

Abstract
For many years obesity was believed to be a condition of overeating that could be resolved through counseling and short term drug treatment. Obesity was not recognized as a chronic disease until 1985 by the scientific community and 2013 by the medical community. Pharmacotherapy for obesity has advanced remarkably since the first class of drugs, amphetamines, were approved for short-term use. Most amphetamines were removed from the obesity market due to adverse events and potential for addiction, and it became apparent that obesity pharmacotherapies were needed that could safely be administered over the long-term. This review of central nervous system (CNS) acting anti-obesity drugs evaluates current therapies such as phentermine/topiramate which act through multiple neurotransmitter pathways to reduce appetite. In the synergistic mechanism of bupropion/ naltrexone, naltrexone blocks the feed-back inhibitory circuit of bupropion to give greater weight loss. Lorcaserin, a selective agonist of a serotonin receptor that regulates food intake, and the glucagon-like-peptide-1 (GLP- 1) receptor agonist liraglutide are reviewed. Future drugs include tesofensine, a potent triple reuptake inhibitor in phase III trials for obesity and semaglutide, an oral GLP-1 analog approved for diabetes and currently in trials for obesity. Another potential new pharmacotherapy, setmelanotide, is a melanocortin-4 receptor agonist which is still in an early stage of development. As our understanding of the communication between the CNS, gut, adipose tissue, and other organs evolves, it is anticipated that obesity drug development will move toward new centrally acting combinations and then to drugs acting on peripheral target tissues.
1.0. Introduction
Obesity is a rapidly expanding disease that results from an imbalance between food intake and energy expenditure. Unfortunately, treatment of obesity is hampered by biological forces that resist maintenance of weight loss. Obesity was recognized by the scientific community as a disease at the National Institutes of Health Consensus Development Conference in 1985, years after recognition of hypertension and diabetes, chronic diseases of blood pressure and blood glucose dysregulation. Prior to 1985, obesity was believed to be a condition caused by overeating, and treatments were designed to retrain behaviors with short-term assistance from anti-obesity medicines[1]. The length of drug treatment required was thought to be about 12 weeks, the length of time needed to break a bad habit or learn to ride a bicycle without training wheels.
Almost a decade after obesity was classified as a disease, leptin was discovered and the idea of obesity being a chronic, physiologically controlled disease began to get traction[2]. Studies of leptin deficient rodents and humans demonstrated that the absence of the leptin hormone resulted in morbid obesity that was reversed by leptin hormone replacement, similar to the disease of type-1 diabetes and its relationship to loss of insulin secretion[3]. A result of the delayed recognition of obesity as a chronic disease is that we have medications approved for short-term use prior to 1985 to treat a disease that is chronic.
The scientific community recognized obesity as a disease in 1985, but it was not until 2013 that obesity was acknowledged as a chronic disease by the American Medical Association. Physicians who trained during the time that leptin was discovered were attuned to the idea of the physiological basis of obesity. Their rise to positions of leadership in the American Medical Association led to acceptance of obesity as a chronic disease. Thus, lack of recognition of the physiological basis of obesity, including the consequent physiological pressures to regain weight after weight loss, has been a major contributor to the delay in promoting pharmacological approaches. In fact, there are physicians who still contend that obesity is a primarily a behavioral problem and are reluctant to prescribe medications to treat it. Poor insurance coverage for antiobesity medications has compounded the problem.
The most effective treatment currently available for obesity and diabetes is bariatric surgery. This is followed by a number of pharmacotherapies, most of which initially act on the central nervous system. Drugs that increase dopamine, norepinephrine, or serotonin activity in the brain can stimulate hypophagia, weight loss and in some cases, energy expenditure. Drug combinations that act on multiple neural pathways can sometimes increase weight loss synergistically. Unfortunately, the experience with obesity medications is littered with many unintended adverse events that have resulted in the withdrawal of many drugs from the market. We begin this review with a journey through the history of centrally acting anti-obesity medications. We will then describe the anti-obesity medications available today that act on the brain, and conclude with a review of the potential of new centrally acting medications in clinical development.
2.0. Past Centrally Acting Anti-Obesity Drugs
The original class of centrally acting anti-obesity medications was based on phenylethylamine, the chemical backbone for noradrenaline and dopamine. Amphetamine and its derivatives act as sympathomimetic stimulants to regulate signaling of reward and locomotor activity. These drugs elevate the activity of a number of neural pathways, and numerous side effects have been documented including dry mouth, insomnia, constipation, headache and dizziness. They are contraindicated in the presence of hyperthyroidism, advanced atherosclerotic vascular disease, moderate to severe hypertension, symptomatic cardiovascular disease, agitated states, a history of drug abuse, glaucoma, pregnancy (due to associated fetal toxicity), lactation, within 14 days of taking a monoamine oxidase inhibitor drug and in the presence of sensitivity to sympathetic amines. Table 1 summarizes past centrally acting drugs that have been withdrawn from the market due to adverse side effects.
Table 1.
Past anti-obesity drugs acting in the central nervous system
| Drug Name | Approved | Mechanism | Reason withdrawn and Date |
|---|---|---|---|
| Amphetamine | 1947 | Central norepinephrine release | DEA-Schedule II, 1970’s |
| Aminorex | 1965 (Ger) | Central Norepinephrine release | Pulmonary HTN, 1968 |
| Fenfluramine | 1973 | Serotonin agonist | Heart valvulopathy, 1997 |
| Phenylpropanolamine | 1976 | Alpha adrenergic agonist | Hemorrhagic stroke, 2000 |
| Caffeine and Ephedra | 1994 (DSHEA) | Non-selective adrenergic agonist | Cardiac and psychiatric AE’s, 2004 |
| Rimonabant | 2005(EU) | Cannabanoid-1 antagonist | Depression and suicide, 2007 |
| Sibutramine | 1997 | Norepinephrine/serotonin reuptake inhibitor | Ml, stroke and death, 2010 |
DEA = Drug Enforcement Administration; GER = Germany; HTN = Hypertension; DSHEA = Dietary Supplements Health and Education Act; AE = Adverse Event; EU =European Union; Mi = Myocardial Infarction
2.1. Amphetamine
Amphetamine (methyl-phenylethylamine) was first synthesized in 1887, and in 1927 its psychopharmacologic properties were described as increased energy, wakefulness, alertness and euphoria. It was noted that subjects lost weight during studies evaluating amphetamine for the treatment of depression and narcolepsy in 1937[4]. A year later, amphetamine was reported as a possible treatment for obesity[5]. It was not until 1947 that a reduction in food intake was proposed as a mechanism for the weight loss observed in dogs and in humans. When humans were given amphetamine or placebo while required to maintain constant food intake, the effect of weight loss was abolished[6]. Amphetamine was subsequently shown to act as a competitive inhibitor of dopamine and noradrenaline reuptake transporter proteins. Amphetamine also induces norepinephrine and dopamine release from nerve storage granules through indirect downstream effects on phosphorylation events[7]. Enhanced dopaminergic signaling is linked to reward circuitry and the potential for drug abuse and addiction. Amphetamine has relatively low affinity for serotonin reuptake transporter proteins.
Because amphetamine use was associated with a number of harmful side effects, several drugs with different modifications of the amphetamine backbone were synthesized in an attempt to reduce stimulatory effects while preserving the reduction in food intake. Substituted amphetamines have stronger inhibitory effects on serotonin reuptake transporters. There are presently four of these drugs that are approved for the treatment of obesity. Phendimetrazine and benzphetamine are in Drug Enforcement Agency (DEA) class III, and phentermine and diethylpropion are in DEA class IV. Amphetamine is in DEA class II and is not approved for the treatment of obesity. The drugs in DEA class III are rarely prescribed due to their greater potential for abuse than those in DEA class IV.
Due to questions about the efficacy of these drugs, Scoville reviewed all double-blind placebo- controlled studies of new drug applications submitted to the United States Food and Drug Administration (FDA) for the treatment of obesity prior to 1973. These trials included 4,543 subjects on drug and 3,100 on placebo. He concluded that all of the drugs based on the amphetamine backbone were equally effective. Subjects on active drugs demonstrated weight loss of approximately 0.5 pounds (0.23 kg) per week more than placebo (p<0.05), which was twice the placebo weight loss [8].
2.2. Aminorex
Aminorex was approved for non-prescription sale as a treatment of obesity in Austria, Switzerland and West Germany in 1965, but was never approved in the United States[9]. Aminorex was a modification of the phenylethylamine backbone that increased the release of norepinephrine in the central nervous system and reduced appetite[10]. From 1967–1968, the prevalence of primary pulmonary hypertension was 20-fold higher than it was in the period from 1955–1966 in those countries. Aminorex was removed from the market in 1968 due to its association with primary pulmonary hypertension and by 1972 the prevalence of primary pulmonary hypertension had fallen to the level prior to the release of aminorex[11]. The symptoms of dyspnea, syncope and chest pain regressed in some cases, but up to half of the individuals exposed were dead by 1980[10]. It was this experience that sensitized the obesity community to the risk of primary pulmonary hypertension with anti-obesity drugs.
2.3. Fenfluramine/dexfenfluramine
Fenfluramine is another derivative of phenylethylamine, but unlike other drugs with this structure, it is a depressant rather than a stimulant and its metabolite nordexfenfluramine acts as an agonist of serotonin receptors. Whereas norepinephrine reduces food intake, serotonin causes early meal termination. Fenfluramine was approved in 1973 but did not become popular until the publication in 1992 of a trial demonstrating additive effects on weight loss when full doses of phentermine and fenfluramine were used in combination[12]. Since both drugs were generic, the cost of using them together was economical. The combination of fenfluramine with phentermine became quite popular and in 1996 more than 18,000,000 prescriptions were filled. Since fenfluramine was a depressant and phentermine was a stimulant, the side effects canceled each other and improved tolerability. Fenfluramine was approved for a few weeks (up to twelve), but the combination was prescribed for much longer time periods. When reports of a low incidence of primary pulmonary hypertension surfaced in 1997, it was felt that the benefits of the drug outweighed the risks[13]. Shortly after, however, 24 women with previously normal hearts developed heart murmurs after taking fenfluramine, and at surgery had left sided valvular lesions that resembled the right sided heart valve lesions commonly associated with carcinoid syndrome[14]. Dexfenfluramine was approved in that year with the aim of improving tolerability, but it was also associated with valvular lesions in the heart. At the request of the FDA, both drugs were removed from the market in 1997. Further investigation showed that both fenfluramine and dexfenfluramine produce a metabolite, nordexfenfluramine, which is an agonist of the serotonin 2B receptor (5-HT2b). Activation of this receptor on the single-cell layer covering heart valves induces replication, and over time, results in the undesirable side effects of heart valve lesions and pulmonary hypertension seen at surgery [15], In 1995, the serotonin 2C (5-HT2c) receptor was identified as the key regulator of satiety in rodent studies and development of selective serotonergic agonists for obesity advanced more rapidly [16].
2.4. Phenylpropanolamine
Phenylpropanolamine is another drug that has an amphetamine, phenylethylamine backbone. It was synthesized in 1910 and was used to treat post-operative hypotension in the 1930’s. It was noted to be a decongestant with a low risk of central nervous system stimulation or elevation of pulse and blood pressure. Its effectiveness in decreasing food intake was recognized in 1939, but it was not approved for the treatment of obesity until 1976 [17]. In 1979 phenylpropanolamine was approved for non-prescription sales. The drug was well tolerated and had similar efficacy to prescription drugs available for obesity at the time[18]. The dose used for obesity was 75mg/d, only half of the 150mg/d dose used as a decongestant. In 2000, a case control study was published that associated phenylpropanolamine with hemorrhagic stroke only in women being treated for obesity, despite the decongestant dose being twice the dose for obesity treatment[19], Phenylpropanolamine was subsequently withdrawn from the market in 2000 for both treatments. A follow-up analysis determined that the women in the phenylpropanolamine group had a statistically significant higher family history of hemorrhagic stroke, personal history of hypertension, and a greater use of tobacco, alcohol and cocaine compared to the control group, which may have influenced the association[20].
2.5. Ecopipam
Ecopipam is a dopamine-1 and dopamine-5 receptor antagonist that was developed for the treatment of obesity. There were 4 randomized, double-blind, multicenter, controlled trials of ecopipam (n=1667) and placebo (n=l118) in obese subjects with type 2 diabetes. Subjects in the phase II trials received 10 mg, 30 mg or 100 mg of oral ecopipam or a placebo for 12 weeks. In the phase III program subjects were given ecopipam 50 mg, 100 mg or placebo for 52 weeks with a weight loss program. In the phase II study 26% of subjects on ecopipam 100 mg/d lost 5% of body weight compared to 6% on placebo (p<0.01). In the phase III trials ecopipam l00mg gave a 3.1% to 4.3% greater weight loss than placebo. In the phase III trials the prevalence of depression, anxiety and suicidal ideation was 31% in the ecopipam 100 mg/d group and 15% in the placebo group. These adverse effects on mood excluded it from its projected use in weight management[21].
