

Abstract
An effective method of combating infectious diseases is the deployment of hand-held devices at the point-of-care (POC) for screening or self-monitoring applications. There is a need for very sensitive, low-cost and quantitative diagnostic devices. In this study, we present a low-cost, multiplexed fluorescence detection platform that has a high sensitivity and wide dynamic range. Our system features inexpensive 3×3 mm interference filter with a high stopband rejection, sharp transition edges, and greater than 90% transmission in the passband. In addition to the filters we improve signal-to-noise ratio by leveraging time for accuracy using a charge-integration-based readout. The fluorescence sensing platform provides a sensitivity to photon flux of ∼1×104 photons/mm2sec and has the potential for 2 to 3 orders of magnitude improvement in sensitivity over standard colorimetric detection that uses colored latex microspheres. We also detail the design, development, and characterization of our low-cost fluorescence detection platform and demonstrate 100% and 97.96% reduction in crosstalk probability and filter cost, respectively. This is achieved by reducing fi r dimensions and ensuring appropriate channel isolation in a 2×2 array configuration. Practical considerations with low-cost interference fi system design, analysis, and system performance are also discussed. The performance of our platform is compared to that of a standard laboratory array scanner. We also demonstrate the detection of antibodies to human papillomavirus (HPV16) E7 protein, as a potential biomarker for early cervical cancer detection in human plasma.
Keywords: Point-of-care, diagnostics, multiplexed, fluorescence, colorimetry, limit of detection
1. Introduction
The relatively lower performance of lateral fl w assays (LFAs) compared to central laboratory assay techniques is a major factor limiting the widespread implementation of fluorescent LFAs in hand-held diagnostic devices (Tang et al., 2016). Other issues include the cost and bulkiness of optical and electronic components. The development of low-cost, compact, and disposable devices, most notably those intended for use in point-of-care (POC) settings have received a lot of attention in the last 2 decades (Hu et al., 2014; Kumar et al., 2015; Lee et al., 2013). Several groups have attempted to develop portable and sensitive platforms aimed at improving the sensitivity and limit of detection (LOD) for POC diagnostic devices. However, many high performance fluorescence systems rely on bulky optical components and expensive detectors including cooled charged coupled devices (CCD), complementary metal-oxide semiconductor (CMOS) cameras, or photomultiplier tubes (PMTs). Many systems intended for POC application still use bulky laser light sources, expensive interference filters, and bulky optics such as lenses and mirrors (Lee et al., 2013), which require very precise alignment. High-Q filters with sharp transition edges and high transmission are critical for high-sensitivity fluorescence applications (Dandin et al., 2007); however, these high performance filters are prohibitively expensive.
Many LFA-based devices rely on colorimetric detection usually using an enzyme as in an enzyme-linked immunosorbent assay (ELISA) or colored labels such as latex microspheres, incorporating several signal enhancement or amplification strategies including colloidal gold, silver enhanced gold, cellulose nanobeads, and carbon black nanoparticles (Hu et al., 2013; O’Farrell, 2015). The use of fluorescence t labels toward improving the sensitivity of LFAs has also been investigated and well-reported in the literature (Banerjee et al., 2010; O’Farrell, 2015). In particular, several groups have investigated the use of fluorescent labels for signal enhancement in compact microfluidic detection platforms (Lee et al., 2013; Yetisen et al., 2013). The relatively low sensitivity reported in many systems (Khreich et al., 2010; Lee et al., 2013; Li et al., 2010; Williams et al., 2014), is mostly due to the difficulty in deploying low-cost and efficient filter which significantly impact the signal-to-noise (SNR) ratio (Dandin et al., 2007; Yetisen et al., 2013). In the past decade, several studies have reported the use of gel filter (Venkatraman and Steckl, 2015), absorption filter (Lefevre et al., 2015; Ryu et al., 2011), and cross polarizers (Banerjee et al., 2010). The aforementioned systems show low performance in terms of analytical sensitivity and limit of detection. Others are expensive or very difficult to fabricate, and thus not suitable for low-cost, high volume applications requiring high sensitivity or for applications in resource-poor settings.
Previously, we have reported the use of organic light emitting diodes (OLEDs), adapted from flat panel display technology, as the excitation source in a single-plex detection system (Katchman et al., 2016; Obahiagbon et al., 2016; Smith et al., 2016). We demonstrated the detection of antibodies to human papillomavirus (HPV16) E7 protein using expensive interference filters Compared to single-plex systems, low-cost, multiplexed diagnostic platforms are useful for disease diagnosis, making it possible for simultaneous detection of multiple analytes from a single patient sample volume. Multiplexed systems have the potential to reduce cost per test, increase speed, and minimize both false positives and negatives.
In this work, we present a compact, multiplexed fluorescene detection platform. We detail the design and assembly of our 2×2 array platform, using inexpensive high-Q interference filters and readout electronics.The system leverages time integration of the output signal to improve accuracy with the capability of providing quantitative results. Our platform is suitable for low-cost applications as the optoelectronic and optomechanic parts are designed from inexpensive off-the-shelf components, making it amenable to mass manufacturing.Visual colorimetric methods are still the dominant means of signal readout in most lateral flow assays.We compare labels used in a typical colorimetric assay to fluorescent labels with the aim of highlighting the benefits and properly quantifying the advantages of fluorescent without compounding the interpretation with assay dependencies including assay efficiency, repeatability, and reproducibility. By estimating the number of colored latex microspheres required for test line visibility (Mansfield, 2015), we make a comparison with fluorescent latex microspheres of similar size. We quantify the LOD of our fluorescence-based approach, and benchmark our platform against a clinical laboratory scale fluorescence detection device. Our platform is designed to read LFA test strips with 2 to 3 orders of magnitude enhancement in sensitivity over visual or reflected-light colorimetric readouts. Our platform delivers clinical-level-sensitivity in point-of-care fluorescence based detection devices.
