

SUMMARY
Protein phosphatase 2A (PP2A) inhibition causes hy-perphosphorylation of tau and APP in Alzheimer’s disease (AD). However, the mechanisms underlying the downregulation of PP2A activity in AD brain remain unclear. We demonstrate that Cancerous Inhibitor of PP2A (CIP2A), an endogenous PP2A inhibitor, is overexpressed in AD brain. CIP2A-mediated PP2A inhibition drives tau/APP hyperphosphorylation and increases APP β-cleavage and Aβ production. Increase in CIP2A expression also leads to tau mislocalization to dendrites and spines and synaptic degeneration. In mice, injection of AAV-CIP2A to hippocampus induced AD-like cognitive deficits and impairments in long-term potentiation (LTP) and exacerbated AD pathologies in neurons. Indicative of disease exacerbating the feedback loop, we found that increased CIP2A expression and PP2A inhibition in AD brains result from increased Aβ production. In summary, we show that CIP2A overexpression causes PP2A inhibition and AD-related cellular pathology and cognitive deficits, pointing to CIP2A as a potential target for AD therapy.
In Brief
PP2A inactivation plays a key role in AD pathogenesis. Shentu et al. report that endogenous PP2A inhibitor CIP2A is upregulated in AD brains. CIP2A overexpression promotes β-cleavage of APP and tau hyperphosphorylation through PP2A inhibition, leading to impairments in synapse, hippocampal LTP, and memory.
Graphical Abstract

INTRODUCTION
Alzheimer’s disease is a neurodegenerative disorder characterized by the progressive development of cognitive deficits. Its neuropathology is defined by extracellular accumulation of amyloid-β peptides into amyloid plaques and intraneuronal fibrillar aggregates of hyper- and abnormally phosphorylated tau proteins (Masters et al., 1985; Grundke-Iqbal et al., 1986). An increasing number of studies suggest that protein phosphatase 2A (PP2A) plays a key role in tau hyperphosphorylation and aggregation. In Alzheimer’s disease (AD) patient brains, PP2A activities were decreased (Gong et al., 1993, 1995). In vitro analysis of the dephosphorylation ability of different phosphatases toward tau showed that PP2A contributed 71% of the dephosphorylation of human hyperphosphorylated tau isolated from AD patient brains (Liu et al., 2005). Incubation of PP2A with hyperphosphorylated tau resulted in tau dephosphorylation and restoration of tau function in promoting microtubule assembly (Wang et al., 1995). Inhibition of PP2A in cultured brain slices and rat brains induced tau hyperphosphorylation at multiple sites known to be related to AD (Gong et al., 2000; Sun et al., 2003). Furthermore, PP2A inactivation is also implicated in promoting phosphorylation of amyloid precursor protein (APP) at T668 and Aβ over-production (Sontag et al., 2007). However, even though modulation of PP2A activity in transgenic mouse models (Nicholls et al., 2016) further supports the causative role for PP2A inhibition in AD pathologies, the molecular mechanisms underlying PP2A inhibition in AD remain less clear.
Cancerous Inhibitor of PP2A (CIP2A) has been shown to be upregulated in a variety of peripheral tumors, leading to an inhibition of PP2A activity (Junttila et al., 2007; Soo Hoo et al., 2002; Khanna et al., 2013a). The robust tumor-promoting effects of CIP2A overexpression has been linked to selective regulation of well-established cancer drivers such as MYC and E2F1 (Junttila et al., 2007; Laine et al., 2013). Interestingly, in addition to the peripheral tissues, CIP2A is also expressed in normal adult human and mouse brains, especially in olfactory bulb and hippocampus (Kerosuo et al., 2010). One study reported that CIP2A could promote self-renewal and proliferation of mouse neural progenitor cells in vitro (Kerosuo et al., 2010). Another study suggested a role of CIP2A in the development of astrocytoma (Yi et al., 2013). However, thus far CIP2A has not been implicated in any neurodegenerative disorder.
In the present study, we report that the CIP2A levels are increased in AD patient brains and neurons exposed to Aβ. We show that CIP2A enhances APP phosphorylation at T668 by inhibiting PP2A, and thus promotes the p-secretase processing of APP and Aβ production. Also, in a PP2A-dependent manner, CIP2A expression results in AD-like tau hyperphosphorylation and abnormal localization of tau in the soma and dendrites, as well as synaptic degeneration. Overexpression of CIP2A in mice led to aberrant synaptic plasticity and deficits in learning and memory. Our findings strongly indicate a pathophysiological role for CIP2A in the development of AD and its potential as a novel AD therapy target.
RESULTS
CIP2A Expression Levels Are Increased in AD Human Brains and Aβ-Treated Cells
To evaluate a potential pathophysiological role of CIP2A in human AD, we detected the protein levels of CIP2A in AD patient brains. Notably, we found that CIP2A expression levels were increased in the frontal and temporal cortex in AD human brains compared with age-matched controls (Figures 1A and 1B).
Figure 1. CIP2A Expression Levels Are Increased in AD Human Brains and in Aβ-Treated Cells.

(A) Left: brain tissues (temporal cortex) from AD and age-matched normal control (Con) humans were homogenized, and CIP2A expression levels were detected by immunoblotting. Glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Right: quantitative analysis of the CIP2A levels. *p < 0.05 versus control group. n = 6, control group; n = 7, AD group.
(B) Left: brain tissues (frontal cortex) from AD and age-matched normal control (Con) humans were homogenized, and CIP2A expression levels were detected by immunoblotting. β-Actin was used as a loading control. Right: quantitative analysis of the CIP2A levels is shown. *p < 0.05 versus control group. n = 4, control group; n = 5, AD group.
(C) Rat primary hippocampal neurons (DIV 12) were incubated with Aβ oligomer for 48 hr. Immunofluorescence staining of MAP-2 (red) and CIP2A (green).
(D) Cell viability was detected by LDH cytotoxicity assay kit. n = 9 wells (in 96-well plate) per group.
(E) PP2A activities were detected by Promega Serine/Threonine Phosphatase Assay kit. n = 9 per group.
(F) Left: CIP2A and neuronal nuclei (NeuN) expression levels were detected by immunoblotting. GAPDH was used as a loading control. Right: quantitative analysis of the protein levels is shown. n = 3 per group.
(G) Left: SH-SY5Y cells were transfected with vector or APP plasmids for 48 hr. Representative immunoblots of CIP2A, PP2Ac, and DM1A. Right: quantitative analysis of the protein levels; n = 3 per group.
*p < 0.05; **p < 0.01; ***p < 0.001 versus Con. Data are mean ± SEM.
In cancer cells, CIP2A expression is driven by constitutive DNA-damage signaling (Khanna et al., 2013b). In AD neurons, oxidative DNA damage signaling is induced by exposure of cells to Aβ (Suberbielle et al., 2013; Wu et al., 2014; Yan et al., 2014). To test whether Aβ could contribute to overexpression of CIP2A observed in human AD samples, we treated primary neurons with increasing doses of Aβ. Notably, Aβ treatment induced a dose-dependent increase in CIP2A protein expression in cultured neurons, with no obvious signs of toxicity. Importantly, the Aβ-induced CIP2A protein expression was reflected in similarly dose-dependent inhibition of PP2A catalytic activity in cultured neurons (Figures 1C–1F). We also demonstrate that the CIP2A-inducing effects of recombinant Aβ can be recapitulated by endogenous APP overexpression in SH-SY5Y cells (Figure 1G).