2.6. Rimonabant
Rimonabant is a cannabinoid-1 (CB-1) receptor antagonist that was developed for the treatment of obesity based on the observation that stimulation of the CB-1 receptor by marijuana increased appetite and intake of sweet and fatty foods[22]. In phase III of drug development, rimonabant was studied in 6,000 subjects and received a letter for approval from the FDA in 2006. Rimonabant gave a 4.8 kg greater weight loss than placebo, improved cardiovascular risk factors and appeared to be well-tolerated. The most common side effects were nausea and diarrhea, but anxiety, depression, insomnia and dizziness were also noted[23]. Rimonabant was approved in Europe in 2006, but the FDA advisory panel that reviewed Rimonabant in 2007 recommended against its approval. The European Medicines Agency withdrew rimonabant from the market citing risks that were greater than the benefits due to subjects developing depression, anxiety and suicidal ideation. Since these side effects are the opposite of the side effects of marijuana (relaxation and euphoria) which stimulates the CB-1 receptor, the anxiety and depression did not come as a complete surprise. The development of anxiety and depression were effects of the entire class of CB-1 antagonists, ultimately causing several pharmaceutical companies that were developing CB-1 antagonists for the treatment of obesity to abandon their CB-1 development programs. Cannabanoid-1 receptors are present outside of the brain in addition to being present in the CNS. Since the side effects that led to the withdrawal of rimonabant were associated with the central nervous system and all of the CB-1 antagonists in development were designed to cross the blood brain barrier, Jenrin Discovery has developed CB-1 antagonists that are excluded from the CNS. Preclinical studies have demonstrated that these CB-1 antagonists have efficacy in treating obesity in diet-induced obese mice, and Jenrin filed an IND application which was cleared for phase 1 trials in 2017. CB-1 antagonists that do not cross the blood brain barrier are not anticipated to have dose limiting side effects on anxiety and depression when translated into humans and further development in this area has the potential for future clinical trials [24, 25].
2.7. Sibutramine
Sibutramine, a norepinephrine and serotonin reuptake inhibitor that acts by decreasing food intake, was approved in 1997 for the long-term treatment of obesity. Sibutramine had efficacy similar to rimonabant, giving approximately 5 kg more weight loss than placebo and improved cardiovascular risk factors with the exception of blood pressure and pulse rate[26]. The side effects were dry mouth, insomnia, constipation, headache and dizziness, typical of norepinephrine agonists[27]. Despite there being no evidence of abuse, sibutramine was classified in DEA schedule IV due to structural similarities with amphetamine[28]. The increase in pulse and blood pressure were of concern to the regulators, and contingent on approval, the sponsor agreed to do a cardiovascular safety study. That study, called the SCOUT study, enrolled subjects with diabetes and heart disease, conditions for which the drug was not approved. All subjects, including those who did not experience weight loss, were kept on the drug which would not have been done in normal practice. Individuals in the SCOUT trial showed a 16% increase in cardiovascular endpoints like heart attack, stroke and death[29]. The European authorities removed sibutramine from the market following the results of the SCOUT trial. The FDA initially added a black box warning, but in 2010 followed the European authorities and withdrew sibutramine from the market. This experience led to re-evaluation of the design of future cardiovascular safety trials.
2.8. Caffeine and Ephedrine
Ephedra has been used in Chinese medicine for over 2,000 years and has 4 isomers, the most potent of which is ephedrine. Chen introduced ephedrine into the United States in 1930 and described its pharmacology and medicinal uses [30]. The combination of ephedrine with a xanthine has been used for the treatment of asthma in adults and children since the 1930’s, and during the 1970’s, a physician in Denmark noted that the combination of caffeine and ephedrine caused weight loss. Caffeine 200mg with ephedrine 20mg taken three times a day was approved as a prescription drug in Denmark and held 80% of the market share in that country even when fenfluramine was available[31]. The caffeine with ephedrine combination was removed from the Danish market a decade later when stimulants for the treatment of obesity were removed from the European market.
The Dietary Supplement Health and Education Act (DSHEA) was approved in the United States in 1994, classifying dietary supplements as foods if they had been in the food supply before 1994. This legislation gave rise to wide spread use of ephedra and caffeine sold as a dietary supplement for weight loss. The FDA received reports of cardiovascular and neuropsychiatric adverse events and attempted to take ephedra with caffeine off the market [32]. The supplement industry fought this ban legally and was successful. An extensive meta-analysis of ephedra and ephedrine with and without caffeine for weight loss and improving athletic performance showed a 2.2 to 3.6 fold increase in the odds of psychiatric, autonomic, or gastrointestinal symptoms and heart palpitations. As a consequence, it became difficult for the supplement makers of caffeine with ephedrine to obtain liability insurance and the supplement manufacturers stopped contesting the FDA imposed ban on the combination[33].
3.0. Current Centrally Acting Anti-Obesity Drugs
Anti-obesity medications that were approved before 1985 were indicated for short-term use over a period of weeks. Those that are still on the market will be described first. Four centrally acting medications are presently approved for the long-term treatment of obesity (see Tables 2 and 3). A recent review of 50 reports involving 43,443 subjects compared the efficacy of these medications quantitatively (29). They found that the maximal mean weight loss relative to placebo for lorcaserin, naltrexone-bupropion, phentermine-topiramate (7.5/46mg) and liraglutide was −3.06 kg, −6.15 kg, −7.45 kg and −5.5 kg, at weeks 54, 67, 59 and 65, with mean rates of regain of 0.48 kg, 0.91 kg, 1.27 kg and 0.43 kg per year, respectively. The one year dropout rates for lorcaserin, naltrexone-bupropion, phentermine-topiramate and liraglutide were 40.9%, 49.1%, 34.9% and 24.3%, respectively. These four medications will be discussed in more detail below, but a recent review puts their relative efficacy and tolerability into context [34]
Table 2.
Approved anti-obesity drugs acting in the central nervous system
| Drug name | Approved | Wt. loss - placebo | Common AE | Concerning AE |
|---|---|---|---|---|
| Phentermine et al. | 1959 −1960 | 0.23 kg/week above placebo | Stimulation | HTN |
| Topiramate/phentermine | 2012 | 6.6 kg (7.5/46mg/d) | Paresthesia | Cleft lip/palate |
| Lorcaserin | 2012 | 4% | Headache | Heart valvulopathy |
| Bupropion/naltrexone (BN) | 2014 | 4.8 kg (BN 360/32/d) | Nausea | HTN |
| Liraglutide | 2014 | 5.6% | Nausea | Pancreatitis |
| Lorcaserin-ER | 2016 | 4% | Headache | Heart valvulopathy |
| Drugs in Phase III | ||||
| Tesofensine | NA | 2% −10.6% /4.7bpm - 8.5bpm (.25-lmg/d) | Dry mouth | HTN/pulse rate |
HTN = Hypertension; ER = Extended Release; BN = Bupropion/Naltrexone
Table 3.
Mechanism of anti-obesity drugs acting in the central nervous system
| Drug name | Mechanism |
|---|---|
| Phentermine, diethylpropion, benzphetamine, and phendimetrazine | norepinephrine release with minor dopamine release |
| Bupropion/naltrexone | Stimulates dopamine and POMC neurons and blocks inhibitory feedback to mu opioid receptor on POMC neurons |
| Topiramate/phentermine | norepinephrine release, GABA modulation, voltage-gated ion channel modulation, inhibition of AMPA/kainite excitatory glutamate receptors and inhibition of carbonic anhydrase |
| Lorcaserin | Serotonin 2C receptor agonist |
| Lorcaserin-ER | Serotonin 2C receptor agonist |
| Liraglutide | Glucagon-like peptide-1 agonist |
| Drugs in Phase III | |
| Tesofensine | Reuptake inhibitor of serotonin, norepinephrine and dopamine |
ER = Extended Release; POMC = Proopiomelanocortin; GABA = Gamma-Aminobutyric Acid; AMPA = α- amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
3.1. Phentermine
Phentermine, an appetite-suppressant, is an amphetamine derivative with an α-methyl substitution on the phenylethylamine side chain that causes a reduction in CNS stimulation. It is approved for up to 12 weeks and can have side effects such as increased blood pressure and pulse rate, insomnia and dry mouth. Alcohol consumption is not recommended with this drug. Phentermine is the most commonly prescribed anti-obesity medication due in large measure to its low potential for CNS stimulation and abuse, and its low price as a generic drug, approved in 1959. Phentermine is available in 15 mg and 30 mg capsules in a resin form, and it is available as phentermine HC1 in 37.5 mg and 8 mg tablets. As obesity is now universally recognized as a chronic disease for which long term medication is appropriate, using the drug within the recommended 12 weeks is a challenge. A trial comparing phentermine treatment in alternating months with continuous treatment showed the same weight loss at the 6 month plateau[35] .Thus, alternate month therapy with phentermine may be a way to use phentermine for the long-term treatment of obesity and stay within the package insert recommendations.
3.2. Diethylpropion
Diethylpropion is available in 25 mg immediate release and 75mg sustained release tablets that are taken three times or once a day respectively. CNS stimulation has been reduced by a keto substitution on the beta carbon of the phenethylamine backbone. Diethylpropion is the popular amphetamine-related anti-obesity drug in Brazil, as phentermine is in the United States. Diethylpropion is to be used with caution below the age of 12 years and in people with epilepsy due to the initiation of seizures in patients with epilepsy.
3.3. Phendimetrazine
Phendimetrazine is sympathomimetic amine available as 35mg tablets. It has side effects that are similar to, but less frequent than those associated with amphetamine.
3.4. Benzphetamine
Benzphetamine was approved in 1969 and is available in 25mg tablets. Like phendimetrazine, benzphetamine has side effects similar to, but less frequent than those associated with amphetamine.
3.5. Bupropion and naltrexone (Contrave)
The discovery of the synergistic activity of the bupropion/naltrexone combination was based on an understanding of the physiological responses to the neural circuitry affected by each drug, unlike most other obesity medications which were developed following observations of weight loss during therapeutic intervention. Bupropion is a monoamine reuptake inhibitor that increases the synaptic activity of dopamine and norepinephrine. Bupropion also stimulates the hypothalamic pro-opiomelanocortin (POMC) neurons to decrease appetite and increase energy expenditure. Subsequent to its release, POMC is cleaved into α-melanocyte-stimulating hormone (α-MSH) and β-endorphin to activate neural pathways with opposing effects on appetite[36]. Naltrexone, an opioid antagonist, blocks the orexigenic effects of P-endorphin activity, which in theory should enhance the hypophagic effect of a-MSH. Experimental data from preclinical studies demonstrated that compared to buproprion alone, bupropion plus naltrexone increased the firing rate of green fluorescent-labeled POMC neurons in murine brain slices and reduced food intake in lean and obese mice, confirming the synergy for the drug combination [3 7]. Next, a double-blind, proof-of-concept clinical trial was conducted which randomized 238 obese subjects to bupropion 300mg/d, naltrexone 50 mg/d, bupropion 300mg/d and naltrexone 50mg/d or placebo for treatment over 24 weeks. The combination was more effective in inducing weight loss than either individual component or the placebo (p<0.05–0.001). These results supported the hypothesis that naltrexone could block the feed-back inhibition of POMC neurons by β- endorphin and thereby reverse the weight loss plateau seen with bupropion alone[37].
Since the major adverse events leading to discontinuation in the proof-of-concept trial were nausea and vomiting attributable to naltrexone, a 24-week phase II trial evaluated three doses of naltrexone with bupropion to find the most tolerable dose with sufficient efficacy. The trial randomized 419 obese subjects to bupropion alone 400 mg/d, three combination doses of naltrexone/bupropion (NB) with naltrexone at 16 mg/d, 32 mg/d, or 48 mg and bupropion 400 mg/d, or placebo[38]. The placebo subtracted weight loss was greatest (4.65% of body weight) in the NB 32 mg/d group by last observation carried forward (LOCF) analysis due to higher drop outs in the NB 48 mg/d group from nausea and vomiting[38]. In a sub-study of this trial, total and visceral fat was measured by dual energy x-ray absorptiometry (DXA) in a subset of 107 participants. In the eighty subjects that completed the sub-study, there was a greater reduction in total body fat (NB 14% vs. placebo 4%) and visceral fat (NB 15% vs. 4.6%) in the NB combination group compared to placebo or bupropion alone [39].
