

This case report describes an Italian female who was born at full term via Caesarean delivery, performed for breech presentation. She was the first child of healthy, unrelated parents. At birth, she weighed 2600 g (<3rd centile; −2 sd) and measured 45 cm in length (<3rd centile; −2 sd). Psychomotor development was normal for her age. The recommended immunisation schedule was administered. Newborn screening tests (hip ultrasound and acoustic reflexes) as well as echocardiography were normal. There was no known maternal exposure to teratogens or infections. The family history was unremarkable, except for retinitis pigmentosa and hypothyroidism (in the mother).
Short abstract
Can you diagnose this child with minor pulmonary malformations and recurrent pulmonary symptoms? http://ow.ly/6zQB30jHZAP
This case report describes an Italian female who was born at full term via Caesarean delivery, performed for breech presentation. She was the first child of healthy, unrelated parents. At birth, she weighed 2600 g (<3rd centile; −2 sd) and measured 45 cm in length (<3rd centile; −2 sd). Psychomotor development was normal for her age. The recommended immunisation schedule was administered. Newborn screening tests (hip ultrasound and acoustic reflexes) as well as echocardiography were normal. There was no known maternal exposure to teratogens or infections. The family history was unremarkable, except for retinitis pigmentosa and hypothyroidism (in the mother).
The parents reported that the child had been well up to the age of 18 months. After that time, she had been hospitalised for failure to thrive (weight 9600 g, <3rd centile; length 76 cm, <3rd centile). Laboratory findings were negative (first-line: complete blood count, electrolyte measurement and urinalysis; second-line: thyroid tests, testing for food allergies, coeliac disease, nutritional deficits, infectious disease, immune and metabolic disorders), as were the electrocardiogram, echocardiogram and electroencephalogram. She was discharged home but, at the end of the same month, returned for right paracardiac pneumonia, which was treated successfully with antibiotics.
At the age of 24 months, the child was admitted to hospital for a bronchiolitis episode. She was then reported as being well until the age of 2.5 years, when she was affected by a second episode of bronchopneumonia. A sweat test was negative and chest radiography showed opacities in the right lung base and left hilum, also involving the upper lobe. By 24 months of age, the patient experienced recurrent episodes of febrile pharyngotonsillitis, cough and otitis media, requiring treatment with antibiotics or nonsteroidal anti-inflammatory drugs. At the age of 3.5 years, a third episode of bronchopneumonia was reported.
Task 1
What further investigations should be undertaken in a child with these features?
Answer 1
In this case, in light of the recurrent pulmonary symptoms and of the findings on chest radiography, a chest computed tomography (CT) scan was performed.
The chest CT showed pulmonary consolidation both in the medial segment of the middle lobe and in the left lower lobe, bilateral ground glass opacities in the subpleural area of the lower lobes, and decreased calibre of the bronchi as well as bronchial wall thickening (figure 1). The following tests were within normal range: serum total and specific IgE levels; lymphocyte subpopulations; analysis of the sputum; Bordetella pertussis, Mycoplasma pneumoniae and Chlamydia pneumoniae serology; electrocardiogram and echocardiogram.
Figure 1.

Spiral CT: pulmonary consolidation both in the medial segment of the middle lobe and in the left lower lobe, bilateral ground glass opacities in the subpleural area of the lower lobes, and decreased calibre and bronchial wall thickening.
At the age of 3 years and 8 months, a fourth episode of bronchopneumonia was reported. A second sweat test was negative, genetic testing for cystic fibrosis was negative, and chest radiography showed indistinct opacities in the upper fields and the right paracardiac areas.
A fifth episode of bronchopneumonia was reported at the age of 4 years. Cefotaxime and amikacin were administered and a CT scan of the thorax was performed, which showed consolidation and atelectasis in the medial and basal segments of the middle lobe, bilateral ground glass opacities, and mediastinal lymph nodes in the subcarinal, right hilar, paratracheal and aortopulmonary window areas.
Finally, at the age of 4 years and 2 months, she underwent flexible fibre-optic bronchoscopy (figure 2).
Figure 2.
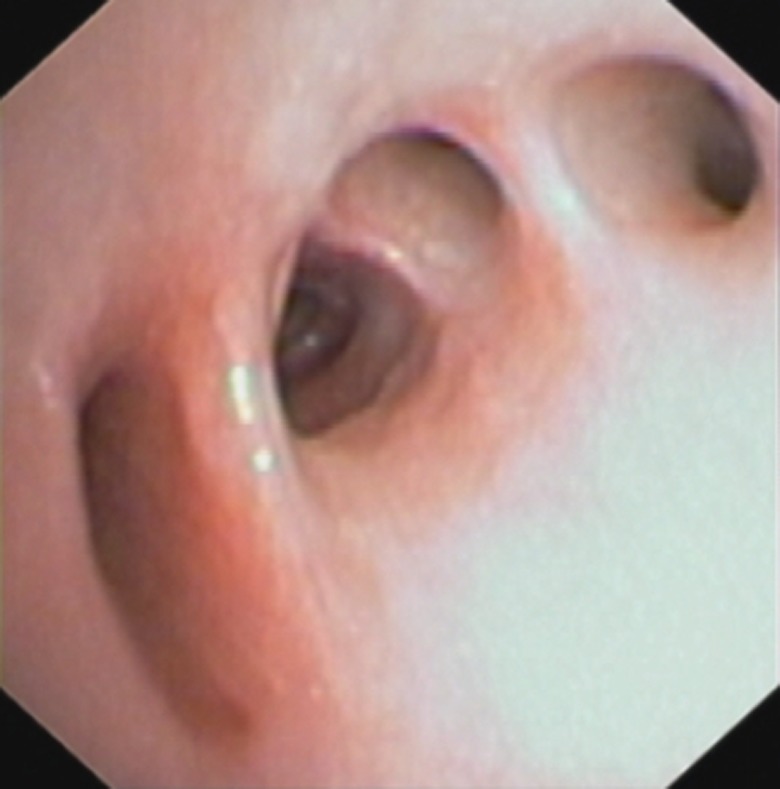
Flexible fibre-optic bronchoscopy.