CIP2A Promotes β-Cleavage of APP and tau Hyperphosphorylation through PP2A Inhibition
To examine whether CIP2A participates in PP2A inhibition and PP2A-regulated amyloidosis, we co-transfected HEK293-T cells with CIP2A (Junttila et al., 2007) and APP plasmids, and detected the PP2A activity and APP processing. Indeed, we found that CIP2A overexpression resulted in PP2A inhibition, indicated by PP2A activity assay via measuring the dephosphorylation of a synthesized general substrate of PP2A (Figure 2A). In addition, interaction of CIP2A with PP2A and APP was confirmed by co-immunoprecipitation (CoIP) assays (Figure 2B). Furthermore, CIP2A promoted APP T668 phosphorylation and β-cleavage (increased sAPPβ), but did not affect the expression of PS1 and BACE1 (Figures 2C and 2D). CIP2A also increased Aβ production (Figures 2E and 2F). Consistent with the involvement of PP2A, all of these changes were reversed by overexpressing PP2A (Figures 2C–2F). These findings suggest that CIP2A promotes β-cleavage of APP by inhibiting PP2A.
Figure 2. CIP2A Promotes β-Cleavage of APP and tau Hyperphosphorylation through PP2A Inhibition.
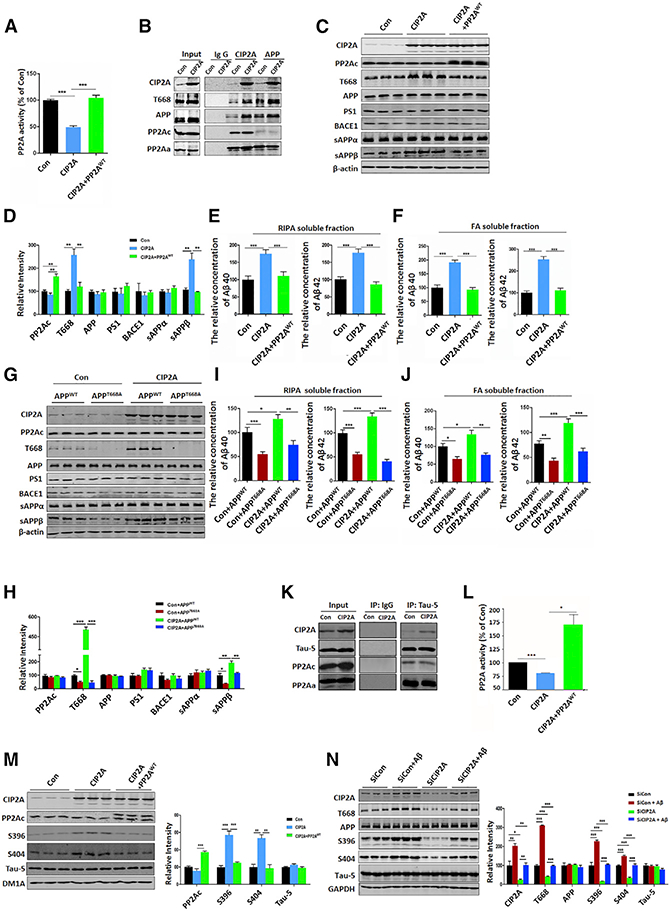
(A) Cell lysates of HEK293-T cells co-transfected with vector + APP, CIP2A + APP, or CIP2A + APP + PP2AWT plasmidsfor48 hr were subjected to PP2A activity assay.
(B) The interaction of CIP2A, APP, PP2Ac, and PP2Aa was examined by CoIP assay.
(C) Representative immunoblots ofCIP2A, PP2Ac, T668, APP, PS1, BACE1, sAPPα, Sappβ, and β-actin in lysates of the three groups in (A).
(D) Quantitative analysis ofthe protein levels in (C).
(E) Aβ40 and Aβ42 levels in RIPA soluble fraction of cell lysates were detected by ELISA.
(F) Aβ40 and Aβ42 levels in formic acid soluble fraction (pellets dissolved in formic acid [FA]) were detected by ELISA.
(G) Representative immunoblots using cell lysates from HEK293-T cells co-transfected with vector + APPWT, CIP2A + APPWT, vector + APPT668A, or CIP2A + APPT668A plasmids for 48 hr.
(H) Quantitative analysis ofthe protein levels in (G).
(I) Aβ40 and Aβ42 levels in RIPA soluble fraction of the four groups in (G) were detected by ELISA.
(J) Aβ40 and Aβ42 levels in FAsolublefraction ofthe four groups in (G) were detected by ELISA.
(K) The interaction of CIP2A, tau, PP2Ac, and PP2Aawas revealed by CoIP in lysates of HEK293-tau cellstransfected with vectoror CIP2A plasmids for 48 hr.
(L) HEK293-tau cells were transfected with vector, CIP2A, or CIP2A + PP2AWT plasmids for 48 hr. PP2A activities in the three groups were detected.
(M) Left: representative immunoblots of CIP2A, PP2Ac, S396, S404, and total tau (Tau-5) in lysates of the three groups in (L). Right: quantitative analysis of the proteins levels is shown.
(N) Rat primary hippocampal neurons (DIV 9) were infected with siCIP2A lentivirus and were incubated with Aβ oligomer (at DIV 12) for 48 hr. Left: representative immunoblots of CIP2A, T668, APP, S396, S404, tau, and GAPDH in lysates of the primary neurons are shown. Right: quantitative analysis of the proteins levels is shown.
*p < 0.05; * **p < 0.01; ***p < 0.001. n = 3 per group. N = 3 independent experiments. Data are mean ± SEM.
To examine whether the CIP2A-induced enhancement of APP β-cleavage is dependent on the phosphorylation of APP-T668, we co-transfected HEK293-T cells with CIP2A and APP-T668A mutant, which is unable to undergo phosphorylation. Under this condition, CIP2A failed to promote β-secretase processing of APP (Figures 2G–2J). Thus, the stimulating effect of CIP2A on APP β-cleavage requires APP-T668 phosphorylation induced by PP2A inhibition.
Because PP2A is the most important tau phosphatase, CIP2A may play a role in tau phosphorylation. To explore whether CIP2A promotes tau pathology through inhibiting PP2A, we co-transfected CIP2A and tau plasmids into HEK293-T cells and revealed the interaction of CIP2A, PP2A, and tau by CoIP (Figure 2K). Furthermore, CIP2A-induced PP2A inhibition (Figure 2L) was accompanied by tau hyperphosphorylation at S396 and S404 (Figure 2M), both typical phosphorylation sites involved in AD. The effect of CIP2A-induced tau hyperphosphorylation was reversed by overexpression of PP2A (Figure 2M). On the contrary, knockdown of CIP2A by short hairpin RNA (shRNA) resulted in PP2A activation and tau dephosphorylation (Figures S1A-S1C). These results indicate that CIP2A promotes tau hyperphosphorylation through PP2A inhibition.