The phase III trials consisted of four studies: Contrave Obesity Research 1 (COR-1), a study comparing sustained-release (SR) naltrexone 32 mg plus SR bupropion 360 mg/d (NB-32), SR naltrexone 16 mg plus SR bupropion 360mg/d (NB-16) and placebo in 1,742 healthy, dyslipidemic or controlled hypertensive obese and overweight subjects (BMI 27–45kg/m2) over 56 weeks, randomized in a 1:1:1 ratio. The study was analyzed using the last observation carried forward (LOCF), as requested by the FDA. At 56 weeks, the placebo group had a weight loss of 1.3% versus 5% in NB-16 and 6.1% in NB-32 groups (p<0.0001 drug groups vs placebo). The percentage of participants who lost at least 5% of body weight was 16% in the placebo group versus 39% and 48% in the NB-16 and NB-32 groups. The percentage of responders who lost at least 10% of body weight was 7%, 20%, and 25% in the same groups, respectively. The most common side effect was nausea affecting 30%, 27% and 5% in the NB-32, NB-16 and placebo groups respectively followed by headache, constipation, dizziness, vomiting and dry mouth. The number of serious adverse events did not differ between groups, and none were believed to be related to the treatment. There was a 1.5 mmHg increase in systolic blood pressure above baseline for 8 weeks followed by a return to baseline by 12 weeks, and no increase in depression or suicidality was observed. [40].
Cor-2 was a similar trial to Cor-1 but the 1,496 obese or overweight subjects were randomized in a 2:1 ratio to SR NB-32 or placebo and those in the NB-32 group who did not lose 5% of their body weight at week 28 were randomized in a 1:1 ratio to continue NB-32 or have NB increased to 48 mg (NB-48). Weight loss was 6.5% NB-32 vs. 1.9% placebo and 6.4% NB-32 vs. 1.2% placebo group at weeks 28 and 56, respectively. There were improvements in cardio-metabolic risk factors and participant-reported weight-related quality of life. Additionally, food craving was reduced[41].
Cor-BMOD randomized 793 obese and overweight subjects to placebo or SR NB-32 in a 1:3 ratio for 56 weeks accompanied by an intensive behavioral modification program. The behavior modification program consisted of a calorie-restricted diet, brisk walking for 180 min/week, strength straining and weekly meetings in groups of 10–20 lasting for 90 minutes and run by clinical psychologists, exercise specialists and registered dietitians at academic medical centers for the first 16 weeks, followed by bi-weekly meetings for the next 12 weeks and then monthly meetings for 28 weeks. Weight loss at 56 weeks was 5.1% in the placebo group vs. 9.3% in the NB-32 group[42].
Cor-DM randomized 505 obese and overweight subjects with type 2 diabetes to SR NB-32 or placebo in a 1:1 ratio for 56 weeks of treatment. Weight loss at 56 weeks was 5% in the NB-32 group vs. 1.8% in the placebo group, and hemoglobin Alc (HbAlc) dropped more in the NB-32 group (0.6% vs. 0.1%). Cardio-metabolic risk factors improved and side effects reflected those seen in the other phase III studies[43].
Several NB trials evaluated quality of life. A trial of SR NB-32 in 25 patients with major depressive disorder for 24 weeks gave significant improvements in the Montgomery-Asberg Depression Rating scale[44]. A study of weight-related quality of life at 56 weeks across four phase III trials showed a clinically and statistically meaningful improvement in the Impact of Weight on Quality of Life scale. The greatest improvement occurred in subjects that lost the most weight[45]. Since nausea is the most common side effect seen with NB-32, a causal relationship between nausea and weight loss was evaluated across three phase III, 56-week clinical trials. Nausea was found not to be a contributor to weight loss and was independent of the magnitude of weight loss[46]. Another analysis of data from four phase III trials found that the improvement in food cravings at week 8 was associated with weight loss at week 56[47].
NB-32 SR (Contrave) was approved for the treatment of obesity in 2014 and carries the black box warning about suicidal ideation and actions typical of anti-depressant medications. It is indicated for subjects with a BMI greater than 30 kg/m2 and for subjects with a BMI greater than 27 kg/m2 and weight-related co-morbidities. The medication comes as tablets containing 8 mg naltrexone SR and 90 mg of bupropion SR. The dosing begins with one tablet every morning for the first week, one tablet twice a day for the next week, two tablets in the morning and one in the evening for the next week and then two tablets twice a day. The escalation in dosing is to minimize nausea and dose escalation can be slowed, if nausea has not abated by the permissible time to make a dose increase. Contraindications to the use of NB-32 include uncontrolled hypertension, seizure disorders, anorexia nervosa, bulimia, during withdrawal from alcohol, benzodiazepines, antiepileptic drugs or barbiturates, use of a monoamine oxidase inhibitor within 14 days and chronic use of opioid medication, since naltrexone blocks the effect of opioids[48].
As part of the approval process, the FDA requested that Orexigen, the sponsor, perform a cardiovascular safety study to demonstrate that NB-32 doesn’t increase major events as determined by a non-inferiority hazard ratio of less than 1.4. Orexigen enrolled 8,910 overweight and obese subjects in an outcome study, LIGHT, driven by the number of major cardiovascular events including non-fatal stroke, non-fatal myocardial infarction, and cardiovascular death. The trial confirmed that after the 25% and 50% interim analyses of events, the non-inferiority hazard ratio was less than 2.0. The sponsor broke the blind and released confidential information halfway through the trial and invalidated the results before the noninferiority hazard ratio of 1.4 or less was reached, creating a need to repeat the trial under properly blinded conditions[49]. A second large-scale trial to evaluate major cardiovascular events in obese patients, CONVENE, began in 2015. This trial was terminated in 2016, and Orexigen released a statement that they plan to conduct a new study to satisfy the FDA requirement. The package insert for Contrave recommends that therapy should be evaluated after 12 weeks at the maintenance dose and discontinued, if the patient has not lost 5% of their body weight. A follow-up trial conducted according to these instructions showed that individuals with a weight loss of at least 5% at 16 weeks on NB-32 had a weight loss at one year of 11.7% of body weight[50].
3.6. Topiramate and phentermine (Qsymia)
Topiramate, a sulfamate derivative of fructose, is approved for the treatment of epilepsy and migraine headache prophylaxis. The actions on the CNS by topiramate are not completely understood, and rodent studies suggest that it acts as a neurostabilizer and may enhance thermogenesis[51–55]. The weight loss observed when it was used in the treatment of epilepsy led to clinical trials as a treatment for obesity [56]. In a dose escalation trial of 2 doses per day, the topiramate dose was increased biweekly by 16 mg to doses of 64, 96, 192, and 384 mg/d and the resulting weight losses were 5%, 4.8%, 6.3%, and 6.3%, respectively with the placebo group losing 2.6%. The adverse events included paresthesia, somnolence and difficulty with memory, concentration and attention such that 21% of the topiramate groups withdrew due to adverse events[57]. Topiramate development as a medication for the treatment of obesity was discontinued due to the adverse events. Based on clinical observations in a private practice, topiramate adverse events were mitigated and weight loss efficacy increased by the addition of phentermine, which led to clinical trials to approve the combination as a treatment for obesity. A 28-week trial randomized 755 obese subjects equally to placebo (Po), phentermine 7.5mg (Ph7.5), Phentermine 15mg (Ph-15), topiramate extended release (ER) 46 mg (T-46), topiramate ER 92 mg (T-92), Ph-7.5/T-46, and Ph15/T-92 for 28 weeks. At 28 weeks, subjects lost 1.7%, 5.13, 5.45, 6.06, 6.44, 8.46, and 9.21 in the Po, Ph-7.5, Ph-15, T-46, T-92, Ph-7.5/T-46, and Ph15/T-92 groups respectively. The cognitive battery showed only an impairment in the attention domain[58].
The phase III clinical trials of phentermine and topiramate consisted of two studies. The CONQUER study randomized 2,487 subjects with a BMI of 27–45 kg/m2 with 2 or more metabolic risk factors to placebo, or controlled release phentermine/topiramate at 7.5/46 mg, or phentermine/topiramate at 15/92 mg in a 2:1:2 ratio given once daily over 56 weeks. Weight loss was 1.2%, 7.8% and 10.2% in the placebo, 7.5/46 mg and 15/92 mg groups, respectively (p<0.0001 for drug groups vs placebo). The percentage of responders with > 5% weight loss_in the groups was 21%, 62% and 70%, respectively The percentage of responders with > 10% weight loss_in the groups was 7%, 37% and 48%, respectively. The most common adverse events in the 7.5/46 mg and 15/92 mg were dry mouth (11%, 19%), paresthesia (12%, 19%), constipation (9%, 11%), insomnia (1%, 5%), dizziness (4%, 7%), dysgeusia (6%, 10%), depression (0%, 4%) and anxiety (2%, 5%) greater than placebo, respectively. Discontinuations due to adverse events increased with dose and were 9% for placebo versus 12% and 19% for the 7.5/46 mg and 15/92 mg groups, respectively[59]. This trial had an optional extension called SEQUEL which continued 676 of the 866 eligible subjects on the same treatment for 108 weeks. Weight loss at 108 weeks was 1.8%, 9.3% and 10.5% in the placebo, 7.5/46 mg and 15/92 mg groups, respectively. The percentage of subjects discontinuing the study by week 108 was similar across groups, 3.1%, 4.5% and 4.4% respectively, and the drug was well tolerated[60].
A sub-study of the CONQUER trial evaluated 475 subjects with pre-diabetes and metabolic syndrome for up to 108 weeks who lost 2.5%, 10.9% and 12.1% in the placebo, 7.5/46 mg and 15/92 mg groups, respectively. The reduction in the annual incidence of conversion to diabetes was 70.5% and 78.7% in the 7.5/46 mg and 15/92 mg groups, respectively, compared to placebo. The ability to prevent diabetes and improvements in cardiovascular risk factors was related to the degree of weight loss[61]. A second sub-study of the CONQUER trial evaluated the 388 subjects with type 2 diabetes at 56 weeks who had an HbA1c of 6.8%. Weight loss was 2.7% in the placebo group and 9.4% in the 15/92 mg group, and HbA1c dropped 1.2% and 1.6%, respectively[62].
The EQUIP trial randomized 1,267 severely obese subjects with a BMI >35kg/m2 to placebo or controlled-release phentermine/topiramate at 3.75/23mg or 15/92mg in a 2:1:2 ratio given once daily over 56 weeks. Weight loss was 1.6%, 5.18% and 10.92% in placebo, 3.75/23mg and 15/92mg groups, respectively (p<0.0001 for all comparisons). In these groups, the percentage of subjects that lost 10% of baseline bodyweight was 13%, 27.7%, 67.7% respectively. At the highest dose, there was a significantly greater reduction of waist circumference, systolic blood pressure, diastolic blood pressure, fasting glucose, triglycerides, total cholesterol, low density lipoprotein (LDL) cholesterol and a significant increase in high density lipoprotein cholesterol (HDL). The drug-related adverse events that occurred most frequently were dry mouth, paresthesia, constipation, insomnia, dizziness and dysgeusia and some were more prevalent in the 15/92mg group. Adverse events caused fewer than 1% of patients to withdraw from the study and there were no drug-related serious adverse events[63].
The effect of phentermine/topiramate at 15/92mg was evaluated against placebo over 28 weeks in 45 subjects with sleep apnea. The apnea/hypopnea index declined by 31.5 events/hr in the phentermine/topiramate 15/92mg group compared to 16.6 events/hr in the placebo group and this was associated with a weight loss of 10.2% and 4.3% respectively. The overnight oxygen saturation improved significantly and blood pressure declined[64]. One of the most concerning potential side effects of topiramate is the association with oral cleft defects in infants when taken by birth mothers during early pregnancy. This outcome was evaluated in a study that compared the infants of women exposed to topiramate in the first trimester, women who had formerly taken topiramate and women who had never taken topiramate. The prevalence of oral cleft defects in these groups was 0.36%, 0.14% and 0.07%, respectively confirming the relationship of topiramate with oral cleft defects[65].
The two phase III trials of phentermine/topiramate were reviewed for their impact on health related quality of life as measured by the Impact of weight on Quality of Life-Lite (IWQOL- Lite) questionnaire and the SF-36 Physical Component Summary. Both questionnaires showed statistically significant improvements in quality of life with phentermine/topiramate in comparison to placebo that were mostly mediated by weight loss with an additional improvement in depression[66]. Two studies, both based on the phase III clinical trials, have evaluated the cost effectiveness of phentermine/topiramate. One evaluated the 4-year cost trajectories of real- world patients matched by age, gender and the metabolic profiles of the trial subjects before and after treatment with phentermine-topiramate. The costs of outpatient visits, emergency visits and medications were $2,292 to $3,378 lower per subject after treatment with phentermine- topiramate when treatment cost and potential side effects were excluded from the analysis[67]. The other analysis concluded that phentermine-topiramate is cost-effective, but that conclusion is dependent on the extent to which benefits are maintained post-medication cessation and that further studies are indicated[68].