Task 2
Describe the findings that can be noted from the flexible fibre-optic bronchoscopy image.
Answer 2
On bronchoscopy, an extrinsic, compressive pulsating mass was identified at the transitional region between the middle and distal third of the trachea. At the middle lobe bronchus, two additional bronchial segments were identified. Additionally, a depression in the mucosa was identified that corresponded with an additional segmental bronchus (figure 2).
Cytological and microscopic analyses of bronchoalveolar lavage fluid were normal. The findings of the flexible fibre-optic bronchoscopy should be interpreted as normal variants of the airways, which could interfere with mucociliary clearance, both altering the coughing pattern and leading to retention of airway secretions and development of chronic airway infections.
At the age of 4 years and 5 months, the child was admitted to our department for a sixth episode of bronchopneumonia. Systemic physical examination revealed mild deformities (epicanthal folds and a flattened nasal bridge). Chest radiography showed mild alveolar consolidation around the paracardiac area, and bilateral thickening of the pulmonary hilum.
Task 3
What would be your next step?
Answer 3
In this case, both humoral and cell-mediated immune responses were evaluated in order to detect any suspicious immunological abnormalities. Also, due to the mild deformities revealed on systemic physical examination, array comparative genomic hybridisation (array-CGH) was performed.
Quantitative serum immunoglobulin levels, absolute lymphocyte count, lymphocyte phenotyping and proliferative response to common mitogens were within normal limits. After genetic counselling, array-CGH was performed, which detected an interstitial deletion affecting the long arm of chromosome 22 (∼2.5 MB) in the q11.21 region, consistent with DiGeorge syndrome (DGS).
Once diagnosed, we treated the patient conservatively, starting chest physiotherapy with a positive expiratory pressure mask to improve clearance of trapped secretions [1–3]. Inhaled corticosteroids were also administered to reduce airway inflammation.
Discussion
22q11.2 deletion syndrome, also known as DGS, is a chromosomal anomaly usually due to monosomic deletion on the long arm of chromosome 22, caused by non-allelic meiotic recombination, occurring during spermatogenesis or oogenesis [4, 5]. The worldwide DGS incidence has been estimated to be between one in 2000 and one in 4000 live births [2]. Although several candidate genes have been identified as causing the DGS phenotype, the lack of a clear correlation between type or size of deletion and specific phenotype can often make the diagnosis difficult [6, 7]. The term “umbrella” has been coined to better describe the clinical variability and heterogeneity of DGS. In fact, DGS patients can potentially be affected by a very wide phenotypic spectrum, including: facial dysmorphisms (e.g. malar flatness, ptosis, hypertelorism, epicanthal folds, prominent nasal root); congenital cardiac defects (e.g. truncus arteriosus, tetralogy of Fallot and ventricular septal defect); developmental defects of the upper and lower airway (e.g. choanal atresia, laryngotracheomalacia, abnormal epiglottis and trachea, bronchomalacia); velopharyngeal insufficiency with or without cleft palate; thymic and parathyroid hypoplasia; psychiatric disorders and developmental delay; gastrointestinal (intestinal malrotation, imperforate anus), renal (renal agenesis), ocular, and skeletal (butterfly vertebrae, hemivertebrae) malformations; and hearing loss [8]. Secondary to thymic hypoplasia or aplasia, immunodeficiency of variable degree has been documented. This includes defects in T-lymphocyte number and function as well as humoral defects [9]. Additionally, even in the absence of immunodeficiency, complex pulmonary malformations have been described as causative factors of recurrent pulmonary infections [10–13]. However, to the best of our knowledge, no immune-competent DGS patient has been reported with only minor pulmonary defects.
First described in 1968 and in accordance with “selection theory” [14], the structural upper and lower airway abnormalities in DGS have been attributed to defective function of neural crest-derived cells differentiating into mesenchymal cells, which surround the epithelial layer of the tracheobronchial tract, and/or impaired mesenchymal–epithelial interaction occurring during the fourth week of embryonic development [13, 15, 16]. Moreover, among the several genes linked with the DGS phenotype, TBX1, belonging to the family of transcription factors, due to its pleiotropic effects has been recently identified as a candidate gene for DGS-associated pulmonary malformations [17]. Specifically, TBX1, expressed in the ectoderm of the distal pharyngeal apparatus as well as in mesoderm and endoderm of pharyngeal arches, promotes abnormal growth and remodelling in the respiratory and pharyngeal apparatus and related structures [18]. Nevertheless, the pathogenic cause of respiratory tract malformations remains unclear.