The effect of CIP2A on APP-T668 and tau phosphorylation was further validated in primary neurons treated with Aβ. Aβ incubation resulted in an increase in CIP2A protein expression and APP and tau phosphorylation; the latter was reversed when CIP2A was knocked down by siCIP2A lentivirus (Figure 2N). Taken together, CIP2A promotes APP β-cleavage and tau hyperphosphorylation through PP2A inhibition.
CIP2A-Induced tau Hyperphosphorylation Promotes Synaptic Degeneration
Under normal conditions, tau is preferentially distributed in the axon. However, under pathological conditions, hyperphos-phorylated or mutated tau could dissociate from axonal microtubules, leading to its redistribution to non-axonal subcellularcom-partments. Studies have shown that accumulation of tau at the synaptic sites contributes to synaptopathy in AD (Pooler et al., 2014; Zhou et al., 2017b). To explore whether CIP2A-induced tau hyperphosphorylation participates in synaptic degeneration, we cultured rat primary hippocampal neurons and overexpressed CIP2A by lentivirus infection at days in vitro (DIV) 25 for 6 days. By immunostaining, we found that in mature cultured neurons tau phosphorylation at pS404 or pS396 was maintained at a low level. However, CIP2A overexpression induced a marked increase in tau phosphorylation; the intensive AD-like hyperphosphorylated tau was detected not only at axons, but also at the soma and dendrites (Figures 3A–3C). Using synaptosomal lysates isolated from cultured neurons, we found that the level of hyperphosphorylated tau was significantly increased in CIP2A-overexpressing neurons (Figure 3D). Thus, CIP2A promoted translocation of hyperphosphorylated tau into the synapse. To further confirm this hypothesis, cultured hippocampal neurons were transfected with GFP-tau at DIV 11, followed by CIP2A lentivirus infection at DIV 18 for 6 days. Analysis of GFP-tau in the spines indicated that the amount of tau at the spines was significantly increased (Figure 3E). These findings support the idea that CIP2A promotes tau phosphorylation and accumulation in soma and dendritic spines.
Figure 3. CIP2A Induces tau Mislocalization and Synaptic Degeneration in Primary Neurons.

Rat primary hippocampal neurons (DIV 25) were infected with CIP2A lentivirus for 6 days.
(A) Immunofluorescence staining of pS404-tau (red) and CIP2A (green).
(B) Immunofluorescence staining of pS396-tau (red) and CIP2A (green).
(C) Quantitative analysis of the fluorescence intensity of pS404 and pS396 staining.
(D) Representative immunoblots in whole-cell lysates, cytosolic fractions, and synaptosomal fractions of the neurons infected with or without CIP2A lentivirus.
(E) Top: cultured hippocampal neurons (DIV 11) were transfected with GFP-tau plasmid and then subjected to CIP2A lentivirus infection (at DIV 18) for 6 days. GFP-positive neurons were imaged to show the dendrites. Bottom: quantitative analysis of GFP-tau-positive spines is shown.
(F) Rat primary hippocampal neurons(DIV 11)were transfected with surface-EGFP plasmid and then subjected to CIP2A lentivirus infection (at DIV 25) for 6 days. The EGFP-positive neurons (DIV 31) were observed under fluorescence microscopy. Top: representative images of the neurons are shown. Bottom: quantitative analysis of the spine numbers is shown.
(G) Rat primary hippocampal neurons (DIV 25) were infected with CIP2A lentivirus for 6 days. The neurons (DIV 31) were collected and lysed for immunoblotting. Left: representative blots are shown. Right: quantitative analysis of the protein levels in the left panel is shown.
(H) Neurons in (G) were immunostained with antibodies against N-GluA1 (surface, green) and PSD95 (red). Top: representative images of the dendrites are shown. Bottom: quantitative analysis of N-GluA1/PSD95-positive puncta numbers and fluorescence intensity of GluA1 staining is shown.
(I) Rat primary hippocampal neurons (DIV 10) infected with CIP2A lentivirus for 6 days were subjected to whole-cell patch-clamp recording of mEPSCs. Bottom: normalized amplitude (left) and frequency (right) of AMPAR-mediated mEPSCs of control and CIP2A-overexpressing neurons. n = 8 neurons.
*p < 0.05.
**p < 0.01; ***p < 0.001. For immunoblotting, n = 3 per group. N = 3 independent experiments. Data are mean ± SEM.
Aberrant entry of tau into the spine is related to synaptic dysfunction. To examine the role of CIP2A on spine formation and maturation, we transfected primary neurons of DIV 11 with surface-EGFP (Surf-EGFP) to indicate the cell morphology, and the cells were then infected at DIV 25 with CIP2A lentivirus for 6 days. The number of spines was decreased significantly in neurons overexpressing CIP2A (Figure 3F). Furthermore, the post-synaptic proteins including PSD95 and GluA1 were decreased (Figure 3G). Because synaptic transmission is mediated by surface α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) at the spines, we then immunostained surface AMPARs with antibodies against the N terminus of GluA1 (green), together with antibodies against PSD95 to indicate the synaptic sites (red). A significant reduction of spine number and surface GluA1 was observed in CIP2A-overexpressing neurons (Figure 3H). In line with this finding, in CIP2A-transfected neurons, patch-clamp recordings showed a reduction in the frequency and amplitude of the AMPAR-mediated miniature excitatory postsynaptic current (mEPSC) (Figure 3I). These results collectively indicate that elevated expression of CIP2A results in abnormalities in synaptogenesis and synaptic function.
CIP2A Overexpression Results in Learning and Memory Deficits in Mice
To further determine the impact of increased CIP2A on cognitive function, we performed bilateral intrahippocampal injection of AAV-Con or AAV-CIP2A virus to C57/BL6 mice aged 8–10 weeks. Overexpression of CIP2A in the hippocampus was confirmed by fluorescence microscopy 4 weeks after injection (Figure 4A). We also confirmed the increased tau and APP phosphorylation, as well as the increase in Aα40 and Aα42 levels in hippocampus (Figures S2A-SSE). These data indicate that in vivo in the brain, elevated expression of CIP2A induces aberrant alterations that mimic pathologies in AD.
Figure 4. Overexpression of CIP2A Induces Deficits in Visual Episodic Memory and Spatial Learning and Memory in Mice.

(A) AAV-CIP2A was injected into the hippocampal CA3 region of the C57/BL6 mice, and CIP2A expression was observed (green) 4 weeks after injection. Cell nuclei were stained with Hoechst (blue) to show the hippocampus.
(B) The experimental design of novel object recognition test (NOR). The left is the acquisition trial. The test trial was conducted 1 (middle) and 24 hr (right) after the acquisition trial.
(C) The recognition index of objects A and B in the acquisition trial.
(D) The recognition index of objects A and C in the test trial 1 hr after the acquisition trial. ***p < 0.001 unfamiliar object C versus familiar object A.
(E) The recognition index of objects A and D in the test trial 24 hr after the acquisition trial. ***p < 0.001 unfamiliar object D versus familiar object A.