3.7. Lorcaserin (Belviq)
As mentioned previously in section 2.3, a side effect caused by the non-specific serotonin agonists, fenfluramine and dexfenfluramine, was heart valve lesions, due to stimulation of the peripheral serotonin 2B receptor. There are at least 14 serotonin receptor subtypes that modulate diverse physiological functions, ranging from hallucinations to muscle contraction[69]. Development of serotonergic drugs as medications for obesity has advanced more rapidly since the serotonin 5-HT2C receptor was identified as the key regulator of satiety and feeding behavior in studies of mice with targeted receptor deletion[16]. Lorcaserin, a selective 5-HT2C receptor agonist (15-fold and 100-fold selectivity over the 5-HT2A and 5-HT2C receptors, respectively) was approved in 2012[70]. Evidence from a number of studies suggests that Lorcaserin has multiple psychological effects that contribute to weight loss, including elevation of satiety, reduction in craving and reduction in impulsivity[69].
The phase 2 trial compared lorcaserin 10mg/d, 15mg/d, 10 mg twice a day (bid) and placebo in a randomized, double-blind trial lasting 12 weeks in subjects with obesity (BMI 30–45 kg/m2) who were asked not to change their diet or physical activity[71]. The weight loss in trial completers was 1.8kg, 2.6kg, 3.6kg and 0.3kg, respectively. Lorcaserin was well-tolerated with the most frequent side effects being transient headache, nausea and dizziness. Based on this study, the 10mg bid dose was carried into phase 3 trials.
The phase 3 study called BLOOM randomly assigned 3182 obese or overweight subjects (BMI 27–30 kg/m2 with one or more obesity related co-morbidities and 30–45 kg/m2) to lorcaserin 10mg bid or placebo with a diet and exercise program. At the end of 1 year, weight loss was 5.81kg (7%) in the lorcaserin group and 2.16kg (3%) in the placebo group (p<0.001). During the first year, 47.5% of the lorcaserin group had lost at least 5% of their body weight vs 20.3% of the placebo group. More patients lost at least 10% of bodyweight in the lorcaserin group compared to the placebo group, 22.6% versus 7.6% respectively (p<0.001). Due to headache and dizziness, slightly more patients in the lorcaserin group withdrew from the trial compared to placebo, 2% versus 0.8%. [72]. At week 52, all placebo subjects and 2/3 of the lorcaserin subjects were continued on the same treatment and 1/3 of the lorcaserin subjects were switched to placebo for a second year of the trial. The weight of those switched to placebo regained to match the placebo curve and those on lorcaserin maintained a 2kg greater weight loss than placebo at 2 years. The drug was well-tolerated with headache, dizziness and nausea being the most frequent adverse events. At 2 years there was no increase in pulmonary hypertension or cardiac valvulopathy with lorcaserin[72].
The phase 3 trial called BLOSSOM randomized 4,008 obese and overweight subjects (BMI as in the BLOOM trial). Subjects were randomized to lorcaserin at doses of l0mg bid, l0mg/d or placebo in a 2:1:2 ratio for 1 year with a diet and exercise program, and weight loss was 5.8%, 4.7% and 2.8%, respectively. Adverse events were similar to the BLOOM trial and there was no increase in pulmonary hypertension or cardiac valvulopathy in the lorcaserin groups [73].
The phase 3 trial called BLOOM-DM enrolled 604 subjects with a BMI between 27 and 45 kg/m2 and type 2 diabetes on metformin, a sulfonylurea or both and HbAlc of 7%–10%. Subjects were randomized to lorcaserin l0mg/d, l0mg bid or placebo in a 1:1:1 ratio for 1 year with a diet and exercise program. Weight loss was 4.5%, 5% and 1.5% and the drop in HbAlc was 0.9%, 1% and 0.4% in the lorcaserin daily, lorcaserin bid and placebo groups, respectively. Adverse events were headache, back pain, nasopharyngitis and nausea[74].
In an effort to restrict the use of lorcaserin to responders, those who do not achieve a weight loss of 5% by week 12 are advised to stop lorcaserin and consider another medication. Weight loss following those instructions was 10.6 kg without diabetes and 9.3 kg with diabetes[75]. Lorcaserin was put in schedule IV of the DEA suggesting a low, but present potential for abuse. This decision conflicts with other research suggesting that lorcaserin, even at two fold higher doses, has no reinforcing effects in poly drug users and has a low potential for abuse[76]. Lorcaserin in combination with varenicline prolonged smoking abstinence, and in those who remained abstinent, limited weight gain[77]. The FDA, upon approval of lorcaserin, asked the sponsor to perform a safety trial of lorcaserin combined with phentermine. The trial randomized 238 overweight and obese subjects to Lorcaserin l0mg bid alone and with phentermine 15mg/d or phentermine 15mg bid, and weight loss at 12 weeks was 3.3%, 7% and 7.2%, respectively. There was a higher incidence of adverse effects and higher dropout rate in the phentermine 15 mg bid group compared to phentermine 15mg group suggesting that lorcaserin l0mg bid with phentermine 15mg/d had the best risk to benefit ratio[78]. As an exploratory endpoint, the Control of Eating (COE) questionnaire which looks at general cravings and the Food Craving Inventory which looks at cravings for specific foods were administered in the lorcaserin/phentermine clinical trial. The combination of diet and lorcaserin gave a significant reduction in craving that was enhanced dose-dependently by phentermine[79], These findings are consistent with a functional MRI study showing lorcaserin reduces activity in the reward centers in the brain[80].
Lorcaserin’s physiological mechanism was explored using DXA, food intake and energy expenditure (metabolic chamber) measurements for 7 days and 56 days. There was a reduction in body fat and energy intake, and no reduction in respiratory quotient or energy expenditure, indicating that weight loss was due to the drop in energy intake [81]. An analysis of pooled phase III studies showed that lorcaserin gave an equivalent reduction in HbAlc to naltrexone/bupropion and topiramate-phentermine, which gave more weight loss. It also slowed the progression of prediabetes to diabetes and increased the percentage of subjects with prediabetes that reverted to euglycemia, a finding that is associated with a longer prevention of diabetes[82]. Analysis of the three phase 3 clinical trials demonstrated a significant improvement in health related quality of life and depression measured by the Impact of Weight on Quality of Life-Lite questionnaire mostly attributable to weight loss[83]. Analysis of the three phase 3 trials also demonstrated a 2.04% incidence of new cardiac valvulopathy in the placebo group and 2.37% in the lorcaserin group giving a risk 1.15 for those attaining a 5% weight loss[84]. As there was no significant increase in valvulopathy with lorcaserin treatment, these data suggest that this drug does not adversely affect pre-existing valvular disease.
The antipsychotic drug olanzapine can induce weight gain and type 2 diabetes, and a study in mice recently demonstrated that olanzapine-induced weight gain and impaired glucose tolerance can be reversed by lorcaserin [85]. These studies suggest that olanzapine effects are mediated partly by antagonism of the serotonin 5HT-2C receptor, and that lorcaserin has potential to improve these unwanted side effects. These findings will hopefully be explored with future clinical trials in humans.
A time-release formulation 20 mg dose of lorcaserin has now been approved for use, and the pharmacokinetics have demonstrated drug exposure bioequivalency to lorcaserin immediate- release 10mg bid[86].
3.8. Liraglutide
Liraglutide is a glucagon-like peptide-1 (GLP-1) receptor agonist that was initially approved for the treatment of type 2 diabetes at a dose of 1.8mg/d and is now approved for obesity treatment at a dose of 3.0 mg/d. The hypoglycemic effect is insulin dependent and is not associated with significant symptomatic hypoglycemia unless combined with an insulin secretagogue. Liraglutide has a prolonged half-life compared to native GLP-1 and is a peptide that must be administered by subcutaneous injection[87]. Native GLP-1 is secreted by L-cells in the small intestine and acts on the pancreas to increase insulin transcription and inhibit glucagon secretion. It also acts on the intestinal tract to slow gastric emptying and gut motility. The use of liraglutide for the treatment of diabetes was found to be associated with weight loss which involved both a peripheral and a central mechanism. GLP-1 acts on the mesolimbic reward system in addition to hypothalamic and brainstem circuits regulating homeostatic feeding. GLP-1 crosses the blood brain barrier at the area postrema and directly stimulates POMC and other hypothalamic anorexigenic neurons, and may also act through intestinal vagal afferents to activate neurons in the nucleus tractus solitarius. Additionally, GLP-1 activates the reward system involving the ventral tegmental area and the nucleus acumbens[88].
A phase II dose-ranging study of liraglutide was done in obese subjects to examine the effects on food intake and body weight. Five hundred and sixty four obese subjects were randomized to orlistat 120mg tid (included as comparator), placebo and liraglutide 1.2 mg/d, 1.8 mg/d, 2.4 mg/d and 3.0 mg/d. Weight loss was 4.1kg, 2.8kg, 4.8kg, 5.5kg, 6.3kg and 7.2kg respectively at 20 weeks and more than 76% of subjects in the liraglutide 3.0mg group lost more than 5% body weight compared to 30% in the placebo group. Blood pressure was reduced in all liraglutide groups from baseline and the prevalence of pre-diabetes in the 3mg group was reduced by 96%. The most frequent adverse events were nausea and vomiting which were mainly transient and rarely led to discontinuation[89]. At 20 weeks, the trial was unblinded and extended to 2 years in 398 of the subjects, of which 268 completed the study. Subjects in the placebo group were switched to liraglutide 2.4 mg/d at 1 year and to 3.0 mg/d at 70 weeks. From randomization to year one, subjects given the 3.0mg dose of liraglutide lost 5.8 kg more weight than placebo and at year two weight loss was 3.0 kg in excess of placebo [90].
The phase III obesity and prediabetes clinical trial called SCALE randomized 3731 subjects with a BMI over 30 kg/m2, or BMI over 27kg/m2 with untreated hypertension or dyslipidemia, to liraglutide 3mg/d (2487) or placebo (1244) for 1 year with a diet and lifestyle program. Weight loss was 8.4 kg in the liraglutide group, which was 5.6kg more than placebo. A total of 63% of subjects administered liraglutide lost over 5% of bodyweight in comparison to 27% in the placebo group (p<0.001). In the liraglutide group 33.1% lost more than 10% of body weight compared to 10.6% in the placebo group (p<0.001). Of the study population, 78.5% were women and 61.2% had prediabetes. The most common side effects were transient nausea and vomiting and serious adverse events were seen in 6.2% of the liraglutide group and 5% of the placebo group[91]. The subjects with prediabetes remained in the blinded trial for 3 years and 1128 study participants (50%) on liraglutide and 714 placebo subjects (47%) finished the trial. The conversion to diabetes was 2% in the liraglutide group and 6% in the placebo group with the liraglutide group taking 2.7 times longer on average to convert[92].
The SCALE maintenance trial randomized 422 obese or overweight subjects who had lost 5% or more of their body weight with calorie restriction to liraglutide 3mg or placebo. The liraglutide group lost an additional 6.1% body weight compared to 0.2% additional body weight in the placebo group over 56 weeks, and all subjects were given diet, exercise and weight counseling during the trial[93]. The SCALE type 2 diabetes trial randomized 846 obese or overweight subjects with type 2 diabetes taking 0–3 medicines (metformin, sulfonylurea or thazolidinedione) with an HbA1c of 7% to 10% in a 2:1:1 ratio to Liraglutide 3.0mg/d, 1.8mg/d or placebo for 56 weeks. The weight loss was 6%, 4.7% and 2% respectively and the corresponding HbA1c drop was 1.3%, 1.1% and 0.3%[94]. The SCALE sleep apnea trial randomized 359 non-diabetic obese subjects with moderate or severe obstructive sleep apnea who were unwilling to use CPAP to 1:1 liraglutide 3mg/d or placebo for 32 weeks accompanied by diet and exercise instruction. The reduction in the apnea-hypopnea index was greater in the liraglutide group (12.2 vs. 6.1) as was the weight loss (5.7% vs 1.6%)[95].
Several studies evaluated changes across the four SCALE trials. Cardiovascular death, nonfatal myocardial infarction or nonfatal stroke were 1.54 events/1000 person years in the liraglutide group compared to 3.65 events/1000 person years in the placebo group (hazard ratio 0.42 with 95% confidence interval of 0.17 to 1.08), demonstrating that liraglutide did not increase cardiovascular risk[96]. Over the 56 weeks of the trials, more participants in the liraglutide 3mg/d group had elevations of serum amylase (9.4% vs 5.9%), lipase 43.5% vs 15.1%) and acute pancreatitis (0.3% vs. 0.1%) that was associated with gall stones 50% of the time. Pancreatic enzymes returned to normal after liraglutide was discontinued at the end of the trials[97]. The Impact of Weight on Quality of Life-Lite questionnaire showed an improvement in the liraglutide group (11.0) compared to the placebo group (8.1) but this did not reach clinical significance. These changes persisted over 3 years on liraglutide and the SF-36 health survey improved as well[98].