Because minor anomalies of the lower respiratory tract are rarely reported in the literature, having a positive clinical history for recurrent pulmonary infection is easily re-directed to an investigation of immunodeficiency status and/or major pulmonary malformations, leading to an underestimated incidence of lower airways anomalies, which might be more clinically significant than hitherto hypothesised. As in our clinical case, this clearly becomes even more evident in the immune-competent patient. In fact, the negative laboratory findings strongly indicated that the severe respiratory infections documented in our child were exclusively caused by pulmonary anomalies. Therefore, we suggest that minor respiratory tree anomalies should be included in the spectrum of DGS anomalies, in order to alert clinicians to a correct and prompt diagnosis.
Future retrospective and prospective longitudinal studies will estimate the real incidence and clinical significance of minor congenital pulmonary anomalies. However, prompt detection of such minor airway malformations can facilitate early physiotherapy intervention as well as the prevention of more extensive airway injury.
Footnotes
Author contributions: S. Manti and G.F. Parisi wrote the first draft; L. Tardino wrote the second draft; M. Cutrupi performed genetic counselling; C. Salpietro and C. Cuppari performed the immunology test; O. Sacco performed the bronchoscopy; and S. Leonardi conceived the study and revised the manuscript for final submission.
Conflict of interest: None declared.
References
- 1.Lee AL, Button BM, Tannenbaum EL. Airway-clearance techniques in children and adolescents with chronic suppurative lung disease and bronchiectasis. Front Pediatr 2017; 5: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leonardi S, Cuppari C, Lanzafame A, et al. . Exhaled breath temperature in asthmatic children. J Biol Regul Homeost Agents 2015; 29: Suppl. 1, 47–54. [PubMed] [Google Scholar]
- 3.Fagevik Olsén M, Lannefors L, Westerdahl E. Positive expiratory pressure – common clinical applications and physiological effects. Respir Med 2015; 109: 297–307. [DOI] [PubMed] [Google Scholar]
- 4.DiGeorge AM. Discussions on a new concept of the cellular basis of immunology. J Pediatr 1965; 67: 907–908. [Google Scholar]
- 5.Orphanet . The portal for rare diseases and orphan drugs. www.orpha.net Date last updated: May 2, 2018.
- 6.Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev Disabil Res Rev 2008; 14: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hacıhamdioğlu B, Hacıhamdioğlu D, Delil K. 22q11 deletion syndrome: current perspective. Appl Clin Genet 2015; 8: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancrini C, Puliafito P, Digilio MC, et al. . Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr 2014; 164: 1475–1480. [DOI] [PubMed] [Google Scholar]
- 9.Maggadottir SM, Sullivan KE. The diverse clinical features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome). J Allergy Clin Immunol Pract 2013; 1: 589–594. [DOI] [PubMed] [Google Scholar]
- 10.Deerojanawong J, Chang AB, Eng PA, et al. . Pulmonary diseases in children with severe combined immune deficiency and DiGeorge syndrome. Pediatr Pulmonol 1997; 24: 324–330. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham ML, Perry RJ, Eby PR, et al. . Primary pulmonary dysgenesis in velocardiofacial syndrome: a second patient. Am J Med Genet A 2003; 121A: 177–179. [DOI] [PubMed] [Google Scholar]
- 12.Saydam TC, Mychaliska GB, Harrison MR. Esophageal lung with multiple congenital anomalies: conundrums in diagnosis and management. J Pediatr Surg 1999; 34: 615–618. [DOI] [PubMed] [Google Scholar]
- 13.Bertolani MF, Bergamini BM, Predieri B, et al. . Tracheobronchial anomalies in chromosome 22q11.2 microdeletion. Am J Med Genet A 2006; 140: 790–793. [DOI] [PubMed] [Google Scholar]
- 14.Dische MR. Lymphoid tissue and associated congenital malformations in thymic agenesis. Findings in one infant and two severely malformed stillborns. Arch Pathol 1968; 86: 312–316. [PubMed] [Google Scholar]
- 15.Yamagishi H. The 22q11.2 deletion syndrome. Keio J Med 2002; 51: 77–88. [DOI] [PubMed] [Google Scholar]
- 16.Ghaye B, Szapiro D, Fanchamps JM, et al. . Congenital bronchial abnormalities revisited. Radiographics 2001; 21: 105–119. [DOI] [PubMed] [Google Scholar]
- 17.Gao S, Li X, Amendt BA. Understanding the role of Tbx1 as a candidate gene for 22q11.2 deletion syndrome. Curr Allergy Asthma Rep 2013; 13: 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall JG. CATCH 22. J Med Genet 1993; 30: 801–802. [DOI] [PMC free article] [PubMed] [Google Scholar]