAM files using Samtools (Li et al. 2009) (http://samtools.sourceforge.net/, last accessed November 14, 2016) and reads mapping to specific genome features (genes) were counted using htseq-count (Anders et al. 2015) (http://www-huber.embl.de/users/anders/HTSeq/doc/count.html, last accessed November 14, 2016). The read count data table obtained via HTSeq was used for downstream analyses. All pre-processing was conducted using computational resources and services at the Center for Computation and Visualization, Brown University.
RNA-Seq Data Analysis
Differential expression analyses and gene clustering were conducted using three software packages: (i) DESeq (Anders and Huber 2010), (ii) edgeR (Robinson et al. 2010), and (iii) MBCluster.Seq (Si et al. 2014). We specify in the relevant Results sections which analysis program was used.
Between Nuclear Background SNP Estimates (Pair Analysis)
We estimated the number of SNPs that were different between OregonR and AutW132 nuclear backgrounds using the RNA-seq reads that mapped to the D. melanogaster transcriptome. To estimate the number of SNP variants between nuclear backgrounds, we first merged the .bam files from three replicates of each genotype in males at timepoint = 0. The pair of genotypes was OreR;OreR and OreR;AutW132. We used the merged .bam files to achieve greater confidence in SNP calling, along with greater transcriptome-wide coverage. Combined .bam files were aligned with each other and overlapping (intersected) sections were compared for consistency of genotype calls between members of the pair. We used the “pair” function implemented in samtools following user guidelines. Specifically, this was the “bcftools view -T” option (http://www.htslib.org/doc/samtools-0.1.19.html, last accessed November 14, 2016). The genotype caller identified 115,028 variants with CLR > 1 (which included all variable genotypes, even of extremely low call quality). The CLR value is the Phred log ratio of genotype likelihoods with and without the pair constraint. Higher values correspond with higher confidence SNP calls. We filtered the genotype calls for SNPs that had a CLR value >90 on a scale of 1-255. At CLR > 90, we estimate 48,449 SNPs between OregonR and AutW132 transcriptomes. With the most stringent and conservative analysis (CLR > 255), we estimate at least 23,539 SNPs. We confirmed a number of the filtered SNP calls by eye using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011). In the main manuscript we report the intermediate CLR Phred score of >90.
DESeq
We used DESeq (Anders and Huber 2010) to estimate the variance stabilized data (vsd) after normalization as an estimate of expression in the construction of figures 5 and 8I–P. Comparisons were scaled within a row and therefore expression levels are relative and not sensitive to any gene size differences.
edgeR Model Implementation and Interpretation
For each edgeR DE analysis we estimated a common dispersion, followed by trended dispersion, followed by a tagwise dispersion as strongly recommended for multi factor experiments (Robinson et al. 2010; Chen et al. 2016).To estimate first order effects of nDNA, mtDNA and hypoxia in the data, we fit negative binomial generalized linear models (glmFit), as implemented in edgeR. The model matrix included the intercept and the term against the model (e.g. nDNA, mtDNA or hypoxia). We then conducted likelihood ratio tests using the glmLRT function, with the contrast between the baseline (e.g. nDNA OreR), against the nDNA coefficient (e.g. nDNA AutW132). The same analysis was performed for contrasts between mtDNAs, and hypoxia time points. For higher order effects, we fit generalized linear models as before, but included interaction terms in the design matrix. The likelihood ratio tests were performed on the interaction effect against the model intercept (e.g. to identify genes that show significant G × G × E (nDNA × mtDNA × hypoxia) interaction effects, we tested this model term using likelihood ratio tests against the intercept (OreRnDNA; OreRmtDNA; 0minsHypoxia)). We report genes that remain significant with a false discovery rate of less than 10% (FDR < 0.1).