This paper is structured as follows: in section 2, we describe the principle of operation, materials, and techniques used in our inexpensive interference filter based fluorescence system design and fabrication. In section 3, we detail the experimental results and discuss system performance. We present initial tests performed to evaluate the system performance and discuss the characterization results, including crosstalk as a function of concentration. Finally, we conclude with a discussion on the application of our system in disease diagnosis by demonstrating the detection of antibodies to HPV16 E7 protein, as a potential biomarker for early cervical cancer detection in human plasma.
2. Materials and methods
Previously, many groups have reported the use of interference filter in the design of compact fluorescence detection systems (Banerjee et al., 2010; Dandin et al., 2007). However, some of the major challenges with such systems are the tight process control required in fabrication, difficulty in integration, and the prohibitive cost of interference filter (Banerjee et al., 2010). Furthermore, low-cost systems do not favor the use of laser excitation sources, PMTs, CCD, or CMOS based camera systems. Here, we detail the design, construction, assembly, and testing of our prototype 4-site (2×2) fluorescence detection platform. The architecture borrows inspiration from typical laboratory-based fluorescence measurement devices like the epi-fluorescence microscope, array scanners, spectro-photometers, and spectrofluorometers. Common to these instruments is an optical path, which usually includes an excitation light source, focusing optics, filter a photo detector, electronic readout, and signal processing circuits. We adopt a transillumination detection architecture with bottom-side illumination, eliminating expensive focusing optics, substituting laser/tungsten-halogen sources for light emitting diodes (LEDs), and using photodiodes in place of PMTs, CCD or CMOS cameras, thus ensuring a reduction in overall instrument cost and size.
2.1. Principle of operation
A functional diagram for our prototype sensing platform is shown in Fig. 1. The system is divided into 5 modules detailed below. The excitation module (M1) comprises an 11.5 mm pitched 2×2 array of inorganic light emitting diodes (LEDs) that have a peak emission λ = 520 nm (SunLED, XZM2DG45S). The LEDs are mounted on a printed circuit board (PCB) as shown in Fig. 2(B). The LED broadband spectrum is filter using a low-cost 3×3 mm excitation (520/40 nm) interference filter (Chroma Technology Corp.) mounted directly on each LED in the array. Filter mounting and assembly is discussed in section 2.2. The LEDs function as excitation sources, which are sequentially turned on for each position in the array (designated hereafter as sites/channels A0, A1, A2, and A3) to interrogate a fluorescence biorecognition site or test line/spot in the sample chamber module (M2). The sample chamber is capable of holding glass microscope slides with or without mounted nitrocellulose membranes. The emission and signal readout module (M3) captures the weak fluorescence signal re-emitted at a longer wavelength (Stokes shift). The emitted fluorescence passes through an emission filter (Chroma Technologies, 605/70 nm) and is detected using coaxially aligned photodiodes (Advanced Photonix Incorporated, Camarillo, CA, USA, PDB-C139, responsivity, R(λ) of ∼0.2 to 0.3 A/W from 550 to 650 nm and a 59° viewing angle). The photodiodes are connected in photovoltaic mode as part of a low-noise charge-integration amplifier readout (LMC6041IN, National Semiconductor). The readout circuit is shown in Fig. 2(A). The magnitude of the current generated by the photodiode, I, is proportional to the fluorescent power or intensity reaching the photodiode (Banerjee et al., 2010; Guilbault, 1990; Sze, 2007; Thrush, 2004) and could be written as
| (1) |
where, c, is the molar concentration; l, is the optical path length through the sample; є(λ) is the extinction coefficient or molar absorptivity of the fluorescence I0(λ), is the excitation source (LED) intensity; K, is a lumped constant that depends on photodiode responsivity, R(λ); collection efficiency of the system, ηLCE ; excitation, TX (λ) and emission, TM (λ) filter transmission, and flurophore quantum yield (QYfl(λ)). Optical spectra are shown in Fig. 3(E). The photodiode responsivity R(λ), is plotted on the secondary axis. The output of the system is a fluorescence concentration-dependent voltage ramp which is inversely correlated with the slope of the voltage-time profile and written as
| (2) |
where IB represents total noise current (Idark+Ileak+Inoise). Equation 2 models sensor output voltage as a function of system parameters, photodiode output current and time. This equation is useful for predicting the output voltage profile over time and determining system performance during the design stage. Details of the derivation and an example predicting the system response are presented in the supplemental information.(See Table S1 and Figure S3 By leveraging time for accuracy, this technique ensures high sensitivity, as we can detect weak fluorescence signals above the noise fluorescence or. Furthermore, this readout method enables inexpensive electronic processing as high speed circuits and/or high accuracy analog-to-digital converters are not required since the system averages the measured signal over a relatively long time period (30 to 60 sec). At the center of the system, providing sequencing and control is the microcontroller module (M4) implemented with an Arduino Uno, that monitors, acquires, and digitizes the amplifier output voltage, recording time taken for the output voltage to rise to the voltage rail of the amplifier (ramp-time, RT). The RT is inversely correlated with the sample concentration as a higher fluorophore concentration implies more light, a steeper slope, and thus, a smaller RT (Obahiagbon et al., 2016). We define detection time (DT) as the ramp time different between a negative control or reference sample and a sample under test. Detection time is directly correlated with sample concentration or fluorescence intensity. We envision a completely miniaturized configuration that would be paired with a smartphone via bluetooth communication. User instructions and data would be issued, transmitted, and saved on the smartphone or remotely on the cloud. The microcontroller module was programmed to control the ON and OFF sequence of the precisely aligned LED-photodiode pairs. Each LED-photodiode pair is sequentially turned on, and the time taken to reach a predefine threshold voltage is recorded and stored. The display and data connectivity module (M5) comprises an OLED display (optional) and a smartphone or computer that prompts the user, analyzes the data, and displays/reports the results of a test.