The comparative efficacy of liraglutide was evaluated above and below a BMI of 35kg/m2 and found that liraglutide performed equally well in both classes of obesity[99]. Efficacy of liraglutide was compared across racial groups and was shown to give similar weight loss[100]. Due to the concern about neuropsychiatric symptoms from a centrally acting obesity medication, these symptoms were evaluated with validated questionnaires and there was no significant difference detected between the liraglutide and placebo groups in depression, anxiety, suicidal ideation or suicidal behavior[101]. The pooled SCALE data was also used to evaluate early weight loss as a predictor for responders. It was found that a weight loss of greater than 4% at 16 weeks of treatment predicted a weight loss of greater than 5% after 56 weeks. Thus, the recommendations in the liraglutide package insert suggest that subjects with less than a 4% weight loss at 16 weeks discontinue the medication [102].
Other studies have shown that liraglutide slows gastric emptying acutely, and this effect at five and 16 weeks correlates with weight loss and not satiety[103]. Genetic polymorphisms in the GLP-1 receptor explain some of the variability of weight loss in obese women with polycystic ovarian syndrome. Carriers of one particular polymorphic allele of the GLP-1 receptor had a lower response to liraglutide than wild type carriers, while carriers of a different allele had a stronger response[104]. A pilot study evaluating liraglutide in subjects with binge eating disorder found that liraglutide reduced binge eating and increased weight loss compared to a placebo, but increased ghrelin significantly which may have attenuated the weight loss[105]. A study of 20 subjects with type 2 diabetes found that liraglutide decreased food preference for fat, reduced hunger scores and increased serum C-peptide after 20 days[106]. Liraglutide increased bone formation by 16% and prevented bone loss in women after weight loss with a low calorie diet[107]. Treatment for six months with liraglutide in subjects with type 2 diabetes improved arterial stiffness and left ventricular strain by lowering oxidative stress[108]. To evaluate improvement in antipsychotic-induced weight gain, a study randomized 103 subjects with schizophrenia who were overweight or obese, had prediabetes and were treated with olanzapine or clozapine. The liraglutide group lost 5.3 kg more than placebo, 64% developed normal glucose tolerance, and blood pressure and LDL cholesterol were significantly reduced[109].
Liraglutide 3mg is administered subcutaneously on a daily basis, and the dose is started at 0.6 mg and increased by that amount weekly until 3mg is reached. The medication is contraindicated during pregnancy and in people with a personal or family history of medullary thyroid cancer or multiple endocrine neoplasia type 2. There are warnings about thyroid c-cell cancers that are seen in rodents, but whether this applies to humans is not known. Relative to placebo, there is a low but elevated risk of acute pancreatitis, and there is an increase in gall stones and cholecystitis (1.5% vs 0.5%). Hypoglycemia can be severe when liraglutide is combined with insulin secretogogues. Heart rate was increased an average of 2–3 bpm, but tachycardia (heart rate greater than 100 bpm) was seen in 6% vs. 4% in the placebo group. Acute renal failure has occurred, probably in association with dehydration from vomiting or diarrhea. Hypersensitivity reactions including angioedema and anaphylaxis have been reported, and there was a numerical increase in suicidal behavior and ideation (0.2% vs. 0)[110].
Table 4 compares phase III trial data for currently available drugs including percent weight loss, percent of intent to treat (ITT), completers that lost 5% and 10% of body weight, and percent of subjects that dropped out of study. In addition to liraglutide, there are four other GLP-1 receptor agonists available in the United States for the treatment of diabetes, and they all result in weight loss[111], Semaglutide is the first GLP-1 receptor agonist available in an oral formulation, and is currently in phase III trials (reviewed in section 4.2).
Table 4.
Percent weight loss (ITT and Completer), Categorical (5% and 10%) and Drops (%)
| Drug | ITT* (p value) |
Completer (p value) |
5% wt loss (p value) |
10% wt loss (p value) |
Drops (%) |
|---|---|---|---|---|---|
| Amphetamine Congeners [8] | N/A (N/A**) | 0.23 kg/wk (<0.05, >placebo) |
N/A (N/A) | N/A (N/A) | 48.5 |
|
Lorcaserin [71] |
5.81±0.16% (<0.001) |
7.1±0.2% (<0.001) |
45.5% (<0.001) |
44.6% | |
| Placebo | 2.16±0.14% | 3.0±0.2% | 20.3% | 54.9% | |
|
Topiramate/ Phentermine [58,62] |
(<0.0001) | (<0.0001) | (<0.05) | (<0.5) | |
| Placebo | 1.6% | 2.1% | 17.3% | 7.4% | 47.1% |
| 23/3.75 mg | 5.1% | 6.7% | 44.9% | 18.8% | 39% |
| 46/7.5 mg | 7.8% | 9.6% | 62% | 37% | 31% |
| 92/15 mg | 10.9% | 14.4% | 66.7% | 47.2% | 36% |
|
Bupropion/Naltrexone [39] |
(<0.0001) | (<0.0001) | (<0.0001) | (<0.0001) | |
| Placebo | 1.3% | 1.8% | 16% | 7% | 50% |
| 32/360mg | 6.1% | 8.1% | 48% | 25% | 50% |
|
Liraglutide [90] |
8.0±6.7% (<0.0001) |
9% (<0.0001) |
63.2% (<0.001) |
33.1% (<0.001) |
36.6% |
| Placebo | 2.6±5.7% | 3.8% | 27.1% | 10.6% (<0.001) |
28.1% |
ITT = Intent to Treat Analysis
N/A = Not Applicable
4.0. Future Centrally Acting Anti-Obesity Drugs
This section on future anti-obesity drugs focuses on tesofensine, since it is the only CNS acting anti-obesity drug that has reached an advanced stage of development. All other CNS acting drugs are in early in clinical development and other than the limited information on semaglutide and setmelanotide have no published trials for obesity treatment[112].
4.1. Tesofensine
Tesofensine is a unique, presynaptic reuptake inhibitor of dopamine, norepinephrine, and serotonin with a half-life of over 8 days[113, 114], The potent, triple-action mechanism of this drug results in greater weight loss than most other anti-obesity drugs. Although it was originally developed to treat Alzheimer’s and Parkinson’s diseases, efficacy criteria were not met and the unintended side effect of weight loss was observed in multiple clinical trials [115] [116]. A phase II study of tesofensine in obese, nondiabetic subjects was subsequently conducted. After a two week run-in period, 203 obese subjects with a body mass index (BMI) between 30 and 40kg/m2 were randomized to placebo, 0.25mg, 0.5mg or l.0mg per day orally along with an energy restricted diet for 24 weeks. One hundred and one subjects completed the study and the mean weight loss was 4.5%, 9.2% and 10.6% of initial body weight, as shown in Figure 1 (p<0.0001 vs. placebo). Beneficial effects on blood triglycerides, cholesterol, insulin and HbAlc were observed. There was no change in blood pressure at the 0.5mg/d dose, but the lmg dose caused an increase compared to placebo. Pulse rate increased 7.4 bpm in the 0.5mg/d tesofensine group (p=0.0001). Mild side effects included an increase in the incidence of headaches at all doses (>25% of subjects), and an increase in mood alterations at the lmg dose[117].
Figure 1: Mean change from baseline in bodyweight.
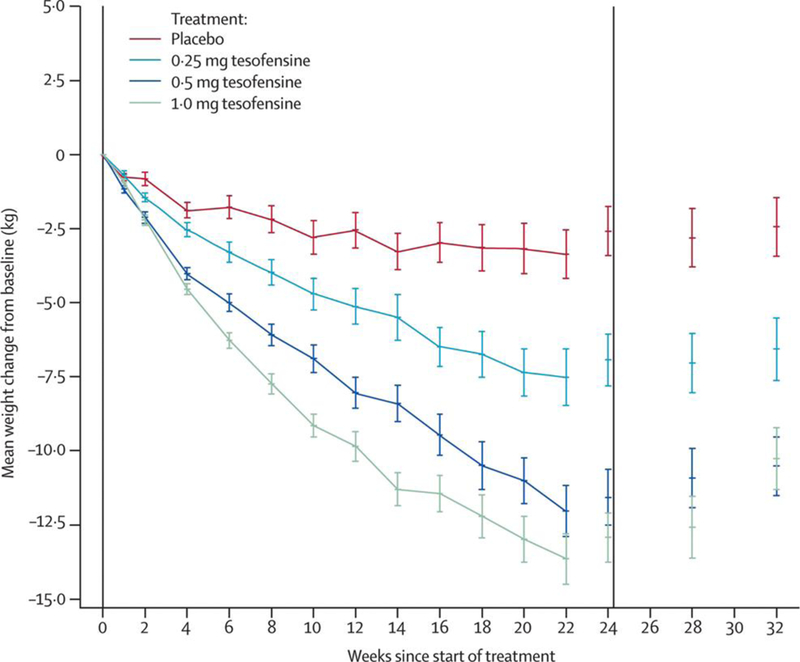
Data are values for patients completing each scheduled visit, and last observation carried forward (values for the full intention-to-treat population with the last observations carried forward). Error bars show SE. Vertical lines indicate start and end of treatment. Reproduced with permission from Elsevier Ltd. The Lancet, 2008;372:1906–1913
In a rat model of diet-induced obesity (DIO), tesofensine treatment produced robust weight loss accompanied by hypophagia. To identify the neural pathways modulating weight loss and hypophagia, reversal of these effects was investigated using various monoaminergic receptor antagonists co-administered with tesofensine. Tesofensine significantly reduced food intake in the first 12 hours of administration in a dose dependent manner, with a maximum effect after 3 days. The hypophagic effect slowly dissipated and returned to control levels by day 15, but the reduction in body weight continued for the duration of the 16 day experiment. Receptor antagonists were added in subsequent experiments that measured acute hypophagia over the first 12 hours of tesofensine treatment. An α1-adrenoreceptor antagonist eliminated most of the hypophagia and a D1 dopamine receptor antagonist showed partial inhibition. Antagonists of the α2-adrenoreceptor, dopamine D2, dopamine D3, and serotonin 2A/C receptors did not reduce tesofensine activity[118].
In an attempt to further define the inhibitory action on monoamine transporters, another study measured dopamine levels in the brains of chow-fed and DIO rats. The dopamine levels in DIO rats were low in the nucleus accumbens and pre-frontal cortex, but levels in the chow-fed rats were not. Tesofensine treatment normalized the dopamine levels in the DIO rats, but had no effect on the chow-fed animals, suggesting that the anti-obesity effects of tesofensine are due, at least in part, to positive modulation of central dopaminergic activity [119]. Results from both studies results suggest that the hypophagic effect of the drug is primarily due to activation of noradrenaline and dopamine circuitry in DIO rats, and other mechanisms such as energy expenditure and motor activity may drive the continued weight loss after food intake returns to normal.
Positron emission tomography (PET) was employed to study dopamine presynaptic transporter occupancy in the human brain after different doses of tesofensine. Between 0.125 and lmg, there was a dose-dependent blockade of binding, and striatal dopamine transporter occupancy varied between 18% and 77%. in a sigmoid- shaped Emax (maximum effect attributable to the drug) relationship. The sigmoid Emax model is a mathematical model that describes the concentration- effect relationship of a drug where the curve gets more sigmoid in shape as the number of molecules binding to the drug receptor increases. The maximal occupancy was 80% and the dose at half occupancy was 0.25mg with a serum level of 4ng/mL. These results suggested that tesofenine-induced reduction in food intake was partially mediated by up-regulation of dopaminergic pathways due to blockade of presynaptic reuptake [120].
A clinical study in humans evaluated the effects of tesofensine on appetite suppression and energy expenditure to clarify the underlying mechanisms. Thirty two healthy males were treated with 2mg/d of tesofensine for 1 week and then randomized to l.0mg/d or placebo for another 7 days. They were asked to maintain their normal levels of food intake and physical activity. Their energy expenditure was assessed in a respiratory chamber before and after treatment, body composition was measured by DXA, appetite was measured using visual analog scales in conjunction with a standardized meal. Even while attempting to maintain food intake, subjects lost 1.8kg over the 2 weeks. Tesofensine treatment increased visual analog scale ratings of satiety and increased 24 hour fat oxidation relative to placebo. Although a change in total energy expenditure was not detected, sleeping energy expenditure was significantly greater. These results suggest that tesofensine induces weight loss primarily by reducing food intake with a small increase in metabolic rate[121], A phase 2 trial focused on long term effects on appetite sensations in subjects given 0.25, 0.5 or 1 mg tesofensine or placebo for 24 weeks. There was a dose-dependent suppression of hunger over the first 12 weeks which correlated with the amount of weight lost over the course of the entire 6 month study, even though the effect on satiety faded as weight loss continued to progress[122].
