

Abstract
The mitis group, a member of the genetically diverse viridans group streptococci, predominately colonizes the human oropharynx. This group has been shown to cause a wide range of infectious complications in humans, including bacteremia in patients with neutropenia, orbital cellulitis and infective endocarditis. Hydrogen peroxide (H2O2) has been identified as a virulence factor produced by this group of streptococci. More importantly, it has been shown that Streptococcus oralis and S. mitis induce epithelial cell and macrophage death via the production of H2O2. Previously, H2O2 mediated killing was observed in the nematode Caenorhabditis elegans in response to S. oralis and S. mitis. The genetically tractable model organism C. elegans is an excellent system to study mechanisms of pathogenicity and stress responses. Using this model, we observed rapid H2O2 mediated killing of the worms by S. gordonii in addition to S. mitis and S. oralis. Furthermore, we observed colonization of the intestine of the worms when exposed to S. gordonii suggesting the involvement of an infection-like process. In response to the H2O2 produced by the mitis group, we demonstrate the oxidative stress response is activated in the worms. The oxidative stress response transcription factor SKN-1 is required for the survival of the worms and provides protection against H2O2 produced by S. gordonii. We show during infection, H2O2 is required for the activation of SKN-1 and is mediated via the p38-MAPK pathway. The activation of the p38 signaling pathway in the presence of S. gordonii is not mediated by the endoplasmic reticulum (ER) transmembrane protein kinase IRE-1. However, IRE-1 is required for the survival of worms in response to S. gordonii. These finding suggests a parallel pathway senses H2O2 produced by the mitis group and activates the phosphorylation of p38. Additionally, the unfolded protein response plays an important role during infection.
Introduction
Members of the mitis group of streptococci, which are also part of the viridans group of streptococci (VGS), are commensals of the oral cavity and the upper respiratory tract of humans [1]. This group of microorganisms includes Streptococcus gordonii, S. mitis, S. oralis, S. sanguinis, and a few related species. Mitis group streptococci have been recognized as causative agents of a variety of human diseases such as infective endocarditis, orbital cellulitis and more recently increased incidences of bacteremia in patients with neutropenia [2–5]. More importantly unique strains of S. mitis and S. oralis that cause bacteremia have been isolated from cancer patients that have undergone chemotherapy [6, 7]. These invasive strains are resistant to more antimicrobial agents than other VGS species, and as a consequence increase the risk of adverse patient outcomes [8]. Despite the clinical significances of these infections, the mechanisms of pathogenesis and the pathophysiology are poorly understood. In addition, due to increased antibiotic resistance, novel therapeutic strategies are required to combat these organisms.
The mitis group is known to produce hydrogen peroxide (H2O2) which has been shown to play important roles in oral microbial communities [9]. H2O2 produced by S. gordonii and S. sanguinis has been reported to reduce the growth of cariogenic S. mutans and several other pathogenic bacteria [10, 11]. In addition, H2O2 stimulates the release of DNA which is important for biofilm formation and enables the exchange of genetic material among bacteria [12]. However, recent studies have highlighted a role for H2O2 as a cytotoxin that induces macrophage, neutrophil and epithelial cell death [13–15]. A more recent study demonstrated H2O2 produced by S. oralis mediates damage of lysosomes resulting in macrophage death [16]. Furthermore, pneumococcal H2O2 induced the activation of stress related cellular signaling pathways confirming the role of H2O2 as an important virulence factor of the mitis group [17].
C. elegans, a microscopic nematode is an excellent model system for studying pathogenesis, immunity and oxidative stress. In its natural habitat, the worms consume bacteria as their food source and thereby encounter numerous threats and insults from the ingested microbes [18]. This has ensured a strong selection pressure to evolve and maintain an immune response within the intestinal cells of the worm that is capable of producing a targeted response [19]. In the worm the mechanisms underlying pathogenesis are mediated through an infection like process or by diffusible toxins. Previously, H2O2 produced by Enterococcus faecium, S. pyogenes, S. pneumonia, S. oralis and S. mitis was shown to be responsible for killing C. elegans [20–22].