(F) The discrimination index of 1-hr test trial and 24-hr test trial.
(G) The experimental design of the Morris water maze test.
(H) Left and middle: the representative searching trace on day 6 of the training. Right: the latency to reach the hidden platform.
(I) Left: swimming speed after removing the platform (1 hr) during the probe trial. Middle: the number of times of crossing the platform is shown. Right: the percentage of time spent in each quadrant is shown. Quadrant 3 is the target quadrant.
(J) The representative searching trace after removing the platform in the probe trial at 48 hr after training.
(K) Left: swimming speed after removing the platform (48 hr) during the probe trial. Middle: the number of times to cross the position of the platform is shown. Right: the percentage of time spent in each quadrant is shown.
*p < 0.05; **p < 0.01; ***p < 0.001 versus Con. n = 10 mice per group. Data are mean ± SEM.
In the novel object recognition test (NOR), the recognition index was comparable between the control and CIP2A mice (Figures 4B and 4C) in the acquisition trial. In the test trial 1 hr after the acquisition trial, mice in both groups showed increased interest to the new object with comparable recognition index (Figure 4D). However, in the test trials 24 hr after the acquisition trial, recognition index to the new object was significantly decreased in the CIP2A mice (Figure 4E), indicating impaired long-term memory. The analysis of the discrimination index at 1- and 24-hr test trials showed the same effects (Figure 4F). These results show that CIP2A overexpression induces deficits in visual episodic memory. We then examined the effect of CIP2A on the reference spatial memory using the Morris water maze (Figure 4G). In the acquisition training phase, CIP2A mice showed impaired learning, which was indicated by significantly longer latency at the last 2 days (days 5 and 6; Figure 4H). In the test trials at both 1 and 48 hr, CIP2A mice showed decreased crossing times and swimming time in the target quadrant compared with the control mice, with no changes in swimming speed (Figures 4I–4K). These results suggest that CIP2A causes disruption in spatial learning and memory. Consistently, in the fear conditioning test, CIP2A mice showed a significant reduction in freezing time in both the contextual and the tone conditioning paradigms (Figure S3). Together, these results indicate that CIP2A induces learning and memory deficits.
CIP2A Induces Impairment of Hippocampal LTP and Synaptic Degeneration In Vivo
Hippocampal synaptic plasticity, manifested primarily as long-term potentiation (LTP) and long-term depression (LTD), is believed to be the cellular basis for learning and memory. Given that CIP2A overexpression led to impairments in memory, we wonder whether CIP2A plays a role in synaptic plasticity. To this end, we performed electrorecordings on hippocampal slices from CIP2A-overexpressing mice to examine LTP of the CA3-to-CA1 circuit. Indeed, compared with the control, expression of CIP2A significantly reduced the magnitude of LTP, indicating a deleterious effect of CIP2A upregulation on synaptic plasticity (Figures 5A-5C).
Figure 5. CIP2A Induces Impairments in Hippocampal LTP and Synaptic Degeneration in Mouse Brains.
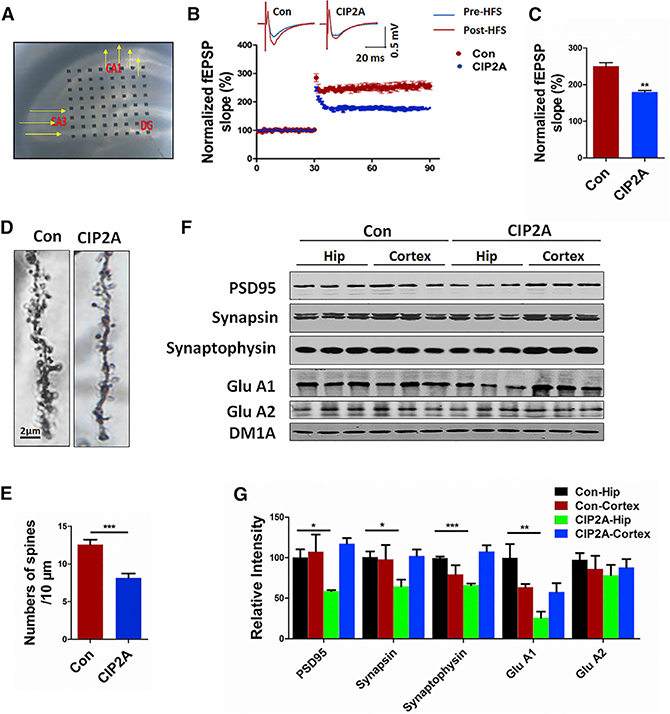
AAV-CIP2A was injected into the hippocampal region, and brain slices were prepared 4 weeks later for electrical recordings.
(A) Hippocampal CA3-CA1 LTP was recorded by using the MED64 system.
(B) Normalized CA3-CA1 fEPSP mean slope recorded from the CA1 dendritic region in acute hippocampal slices.
(C) Quantitative analysis of the normalized fEPSP slope in (B). **p < 0.01 versus Con. n = 3 mice per group; 5 brain slices per mouse were recorded. Data are mean ± SEM.
(D) Representative dendritic spines of neurons from Golgi impregnated hippocampus.
(E) Averaged spine density (mean spine number per 10-mm dendrite segment) was decreased in mice after injection of AAV-CIP2A. ***p < 0.001 versus Con. n = 3 mice per group, 5 brain slices per mouse.
(F) Synaptic proteins in the hippocampus (Hip) and cortex of the mouse brains were detected by immunoblotting.
(G) Quantitative analysis of the synaptic protein levels in (F). *p < 0.05; **p < 0.01; ***p < 0.001 versus Con-Hip. n = 3 per group. N = 3 independent experiments. Data are mean ± SEM.
Synaptic plasticity can be expressed at the functional as well as structural levels. Of the latter, dendritic spines are highly dynamic and can respond to external stimuli with structural remodeling (Oomen et al., 2010). We therefore further examined spine density of hippocampal neurons in vivo. In CIP2A-overexpressed mice, we observed a marked decline in spine density compared with the control mice (Figures 5D and 5E). In addition, a downre-gulation was observed in the levels of pre-synaptic proteins synapsin and synaptophysin, as well as postsynaptic proteins including GluA1 and PSD95 (Figures 5F and 5G; Table 1). Taking together, these results demonstrate that an elevated amount of CIP2A causes synaptic degeneration in mice.
Table 1.