MBCluster.Seq
Model based clustering of gene expression profiles was performed using MBCluster.Seq (Si et al. 2014), with k = 70 clusters in both sexes. Each genotype (×4) × hypoxia condition (×3) (12 in total) was treated as an individual condition. Negative binomial models were used to perform the hierarchical clustering and hybrid tree builds as implemented in the MBCluster.Seq package. Genes in the lowest 30% quantile were removed from the clustering analysis since these had very low or zero read counts, At Θ = 0.3, 9,866 transcripts were clustered in both female and male datasets. Gene clusters are described in the supplementary figures S4–S7, Supplementary Material online. Interaction plots of genotype values per cluster were calculated. The log-fold change is relative to the normalized gene expression across all genotype and hypoxia treatments with each row of the log-fold change matrix having zero sum. The clustering algorithm allocates genes to a group based on their gene expression profile (in this case, the shape of the gene expression–condition relationship).
GO Enrichment
To understand which, if any, functional gene ontology categories were enriched in our DE data sets, we used GOrilla (Eden et al. 2009) to characterize GO process, GO function and GO components of the DE gene lists. We used two-unranked analyses and the background “universe” gene set was the list of genes that could have been significant, e.g., all genes in the statistical analysis, following good practice recommendations (Timmons et al. 2015). This background set was variable between analyses because different datasets contained genes that did not have sufficient read counts to be included in the analyses. For example, our filtering criterion for the edgeR analyses required 1 count per million (cpm) in at least three libraries, removing ∼3,000 genes from the analysis, while ∼11,000 transcripts remained. The background “universe” gene sets therefore reflected these minor differences between datasets.
Pre-Computed Gene Lists
In a number of analyses, we exclusively tested gene expression in known hypoxia-sensitive genes from the literature and known genes in the electron transport chain (ETC) in Drosophila. The purpose of the targeted gene analyses was to explicitly test how these genes were affected by mitonuclear introgression and hypoxia. We hypothesized that these genes would be disproportionately affected due to their intimate relationships with each other, and oxygen availability. We obtained these lists from two main sources: NCBI and Flybase. Genes with the GO biological process term “response to hypoxia” (GO:0001666) were downloaded from Flybase. We also identified additional genes that demonstrated known interactions with sima (HIF-1α in Drosophila). These additional genes were (i) p53, (ii) CG7407, (iii) Rlip, (iv) tgo, (v) Hnf4, and (vi) CngA.
We downloaded the known D. melanogaster genes in the four complexes of the OXPHOS pathway and ATP synthase from Flybase. The Gene Ontology IDs for the genes were: (i) complex I: GO:0005747, (ii) complex II: GO:0005749, (iii) complex III: GO:0005750, (iv) complex IV: GO:0005751, and (v) ATP synthase: GO:0000276.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Author Contributions
JGT prepared flies for the experiment. JAM and JGT extracted RNA and conducted RNA-seq. JAM conducted the qPCR analyses, analyzed the data with input from NL and WZ, prepared figures and wrote the manuscript with comments from DMR. NJ conducted the survival analyses.
Supplementary Material
Acknowledgments
We gratefully thank Christoph Schorl for assistance in the processing of RNA-seq libraries, and Mark Howison for de-multiplexing the samples. Leann Biancani provided assistance with fly husbandry. We thank A.M. Hernandez for critical comments on the manuscript. This work was conducted using computational resources and services at the Center for Computation and Visualization, Brown University. This work was supported by National Institutes of Health grants R01-GM067862 and AG027849 to DMR.