Figure 1:
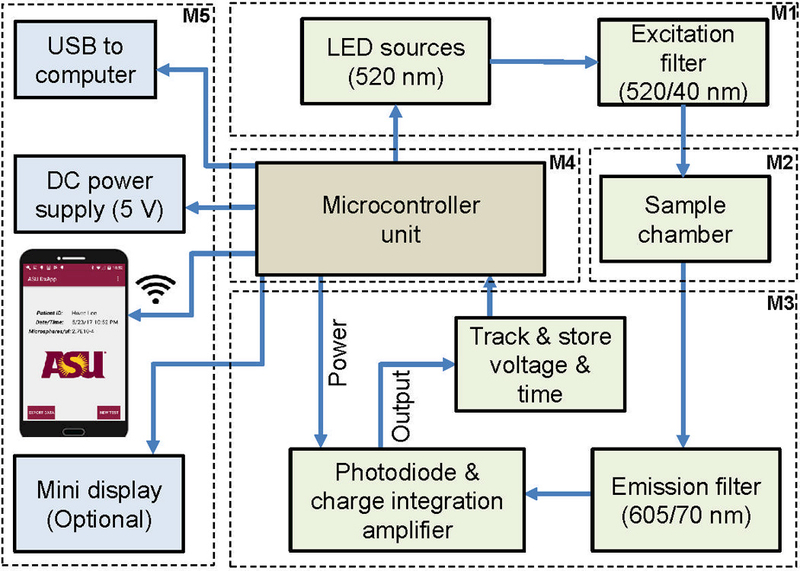
Functional block diagram of the system. The platform includes: an excitation module (M1) consisting of LEDs (520 nm center) and excitation filters (520/40 nm); a sample chamber module (M2, that accommodates a microscope slide, nitrocellulose membrane, or cassette; an emission and signal readout module (M3), consisting of emission filters (605/70 nm), photodiodes, and charge integration readout electronics; a microcontroller module (M4), at the heart of sequence control, power, and data processing; a display and connectivity module (M5) which includes an optional mini display that prompts and presents results to the user. A smartphone with a user interface establishes data connectivity via Bluetooth, shows device status, and displays results with the capability of uploading the information for cloud-based processing and interpretation by a primary care physician.
Figure 2:
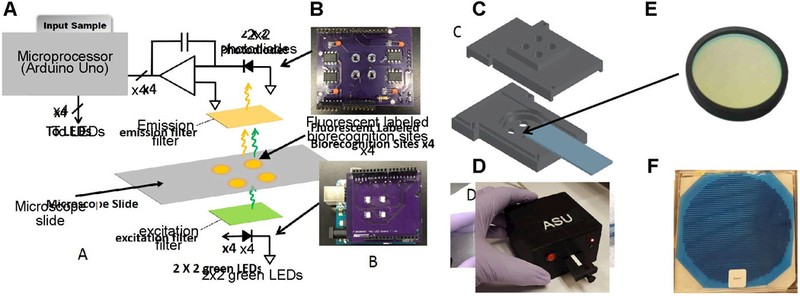
Schematic, prototype board, CAD design, and filters in a 2×2 array fluorescence detection platform. (A) Circuit schematic showing charge-integration amplifier readout circuit, 2×2 biorecognition sites on a microscope slide and LEDs used as the excitation source (B) Printed circuit board (PCB) showing 2×2 photodiode and amplifiers as well as the 2×2 LEDs soldered (C) 3D printed optical assembly showing apertures for excitation sources, holders for filters, slot for microscope slide, and aperture to fit photodiodes on the top piece (emission filter slot on reverse side) (D) Stacked microcontroller board, PCBs, and 3D printed assembly packaged in a 3D printed enclosure (E) 25 mm filter typically used in fluorescence microscopy; arrow points to 25.3 mm diameter slot in 3D printed assembly shown (F) 6 inch filter dielectric stack deposited on a 1 mm thick substrate and diced to ∼3×3 mm (not to scale).