Phase II reactions provide protection against Reactive Oxygen Species (ROS)-induced oxidative stress damage in tissues that are associated with many diseases and aging [23, 24]. Enzymes supporting these detoxification reactions are involved in the biosynthesis of glutathione (GCS-1) and its conjugation of substances such as glutathione-S-transferases (GSTs). The induction of phase II gene expression has been shown to be regulated by the Cap and Collar (CnC) transcription factors. For example Nrf-2 (erythroid-derived-2)-like-2) in mammals and SKN-1 in C. elegans, mainly control transcription of the phase II response [25, 26]. Several studies in C. elegans have shed light into the regulation of SKN-1 an orthologue of Nrf-2 [25, 27]. SKN-1 is activated in response to different stimuli mediated by several regulatory pathways [28–30]. Oxidative stress induced activation is facilitated by the phosphorylation of SKN-1 via the p38 MAPK pathway [31]. This signal pathway is comprised of the MAPKKK, NSY-1 (ASK-1 homologue), the MAPKK, SEK-1 and the MAPK, PMK-1 (p38 homologue). Similarly, ROS produced by the dual oxidase Ce-DUOX-1/BLI-3 in response to pathogens activates SKN-1 through the p38 MAPK pathway [32]. Until recently, the primary trigger that activates the p38 MAPK pathway by oxidative stress in the worms was not discovered. Work published by Hourihan et al demonstrated the endoplasmic reticulum (ER) transmembrane protein IRE-1 initiates the p38/SKN-1(Nrf2) antioxidant response by ROS that are generated at the ER or by the mitochondria [33].
In this study, we demonstrate members of the mitis group of streptococci are able to colonize and cause death of C. elegans via the production of H2O2. We demonstrate SKN-1 is required for the survival of the worms and is protective against these pathogens when overexpressed. Furthermore, the activation of SKN-1 is via the p38 MAPK pathway. However, based on our data the activation of p38 MAPK pathway does not require IRE-1 as previously seen during oxidative stress induced by arsenite. Moreover, in the presence of S. gordonii phosphorylation of PMK-1 is not dependent on Ce-Duox1/BLI-3.
Materials and methods
C. elegans strains
C. elegans strains were grown and maintained as previously described [34]. The strains used in this study are listed in the supplementary data (S1 Table).
Bacterial strains
All streptococci strains were grown in Todd Hewitt media supplemented with 0.3% yeast extract (THY) with or without sheep blood, while E. coli strains were grown in Luria-Bertani (LB) broth in the presence or absence of antibiotics streptomycin or carbenicillin. Strains used in this study were S. gordonii DL1 Challis (WT), S. gordonii Challis DL1 ΔspxB, S. gordonii Challis DL1 ΔspxB;spxB+, S. mitis ATCC, S. oralis ATCC, S. mutans, S. salivarius, and E. coli OP50. Clinical isolates of S. mitis (VGS# 3 and 4) and S. oralis (VGS# 10 and 13) were used in this study.
Killing assays
For streptococcus killing assays, bacterial strains grown in THY medium with or without sheep blood for 16 hours were seeded on THY plates in the presence or absence of 1000U of catalase or 100U of superoxide dismutase and incubated at 37°C for 24 hours in a candle jar. 60–90 L4 larvae were transferred to two replica plates and incubated at 25°C under aerobic conditions. Larvae were scored for survival at various points along the time course. The experiment was repeated 3 times.
Colonization of worms
Overnight cultures of the bacteria were pelleted, resuspended in PBS containing 10mM 5-(and-6)-Carboxy-X-Rhodamine, Succinimidyl Ester (5(6)-ROX, SE) and incubated at room temperature (RT). Thereafter, concentrated stained cultures were plated on to THY plates and incubated in the dark at RT for 30 minutes. L4 larvae were subsequently added to the plates and incubated at 25°C in the dark for 2 hours. Worms were collected using M9, anesthetized in M9 containing 0.1% sodium azide, mounted on 2% agarose pads and imaged using a Nikon C2 plus confocal microscope.
Worms exposed to the bacteria were collected and washed 3 times in M9 containing 25 mM levamisole. Thereafter, worms were exposed to 25 mM levamisole, 1mg/ml ampicillin and 1mg/ml streptomycin in M9 for 1 hour to kill the external bacteria. Worms were washed again in M9 containing 25 mM levamisole and five worms were disrupted in M9 using a motorized pestle. Serial dilutions of the lysate were grown overnight on THY at 37°C under microaerophilic conditions and the colony forming units were counted. At least three replicates for each strain of bacteria were processed.