Dopamine circuitry modulates motivation-related behavior and is associated with addiction, necessitating an analysis of the abuse potential of tesofensine. A study was conducted on 52 recreational stimulant users who received a single dose of placebo, tesofensine, bupropion and atomoxetine in a double-blind, random-order, crossover trial. D-amphetamine was used as a positive control and as expected, a single dose gave indications of abuse potential. Tesofensine, bupropion, atomoxetine and placebo were significantly different from D-amphetamine and did not have significant euphoric or other psychoactive effects, indicating a lack of abuse potential [123].
The dose limiting adverse effects of tesofensine commonly observed in clinical trials were elevations in blood pressure and pulse rate. Postulating that the increase in blood pressure was due to adrenergic stimulation, a study was conducted on tesofensine-treated rats, and acute increases in blood pressure and heart rate were observed. This rise in blood pressure and pulse rate was reversed by a beta-1-adrenergic blocking medication without affecting the reduction in food intake. An angiotensin blocker did not affect the reduction in food intake, but only partially blocked the increase in blood pressure and pulse rate suggesting that tesofensine may increase sympathetic activity [124]. A phase III trial will be completed in 2018 to study change in body weight in 372 adults with obesity treated with placebo, 0.25mg or 0.5mg tesofensine for 24 weeks.
4.2. Semaglutide
Semaglutide is a GLP-1 receptor agonist that was first developed as a weekly injectable agent and now has a unique oral formulation being tested in phase III trials. In the SUSTAIN-6 phase III trial for the injectable formulation, patients on the 1mg weekly dose lost 4.3kg after 104 weeks[125]. A phase II, 26 week trial has been completed using the oral formulation in 630 patients with type 2 diabetes. Tablets were given once daily in doses ranging from 2.5mg to 40mg, and weight loss was dose-dependent with a maximum weight loss of 6.9kg for the 40mg dose (p<0.001). Similar to other injectable GLP-1 receptor agonists, the most common adverse events were gastrointestinal[126]. A trial of semaglutide for the treatment of obesity has been initiated, but the trials presently published relate to the treatment of type 2 diabetes which is usually associated with obesity.
Setmelanotide
Setmelanotide is a melanocortin-4 receptor agonist. Two subjects with a rare genetic deficiency of proopiomelanocortin causing early onset severe obesity with hyperphagia that requires glucocorticoid replacement to sustain life were treated with setmelanotide. Both subjects had a reduction in hunger and food intake. One subject lost 51 kg over 42 weeks and the other lost 20.5 kg over 12 weeks. Setmelanotide is currently being developed for orphan diseases associated with obesity such as a deficiency of proopiomelanocortin[127]
The Path Forward for Obesity Medications
Looking back through the history of obesity treatment, we note that the first low carbohydrate diet was the Banting Diet, published in 1863. This diet became so popular that it went through 10 editions between 1863 and 1902[128]. Diet still plays an important role in weight loss, but longterm pharmacotherapies with limited side effects are critical for maintaining weight loss. The first jejunoileal bypass for obesity was reported in the 1950’s[128], and the operation did not become popular until the 1970’s. More advanced procedures are used now and surgery still has a significant place in the treatment of obesity, giving the largest weight loss, best maintenance of weight loss, and reversal of insulin resistance.
Advances in the clinical development of CNS-acting obesity drugs have resulted in currently available drugs that are capable of reducing food intake, reducing craving, increasing satiety and possibly increasing energy expenditure. We are now in a stage of treating obesity with lower dose drug combinations acting through multiple monoamine pathways. As reviewed in the section on presently available obesity medications, two examples of these combination therapies most recently approved are bupropion/naltrexone and phentermine/topiramate.
Tesofensine is clearly the most effective single agent for obesity treatment to this point, but concerns about its effect on blood pressure and pulse rate may require combining it with a beta-1 adrenergic blocking agent. Will it be possible to achieve even greater long-term efficacy from centrally acting pharmacotherapies with a reduction in side effects? An obesity treatment strategy with potential is the combination of centrally acting and peripherally acting pharmacotherapies to increase efficacy. With a drug that acts on a peripheral target, there is no activity of downstream pathways involving other physiological systems as with drugs that act high in the CNS. Orlistat is an inhibitor of gastrointestinal lipase that reduces absorption of fat[129]. A study was conducted to determine whether orlistat and sibutramine gave greater weight loss than either treatment alone, as both were approved for long-term use. Unfortunately, the resulting weight loss was no different from that of sibutramine[130].
Another central/peripheral drug combination successfully demonstrated significantly greater weight loss than either monotherapy. Canagliflozin, a renal sodium-glucose transporter inhibitor, is a diabetes drug that stimulates weight loss. A 26-week trial combined canagliflozin with phentermine to study percent change in body weight. The drug combination group had an 8% reduction in body weight compared to 4.6% for phentermine, 2.6% for canagliflozin, and 1.1% for placebo[131]. The combination group had greater improvements in blood pressure and heart rate.
Although tesofensine, a drug that acts in the central nervous system by inhibiting the reuptake of norepinephrine, dopamine and serotonin is the only obesity drug currently in development with published clinical trials, it is likely that future drugs for the treatment of obesity will focus more on peripheral mechanisms in an attempt to achieve efficacy while minimizing adverse effects. Hopefully tesofensine will take its place with the other obesity medications now approved and expand the clinical options available to physicians treating obesity. Presumably, future medications for obesity treatment will favor those with peripheral mechanisms of action which will be likely to reduce the adverse events and maximize efficacy.
Key Points.
Obesity medications acting on the central nervous system (CNS) started with the discovery of amphetamine. Many of the previously approved centrally acting drugs have been withdrawn from the market due to unanticipated side effects. By acting high in system, these drugs have a greater potential to affect other physiological systems and engender increased adverse events.
Centrally-acting drugs for chronic diseases like obesity and hypertension have advanced from single drugs to lower dose combinations with better efficacy and fewer side effects than monotherapies. Finally, there is an evolution toward drugs that act on the disease target, like angiotensin blockers acting directly on the blood vessels in hypertension.
Obesity is now entering the era of lower dose combinations of centrally acting drugs with the approval of bupropion/naltrexone (Contrave) and topiramate/phentermine (Qsymia).
Tesofensine has entered into phase III trials for the treatment of obesity. Tesofensine is a centrally acting reuptake inhibitor of serotonin, dopamine and norepinephrine and gives more weight loss than any single agent presently approved for obesity treatment.
Semaglutide, a GLP-1 analog that gave a 6.9 kg weight loss at 24 weeks in trials for treatment of type 2 diabetes, has oral and intravenous formulations in clinical trials for an anti-obesity indication.
Acknowledgements:
This work is supported in part by 1 U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health which funds the Louisiana Clinical and Translational Science Center and in part on work that was supported by the National Institutes of Health under an award (T32 A T004094) from the National Center for Complementary and Integrative Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosures:
Frank Greenway has served on the advisory boards for BAROnova, Eisai Inc., Curves, Novo Nordisk, Microbiome Therapeutics, Orexigen, Pamlab, PlenSat, Zaluvida and Zafgen. He has consulted for Basic Research, General Nutrition Corporation, Neothetics, Takeda, and Tech Enterprises
References
- 1.Health implications of obesity. National Institutes of Health Consensus Development Conference Statement. Ann Intern Med. 1985. July;103(1):147–51. [PubMed] [Google Scholar]
- 2.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994. December 1;372(6505):425–32. [DOI] [PubMed] [Google Scholar]
- 3.Williamson DA, Ravussin E, Wong ML, Wagner A, Dipaoli A, Caglayan S, et al. Microanalysis of eating behavior of three leptin deficient adults treated with leptin therapy. Appetite. 2005. August;45(1):75– 80. [DOI] [PubMed] [Google Scholar]
- 4.Nathanson M The Central Action of beta-aminopropylbenzene (Benzedrine) clinical observations. JAMA. 1937;108():528–31. [Google Scholar]
- 5.Lesses MF, Myerson A. Human autonomic pharmacology. XVI. Benzedrine sulfate as an aid in the treatment of obesity. 1938. Obes Res. 1994. May;2(3):286–92. [DOI] [PubMed] [Google Scholar]
- 6.Harris SC, Ivy AC, Searle LM. The mechanism of amphetamine-induced loss of weight; a consideration of the theory of hunger and appetite. J Am Med Assoc. 1947. August 23;134(17):1468–75. [DOI] [PubMed] [Google Scholar]
- 7.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–98. [DOI] [PubMed] [Google Scholar]
- 8.Scoville BA. Review of amphetamine-like drugs by the Food and Drug Administration. . Washington, DC: US Government Printing Office; 1976. [Google Scholar]
- 9.Byrne-Quinn E, Grover RF. Aminorex (Menocil) and amphetamine: acute and chronic effects on pulmonary and systemic haemodynamics in the calf. Thorax. 1972. January;27(1):127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michelakis ED, Weir EK. Anorectic drugs and pulmonary hypertension from the bedside to the bench. Am J Med Sci. 2001. April;321(4):292–9. [DOI] [PubMed] [Google Scholar]
- 11.Gurtner HP. Pulmonary hypertension, “plexogenic pulmonary arteriopathy” and the appetite depressant drug aminorex: post or propter? Bull Eur Physiopathol Respir. 1979. Sep-Oct;15(5):897–923. [PubMed] [Google Scholar]
- 12.Weintraub M Long-term weight control: the National Heart, Lung, and Blood Institute funded multimodal intervention study. Clin Pharmacol Ther. 1992. May;51(5):581–5. [DOI] [PubMed] [Google Scholar]
- 13.Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996. August 29;335(9):609–16. [DOI] [PubMed] [Google Scholar]
- 14.Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997. August 28;337(9):581–8. [DOI] [PubMed] [Google Scholar]
- 15.Rothman RB, Baumann MH. Serotonergic drugs and valvular heart disease. Expert Opin Drug Saf. 2009. May;8(3):317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995. April 6;374(6522):542–6. [DOI] [PubMed] [Google Scholar]
- 17.Greenway F, Herber D, Raum W, Herber D, Morales S. Double-blind, randomized, placebo- controlled clinical trials with non-prescription medications for the treatment of obesity. Obes Res. 1999. July;7(4):370–8. [DOI] [PubMed] [Google Scholar]
- 18.Greenway FL. Clinical studies with phenylpropanolamine: a metaanalysis. Am J Clin Nutr. 1992. January;55(1 Suppl):203S–5S. [DOI] [PubMed] [Google Scholar]
- 19.Kernan WN, Viscoli CM, Brass LM, Broderick JP, Brott T, Feldmann E, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med. 2000. December 21;343(25):1826–32. [DOI] [PubMed] [Google Scholar]
- 20.Domingue JN. Phenylpropanolamine contained in cold remedies and risk of hemorrhagic stroke. Neurology. 2007. July 17;69(3):320; author reply −1. [DOI] [PubMed] [Google Scholar]
- 21.Astrup A, Greenway FL, Ling W, Pedicone L, Lachowicz J, Strader CD, et al. Randomized controlled trials of the D1/D5 antagonist ecopipam for weight loss in obese subjects. Obesity (Silver Spring). 2007. July;15(7):1717–31. [DOI] [PubMed] [Google Scholar]
- 22.Simiand J, Keane M, Keane PE, Soubrie P. SR 141716, a CB1 cannabinoid receptor antagonist, selectively reduces sweet food intake in marmoset. Behav Pharmacol. 1998. March;9(2):179–81. [PubMed] [Google Scholar]
- 23.Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S, Group RI- ES. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005. April 16– 22;365(9468):1389–97. [DOI] [PubMed] [Google Scholar]