References
- Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. 2001. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 20:1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11(1): 12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askie LM, Henderson-Smart DJ, Irwig L, Simpson JM. 2003. Oxygen-saturation targets and outcomes in extremely preterm infants. N Engl J Med. 349:959–967. [DOI] [PubMed] [Google Scholar]
- Ballard JWO. 2000. Comparative genomics of mitochondrial DNA in Drosophila simulans. J Mol Evol. 51:64–75. [DOI] [PubMed] [Google Scholar]
- Ballard JWO, Melvin RG, Miller JT, Katewa SD. 2007. Sex differences in survival and mitochondrial bioenergetics during aging in Drosophila. Aging Cell 6:699–708. [DOI] [PubMed] [Google Scholar]
- Barreto FS, Burton RS. 2013. Evidence for compensatory evolution of ribosomal proteins in response to rapid divergence of mitochondrial rRNA. Mol Biol Evol. 30:310–314. [DOI] [PubMed] [Google Scholar]
- Baysal BE. 2003. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol. Metab. 14:453–459. [DOI] [PubMed] [Google Scholar]
- Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. 2009. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 37:4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, Kotlikoff M, Avadhani NG. 1999. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 18:522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. 2001. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294:1337–1340. [DOI] [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. 1996. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 76:839–885. [DOI] [PubMed] [Google Scholar]
- Callier V, Hand SC, Campbell JB, Biddulph T, Harrison JF. 2015. Developmental changes in hypoxic exposure and responses to anoxia in Drosophila melanogaster. J Exp Biol. 218:2927–2934. [DOI] [PubMed] [Google Scholar]
- Camus MF, Clancy DJ, Dowling DK. 2012. Mitochondria, maternal inheritance, and male aging. Curr Biol. 22:1717–1721. [DOI] [PubMed] [Google Scholar]
- Chae S, Ahn BY, Byun K, Cho YM, Yu M-H, Lee B, Hwang D, Park KS. 2013. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci Signal. 6:rs4. [DOI] [PubMed] [Google Scholar]
- Chen S, Oliveira MT, Sanz A, Kemppainen E, Fukuoh A, Schlicht B, Kaguni LS, Jacobs HT. 2012. A cytoplasmic suppressor of a nuclear mutation affecting mitochondrial functions in Drosophila. Genetics 192:483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, McCarthy D, Robinson M, Smyth GK. 2016. edgeR: differential expression analysis of digital gene expression data User’s Guide. http://www.bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf. [DOI] [PMC free article] [PubMed]
- Chi J-T, Wang Z, Nuyten DSA, et al. 2006. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 3:e47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom B, Alcolea MP, Valle A, Oliver J, Roca P, García-Palmer FJ. 2007. Skeletal muscle of female rats exhibit higher mitochondrial mass and oxidative-phosphorylative capacities compared to males. Cell Physiol Biochem. 19:205–212. [DOI] [PubMed] [Google Scholar]
- Denko NC. 2008. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713. [DOI] [PubMed] [Google Scholar]
- Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, Giaccia AJ. 2003. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene 22:5907–5914. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. 2003. Mechanisms of disease: Mitochondrial respiratory-chain diseases. N Engl J Med. 348:2656–2668. [DOI] [PubMed] [Google Scholar]
- Eckardt KU, Bernhardt WM, Weidemann A, Warnecke C, Rosenberger C, Wiesener MS, Willam C. 2005. Role of hypoxia in the pathogenesis of renal disease. Kidney Int. 68:46–51. [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. 2010. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 11:446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison CK, Burton RS. 2006. Disruption of mitochondrial function in interpopulation hybrids of Tigriopus californicus. Evolution 60:1382–1391. [PubMed] [Google Scholar]
- Ellison CK, Burton RS. 2008. Genotype-dependent variation of mitochondrial transcriptional profiles in interpopulation hybrids. Proc Natl Acad Sci U S A. 105:15831–15836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth MW. 1965. Oxidative phosphorylation in the minute mutants of Drosophila. J Exp Zool. 160:363–368. [DOI] [PubMed] [Google Scholar]
- Farzin M, Albert T, Pierce N, VandenBrooks JM, Dodge T, Harrison JF. 2014. Acute and chronic effects of atmospheric oxygen on the feeding behavior of Drosophila melanogaster larvae. J Insect Physiol. 68:23–29. [DOI] [PubMed] [Google Scholar]