RNA Isolation and sequencing (RNAseq)
L4 larvae were exposed to bacterial strains for 2–3 hours. Thereafter, RNA was extracted from the worms using Trizol (Invitrogen) as recommended by the manufacturer. Subsequently, samples were treated with Tubro DNase I (Ambion) to remove DNA contamination according to manufacturer’s instructions. RNA samples were sequenced by Genewiz Inc. The average reads/sample was 35,106,625.25, while the average percentage of the bases with ≥Q30 reads was 98.4. Using CLC Genomics Server program the sequences were mapped to the C. elegans genome. After quantile normalization on RKPM values, unsupervised hierarchical clustering and Principal Component Analysis (PCA) were performed. The gene expression comparison between the control samples and the test samples was conducted using proportion based Kal’s test, which generalizes the RPKM as counts for 1 vs 1 comparison. For all the statistical tests, p-values and fold-change values were calculated. A list of differentially expressed genes with a normalized RPKM fold difference greater than 2 or less than -2 that had a FDR corrected p-value < 0.05 were selected as differentially expressed genes.
Quantitative Real Time Polymerase Chain Reaction (qRT-PCR) Analysis
RNA was isolated as described above and qRT-PCR was performed on a Bio-rad CFX96 Touch Real-Time PCR Detection System using an iTaq Universal SYBR Green one-step kit (Bio-rad). Comparative CT method was used to determine fold changes in gene expression normalized to act-1. Three biological replicates were performed. Primers used in this study were previously described [32].
RNA Interference
RNAi was induced by feeding L1 worms through L4 stage with bacteria producing dsRNA to target genes. To induce bli-3 knockdown in the worms, E. coli HT115(DE3) expressing bli-3 RNAi was diluted in a 1:30 ratio with E.coli harboring the vector control as previously described [32]. RNAi expressing clones were obtained from the C. elegans library (Source Bioscience). All clones were verified by sequencing. Clones absent in the library were constructed as previously described [32].
Fluorescence microscopy
To investigate the expression of gcs-1::gfp, worms were exposed to bacterial strains for 2 hours at 25°C, subsequently anesthetized using 0.1% sodium azide, mounted on 2% agarose pads, and imaged using a Nikon C2 plus confocal microscope. The levels of GFP expression were scored as previously described [32]. SKN-1B/C::GFP expression was analyzed by fluorescence microscopy in worms exposed to bacterial strains for 2–3 hours. Imaging was performed using the FITC and DAPI channels. Percentages of worms indicating the degree of nuclear localization of SKN-1B/C::GFP in the intestinal cells were scored as previously described [31]. All experiments were repeated more than three times.
Western blot
Worms exposed to the bacterial strains for 2 hours were washed and collected in a 100 μl pellet using protein extraction buffer containing a protease inhibitor cocktail (Pierce) and a phosphatase inhibitor (Pierce). Pellets were sonicated two times on ice for 10 seconds at level 5 and 35 duty. The resulting suspensions were incubated with 1% SDS for 5 minutes and thereafter centrifuged at 14,000 XG for 10 minutes at 4°C. Supernatants were transferred to fresh Eppendorf tubes and the total protein concentration was estimated using a Bradford assay. Sample buffer was added to protein lysates and boiled for 5 minutes. For phospho-p38 detection, 20μg of total protein for each sample was resolved on a 12% SDS-PAGE gel and subsequently transferred to a PVDF membranes for 45 minutes at Room Temperature (RT) using a semidry transfer apparatus. The membranes were blocked with TBS-T blocking buffer for 1 hour. Thereafter, the membranes were incubated with 1:1000 Phospho-p38 antibodies (cell signaling) overnight at 4°C. The blots were washed 3 times with TBS-T and incubated with 1:5000 secondary antibodies conjugated to horseradish peroxidase for 1 hour at RT. Subsequently, the membranes were washed with TBS-T 3 times, incubated with enhanced chemiluminescent substrate (Pierce) for detection of horseradish peroxidase (HRP) activity for 1 minute and visualized using the Biorad Chemidoc Imaging System. The detection of α-tubulin by anti-α-tubulin antibodies was used as a loading control.