Figure 3:
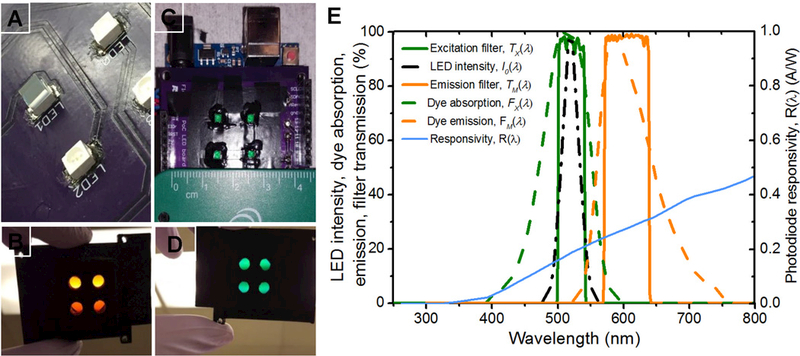
Low-cost filter assembly, alignment on LEDs, apertures, and optical design of a 2×2 array fluorescence detection platform. (A) 3×3 mm 520/40 nm green excitation filter mounted on an LED and cured with a clear UV curing adhesive (top left, marked LED1) (B) 2×2 aperture array in a 3D-printed optical isolation for orange emission filters (a single 25 mm diameter filter ring on reverse side) (C) 3×3 mm, 520/40 nm green excitation filter mounted on LED, cured with UV adhesive and the walls coated with an opaque paint (D) 2×2 aperture array in a 3D-printed optical isolation for green excitation filters (25 mm diameter filter ring on reverse side) (E) LED intensity (left dash dot), fluorophore absorption spectra (left dashed) and fluorophore emission (right dashed), excitation (left solid) and emission filter (right solid) transmission spectra. The filters were chosen to match the excitation light source, peak excitation and emission spectrum of the fluorophore. The fluorophore was chosen to match the LED peak emission.The photodiode responsivity, R(λ) is plotted on the secondary axis
2.2. Low-cost Interference filters
Previously, we reported the performance of a fluorescence detection system that uses relatively expensive 25 mm fi in a single site configuration (Katchman et al., 2016; Obahiagbon et al., 2016; Smith et al., 2016). Here, we discuss the use of 3×3 mm filter The 3×3 mm fi were mounted directly on the LEDs and photodiodes using optically clear UV curing adhesive, (NOA63, Norland Prodcuts Inc., NJ, USA) as shown in Fig. 3(A) and (C), respectively. The top surface of LEDs and photodiode was coated with epoxy, and the 3×3 mm fi were centered using a mask and forceps. The assembly was then exposed to UV light using a hand-held UV lamp (Spectroline EN-180, Spectronics Corp. NY, USA) for 5 minutes. The side walls of the assembly were then coated with an opaque paint to block light leakage through the sides of the photodiode and fi substrate. Additionally, a 0.5 mm margin was coated around the top surface of the fi to block leakage light through the notches and imperfections as a result of the dicing process. The assembly was left to air-dry under ambient conditions for 24 hours. A 2×2 aperture array configuration fi with 25 mm filters was also fabricated, and the configuration is shown in Fig. 3(B) and (D). Apertures were created in the 3D-printed structures for each site, and a fi slot was incorporated into the design. The performance of the 25 mm system was compared to the 3×3 mm fi system, as presented in the supplementary information (see Figure S5). Previously, cost-prohibitive interference fi (25 mm diameter) used in Filter Cubes for fluorescence microscopes, would render the cost of optoelectronic platforms like ours impractical, especially for the deployment of high-performance diagnostic devices, in low and middle-income countries (LMICs). Our low-cost 2×2 array platform addresses this issue. In the 25 mm 4-site system reported here as well as the single site system previously reported (Katchman et al., 2016), the 25 mm diameter fi set (excitation and emission) costs $625, whereas the 3×3 mm individual fi set on each channel costs $3.18 and thus, a total of $12.72 for all four channels (for a production quantity of 1,000 pieces). This represents a significant reduction in fi cost by 97.96% without compromising device performance.
2.3. Multiplexed Platform characterization
Nile red fluorescent microspheres (1 µm diameter, product no. F-8819, lot no. 1154194, specified absorption/emission peak/center wavelength 535/575 nm, molar absorption coefficient, є, 37859 M−1 cm−1, relative quantum yield, QYfl, 0.19) were purchased from Life Technologies, CA, USA. These fluorescent particles were designed by entrapping Nile red fluorescence in latex spheres. The microspheres were intentionally chosen as the dye has a good match for the excitation spectrum of the LEDs. The fi described above in section 2.2, were also designed to match the absorption and emission spectra of the fluorophore. The filter set ensures the dye is excited at peak absorption and that peak emissions are collected, while significantly rejecting excitation light. To reduce the excitation light reaching the detector, we used an orange emission filter with sharp transition edges as shown in Fig. 3(E).
To characterize our platform, samples of spotted fluorophores were prepared. Microscope slides were pre-treated with 2% 3-Aminopropyl-ethoxysilane (APTES) purchased from Sigma Aldrich, and prepared by dissolving 12 mL of 99.5% APTES in 600 mL of acetone. The slides were submerged in APTES solution for 15 mins, rinsed 5x in acetone, followed by a deionized (DI) water rinse, and then blow-dried with compressed air. The microspheres were diluted in DI water (Milli-Q system, Millipore, MA), immobilized, and dried on the pre-treated microscope slides (VWR Microscope slides, 75 mm x 25 mm x 1 mm). To prevent agglomeration, the Nile red microspheres were fi sonicated for 15 minutes before use. A logarithmic dilution series was prepared, and test samples were spotted (N=5) using a silicone-well template cut-out for precise and repeatable location of test spots in the 2×2 array. Four 2.3 mm diameter holes, with an 11.5 mm pitch (same as LED and photodiode pitch) were laser cut (Universal Laser Cutter, VLS 6.60,) in a 3 mm thick Silicone Isolator sheet (GraceBio, OR). Silicone sheets were brought in contact with the surface of the APTES coated glass slides, and fluorophore dilutions were pipetted into wells minimizing sample spreading on the glass slide and preventing misalignment of the samples in the optical path. Five slides for each dilution (1:10 to 1:106) were prepared and spotted at 37°C on a hot plate (Torrey Pines Scientific, CA) to accelerate sample drying. The system photon sensitivity, optical signal-to-noise (SNR) and crosstalk were evaluated as presented in section 3.2 and section 3.3.
2.4. Detection of antibodies to HPV16 E7 protein in human plasma
Glass microscope slides (VWR International) were treated with 2% aminosilane (Pierce) for 15 minutes at room temperature, rinsed with acetone, DI water, dried with compressed air, and in-spected for imperfections. All proteins (E7, BSA, and IgG) were diluted in DI water to 25 ug/mL. HPV16 E7 Protein was spotted on two of four sites on the aminosilane-treated slides at room tem-perature. A template was used to ensure that the sites were aligned to the optical path (channels) in our 2×2 array platform. For controls, 5 µL of BSA (negative control, 25 µg/mL ) and 5 µL of whole human IgG protein (positive control) were spotted on the other two sites on the aminosilane-treated slides at room temperature. Slides were stored at 4 °C overnight and blocked with 5% milk-PBST for one hour at room temperature. Plasma samples were diluted 1:1 in 5% milk-PBST, incubated on the slides for one hour at room temperature and washed with PBST (0.2% Tween in 1XPBS). Two label-ing methods were used as the secondary capture antibody conjugate to detect the presence of human IgG antibodies specific for HPV E7 proteins: Dylight 549-conjugated AffiniPure goat anti-human IgG (Jackson ImmunoResearch Laboratories, Inc.) and 1 µm Nile red microspheres-conjugated AffiniPure goat anti-human IgG. Slides were incubated with secondary antibodies diluted 1:50 for one hour at room temperature in the dark, rinsed 3X with PBST (0.2% Tween in 1XPBS), DI water, dried with compressed air, and analyzed with our 2×2 array platform.