- Fox TD. 2012. Mitochondrial protein synthesis, import, and assembly. Genetics 192:1203–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SA, Hurst LD. 1996. Mitochondria and male disease. Nature 383:224-224.. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J. 2014. Mitochondrial form and function. Nature 505:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. 2004. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899. [DOI] [PubMed] [Google Scholar]
- Gibson G, Riley-Berger R, Harshman L, Kopp A, Vacha S, Nuzhdin S, Wayne M. 2004. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 167:1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Pritchard JK. 2008. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 24:408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJY, et al. 2013. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155:1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grocott MPW, Martin DS, Levett DZH, McMorrow R, Windsor J, Montgomery HE. 2009. Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med. 360:140–149. [DOI] [PubMed] [Google Scholar]
- Guantes R, Rastrojo A, Neves R, Lima A, Aguado B, Iborra FJ. 2015. Global variability in gene expression and alternative splicing is modulated by mitochondrial content. Genome Res. 25:633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. 2005. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1:401–408. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Schumacker PT. 2006. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 91:807–819. [DOI] [PubMed] [Google Scholar]
- Haddad GG. 2006. Tolerance to low O2: lessons from invertebrate genetic models. Exp Physiol. 91:277–282. [DOI] [PubMed] [Google Scholar]
- Haddad GG, Sun Y-a, Wyman RJ, Xu T. 1997. Genetic basis of tolerance to O2 deprivation in Drosophila melanogaster. Proc Natl Acad Sci U S A. 94:10809–10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. 2002. Hypoxia - a key regulatory factor in tumour growth. Nat Rev Cancer 2:38–47. [DOI] [PubMed] [Google Scholar]
- Harrison JF, Haddad GG. 2011. Effects of oxygen on growth and size: synthesis of molecular, organismal, and evolutionary studies with Drosophila melanogaster. Annu Rev Physiol. 73:95–113. [DOI] [PubMed] [Google Scholar]
- Heinrich EC, Farzin M, Klok CJ, Harrison JF. 2011. The effect of developmental stage on the sensitivity of cell and body size to hypoxia in Drosophila melanogaster. J Exp Biol 214:1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra LA, Siddiq MA, Montooth KL. 2013. Pleiotropic effects of a mitochondrial-nuclear incompatibility depend upon the accelerating effect of temperature in Drosophila. Genetics 195:1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmbeck MA, Donner JR, Villa-Cuesta E, Rand DM. 2015. A Drosophila model for mito-nuclear diseases generated by an incompatible interaction between tRNA and tRNA synthetase. Dis Model Mech. 8:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DJ. 2005. Gene-environment interactions in human diseases. Nat Rev Genet. 6:287–298. [DOI] [PubMed] [Google Scholar]
- Innocenti P, Morrow EH, Dowling DK. 2011. Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science 332:845–848. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian Y-M, et al. 2001. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472. [DOI] [PubMed] [Google Scholar]
- Jha AR, Zhou D, Brown CD, Kreitman M, Haddad GG, White KP. 2015. Shared genetic signals of hypoxia adaptation in Drosophila and in high-altitude human populations. Mol Biol Evol 33:501–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justo R, Boada J, Frontera M, Oliver J, Bermúdez J, Gianotti M. 2005. Gender dimorphism in rat liver mitochondrial oxidative metabolism and biogenesis. Am J Physiol Cell Physiol. 289:372–378. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402. [DOI] [PubMed] [Google Scholar]
- King A, Selak MA, Gottlieb E. 2006. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25:4675–4682. [DOI] [PubMed] [Google Scholar]
- Koh MY, Powis G. 2012. Passing the baton: the HIF switch. Trends Biochem Sci. 37:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJH, Willems PHGM, Smeitink JAM. 2012. Monogenic mitochondrial disorders. N Engl J Med. 366:1132–1141. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Lee KL, Yang H, Poellinger L. 2009. Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet. 10:821–832. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data P. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Padmanabha D, Gentile LB, Dumur CI, Beckstead RB, Baker KD. 2013. HIF- and non-HIF-regulated hypoxic responses require the estrogen-related receptor in Drosophila melanogaster. PLoS Genet. 9:e1003230.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795. [DOI] [PubMed] [Google Scholar]