- 24.Tam J, Szanda G, Drori A, Liu Z, Cinar R, Kashiwaya Y, et al. Peripheral cannabinoid-1 receptor blockade restores hypothalamic leptin signaling. Mol Metab. 2017. October;6(10):1113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam J, Hinden L, Drori A, Udi S, Azar S, Baraghithy S. The therapeutic potential of targeting the peripheral endocannabinoid/CB1 receptor system. Eur J Intern Med. 2018. March;49:23–9. [DOI] [PubMed] [Google Scholar]
- 26.Kim SH, Lee YM, Jee SH, Nam CM. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res. 2003. September;11(9):1116–23. [DOI] [PubMed] [Google Scholar]
- 27.James WP, Astrup A, Finer N, Hilsted J, Kopelman P, Rossner S, et al. Effect of sibutramine on weight maintenance after weight loss: a randomised trial. STORM Study Group. Sibutramine Trial of Obesity Reduction and Maintenance. Lancet. 2000. December 23–30;356(9248):2119–25. [DOI] [PubMed] [Google Scholar]
- 28.James WP, Caterson ID, Coutinho W, Finer N, Van Gaal LF, Maggioni AP, et al. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med. 2010. September 2;363(10):905–17. [DOI] [PubMed] [Google Scholar]
- 29.The amphetamine appetite suppressant saga. Prescrire Int. 2004. February;13(69):26–9. [PubMed] [Google Scholar]
- 30.Chen K Ephedrine and related substances. Medicine. 1930;9:1–119. [Google Scholar]
- 31.Greenway FL. The safety and efficacy of pharmaceutical and herbal caffeine and ephedrine use as a weight loss agent. Obes Rev. 2001. August;2(3):199–211. [DOI] [PubMed] [Google Scholar]
- 32.Haller CA, Benowitz NL. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med. 2000. December 21;343(25):1833–8. [DOI] [PubMed] [Google Scholar]
- 33.Shekelle PG, Hardy ML, Morton SC, Maglione M, Mojica WA, Suttorp MJ, et al. Efficacy and safety of ephedra and ephedrine for weight loss and athletic performance: a meta-analysis. JAMA. 2003. March 26;289(12):1537–45. [DOI] [PubMed] [Google Scholar]
- 34.Dong Z, Xu L, Liu H, Lv Y, Zheng Q, Li L. Comparative efficacy of five long-term weight loss drugs: quantitative information for medication guidelines. Obes Rev. 2017. December;18(12):1377–85. [DOI] [PubMed] [Google Scholar]
- 35.Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J. 1968. February 10;1(5588):352–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005. May;8(5):571–8. [DOI] [PubMed] [Google Scholar]
- 37.Greenway FL, Whitehouse MJ, Guttadauria M, Anderson JW, Atkinson RL, Fujioka K, et al. Rational design of a combination medication for the treatment of obesity. Obesity (Silver Spring). 2009. January;17(1):30–9. [DOI] [PubMed] [Google Scholar]
- 38.Greenway FL, Dunayevich E, Tollefson G, Erickson J, Guttadauria M, Fujioka K, et al. Comparison of combined bupropion and naltrexone therapy for obesity with monotherapy and placebo. J Clin Endocrinol Metab. 2009. December;94(12):4898–906. [DOI] [PubMed] [Google Scholar]
- 39.Smith SR, Fujioka K, Gupta AK, Billes SK, Burns C, Kim D, et al. Combination therapy with naltrexone and bupropion for obesity reduces total and visceral adiposity. Diabetes Obes Metab. 2013. September;15(9):863–6. [DOI] [PubMed] [Google Scholar]
- 40.Greenway FL, Fujioka K, Plodkowski RA, Mudaliar S, Guttadauria M, Erickson J, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010. August 21;376(9741):595–605. [DOI] [PubMed] [Google Scholar]
- 41.Apovian CM, Aronne L, Rubino D, Still C, Wyatt H, Burns C, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring). 2013. May;21(5):935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wadden TA, Foreyt JP, Foster GD, Hill JO, Klein S, O’Neil PM, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring). 2011. January;19(1):110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hollander P, Gupta AK, Plodkowski R, Greenway F, Bays H, Burns C, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013. December;36(12):4022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McElroy SL, Guerdjikova AI, Kim DD, Burns C, Harris-Collazo R, Landbloom R, et al. Naltrexone/Bupropion combination therapy in overweight or obese patients with major depressive disorder: results of a pilot study. Prim Care Companion CNS Disord. 2013;15(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolotkin RL, Chen S, Klassen P, Gilder K, Greenway FL. Patient-reported quality of life in a randomized placebo-controlled trial of naltrexone/bupropion for obesity. Clin Obes. 2015. October;5(5):237– 44. [DOI] [PubMed] [Google Scholar]
- 46.Hong K, Herrmann K, Dybala C, Halseth AE, Lam H, Foreyt JP. Naltrexone/Bupropion extended release-induced weight loss is independent of nausea in subjects without diabetes. Clin Obes. 2016. October;6(5):305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalton M, Finlayson G, Walsh B, Halseth AE, Duarte C, Blundell JE. Early improvement in food cravings are associated with long-term weight loss success in a large clinical sample. Int J Obes (Lond). 2017. August;41(8):1232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.2017 [cited; Contrave Prescribing Instructions]. Available from: https://contrave.com/wp- content/uploads/2017/05/Contrave PI.pdf
- 49.Nissen SE, Wolski KE, Prcela L, Wadden T, Buse JB, Bakris G, et al. Effect of Naltrexone- Bupropion on Major Adverse Cardiovascular Events in Overweight and Obese Patients With Cardiovascular Risk Factors: A Randomized Clinical Trial. JAMA. 2016. March 8;315(10):990–1004. [DOI] [PubMed] [Google Scholar]
- 50.Fujioka K, Plodkowski R, O’Neil PM, Gilder K, Walsh B, Greenway FL. The relationship between early weight loss and weight loss at 1 year with naltrexone ER/bupropion ER combination therapy. Int J Obes (Lond). 2016. September;40(9):1369–75. [DOI] [PubMed] [Google Scholar]
- 51.Coomans CP, Geerling JJ, van den Berg SA, van Diepen HC, Garcia-Tardon N, Thomas A, et al. The insulin sensitizing effect of topiramate involves KATP channel activation in the central nervous system. Br J Pharmacol. 2013. October;170(4):908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia. 2000;41 Suppl 1:S17–20. [PubMed] [Google Scholar]
- 53.Gibbs JW,, Sombati S, DeLorenzo RJ, Coulter DA. Cellular actions of topiramate: blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia. 2000; 41 Suppl 1:S10–6. [DOI] [PubMed] [Google Scholar]
- 54.Picard F, Deshaies Y, Lalonde J, Samson P, Richard D. Topiramate reduces energy and fat gains in lean (Fa/?) and obese (fa/fa) Zucker rats. Obes Res. 2000. December;8(9):656–63. [DOI] [PubMed] [Google Scholar]
- 55.Richard D, Picard F, Lemieux C, Lalonde J, Samson P, Deshaies Y. The effects of topiramate and sex hormones on energy balance of male and female rats. Int J Obes Relat Metab Disord. 2002. March;26(3):344–53. [DOI] [PubMed] [Google Scholar]
- 56.Ben-Menachem E, Axelsen M, Johanson EH, Stagge A, Smith U. Predictors of weight loss in adults with topiramate-treated epilepsy. Obes Res. 2003. April;11(4):556–62. [DOI] [PubMed] [Google Scholar]
- 57.Bray GA, Hollander P, Klein S, Kushner R, Levy B, Fitchet M, et al. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res. 2003. June;11(6):722–33. [DOI] [PubMed] [Google Scholar]
- 58.Aronne LJ, Wadden TA, Peterson C, Winslow D, Odeh S, Gadde KM. Evaluation of phentermine and topiramate versus phentermine/topiramate extended-release in obese adults. Obesity (Silver Spring). 2013. November;21(11):2163–71. [DOI] [PubMed] [Google Scholar]
- 59.Gadde KM, Allison DB, Ryan DH, Peterson CA, Troupin B, Schwiers ML, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011. April 16;377(9774): 1341–52. [DOI] [PubMed] [Google Scholar]
- 60.Garvey WT, Ryan DH, Look M, Gadde KM, Allison DB, Peterson CA, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr.2012. February;95(2):297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garvey WT, Ryan DH, Henry R, Bohannon NJ, Toplak H, Schwiers M, et al. Prevention of type 2 diabetes in subjects with prediabetes and metabolic syndrome treated with phentermine and topiramate extended release. Diabetes Care. 2014. April;37(4):912–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garvey WT, Ryan DH, Bohannon NJ, Kushner RF, Rueger M, Dvorak RV, et al. Weight-loss therapy in type 2 diabetes: effects of phentermine and topiramate extended release. Diabetes Care. 2014. December;37(12):3309–16. [DOI] [PubMed] [Google Scholar]
- 63.Allison DB, Gadde KM, Garvey WT, Peterson CA, Schwiers ML, Najarian T, et al. Controlled- release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012. February;20(2):330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winslow DH, Bowden CH, DiDonato KP, McCullough PA. A randomized, double-blind, placebo- controlled study of an oral, extended-release formulation of phentermine/topiramate for the treatment of obstructive sleep apnea in obese adults. Sleep. 2012. November 1;35(11):1529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mines D, Tennis P, Curkendall SM, Li DK, Peterson C, Andrews EB, et al. Topiramate use in pregnancy and the birth prevalence of oral clefts. Pharmacoepidemiol Drug Saf. 2014. October;23(10):1017– 25. [DOI] [PubMed] [Google Scholar]
- 66.Kolotkin RL, Gadde KM, Peterson CA, Crosby RD. Health-related quality of life in two randomized controlled trials of phentermine/topiramate for obesity: What mediates improvement? Qual Life Res. 2016. May;25(5):1237–44. [DOI] [PubMed] [Google Scholar]
- 67.Li J, Reaven NL, Funk SE, McGaughey K, Neovius M. 4-Year Cost Trajectories in Real-World Patients Matched to the Metabolic Profiles of Trial Subjects Before/After Treatment with Phentermine- Topiramate. Drugs Real World Outcomes. 2015. June;2(2):143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finkelstein EA, Kruger E, Karnawat S. Cost-Effectiveness Analysis of Qsymia for Weight Loss. Pharmacoeconomics. 2015. July;33(7):699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roth BL, Willins DL, Kristiansen K, Kroeze WK. 5-Hydroxytryptamine2-family receptors (5- hydroxytryptamine2A, 5-hydroxytryptamine2B, 5-hydroxytryptamine2C): where structure meets function. Pharmacol Ther. 1998. September;79(3):231–57. [DOI] [PubMed] [Google Scholar]
- 70.[cited; Available from: http://www.diabetesincontrol.com/fda-approves-lorcaserin-belviq-for- treatment-of-obesity/
- 71.Smith SR, Prosser WA, Donahue DJ, Morgan ME, Anderson CM, Shanahan WR, et al. Lorcaserin (APD356), a selective 5-HT(2C) agonist, reduces body weight in obese men and women. Obesity (Silver Spring). 2009. March;17(3):494–503. [DOI] [PubMed] [Google Scholar]
- 72.Smith SR, Weissman NJ, Anderson CM, Sanchez M, Chuang E, Stubbe S, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010. July 15;363(3):245–56. [DOI] [PubMed] [Google Scholar]
- 73.Fidler MC, Sanchez M, Raether B, Weissman NJ, Smith SR, Shanahan WR, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011. October;96(10):3067–77. [DOI] [PubMed] [Google Scholar]
- 74.O’Neil PM, Smith SR, Weissman NJ, Fidler MC, Sanchez M, Zhang J, et al. Randomized placebo- controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring). 2012. July;20(7):1426–36. [DOI] [PubMed] [Google Scholar]
- 75.Smith SR, O’Neil PM, Astrup A, Finer N, Sanchez-Kam M, Fraher K, et al. Early weight loss while on lorcaserin, diet and exercise as a predictor of week 52 weight-loss outcomes. Obesity (Silver Spring). 2014. October;22(10):2137–46. [DOI] [PubMed] [Google Scholar]
- 76.Shram MJ, Schoedel KA, Bartlett C, Shazer RL, Anderson CM, Sellers EM. Evaluation of the abuse potential of lorcaserin, a serotonin 2C (5-HT2C) receptor agonist, in recreational polydrug users. Clin Pharmacol Ther. 2011. May;89(5):683–92. [DOI] [PubMed] [Google Scholar]
- 77.Hurt RT, Croghan IT, Schroeder DR, Hays JT, Choi DS, Ebbert JO. Combination Varenicline and Lorcaserin for Tobacco Dependence Treatment and Weight Gain Prevention in Overweight and Obese Smokers: A Pilot Study. Nicotine Tob Res. 2017. August 1;19(8):994–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith SR, Garvey WT, Greenway FL, Zhou S, Fain R, Pilson R, et al. Coadministration of lorcaserin and phentermine for weight management: A 12-week, randomized, pilot safety study. Obesity (Silver Spring). 2017. May;25(5):857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rebello CJ, Nikonova EV, Zhou S, Aronne LJ, Fujioka K, Garvey WT, et al. Effect of Lorcaserin Alone and in Combination with Phentermine on Food Cravings After 12-Week Treatment: A Randomized Substudy. Obesity (Silver Spring). 2018. February;26(2):332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farr OM, Upadhyay J, Gavrieli A, Camp M, Spyrou N, Kaye H, et al. Lorcaserin Administration Decreases Activation of Brain Centers in Response to Food Cues and These Emotion- and Salience- Related Changes Correlate With Weight Loss Effects: A 4-Week-Long Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Diabetes. 2016. October;65(10):2943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martin CK, Redman LM, Zhang J, Sanchez M, Anderson CM, Smith SR, et al. Lorcaserin, a 5- HT(2C) receptor agonist, reduces body weight by decreasing energy intake without influencing energy expenditure. J Clin Endocrinol Metab. 2011. March;96(3):837–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nesto R, Fain R, Li Y, Shanahan W. Evaluation of lorcaserin on progression of prediabetes to type 2 diabetes and reversion to euglycemia. Postgrad Med. 2016. May;128(4):364–70. [DOI] [PubMed] [Google Scholar]