Statistical analysis
All statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA). Statistical differences for scored fluorescent micrographs were determined by Chi square and Fisher's exact tests. Each experimental condition was compared to the control condition. P-values of <0.05 were considered to be statistically significant. Statistically significant differences are indicated in the figures with asterisks next to the experimental condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
Results and discussion
Killing of the worms was mediated via H2O2 production by the mitis group of streptococci
A previous study by Bolm, et al established that S. oralis and S. mitis caused the death of the worms via the production of H2O2 in liquid killing assays [20]. In addition to these strains, we compared the survival of worms on S. gordonii, S. mutans, and S. salivarius grown on Todd Hewitt agar supplement with yeast extract relative to non-pathogenic E. coli OP50. As observed previously, members of the mitis group rapidly killed C. elegans as opposed to S. mutans, S. salivarius and E. coli (Fig 1A). The killing was abolished in the presence of 1,000 U/ml of catalase, but not on media supplemented with 100 U/ml of superoxide dismutase (S1 and S2 Figs). These data suggest the death of the worms is mediated by H2O2. Catalase and superoxide dismutase had no impact on the growth of the bacteria nor did they have an effect on the survival of the worms (data not shown). The production of H2O2 is mainly attributed to the pyruvate oxidase SpxB enzyme activity, which is conserved among the mitis group streptococci [35]. Therefore, we investigated the survival of the worms on S. gordonii ΔspxB mutant strain compared to the wild-type (WT) and complemented strain ΔspxB;spxB+. We observed no death of the worms on the ΔspxB mutant strain, whereas on the complement strain, the killing phenotype was restored as seen with the WT strain (Fig 1B). We did not follow the survival of the worms on the ΔspxB beyond 24 hours. Observing the survival of worms on ΔspxB mutant strain may provide insights if the mutant bacteria are completely avirulent or attenuated. In addition, continuous scoring of the WT worms on the ΔspxB mutant could potential identify other virulence factors of this group of bacteria. We also observed similar killing kinetics when worms were exposed to mitis group streptococci obtain from the blood of cancer patients that had undergone chemotherapy (M D. Anderson) (S3 Fig). Based on these data we can conclude that the mitis group streptococci mediate the killing of the worms via the production of H2O2.
Fig 1. H2O2 mediated killing of C. elegans by mitis group streptococci.

(A) Survival of N2 L4 larvae exposed to S. gordonii, S. oralis, S. mitis, S. salivarius S. mutans and E. coli OP50 on THY plates. (B) Survival of S. gordonii WT, ΔspxB mutant and ΔspxB;spxB+ complemented strains on N2 L4 larvae containing THY plates. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant. (C) Fluorescently labelled WT, ΔspxB mutant and ΔspxB;spxB+ complemented strains of S. gordonii were exposed to L4 larvae for 2 hours and visualized by confocal microscopy. The streptococcal strains are labelled in red and are located within the intestinal lumen of the worms. Close-ups are shown in the upper right-hand corner of each image. No difference was observed in the levels of colonization in the worms between the strains. The experiment was repeated three times. (D) No significant difference in intestinal accumulation of WT, ΔspxB mutant and ΔspxB;spxB+ complemented strains were observed in L4 worms following a 2 hour exposure. The experiment was repeated 3 times and a total of 25 worms were disrupted for each strain of bacteria.
Worms are colonized with viable strains of the mitis group
We utilized two approaches to determine if the worms were colonized with streptococci. First, we visualized worms colonized with fluorescently labelled bacteria using confocal microscopy. Significant accumulation of bacteria was observed in the intestine of the worms exposed to the WT, ΔspxB mutant and ΔspxB;spxB+ complemented strains of S. gordonii (Fig 1C). However, this method did not confirm if the colonized bacteria were viable within the intestine of the worms. As second approach we homogenized the worms colonized with the bacteria and estimated the number of viable bacterial cells per a worm by plating. Similar levels of colonization in worms was observed for all 3 strains of S. gordonii (Fig 1D). These observations suggest the intestine of the worms is colonized with viable bacteria and the loss spxB did not cause a metabolic burden on the mutant strain as represented by the similar levels of intestinal colonization of the worms by the three strains of bacteria. More importantly, there is accumulation of the mitis group in the intestine of the worms as opposed to a previous study that showed no colonization of the worms by S. pyrogenes [22]. In this study the worms were exposed to the bacteria grown on solid media compared to S. pyrogenes grown in liquid media. This difference in growth conditions may contribute to the levels of colonization of the worms.