- Liu G, Roy J, Johnson EA. 2006. Identification and function of hypoxia-response genes in Drosophila melanogaster. Physiol Genomics 25:134–141. [DOI] [PubMed] [Google Scholar]
- Liu Z, Butow RA. 2006. Mitochondrial retrograde signaling. Annu Rev Genet. 40:159–185. [DOI] [PubMed] [Google Scholar]
- Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E. 2010. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 81:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lőw P, Varga Á, Pircs K, Nagy P, Szatmári Z, Sass M, Juhász G. 2013. Impaired proteasomal degradation enhances autophagy via hypoxia signaling in Drosophila. BMC Cell Biol. 14:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Moore JH. 2014. Why epistasis is important for tackling complex human disease genetics. Genome Med. 6:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Richards S, Stone EA, et al. 2012. The Drosophila melanogaster genetic reference panel. Nature 482:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18(9):1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massouras A, Waszak SM, Albarca-Aguilera M, et al. 2012. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 8:e1003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Holmbeck MA, Siddiq MA, Abt DN, Rand DM, Montooth KL. 2013. An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 9:e1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montooth KL, Meiklejohn CD, Abt DN, Rand DM. 2010. Mitochondrial-nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution 64:3364–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman JA, Biancani LM, Zhu C-T, Rand DM. 2016. Mitonuclear epistasis for development time and its modification by diet in Drosophila. Genetics 203:463–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman JA, Tross JG, Li N, Wu Z, Rand DM. 2016. Mitochondrial-nuclear interactions mediate sex-specific transcriptional profiles in Drosophila. Genetics 204:613–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir R, Diot A, Poulton J. 2016. Mitochondrial content is central to nuclear gene expression: profound implications for human health. Bioessays 38:150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ. 2016. Energy metabolism and the high-altitude environment. Exp Physiol. 101:23–27. [DOI] [PubMed] [Google Scholar]
- Nambu JR, Chen W, Hu S, Crews ST. 1996. The Drosophila melanogaster similar bHLH-PAS gene encodes a protein related to human hypoxia-inducible factor 1 alpha and Drosophila single-minded. Gene 172:249–254. [DOI] [PubMed] [Google Scholar]
- Peck LS, Maddrell SHP. 2005. Limitation of size by hypoxia in the fruit fly Drosophila melanogaster. J Exp Zool A Comp Exp Biol. 303A:968–975. [DOI] [PubMed] [Google Scholar]
- Pollard P, Wortham N, Tomlinson I. 2003. The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Ann Med. 35:634–635. [DOI] [PubMed] [Google Scholar]
- Rand DM. 2001. The units of selection on mitochondrial DNA. Annu Rev Ecol Syst. 32:415–448. [Google Scholar]
- Rand DM, Haney RA, Fry AJ. 2004. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol. 19:645–653. [DOI] [PubMed] [Google Scholar]
- Renolleau S, Fau S, Charriaut-Marlangue C. 2008. Gender-related differences in apoptotic pathways after neonatal cerebral ischemia. Neuroscientist 14:46–52. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotech. 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero NM, Dekanty A, Wappner P. 2007. Cellular and developmental adaptations to hypoxia: a Drosophila perspective. Methods Enzymol: 435:123–144. [DOI] [PubMed] [Google Scholar]
- Rytkönen KT, Williams TA, Renshaw GM, Primmer CR, Nikinmaa M. 2011. Molecular evolution of the metazoan PHD–HIF oxygen-sensing system. Mol Biol Evol. 28:1913–1926. [DOI] [PubMed] [Google Scholar]
- Sanz A, Hiona A, Kujoth GC, Seo AY, Hofer T, Kouwenhoven E, Kalani R, Prolla TA, Barja G, Leeuwenburgh C. 2007. Evaluation of gender differences on mitochondrial bioenergetics and apoptosis in mice. Exp Gerontol 42:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. 2004. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 5:343–354. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. 2005. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7:77–85. [DOI] [PubMed] [Google Scholar]
- Shen C, Nettleton D, Jiang M, Kim SK, Powell-Coffman JA. 2005. Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem. 280:20580–20588. [DOI] [PubMed] [Google Scholar]
- Si Y, Liu P, Li P, Brutnell TP. 2014. Model-based clustering for RNA-seq data. Bioinformatics 30:197–205. [DOI] [PubMed] [Google Scholar]