- 83.Kolotkin RL, Crosby RD, Wang Z. Health-related quality of life in randomized controlled trials of lorcaserin for obesity management: what mediates improvement? Clin Obes. 2017. December;7(6):347–53. [DOI] [PubMed] [Google Scholar]
- 84.Weissman NJ, Smith SR, Fain R, Hall N, Shanahan WR. Effects of lorcaserin on pre-existing valvulopathy: A pooled analysis of phase 3 trials. Obesity (Silver Spring). 2017. January;25(1):39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lord CC, Wyler SC, Wan R, Castorena CM, Ahmed N, Mathew D, et al. The atypical antipsychotic olanzapine causes weight gain by targeting serotonin receptor 2C. J Clin Invest. 2017. September 1;127(9):3402– 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hurren KM, Dunham MW. Pharmacokinetic drug evaluation of extended release lorcaserin for the treatment of obesity. Expert Opin Drug Metab Toxicol. 2017. August;13(8):891–6. [DOI] [PubMed] [Google Scholar]
- 87.Madsbad S Exenatide and liraglutide: different approaches to develop GLP-1 receptor agonists (incretin mimetics)--preclinical and clinical results. Best Pract Res Clin Endocrinol Metab. 2009. August;23(4):463–77. [DOI] [PubMed] [Google Scholar]
- 88.van Bloemendaal L, Ten Kulve JS, la Fleur SE, Ijzerman RG, Diamant M. Effects of glucagon-like peptide 1 on appetite and body weight: focus on the CNS. J Endocrinol. 2014. April;221(1):T1–16. [DOI] [PubMed] [Google Scholar]
- 89.Astrup A, Rossner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009. November 7;374(9701):1606–16. [DOI] [PubMed] [Google Scholar]
- 90.Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME, et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond). 2012. June;36(6):843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N Engl J Med. 2015. July 2;373(1):11–22. [DOI] [PubMed] [Google Scholar]
- 92.le Roux CW, Astrup A, Fujioka K, Greenway F, Lau DCW, Van Gaal L, et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double-blind trial. Lancet. 2017. April 8;389(10077):1399–409. [DOI] [PubMed] [Google Scholar]
- 93.Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013. November;37(11):1443–51. [DOI] [PubMed] [Google Scholar]
- 94.Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjoth TV, et al. Efficacy of Liraglutide for Weight Loss Among Patients With Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. JAMA. 2015. August 18;314(7):687–99. [DOI] [PubMed] [Google Scholar]
- 95.Blackman A, Foster GD, Zammit G, Rosenberg R, Aronne L, Wadden T, et al. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: the SCALE Sleep Apnea randomized clinical trial. Int J Obes (Lond). 2016. August;40(8):1310–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davies MJ, Aronne LJ, Caterson ID, Thomsen AB, Jacobsen PB, Marso SP, et al. Liraglutide and cardiovascular outcomes in adults with overweight or obesity: A post hoc analysis from SCALE randomized controlled trials. Diabetes Obes Metab. 2017. September 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Steinberg WM, Buse JB, Ghorbani MLM, Orsted DD, Nauck MA, Committee LS, et al. Amylase, Lipase, and Acute Pancreatitis in People With Type 2 Diabetes Treated With Liraglutide: Results From the LEADER Randomized Trial. Diabetes Care. 2017. July;40(7):966–72. [DOI] [PubMed] [Google Scholar]
- 98.Kolotkin RL, Gabriel Smolarz B, Meincke HH, Fujioka K. Improvements in health-related quality of life over 3 years with liraglutide 3.0 mg compared with placebo in participants with overweight or obesity. Clin Obes. 2018. February;8(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.le Roux C, Aroda V, Hemmingsson J, Cancino AP, Christensen R, Pi-Sunyer X. Comparison of Efficacy and Safety of Liraglutide 3.0 mg in Individuals with BMI above and below 35 kg/m(2): A Post-hoc Analysis. Obes Facts. 2017;10(6):531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ard J, Cannon A, Lewis CE, Lofton H, Vang Skjoth T, Stevenin B, et al. Efficacy and safety of liraglutide 3.0 mg for weight management are similar across races: subgroup analysis across the SCALE and phase II randomized trials. Diabetes Obes Metab. 2016. April;18(4):430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O’Neil PM, Aroda VR, Astrup A, Kushner R, Lau DCW, Wadden TA, et al. Neuropsychiatric safety with liraglutide 3.0 mg for weight management: Results from randomized controlled phase 2 and 3a trials. Diabetes Obes Metab. 2017. November;19(11):1529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fujioka K, O’Neil PM, Davies M, Greenway F, D CWL, Claudius B, et al. Early Weight Loss with Liraglutide 3.0 mg Predicts 1-Year Weight Loss and is Associated with Improvements in Clinical Markers. Obesity (Silver Spring). 2016. November;24(11):2278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Halawi H, Khemani D, Eckert D, O’Neill J, Kadouh H, Grothe K, et al. Effects of liraglutide on weight, satiation, and gastric functions in obesity: a randomised, placebo-controlled pilot trial. Lancet Gastroenterol Hepatol. 2017. December;2(12):890–9. [DOI] [PubMed] [Google Scholar]
- 104.Jensterle M, Pirs B, Goricar K, Dolzan V, Janez A. Genetic variability in GLP-1 receptor is associated with inter-individual differences in weight lowering potential of liraglutide in obese women with PCOS: a pilot study. Eur J Clin Pharmacol. 2015. July;71(7):817–24. [DOI] [PubMed] [Google Scholar]
- 105.Robert SA, Rohana AG, Shah SA, Chinna K, Wan Mohamud WN, Kamaruddin NA. Improvement in binge eating in non-diabetic obese individuals after 3 months of treatment with liraglutide - A pilot study. Obes Res Clin Pract. 2015. May-Jun;9(3):301–4. [DOI] [PubMed] [Google Scholar]
- 106.Inoue K, Maeda N, Kashine S, Fujishima Y, Kozawa J, Hiuge-Shimizu A, et al. Short-term effects of liraglutide on visceral fat adiposity, appetite, and food preference: a pilot study of obese Japanese patients with type 2 diabetes. Cardiovasc Diabetol. 2011. December 1;10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iepsen EW, Lundgren JR, Hartmann B, Pedersen O, Hansen T, Jorgensen NR, et al. GLP-1 Receptor Agonist Treatment Increases Bone Formation and Prevents Bone Loss in Weight-Reduced Obese Women. J Clin Endocrinol Metab. 2015. August;100(8):2909–17. [DOI] [PubMed] [Google Scholar]
- 108.Lambadiari V, Pavlidis G, Kousathana F, Varoudi M, Vlastos D, Maratou E, et al. Effects of 6- month treatment with the glucagon like peptide-1 analogue liraglutide on arterial stiffness, left ventricular myocardial deformation and oxidative stress in subjects with newly diagnosed type 2 diabetes. Cardiovasc Diabetol. 2018. January 8;17(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Larsen JR, Vedtofte L, Jakobsen MSL, Jespersen HR, Jakobsen MI, Svensson CK, et al. Effect of Liraglutide Treatment on Prediabetes and Overweight or Obesity in Clozapine- or Olanzapine-Treated Patients With Schizophrenia Spectrum Disorder: A Randomized Clinical Trial. JAMA Psychiatry. 2017. July 1;74(7):719–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.FDA. 2014 [cited; Liraglutide label]. Available from: https://www.accessdata.fda.gov/drugsatfda docs/label/2014/206321Qrig1s000lbl.pdf
- 111.Tran KL, Park YI, Pandya S, Muliyil NJ, Jensen BD, Huynh K, et al. Overview of Glucagon-Like Peptide-1 Receptor Agonists for the Treatment of Patients with Type 2 Diabetes. Am Health Drug Benefits. 2017. June;10(4):178–88. [PMC free article] [PubMed] [Google Scholar]
- 112.Bessesen DH, Van Gaal LF. Progress and challenges in anti-obesity pharmacotherapy. Lancet Diabetes Endocrinol. 2018. March;6(3):237–48. [DOI] [PubMed] [Google Scholar]
- 113.Lehr T, Staab A, Tillmann C, Nielsen EO, Trommeshauser D, Schaefer HG, et al. Contribution of the active metabolite M1 to the pharmacological activity of tesofensine in vivo: a pharmacokinetic- pharmacodynamic modelling approach. Br J Pharmacol. 2008. January;153(1):164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Thatte U NS-2330 (Neurosearch). Curr Opin Investig Drugs. 2001. November;2(11):1592–4. [PubMed] [Google Scholar]
- 115.Hauser RA, Salin L, Juhel N, Konyago VL. Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson’s disease. Mov Disord. 2007. February 15;22(3):359–65. [DOI] [PubMed] [Google Scholar]
- 116.Astrup A, Meier DH, Mikkelsen BO, Villumsen JS, Larsen TM. Weight loss produced by tesofensine in patients with Parkinson’s or Alzheimer’s disease. Obesity (Silver Spring). 2008. June;16(6):1363–9. [DOI] [PubMed] [Google Scholar]
- 117.Astrup A, Madsbad S, Breum L, Jensen TJ, Kroustrup JP, Larsen TM. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet. 2008. November 29;372(9653):1906–13. [DOI] [PubMed] [Google Scholar]
- 118.Axel AM, Mikkelsen JD, Hansen HH. Tesofensine, a novel triple monoamine reuptake inhibitor, induces appetite suppression by indirect stimulation of alpha1 adrenoceptor and dopamine D1 receptor pathways in the diet-induced obese rat. Neuropsychopharmacology. 2010. June;35(7):1464–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hansen HH, Jensen MM, Overgaard A, Weikop P, Mikkelsen JD. Tesofensine induces appetite suppression and weight loss with reversal of low forebrain dopamine levels in the diet-induced obese rat. Pharmacol Biochem Behav. 2013. September;110:265–71. [DOI] [PubMed] [Google Scholar]
- 120.Appel L, Bergstrom M, Buus Lassen J, Langstrom B. Tesofensine, a novel triple monoamine reuptake inhibitor with anti-obesity effects: dopamine transporter occupancy as measured by PET. Eur Neuropsychopharmacol. 2014. February;24(2):251–61. [DOI] [PubMed] [Google Scholar]
- 121.Sjodin A, Gasteyger C, Nielsen AL, Raben A, Mikkelsen JD, Jensen JK, et al. The effect of the triple monoamine reuptake inhibitor tesofensine on energy metabolism and appetite in overweight and moderately obese men. Int J Obes (Lond). 2010. November;34(11):1634–43. [DOI] [PubMed] [Google Scholar]
- 122.Gilbert JA, Gasteyger C, Raben A, Meier DH, Astrup A, Sjodin A. The effect of tesofensine on appetite sensations. Obesity (Silver Spring). 2012. March;20(3):553–61. [DOI] [PubMed] [Google Scholar]
- 123.Schoedel KA, Meier D, Chakraborty B, Manniche PM, Sellers EM. Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin Pharmacol Ther. 2010. July;88(1):69–78. [DOI] [PubMed] [Google Scholar]
- 124. Bentzen BH, Grunnet M, Hyveled-Nielsen L, Sundgreen C, Lassen JB, Hansen HH. Antihypertensive treatment preserves appetite suppression while preventing cardiovascular adverse effects of tesofensine in rats. Obesity (Silver Spring). 2013. May;21(5):985–92. [DOI] [PubMed] [Google Scholar]
- 125.Marso SP, Holst AG, Vilsboll T. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2017. March 2;376(9):891–2. [DOI] [PubMed] [Google Scholar]
- 126.Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA. 2017. October 17;318(15):1460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kuhnen P, Clement K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. N Engl J Med. 2016. July 21;375(3):240–6. [DOI] [PubMed] [Google Scholar]
- 128.Banting W Letter on corpulence, addressed to the public. London, UK: Harris and Sons; 1863. [DOI] [PubMed] [Google Scholar]
- 129.Sjostrom L, Rissanen A, Andersen T, Boldrin M, Golay A, Koppeschaar HP, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998. July 18;352(9123):167–72. [DOI] [PubMed] [Google Scholar]
- 130.Sari R, Balci MK, Cakir M, Altunbas H, Karayalcin U. Comparison of efficacy of sibutramine or orlistat versus their combination in obese women. Endocr Res. 2004. May;30(2):159–67. [DOI] [PubMed] [Google Scholar]
- 131.Hollander P, Bays HE, Rosenstock J, Frustaci ME, Fung A, Vercruysse F, et al. Coadministration of Canagliflozin and Phentermine for Weight Management in Overweight and Obese Individuals Without Diabetes: A Randomized Clinical Trial. Diabetes Care. 2017. May;40(5):632–9. [DOI] [PubMed] [Google Scholar]