Phase II response is activated in response to the pathogens
In light of our findings that H2O2 mediates killing of the worms, we hypothesized that oxidative stress response genes will be upregulated against this cytotoxin. We performed RNAseq on RNA extracted from worms exposed for 2 hours to WT and ΔspxB mutant strains of S. gordonii. Indeed, we observed significant increase in the expression of phase II detoxification genes glutathione-S-transferases (gst) and γ-glutamine cysteine synthase heavy chain (gcs-1) in worms exposed to WT S. gordonii compared to ΔspxB mutant strain (Table 1 and S1 Table). Expression of two important phase II genes gst-4 and gcs-1 was examined using quantitative real-time PCR (qRT-PCR) following exposure for 2 hours to WT and ΔspxB mutant strains of S. gordonii. The expression of these genes was significantly higher in the WT strain compared to the ΔspxB mutant strain further confirming the RNAseq data (Fig 2A). Furthermore, we observed similar levels of expression of these genes in L4 larvae exposed to the strains obtained from cancer patients when compared to non-pathogenic E. coli OP50 (S4 Fig). To further examine the expression of phase II response genes, we used a transgeneic worm strain expressing green fluorescent protein (GFP) fused to gcs-1. This strain was exposed to the WT, ΔspxB mutant and ΔspxB;spxB+ complement strains of S. gordonii and the levels of GFP expression was observed using a confocal microscope. Significantly higher levels of gcs-1::gfp expression was observed in the intestines of the worms exposed to the WT and complemented strains (Fig 2B and 2C). Whereas on the ΔspxB mutant, little or no expression of GFP was detected in the intestine of the worms. In addition, gcs-1::gfp levels was increased in worms fed on the strains acquired from cancer patients compared to E. coli OP50 (S5 Fig). Together these observations suggest the phase II genes in C. elegans are induced in response to H2O2 produced by the mitis group.
Table 1. Expression of phase II genes determined by RNAseq on S. gordonii WT relative to ΔspxB mutant strain.
| Gene | Fold change in expression | p-value | FDR-corrected p-value |
|---|---|---|---|
| gcs-1 | 6.9 | 0 | 0 |
| gst-1 | 2.1 | 0 | 0 |
| gst-4 | 15.9 | 0 | 0 |
| gst-5 | 4.1 | 4.44E-16 | 5.05574E-14 |
| gst-7 | 1.7 | 5.99E-09 | 3.67613E-07 |
| gst-9 | 3.0 | 0.001314 | 0.031406634 |
| gst-10 | 1.7 | 6.46E-09 | 3.94E-07 |
| gst-12 | 9.8 | 1.42E-06 | 6.33E-05 |
| gst-13 | 4.2 | 0 | 0 |
| gst-19 | 21.5 | 0.000108 | 0.003379 |
| gst-30 | 19.9 | 3.23E-07 | 1.57E-05 |
| gst-35 | 11.3 | 7.12E-09 | 4.31E-07 |
| gst-38 | 5.3 | 4.34E-07 | 2.08E-05 |
Fig 2. Phase II genes are upregulated in response to H2O2 produced by the mitis group streptococci.

(A) qRT-PCR analysis of Phase II genes gst-4 and gcs-1 induced in worms fed for 2 hours on S. gordonii WT and ΔspxB mutant strains. Experiments were performed with three separate replicates; each replicate was measured in duplicate and standardized to the control gene act-1. Error bars represent the standard error of the mean (SEM), P<0.001. (B) Representative images of gcs-1::gfp expression in worms exposed to WT, ΔspxB mutant and ΔspxB;spxB+ complemented strains of S. gordonii for 2 hours. (C) The level of gcs-1::gfp expression and the percentage of worms in each category fed on WT, ΔspxB mutant and ΔspxB;spxB+ complemented strains of S. gordonii. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly low levels of gcs-1::gfp expression was observed in the ΔspxB mutant (P<0.0001) compared to the WT and ΔspxB;spxB+ complement strains of S. gordonii.