Antibodies Used in the Study
| Antibodies | Epitopes | Dilution | References and Sources |
|---|---|---|---|
| CIP2A (poly) | CIP2A | 1/1,000 | ABclonal (Boston, MA, USA) |
| CIP2A (poly) | CIP2A | 1/200–1,000 | Cell Signaling (Danvers, MA, USA) |
| pT668 (poly) | APP (Thr668) | 1/1,000 | Cell Signaling (Danvers, MA, USA) |
| DM1A (mAb) | α-tubulin | 1/500 | Cell Signaling (Danvers, MA, USA) |
| PSD95 (poly) | PSD95 | 1/1,000 | Cell Signaling (Danvers, MA, USA) |
| GAPDH (poly) | GAPDH | 1/1,000 | Cell Signaling (Danvers, MA, USA) |
| PP2Aa (poly) | P2A A subunit | 1/1,000 | Cell Signaling (Danvers, MA, USA) |
| APP (mAb) | APP | 1/1,000 | Droteintech (Chicago, IL, USA) |
| BACE1 (poly) | BACE1 | 1/500 | Droteintech (Chicago, IL, USA) |
| PS1 (poly) | PS1 | 1/500 | Proteintech (Chicago, IL, USA) |
| sAPPα (mAb) | sAPPα/fragments | 1/500 | Immuno-Biological Laboratories (Gunma, Japan) |
| sAPPβ (poly) | Sappβ/fragments | 1/500 | Immuno-Biological Laboratories (Gunma, Japan) |
| PP2Ac (mAb) | PP2AC | 1/1,000 | Millipore (Billerica, MA, USA) |
| GluA1 (poly) | GluA1 protein | 1/500 | Millipore (Billerica, MA, USA) |
| GluA2 (poly) | GluA2 (aa 175–430) | 1/500 | Millipore (Billerica, MA, USA) |
| N-GluA1 (mAb) | GluA1 N terminus | 1/100 | Millipore (Billerica, MA, USA) |
| Synapsin 1 (poly) | Synapsin 1 | 1/1,000 | Millipore (Billerica, MA, USA) |
| Tau-5 (mAb) | Tau-5 (aa 210–241) | 1/1,000 | Millipore (Billerica, MA, USA) |
| Tau-5 (mAb) | - | 1/1,000 | Thermo Fisher (Waltham, MA, USA) |
| Synaptophysin (poly) | Synaptophysin (aa 250–350) | 1/1,000 | Abcam (Cambridge, UK) |
| β-Actin (mAb) | β-actin | 1/1,000 | Abcam (Cambridge, UK) |
| pS396 (mAb) | Tau-5(aa 352–441) | 1/1,000 | Anaspec (Fremont, CA, USA) |
| pS396 (poly) | Tau (Ser 396) | 1/200–1,000 | Signalway Antibody (Pearland, TX, USA) |
| pS404 (poly) | Tau (Ser 404) | 1/200–1,000 | Signalway Antibody (Pearland, TX, USA) |
aa, amino acids; mAb, mouse monoclonal antibody; p, phosphorylated; poly, rabbit polyclonal antibody.
DISCUSSION
PP2A inactivation is regarded as the most important molecular event that causes abnormal tau hyperphosphorylation in the AD brain (Iqbal et al., 2016). PP2A suppression also induces APP phosphorylation at the T668 site, leading to enhanced β-secretase processing of APP (Sontag et al., 2007). Thus, abnormal PP2A activity is a key causal factor in the development of the two primary pathological alterations in AD. However, the endogenous pathophysiological mechanisms that inhibit PP2A activity in human AD have not been resolved.
PP2A is composed of scaffolding A, regulatory B, and catalytic C subunits. The composition of the three subunits determines the activity of PP2A holoenzyme toward the substrates (Janssens and Goris, 2001). Sontag et al. (2004a, 2004b) reported that, in AD brains, impaired methylation of the Leu309 site of the PP2A catalytic subunit (PP2Ac) may decrease the interaction of PP2Ac with PP2A Bα and reduce the level of PP2ABαC holoenzyme, which is the main form of PP2A for tau dephosphorylation. We have reported that Y307 phosphorylation of PP2A was increased in AD and contributed to PP2A inhibition (Liu et al., 2008). In addition, an altered cellular location of endogenous PP2A inhibitors was reported to induce PP2A inactivation in cytoplasm (Tanimukai et al., 2005). Furthermore, PP2A participates in dephosphorylation of a large number of substrates in addition to tau and APP, such as p53, mitogen-activated protein kinase (MEK), extracellular signal-regulated kinase (ERK), and Akt (Dennis et al., 2014; Junttila et al., 2007; Ugi et al., 2002). Small molecules aimed at directly elevating PP2Ac catalytic activity will likely result in dephosphorylation of all of these substrate proteins and induce cell dysfunction. Thus, elucidating the mechanism that contributes to tau and APP-related PP2A inhibition in AD will better help us to understand AD pathogenesis in order to identify potential drug targets.
We focus our attention on CIP2A for a few reasons. First, the inhibitory effect of CIP2A on PP2A is highly specific. A large number of studies especially on cancer have shown that PP2A is the only target of CIP2A (Junttila et al., 2007; Soo Hoo et al., 2002). Second, the inhibitory effect of CIP2A on PP2A is substrate dependent. CIP2A inhibits the PP2A activity only toward a certain number of substrates without influencing the phosphorylation of other substrates. For example, in tumor tissues, CIP2A inhibits the dephosphorylation of c-Myc at the Ser62 site by PP2A, and thus protects c-Myc from degradation, without affecting p53 and MEK phosphorylation (Junttila et al., 2007). Third, the first crystal structure of CIP2A was recently published and revealed potential druggable structures (Wang et al., 2017). Actually, drugs have been identified aiming to inhibit CIP2A in cancer therapy (De et al., 2014). Therefore, it is becoming intriguing and urgent to understand whether CIP2A also participates in tau/APP-related PP2A inhibition in AD pathogenesis and serves as a novel AD therapy target protein.
We first explored the effects of CIP2A on amyloidosis. Aβ toxicity is regarded as the central and initial event in AD development. Aβ induces tau filament formation in mouse brain (Götz et al., 2001). Mice expressing mutated human APP and mutated human tau develop several-fold increased neurofibrillary tangles, compared with mice expressing only mutated human tau (Lewis et al., 2001). We identified the interaction of CIP2A, PP2A, and APP, and found that CIP2A promoted Aβproduction by enhancing β-cleavage of APP. This enhancement was dependent on APP-T668 phosphorylation, because the mutation of Thr to Ala (nonphosphorylated) blocked the CIP2A-induced Aβ overproduction. Furthermore, CIP2A did not change PS1 and BACE1 levels, nor the α-cleavage of APP. CIP2A-induced APP β-cleavage could be reversed by PP2A re-activation, indicating that the effect was mediated by PP2A. Thus, via inhibiting PP2A, CIP2A causes an increase in APP-T668 phosphorylation, which in turn promotes β-cleavage of APP and Aβ production.
An increasing amount of evidence suggests that Aβ and tau interact to either cause or facilitate the pathogenesis of AD (Ittner and Götz, 2011). Neurons without tau are resistant to Aβ toxicity, and APP transgenic mice lacking tau develop Aβ pathology but remain cognitively normal (Rapoport et al., 2002; Roberson et al., 2007). A combined reduction of both Aβ and tau, but not Aβ alone, was found to improve cognition in a mouse model with both plaques and tangles (Oddo et al., 2004). These observations suggest that tau pathology may combine with or downstream of Aβ to cause cognitive deficits. We thus explored the effects of CIP2A on tau. Our results showed that CIP2A, PP2A, and tau were associated with each other, and CIP2A induced tau hyperphosphorylation at the AD-related phosphorylation sites through inhibiting PP2A. In primary neurons, we further observed that CIP2A promoted tau translocalization to dendrites and spines, which is considered to be the key event mediating the synaptic toxicity of tau (Pooler et al., 2014; Zempel et al., 2013). Also, we found that the mislocalization of tau (especially hyperphosphorylated tau) was accompanied by synaptic degeneration indicated by a decrease in spine numbers and synaptic proteins including AMPARs. In line with the role for AMPAR synaptic accumulation in synaptic plasticity, we found impairments in LTP expression in mice infected with AAV-CIP2A. Given the important role of synaptic plasticity in learning and memory, the observed abnormalities in synapse formation and function may serve as the mechanisms underlying CIP2A/PP2A-dependent tau pathology and cognitive deficits.