- Sims NR, Muyderman H. 2010. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta Mol Basis Dis. 1802:80–91. [DOI] [PubMed] [Google Scholar]
- Smeitink J, van den Heuvel L, DiMauro S. 2001. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. 2:342–352. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. 2005. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 6:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Szkop KJ, Gallagher IJ. 2015. Multiple sources of bias confound functional enrichment analysis of global -omics data. Genome Biol. 16:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J. 2006. Sex-specific regulation of aging and apoptosis. Mech Ageing Dev. 127:705–718. [DOI] [PubMed] [Google Scholar]
- Tower J, Arbeitman M. 2009. The genetics of gender and life span. J Biol. 8:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle A, Guevara R, García-Palmer FJ, Roca P, Oliver J. 2007. Sexual dimorphism in liver mitochondrial oxidative capacity is conserved under caloric restriction conditions. Am J Physiol Cell Physiol. 293:1302–1308. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne P, Frelin C. 2010. Hypoxia modifies the feeding preferences of Drosophila. Consequences for diet dependent hypoxic survival. BMC Physiol. 10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne P, Tauc M, Frelin C. 2009. Strong dietary restrictions protect Drosophila against anoxia/reoxygenation injuries. PLoS One 4:e5422.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Cuesta E, Holmbeck MA, Rand DM. 2014. Rapamycin increases mitochondrial efficiency by mtDNA-dependent reprogramming of mitochondrial metabolism in Drosophila. J Cell Sci. 127:2282–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, Haigis MC. 2016. Mitochondria and cancer. Cell 166:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DW, Hájek P, Muffat J, Knoepfle D, Cornelison S, Attardi G, Benzer S. 2006. Hypersensitivity to oxygen and shortened lifespan in a Drosophila mitochondrial complex II mutant. Proc Natl Acad Sci U S A. 103:16382–16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. 1999. Mitochondrial diseases in man and mouse. Science 283:1482–1488. [DOI] [PubMed] [Google Scholar]
- Wallace DC. 2012. Mitochondria and cancer. Nat Rev Cancer 12:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yang E, Mandhan I, Brinkmeyer-Langford CL, Cai JJ. 2014. Population-level expression variability of mitochondrial DNA-encoded genes in humans. Eur J Hum Genet. 22:1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. 1995. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 92:5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. 1956. On the origin of cancer cells. Science 123:309–314. [DOI] [PubMed] [Google Scholar]
- Weinreich DM, Lan Y, Wylie CS, Heckendorn RB. 2013. Should evolutionary geneticists worry about higher-order epistasis? Curr Opin Genet Dev. 23:700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. 2011. Mitochondria in innate immune responses. Nat Rev Immunol. 11:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CS, Burton RS. 2003. Environmental influences on epistatic interactions: viabilities of cytochrome c genotypes in interpopulation crosses. Evolution 57:2286–2292. [DOI] [PubMed] [Google Scholar]
- Wouters BG, Koritzinsky M. 2008. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8:851–864. [DOI] [PubMed] [Google Scholar]
- Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, Finley LW, Lu C, Lindsten T, Cross JR, et al. 2014. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4:1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HW, Haddad GG. 2011. Hypoxic and oxidative stress resistance in Drosophila melanogaster [Review]. Placenta 32:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Guo Y, Sheng Q, Shyr Y. 2014. Advanced heat map and clustering analysis using Heatmap3. Biomed Res Int. 2014:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Xue J, Chen J, Morcillo P, Lambert JD, White KP, Haddad GG. 2007. Experimental selection for Drosophila survival in extremely low O2 environment. PLoS One 2:e490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Xue J, Lai JCK, Schork NJ, White KP, Haddad GG. 2008. Mechanisms underlying hypoxia tolerance in Drosophila melanogaster: hairy as a metabolic switch. PLoS Genet. 4:e1000221.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C-T, Ingelmo P, Rand DM. 2014. GxGxE for lifespan in Drosophila: mitochondrial, nuclear, and dietary interactions that modify longevity. PLoS Genet. 10:e1004354.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. 2012. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 109:1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.