3. Results and Discussion
3.1. Performance characterization: Analytical sensitivity and lower limit of detection (LLOD)
To evaluate the performance of our multiplexed fluorescence 2×2 array platform, microscope slides prepared as described above in section 2.3 were inserted into the sample chamber and analyzed. To ensure that performance of each channel was identical, the 2×2 array was characterized with all four sites containing the same concentration per microscope slide (N=5), for the entire dilution series. The results were then compared with a top-of-the-line laboratory array scanner. The representative images shown in Fig. 4(A) were acquired using microarray scanner, (PowerScanner, Tecan), typically used in the analysis of high-density DNA and protein microarrays. The samples were scanned (PMT gain = 50%; green laser power = 50%) and the images analyzed and quantified with Array-Pro Analyzer software. The system response/signal intensity expressed as a detection time (DT) for each channel for all concentrations is compared as shown in Fig. 4(B), with a blank slide used as the reference. The detection time was obtained as described previously in section 2.1. Integration time for the reference was 32 sec per channel. All channels (A0 to A3) produced identical performance for each set of slides and slight variations were primarily due to spotting variability, accounting for 500 msec maximum deviation (not visible on the scale of the plot) in Fig. 4(B). The counts (a.u.) from the array scanner and the detection time, DT (sec), from our low cost 2×2 array platform is compared in Fig. 4(C). Results indicate that the low-cost platform has a nonlinear response at higher concentrations and that the performance is within an order of magnitude of the laboratory microarray scanner. The different between the POC and conventional laboratory instrument response result from the fact that the laboratory array scanner has extensive signal acquisition and processing tools that enables “auto-gain”, such that the system adaptively adjusts the gain in the saturation regions and normalizes the result to an internal standard. System parameters, including the gain-capacitor size (integration time), voltage range, overall noise, and background in the optical path, play an important role in the performance of the array platform. Our low-cost 2×2 array fluorescence detection platform can reliably detect 2×104 0.5 µm diameter fluorescent microspheres on a glass substrate with a signal-to-noise (SNR) ratio of 2:1 at a 99.73% confidence level. The LOD was determined as the concentration corresponding to the mean plus six standard deviations (details provided in the supplementary information, Figure S1). Instrument errors (a few tens of msec), errors due to pipetting, and variations in glass slides are included in the relative standard error as computed and shown in Fig. 4(C). Applying a simple and low cost integration technique, the 2×2 array platform achieves high sensitivity by separating out the low intensity or weak fluorescence signal over an established, relatively higher background intensity.
Figure 4:
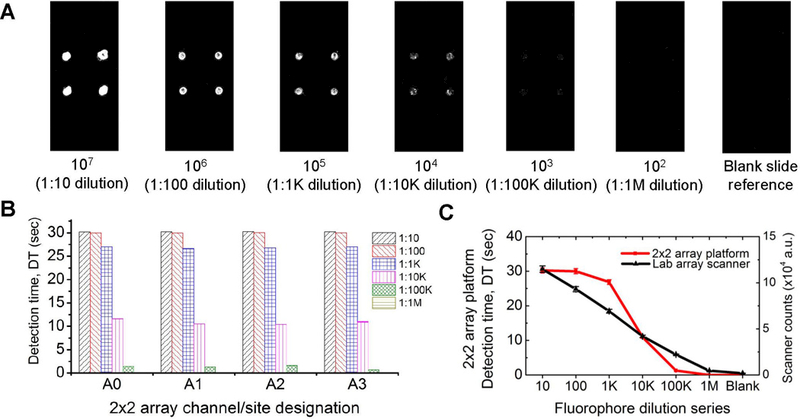
A comparison of our low-cost 2×2 array platform and a top-of-the-line laboratory array scanner. (A) Representative laboratory array scanner images of Nile red fluorescent microspheres spotted with a silicone well. Below each image is the estimated number of microspheres on each spot and the dilution (in parenthesis) (B) Detection time (DT) response to the fluorophore dilution series from 1:10 to 1:1M, measured by the 2×2 array platform. Each set of bars show all four sites/channels (A0, A1, A2 and A3) and their responses (C) Comparison of the responses of laboratory array scanner and our 2×2 array fluorescence detection platform. Dilution series: 1:10, 1:100, 1:103, 1:104, 1:105 and 1:106 are denoted as 10, 100, 1k, 10k, 100k, and 1M; corresponding to 107, 106, 105, 104, 103, and 102 microspheres respectively.
3.2. Photon sensitivity and Optical Signal-to-noise Ratio
Photon sensitivity was determined by measuring power density of the lowest detectable sample concentration. The readout circuit was replaced by an optical power meter (Newport, 1830-C) fitted with a calibrated photodiode (818-SL). A power density value of 27.5 pW/cm2 was measured (after correcting for background and ambient light). The photon flux (photons/m2sec), was calculated by dividing the power density by the available photon energy (Joules or Wsec) due to fluorescence emissions (taking into account necessary unit conversions and background noise). Our system is sensitive to a photon fluorescence of ∼1×104 photons/mm2sec at a signal to noise ratio of 2:1. Here, we define the SNR as the ratio of the emitted fluorescence power (F) to the total noise power level (N). The SNR decreases from 1000:1 at the highest concentration to an SNR of 1:1 at 1:105 dilution. The relatively weak optical signals from fluorophores make SNR analysis very important. As with any fluorescence sensing platform, the amount of fluorescence signal is usually measured alongside undesired sources of light, which contribute to the noise or background level. These include: excitation source (LED) leakage light through the fi (imperfect absorption edges from tiny variations in thin fi layer depositions and shift in absorption edge as angle of incidence varies), ambient light leakage and stray light within the enclosure, background and auto fluorescence, and electronic contributions such as fluorescence noise (shot noise) and dark current in the photodiode/readout circuit.