Consistent with our findings in vitro, the learning and memory deficits were identified in CIP2A-overexpressed mice by a line of behavior tests including novel objective recognition, Morris water maze, and fear conditioning. Furthermore, the two typical AD pathologic hallmarks, tau hyperphosphorylation and Aβ over-production, were also detected in the CIP2A-overexpressing mouse brains. Using the brain tissues, we showed that increased CIP2A expression promoted AD pathogenesis indicated by tau phosphorylation, Aβ over-production, synapse loss, and cognitive impairments.
The upstream mechanism for CIP2A upregulation in AD brains needs further exploration. In cancer cells, CIP2A expression is induced by constitutive DNA-damage signaling (Khanna et al., 2013b). With the recognized knowledge that Aβ induces oxidative DNA damage (Suberbielle et al., 2013; Wu et al., 2014; Yan et al., 2014), we observed increased CIP2A expression together with retarded PP2A activity both in Aβ-treated primary rat neurons and APP-overexpressed SH-SY5Ycells; in Aβ-treated primary neurons, CIP2A knockdown effectively reversed Aβ-induced APP and tau hyperphosphorylation. These data strongly indicate that Aβ is an initial factor that induces CIP2A upregulation in AD brains. Furthermore, aging is the most important risk factor for AD. With DNA damage as a common event in aging (Lu et al., 2004), increased CIP2A expression is very possibly induced during the aging process. We suspected that Aβ accumulation and DNA damage during aging triggers the overexpression of CIP2A, thus forming a vicious cycle to promote AD pathogenesis.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
For all primary antibodies used in this study, see details in Table 1.
Wild-type APP770 plasmid and EGFP-tau (human tau441) plasmid were generous gifts from Prof. Angela Ho (Boston University, Boston, MA, USA) and Prof. Fei Liu (Jiangsu Key Laboratory of Neuroregeneration, China), separately. The APPT668A was a mutant plasmid based on APP770 in which Thr742 (corresponding to the Thr668 site in APP695) was mutated to Ala. Wide-type PP2A plasmid was from Dr. Haendeler (University of Frankfurt, Germany). The surface-EGFP plasmid was constructed by inserting a Lyn before the EGFP sequence to enable EGFP to have the membrane-binding ability. pAAV-SYN-CIP2A-EGFP-3FLAG and control vector were from Obio Technology (Shanghai, China). CIP2A overexpression lentivirus, shRNA-CIP2A, and control shRNA lentivirus were constructed and packaged by Genechem (Shanghai, China). PCMV-u6-shRNA-CIP2A and control plasmids were constructed by Neuron Biotech (Shanghai, China); the target sequence of shRNA-CIP2A is 5’-GCACAATCTTTCTGTTCAA-3’.
Human Brain Tissue and Animals
The human brain tissues were obtained from the NIH NeuroBioBank. For animal experiments, Sprague-Dawley rats and C57 mice were from the Experimental Animal Center of Tongji Medical College, Huazhong University of Science and Technology. Rats and mice were kept under standard laboratory conditions: 12-hr light and 12-hr dark with water and food ad libitum. All animal experiments were approved by the Animal Care and Use Committee of Huazhong University of Science and Technology, and performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Cell Culture and Transfection
For HEK293-T cell culture, the cells were cultured in DMEM-high medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 0.1 mg/mL streptomycin at 37°C in the presence of 5% CO2. The cells were cultured to 60%−70% confluence in six-well plates and changed to fresh medium, then transfected with relevant plasmids by Neofect DNA transfection reagent (Neofect Biological Technology, Beijing, China). After 48 hr, cells and culture media were collected for further detections.
For primary neuron culture, cortical neurons were isolated from embryonic day (E) 18 Sprague-Dawley rats and cultured as previously described (Sun et al., 2016). At the end of treatments, cells were collected and lysed in radio immunoprecipitation assay (RIPA) buffer for further biological detections or fixed with 4% paraformaldehyde for immunofluorescence imaging.
Western Blotting and CoIP
Western blotting was performed as previously described (Zhou et al., 2017a). Blots were visualized using the Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE, USA) or the enhanced chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK). The protein bands were quantitatively analyzed by ImageJ software (Rawak Software, Germany).
To analyze protein-protein interactions, we performed CoIP experiments using cell lysates. Specified antibody and protein G agarose were incubated with the samples overnight at 4°C. The resins were washed three times with PBS. After elution by 2× loading buffer, and boiled at 95°C for 5 min, the bound proteins were analyzed by western blotting.
Aβ40/42 Assay by ELISA
Cells were lysed in RIPA buffer and centrifuged for 5 min at 3,000 × g at 40C, the supernatants containing soluble Aβ40 and Aβ42 were collected. The pellets werefurtherdissolved in formic acid (FA) fordetection ofAβ. The levelsofAβ40 and Aβ42 in RIPA-soluble and FA-soluble fractions were detected by ELISA following the construction offered by the assay kit manufacturer (Elabscience Biotechnology, Wuhan, China).
Fluorescence Imaging and Confocal Microscopy
For immunofluorescence, neurons were fixed with 4% paraformaldehyde for 15 min, permeabilized in 0.5% Triton X-100 for 10 min, followed by incubation with 3% BSAto block nonspecific sites. Primary antibody incubation was performed overnight at 40C. Alexa 488- and 543-conjugated secondary antibodies (1:200, A-21206, A-10036; Invitrogen) were used for double-fluorescence labeling. For labeling of the surface GluA1, at the end of treatment, antibody against the N-terminal GluA1 (1:100) was added intothe culture media for 45 min. Cells were then washed with cold PBS two times, fixed, and blocked, followed with Alexa 488-conjugated secondary antibody incubation for 1 hr. Cells were washed again and permeabilized inTritonX-100forimmu-no fluorescence staining with anti-PSD95 primary antibody and Alexa 543-conjugated secondary antibody. For observation of the cell morphology, surface-EGFP-expressed neurons were fixed and observed under the microscope directly. All of the images were observed with the LSM710 confocal microscope (Zeiss, Germany). The spines were quantitatively analyzed by ImageJ software (Rawak Software, Germany).
PP2A Activity and LDH Assay
PP2A activity was measured according to the protocol provided by the manufacturer (V2460 kit; Promega). The LDH assay was performed using an LDH cytotoxicity assay kit (Promega) according to the manufacturer’s instructions.