3.3. Crosstalk characterization and system stability
Crosstalk is caused by light leakage in the system housing, off-axis excitation light transmission through the fi due to oblique incidence, and scattering in the sample chamber. The high performance of interference filter is limited to normal incidence, and undesirable off-axis light at oblique angles will go through the filter with almost no attenuation. This is due to the fact that there is a significant shift in the absorption edge as the incidence angle is varied (Chediak et al., 2004). Our previously published work used 25 mm filter sets. Although the light collection area is larger and alignment is easier, the 3×3 mm filter s perform equally well in terms of the LOD and even better as the crosstalk is eliminated by reducing the filter size and using aperture stops integrated into the optomechanical design. To perform the crosstalk measurements, the LED at site, A0 was used as the reference excitation source, the readout circuit for the other channels were sequentially turned on and ramp time was recorded. Here, we compute the crosstalk probability (CP) as the diff between the ramp times for the active readout channel (Ai) and the reference excitation channel (A0) divided by the ramp time of the reference excitation channel as shown in Equation 3
| (3) |
where Ai denotes the channel in the 2×2 array for i = 0, 1, 2, 3. One method to reduce crosstalk is to ensure normal incidence or limit off-axis light by constricting the aperture size or by increasing separation between detection sites (array pitch). The latter is not desirable as it leads to larger and more expensive fi for single fi systems and lower array density. We observed that crosstalk is eliminated with the 3×3 mm filter More discussion on crosstalk is presented in the supplementary information (see Figure S5).
3.4. Fluorescence immunoassay for the detection of antibodies to HPV16 E7 protein in human plasma
The Center for Disease Control and Prevention and the World Health Organization (World Cancer Report) indicates that HPV is the most common sexually transmitted infection (STI) with HPV16 and 18 strains of the virus being responsible for more than 70% of all cervical cancer cases. HPV is an infectious disease with almost 70% of the global burden occurring in developing countries. It is the fourth most common cause of cancer deaths in women accounting for 266,000 deaths worldwide in 2012. Although preventable, vaccines are not a treatment for pre-existing infections. Screening in the developed world is currently performed by cytology requiring access to central laboratory facilities. To deliver clinical-level sensitivity at the point-of-need, a multiplexed, high-sensitivity platform is required, especially for early detection. In this section, we demonstrate the utility of the 2×2 array platform for detection of antibodies specific to HPV16 E7 protein in patient plasma. Details of the microsphere conjugation protocol is included in the supplementary information. HPV protein/antigen, E7, was printed on APTES functionalized glass. Human IgG Abs from patient sera (primary antibody) was incubated with the immobilized protein. HPV16 E7-specific IgG antibodies in patient plasma samples were detected using a goat anti-human IgG antibody. Goat anti-human IgG antibody was conjugated to the 1 µm fluorescent microspheres and DyLight549 (secondary antibody conjugate) and incubated with the captured E7-specific plasma antibodies. The secondary antibody conjugate was used as the fluorescent detector label/probe. The goat anti-human IgG antibody binds specifically to the constant region of human IgG antibodies. Thus, during the patient sample incubation, E7-specific IgG antibodies in the plasma will bind to the E7 antigen printed on the substrate. Other IgG antibodies present in the sample will get washed away during the wash steps so that during the secondary antibody incubation, the E7-specific IgG antibodies bound to E7 antigen are labeled with the goat anti-human IgG antibodies conjugated to fluorescent microspheres. Results are shown in Fig. 5(A) - (D). Triplicate slides were incubated with selected plasma samples from 2 patients, one with known antibodies to the HPV16 E7 protein and one negative, respectively. These samples were probed with DyLight549 and microsphere conjugates respectively, and tested on the 25 mm and 2×2 array systems. The signal intensity for the microsphere conjugate was relatively higher than the Daylight conjugate, although the microsphere conjugates indicate a significantly higher background level due to non-specific binding (NSB) of the microspheres to the glass substrate. The channels on the 25 mm and 3×3 mm filter systems were run sequentially and hence, crosstalk was minimized. Therefore, the performance of both systems are comparable in detecting both labels. This further supports our hypothesis that reducing the filter dimensions will not only reduce cost, but also improve the overall performance of our platform. Hand-spotting and non-specific adsorption are the most likely causes of variability indicated by the error bars. To resolve these problems, we recently procured a pressure-time dispensing unit even as we continue to explore effective blocking strategies to reduce NSB and intrinsic fluorescence of substrates.
Figure 5:
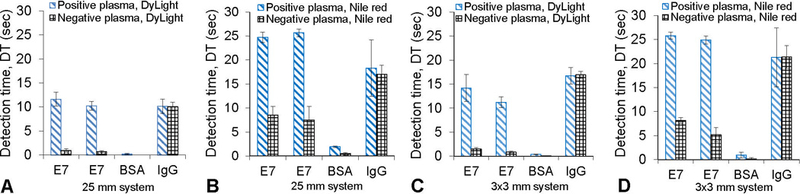
Low-cost array platform applied in the detection of antibodies to HPV16 E7 protein, using two secondary labels. Patient plasma positive for antibodies to HPV 16 E7 and a negative control plasma were used. (A) Positive and negative patient plasma probed with DyLight549 and analyzed with 25 mm filter system (B) Positive and negative patient plasma probed with Nile red microspheres and analyzed with 25 mm filter system (C) Positive and negative patient plasma, probed with DyLight549 and analyzed with 3×3 mm filter system (D) Positive and negative patient plasma probed with Nile red microspheres and analyzed with 3×3 mm filter system.