Crude Synaptosome Fractionation
Cells in a six-well plate were washed with cool artificial cerebral spinal fluid (aCSF) and suspended in 150 μL/well solution A containing 0.32 M sucrose, 1 mM NaHCO3,1 mM MgCl2, 0.5 mM CaCl2*2H2O, 10 mM sodium pyrophosphate, and protease inhibitors on ice. The cell suspension was collected and blown up and down with a pipette for up to 12 times, centrifuged at 710 × g for 10 min, and the supernatant was collected. The pellet (P1) was re-suspended in 50 mL of solution A, following pipette blowing up and down three times, centrifuged at 1,400 × g for 10 min. The supernatant was collected together with the previously collected supernatant as whole-cell lysate (S1, whole). S1 was further centrifuged at 13,800 × g for 10 min; supernatant (S2, cytosolic fraction) and pellet (P2, containing synaptosome and mitochondria) were separated. Pellet was resuspended in RIPA buffer, sonicated on ice, and rotated at 40C for 30 min. The protein concentrations in whole, cytosolic, and crude synaptosomal fractions were measured by BCA assay. Samples were then boiled with 2× loading buffer for western blot detection.
Acute Slice Preparation and LTP Recording
Mice were decapitated under anesthesia with isoflurane, and the brain was quickly removed and placed in ice-cold oxygenated aCSF: 119 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1 mM NaH2PO4, 11 mM glucose, 1.3 mM MgSO4, and 2.5 mM CaCl2 (pH 7.4). Horizontal 350-μm-thick brain slices were cut in ice-cold aCSF using a vibrating microtome (Leica, VT1000S; Germany). Slices were transferred to a recovery chamber for at least 1.5 hr with oxygenated aCSF at room temperature until recordings were performed.
For LTP, acute brain slices were transferred to a recording chamber and submerged in aCSF. Slices were laid down in a chamber with an 8 × 8 microelectrode array (Parker Technology, Beijing, China) in the bottom planar (each 50 × 50 mm in size, with an interpolar distance of 150 μm) and kept submerged in aCSF. Signals were acquired using the MED64 System (Alpha MED Sciences, Panasonic). The field excitatory postsynaptic potentials (fEPSPs) in CA1 neurons were recorded by stimulating CA3 neurons. LTP was induced by applying three trains of high-frequency stimulation (HFS; 100 Hz, 1-s duration). The LTP magnitude was quantified as the percentage change in the fEPSP slope (10%−90%) taken during the 60-min interval after LTP induction.
Electrophysiology
Rat hippocampal neurons (DIV 10) cultured on coverslip were infected with CIP2A or control lentivirus for 6 days. For whole-cell patch-clamp recording, a coverslip of neurons was transferred to a recording chamber with the extracellular solution containing 140 mM NaCl, 3 mM KCl, 1.5 mM MgCl2, 2.5 mM CaCl2, 11 mM glucose, and 10 mM HEPES (pH 7.4), which was supplemented with tetrodotoxin (1 μM) to block action potentials, 2-amino-5-phosphonopetanoate (50 μM) to block NMDARs, and bicuculline (20 mM) to block g-aminobutyric acid A (GABAA) receptor-mediated inhibitory synaptic currents. Whole-cell voltage-clamp recordings were made with patch pipettes filled with an intracellular solution containing 100 mM Cs-methanesulfonate, 10 mM CsCl, 10 mM HEPES, 0.2 mM EGTA, 4 mM Mg-ATP, 0.3 mM Na-GTP, 5 mM QX-314, and 10 mM Na-phosphocreatine (pH 7.4), with the membrane potential clamped at —70 mV.
Golgi Staining
The mice were anesthetized by isoflurane and perfused intracardially with 400 mL of normal saline containing 0.5% sodium nitrite, followed with 400 mL 4% formaldehyde and the Golgi dye solution containing 5% chloral hydrate, 4% formaldehyde, and 5% potassium dichromate. After being perfused, the brains were dissected into 5 mm x 5 mm sections and transferred to a vial containing Golgi dye solution for 3 days in the dark, then immersed in solution containing 1% silver nitrateforanother3days. The brains were serially sectioned into 100-μm-thick slices using a vibrating microtome (Leica, VT1000S, Germany). Images were observed under the microscope (Nikon, Tokyo, Japan).
Behavior Tests
Novel Objective Recognition Test
The mice were habituated to the arenas (50 cm x 50 cm x 50 cm plastic container) for 5 min without objects 24 hr prior to the test. Arenas were cleaned with 70% ethanol between each habituation period. The day after the mice reentered the arenas from the same starting point, they were granted 5 min to familiarize themselves with A object and B object. After each familiarization period the arena and objects were cleaned with 70% ethanol. Exactly 1 hr after the familiarization period, B object was replaced with C object, and the mice were granted 5 min to explore both objects. After 24 hr, C object was replaced with D object, and the mice were granted 5 min to explore both objects. The behavior was recorded by a video camera positioned above the arena. The recognition index was calculated by TA/(TA + TB), TB/(TA + TB), TC/(TA + TC), and TD/(TA + TD). The discrimination index was calculated by (TC — TA)/(TA + TC), (TD — TA)/(TA + TD). TA, TB, TC, and TD were, respectively, the time mice explored the objects A, B, C, and D.
Fear Conditioning Test
The fear conditioning test paradigm was performed following methods previously described. In brief, the test was conducted in a conditioning chamber (33 cm x 33 cm x 33 cm) equipped with white board walls, a transparent front door, a speaker, and a grid floor. On day 1, mice were placed in to the conditioning chamber and allowed free exploration for 2 min before the delivery of the conditioned stimulus (CS) tone (20 s, 80 dB, 2,000 Hz) paired with a foot-shock unconditioned stimulus(US; 2 s, 0.95 mA) through a grid floor at the end of the tone. A total of five CS-US pairs with a 60-s intertrial interval (ITI) were presented to each animal in the training stage. The mouse was removed from the chamber 1 min after the last foot-shock and placed back in its home cage. The contextual fear conditioning stage started 48 hr after the training phase, when the animal was put back inside the conditioning chamber for 5 min. The animal’s freezing responses to the environmental context were recorded. The animal was placed back into the same chamber with different contextual cues, including green wall, smooth plastic floor, and vinegar drops condition for 5 min, and the animal’s freezing responses to the altered context were recorded. The tone fear conditioning stage started 2 hr after the different contextual stage. After 2minoffreeexploration, the mouse was exposed to the exact same3-CStones with 20-s ITI in the training stage without the foot-shock, and its freezing responses to the tones were recorded.
Morris Water Maze Test
Spatial reference memory was detected by the Morris water maze (MWM) test. The Morris water maze task was performed as previously described (Zhou et al., 2017a). Learning consisted of six consecutive daily acquisition sessions, each of them consisting of four trials. Probe tests were carried out 1 or 48 hr after the last acquisition session.
Statistical Analysis
Data are expressed as mean ± SEM and analyzed using GraphPad Prism 5 statistical software (GraphPad Software). The one-way ANOVA was used to determine the differences among groups. For the comparison between two groups, the Student’s t test was used. The significance was assessed at p < 0.05. All results shown correspond to individual representative experiments.
Supplementary Material
Highlights.