3.5. Predicted Sensitivity Gains Using our Fluorescent Biorecognition Platform in Lateral Flow Immunoassay Tests.
In this section, we evaluate the potential improvement of colorimetric assays using our low-cost 2×2 array fluorescence platform, by directly testing the system with colored and fluorescent labels on nitrocellulose strips and comparing the results. This approach compares labels used in a typical colorimetric assay to a fluorescent label with the aim of highlighting the benefits and properly quantifying the advantages of fluorescence without compounding the interpretation with assay dependencies including assay efficiency, repeatability and reproducibility. A very common label used in colorimetry is 0.5 µm diameter blue colored polystyrene/latex microspheres. To match the size of the colored latex microspheres, we used 0.5 µm diameter fluorescent polystyrene microspheres (F1-Y 050, Merck Millipore). Many lateral flow assays use 0.3 µm to 0.5 µm diameter microspheres to ensure good mobility in porous membrane structures with average pore size of 8 to 15 µm (Mansfield, 2015; Wong and Y., 2009). Nitrocellulose sheets (HF18004XSS, Merck Millipore) were laser cut to 5×50 mm strips. The strips were mounted on microscope slides with a transparent adhesive and aligned to the optical path using a pre-printed template. For this study, we designed a single site/channel fluorescence detection system to accommodate the rectangular nitrocellulose strips. A circular aperture (5 mm diameter) was used to precisely align the LED-photodiode pair. Filters were mounted on the LEDs and photodiodes as described previously in section 2.2. A dilution series of the blue latex microspheres (K1 050 Blue, Merck Millipore) was prepared by diluting 10 µL of the stock (1010 microspheres) from 1:10 to 1:100k (105 microspheres) at which point the blue color was no longer visible to the eye as shown in Fig. 6(A). A similar dilution series of fluorescent polystyrene microspheres (F1-Y 050, Merck Millipore) was also prepared but this time diluting 10 µL of the stock concentration (109 microspheres) down to 1:1M (103 microspheres). The 0.5 µm diameter blue polystyrene/latex microspheres and fluorescent polystyrene microspheres were pipetted on nitrocellulose strips mounted on glass slides (N=5). The blue latex microspheres become invisible to the naked eye when the number of microspheres is below 109.
Figure 6:
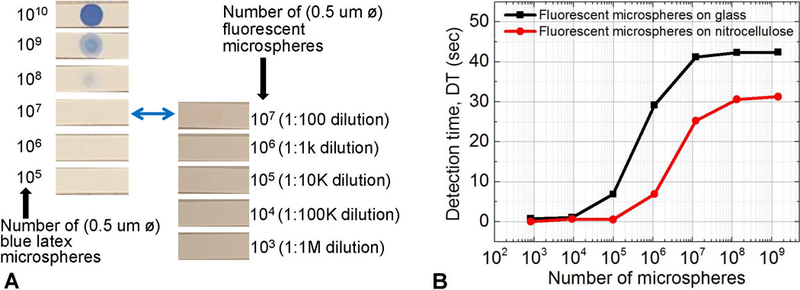
Potential improvement of colorimetric assays by using fluorescent labels. (A) Blue polystyrene/latex col-orimetry microspheres and fluorescent latex microspheres spotted on nitrocellulose. 10 µL of dilutions were spotted, and the number of microspheres at each dilution is shown. (B) Detection time for fluorescent latex dilutions spotted on nitrocellulose and glass analyzed by the low-cost array platform. Our platform can reliably detect 5×105 0.5 µm microspheres on nitrocellulose membranes. This represents 2 to 3 orders of magnitude decrease in the number of microspheres detected, when compared to the number of colored latex microspheres required for a visible (positive) test line. We predict that actual assay variables could limit this number to 2 orders of magnitude.
Our low-cost platform can reliably detect 5×105 0.5 µm diameter fluorescent microspheres at a 99.73% confidence level on a nitrocellulose substrate as shown in Fig. 6(B). In the discussion that follows, we consider a direct comparison to standard colorimetry. To reliably determine if a test is positive in visual colorimetric assays, a test line, 5 mm length, 0.5 mm width, and 10 µm depth requires 3×108 colored microspheres (0.5 µm diameter) on a white nitrocellulose membrane (Mansfield 2015). Our ability to detect 5×105 0.5 µm diameter microspheres represents a 2 to 3 orders of magnitude improvement (over standard latex colorimetry microspheres) in the number of micro-spheres that can be detected, or the number of biorecognition elements that need to be present in the analyte/patient sample. Hence, better sensitivity can be achieved using our fluorescence platform. However, we estimate conservatively, that signal losses due to assay efficiency, non-specific binding, and losses due to intrinsic background fluorescence may limit this improvement to 2 orders of magnitude. Generally, there is an observed attenuation of fluorescence emissions with polymeric substrates like nitrocellulose and thus an overall decrease in the measured signal compared to microscope slides as shown in Fig. 6(B). Also observed is an early signal cutoff at low concentrations with nitrocellulose.