PP2A inhibitor CIP2A is increased in AD human brains
CIP2A promotes β-cleavage of APP and Aβ production through APP-T668 phosphorylation
CIP2A induces tau hyperphosphorylation and mislocalization into dendritic spines
CIP2A causes LTP impairment, synaptic degeneration, and memory deficits in mice
ACKNOWLEDGMENTS
We thank members of the Liu and Man labs for helpful comments on the manuscript and the NIH NeuroBioBank for providing human brain tissues. This work was supported by the National Natural Science Foundation of China (grants 81471304 and 31771189) (to R.L.), Natural Science Foundation of Hubei Province, China (2017CFA065) (to R.L.), Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST (to J.-Z.W.), and NIH grant R01MH079407 (to H.-Y.M.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.009.
REFERENCES
- De P, Carlson J, Leyland-Jones B, and Dey N (2014). Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A (CIP2A): an oncoprotein with many hands. Oncotarget 5, 4581–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MD, Coleman CS, Berg A, Jefferson LS, and Kimball SR (2014). REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci. Signal 7, ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, and Iqbal K (1993). Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem 61, 921–927. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, and Iqbal K (1995). Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J. Neurochem 65, 732–738. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, and Iqbal K (2000). Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J. Biol. Chem 275, 5535–5544. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, and Nitsch RM (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M,Tung YC, Zaidi MS, and Wisniewski HM (1986). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem 261, 6084–6089. [PubMed] [Google Scholar]
- Iqbal K, Liu F, and Gong CX (2016). Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol 12, 15–27. [DOI] [PubMed] [Google Scholar]
- Ittner LM, and Götz J (2011). Amyloid-β and tau—atoxic pasdedeux in Alzheimer’s disease. Nat. Rev. Neurosci 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Janssens V, and Goris J (2001). Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J 353, 417–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemelä M, Ahola R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et al. (2007). CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62. [DOI] [PubMed] [Google Scholar]
- Kerosuo L, Fox H, Perälä N, Ahlqvist K, Suomalainen A, Westermarck J, Sariola H, and Wartiovaara K (2010). CIP2A increases self-renewal and is linked to Myc in neural progenitor cells. Differentiation 80, 68–77. [DOI] [PubMed] [Google Scholar]
- Khanna A, Pimanda JE, and Westermarck J (2013a). Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 73, 6548–6553. [DOI] [PubMed] [Google Scholar]
- Khanna A, Kauko O, Böckelman C, Laine A, Schreck I, Partanen JI, Szwajda A, Bormann S, Bilgen T, Helenius M, et al. (2013b). Chk1 targeting reactivates PP2A tumor suppressor activity in cancer cells. Cancer Res. 73, 6757–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemelä M, Khanna A, Chan EK, Kähäri VM, Kellokumpu-Lehtinen PL, et al. (2013). Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov. 3, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, and Gong CX (2005). Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation oftau phosphorylation. Eur. J. Neurosci 22, 1942–1950. [DOI] [PubMed] [Google Scholar]
- Liu R, Zhou XW, Tanila H, Bjorkdahl C, Wang JZ, Guan ZZ, Cao Y, Gustafsson JA,Winblad B, and Pei JJ (2008). Phosphorylated PP2A(tyro-sine 307) is associated with Alzheimer neurofibrillary pathology. J. Cell. Mol. Med 12, 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, and Yankner BA (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, and Beyreuther K (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 82, 4245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RE, Sontag JM, Zhang H, Staniszewski A, Yan S, Kim CY, Yim M, Woodruff CM, Arning E, Wasek B, et al. (2016). PP2A methylation controls sensitivityand resistance to β-amyloid-induced cognitiveand electro-physiological impairments. Proc. Natl. Acad. Sci. USA 113, 3347–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, and LaFerla FM (2004). Abeta immunotherapy leads to clearance of early, but not late, hyperphos-phorylated tau aggregates via the proteasome. Neuron 43, 321–332. [DOI] [PubMed] [Google Scholar]
- Oomen CA, Soeters H, Audureau N, Vermunt L, van Hasselt FN, Man-ders EM, Joëls M, Lucassen PJ, and Krugers H (2010). Severe early life stress hampers spatial learning and neurogenesis, but improves hippocampal synaptic plasticity and emotional learning under high-stress conditions in adulthood. J. Neurosci 30, 6635–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Noble W, and Hanger DP (2014). A role fortau atthe synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 76 (Pf A), 1–8. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, and Ferreira A (2002). Tau is essential to beta-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 99, 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, and Mucke L (2007). Reducing endogenous tau ame-lioratesamyloid beta-induced deficits in an Alzheimer’sdisease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, and White CL 3rd. (2004a). Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 63, 1080–1091. [DOI] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, and White CL 3rd. (2004b). Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 63, 287–301. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, Dayal S, Lentz SR, Arning E, and Bottiglieri T (2007). Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci 27, 2751–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo Hoo L, Zhang JY, and Chan EK (2002). Cloning and characterization of a novel 90 kDa ‘companion’ auto-antigen of p62 overexpressed in cancer. Oncogene 21, 5006–5015. [DOI] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, De-vidze N, Kreitzer AC, and Mucke L (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci 16, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, and Wang JZ (2003). Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118, 1175–1182. [DOI] [PubMed] [Google Scholar]
- Sun XY, Tuo QZ, Liuyang ZY, Xie AJ, Feng XL, Yan X, Qiu M, Li S, Wang XL, Cao FY, et al. (2016). Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation. Cell Death Dis. 7, e2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimukai H, Grundke-Iqbal I, and Iqbal K (2005). Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am. J. Pathol 166, 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugi S, Imamura T, Ricketts W, and Olefsky JM (2002). Protein phosphatase 2A forms a molecular complex with Shc and regulates Shc tyrosine phosphorylation and downstream mitogenic signaling. Mol. Cell. Biol 22, 2375–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, and Iqbal K (1995). Dephosphorylation of Alzheimer paired helical filaments by protein phospha-tase-2A and-2B. J. Biol. Chem 270, 4854–4860. [DOI] [PubMed] [Google Scholar]
- Wang J, Okkeri J, Pavic K, Wang Z, Kauko O, Halonen T, Sarek G, Ojala PM, Rao Z, Xu W, and Westermarck J (2017). Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. EMBO Rep. 18, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, Yin JH, Hwang CS, Tang CM, and Yang DI (2014). NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res 48, 794–805. [DOI] [PubMed] [Google Scholar]
- Yan S, Sorrell M, and Berman Z (2014). Functional interplay between ATM/ ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci 71, 3951–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Ni W, Liu W, Bai J, and Li W (2013). Expression and biological role of CIP2A in human astrocytoma. Mol. Med. Rep 7, 1376–1380. [DOI] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, and Mandelkow EM (2013). Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32, 2920–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Guo Y, Li X, Liuyang ZY, Shentu YP, Jing XP, Liang JW, Zhou XW, Wang XC, Wang JZ, et al. (2017a). Long-term Helicobacter pylori infection does not induce tauopathy and memory impairment in SD rats. J. Huazhong Univ. Sci. Technolog. Med. Sci 37, 823–827. [DOI] [PubMed] [Google Scholar]
- Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, Wang YC, Swerts J, Beyens J, Miskiewicz K, et al. (2017b). Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun 8, 15295–15308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.