4. Conclusion
A low-cost multiplexed (2×2 array) and highly sensitive fluorescence platform has been presented with 2 to 3 orders of magnitude improvement over colorimetry-based assays. The performance of our array platform is enabled by high quality, inexpensive 3×3 mm interference fi and charge integration readout electronics. The limit of detection of our 2×2 array platform was determined to be 2.1×104 and 5×105 0.5 µm microspheres on glass and nitrocellulose substrates, respectively. These numbers are within an order of magnitude relative to the performance of a top-of-the-line clinical laboratory fluorescence array scanner. Although the array density of our platform is limited by the physical constraints in our discrete component layout and assembly, this system filter well with the form factor, performance, and cost requirements for compact hand-held devices. Such devices become especially useful for personal health monitoring and mobile or decentralized applications in resource-limited areas. We envision that our multiplexed platform will be used in fi trials, for screening patients and detecting antibodies to HPV16 and 18 proteins. Our system has been engineered and adapted to meet the demands of detecting multiple analytes from a single patient sample at the point-of-need.
Supplementary Material
Acknowledgement
Funding: This work was supported in part by the National Science Foundation Smart and Connected Health [grant number IIS-1521904] SCH: INT: “Disposable high sensitivity point of care immunosensor for multiple disease and pathogen detection” and National Cancer Institute Cancer Detection, Diagnosis, and Treatment Technologies for Global Health [grant number CA211415]: “Rapid Point-of-Care Detection of HPV-associated Malignancies”. Thanks to Chroma Technology Corp. for working with us to produce high quality interference filter used in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing financial interest.
Appendix A. Supporting information
References
- Banerjee A, Shuai Y, Dixit R, Papautsky I, Klotzkin D, 2010. Journal of Luminescence 130 (6), 1095 – 1100. [Google Scholar]
- Chediak J, Luo Z, Seo J, Cheung N, Lee LP, Sands TD, 2004. Sensors and Actuators A: Physical 111 (1), 1 – 7, micromechanics section of Sensors and Actuators, based on contributions revised from the Technical Digest of the 16th {IEEE} International conference on Micro Electro mechanical Systems (MEMS 2003). [Google Scholar]
- Dandin M, Abshire P, Smela E, 2007. Lab Chip 7, 955–977. [DOI] [PubMed] [Google Scholar]
- Guilbault GG, 1990. Practical Fluorescence, 2nd Ed. Marcel Dekker, Inc, Madison Avenue, New York. [Google Scholar]
- Hu J, Wang L, Li F, Han YL, Lin M, Lu TJ, Xu F, 2013. Lab Chip 13, 4352–4357. [DOI] [PubMed] [Google Scholar]
- Hu J, Wang S, Wang L, Li F, Pingguan-Murphy B, Lu TJ, Xu F, 2014. Biosensors and Bioelectronics 54, 585 – 597. [DOI] [PubMed] [Google Scholar]
- Katchman BA, Smith JT, Obahiagbon U, Kesiraju S, Lee Y-K, O’Brien B, Kaftanoglu K, Blain Christen J, Anderson KS, Jul. 2016. Scientific Reports 6, 29057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khreich N, Lamourette P, Lagoutte B, Ronco C, Franck X, Cr´eminon C, Volland H, 2010. Analytical and Bioanalytical Chemistry 397 (5), 1733–1742. [DOI] [PubMed] [Google Scholar]
- Kumar AA, Hennek JW, Smith BS, Kumar S, Beattie P, Jain S, Rolland JP, Stossel TP, Chunda-Liyoka C, Whitesides GM, 2015. Angewandte Chemie International Edition 54 (20), 5836–5853. [DOI] [PubMed] [Google Scholar]
- Lee LG, Nordman ES, Johnson MD, Oldham MF, 2013. Biosensors 3 (4), 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre F, Juneau P, Izquierdo R, 2015. Sensors and Actuators B: Chemical 221, 1314 – 1320. [Google Scholar]
- Li Z, Wang Y, Wang J, Tang Z, Pounds JG, Lin Y, 2010. Analytical Chemistry 82 (16), 7008–7014, pMID: 20704391. [DOI] [PubMed] [Google Scholar]
- Mansfield MA , 2015. Tech. rep, OEM Diagnostics Group, EMD Millipore, Bedford, MA. [Google Scholar]
- Obahiagbon U, Kullman D, Smith JT, Katchman BA, Arafa H, Anderson KS, Christen JB, November 2016. In: 2016 IEEE Healthcare Innovation Point-Of-Care Technologies Conference (HI-POCT) pp. 117–120. [Google Scholar]
- O’Farrell B, 2015. Topics in Companion Animal Medicine 30 (4), 139 – 147. [DOI] [PubMed] [Google Scholar]
- Ryu G, Huang J, Hofmann O, Walshe CA, Sze JYY, McClean GD, Mosley A, Rattle SJ, deMello C, deMello AJ, Bradley DDC, 2011. Lab Chip 11, 1664–1670. [DOI] [PubMed] [Google Scholar]
- Smith JT, Katchman BA, Kullman DE, Obahiagbon U, Lee YK, OBrien BP, Raupp GB, Anderson S, Christen JB, March 2016. Journal of Display Technology 12 (3), 273–280. [Google Scholar]
- Sze SM, 2007. Physics of semiconductor devices, 3rd Ed. Wiley Interscience, Hoboken, New Jersey. [Google Scholar]
- Tang R, Yang H, Choi JR, Gong Y, Hu J, Feng S, Pingguan-Murphy B, Mei Q, Xu F, 2016. Talanta 152, 269 – 276. [DOI] [PubMed] [Google Scholar]
- Thrush EP , 2004. Ph.D. thesis. Venkatraman V, Steckl AJ, 2015. Biosensors and Bioelectronics 74, 150 – 155. [DOI] [PubMed] [Google Scholar]
- Williams G, Backhouse C, Aziz H, 2014. Electronics 3 (1), 43–75. [Google Scholar]
- Wong RC, Y., T. H. (Eds.), 2009. Lateral flow immunoassay. Springer. [Google Scholar]
- Yetisen AK, Akram MS, Lowe CR, 2013. Lab Chip 13, 2210–2251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.