SKN-1 is required for the survival of the worms during infection
The oxidative stress response transcription factor SKN-1 has been shown to regulate phase II genes and is required for the survival on pathogens Enterococcus faecalis and Pseudomonas aeruginosa [32, 36]. Phase II genes gcs-1 and gst-4 are predominantly regulated by SKN-1 and we envisioned it is required for the survival of C. elegans on the mitis group streptococci. Knockdown of skn-1 resulted a significant decrease in survival of the worms compared to the vector control treated worms on WT S. gordonii (Fig 3A). To validate this data, we compared the survival of WT worms to a skn-1 loss of function mutant strain, skn-1(zu67). As seen in the RNAi experiment, we observed a similar killing phenotype with the skn-1 mutant demonstrating SKN-1 influences the survival of the worms on these pathogens (Fig 3B). Since the loss of SKN-1 resulted in a susceptibility phenotype, we envisioned overexpression or constitutively active SKN-1 will render the worms resistant to the H2O2 produced by the mitis group. Indeed, strain SKN-1B/C::GFP carrying extra copies of skn-1 and strain SKN-1B/C S393A::GFP expressing a constitutively active form of SKN-1 [32, 37] exhibited increased resistance compared to the N2 worms in the presence of the WT S. gordonii (Fig 3C). There was no significant difference between the survival of the SKN-1B/C::GFP and the SKN-1B/C S393A::GFP strains on the pathogen. Taken together, these data show loss of skn-1 is detrimental to the survival of the worms, whereas increase of SKN-1 activity provides a protective function against these pathogens.
Fig 3. SKN-1 influences the susceptibility of the worms during infection.
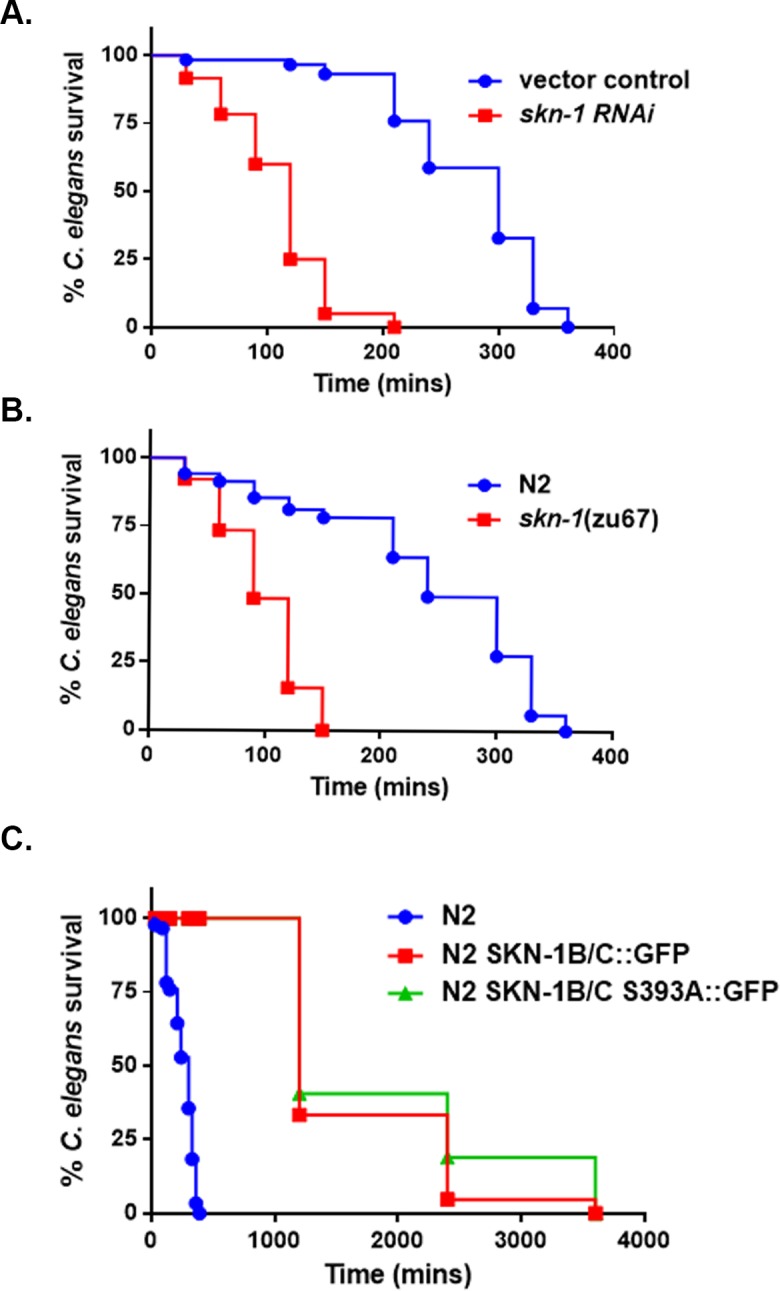
(A) Survival of vector control treated and skn-1 knockdown worms exposed to S. gordonii on THY plates. (B) Survival of N2 and skn-1(zu67) mutant worms fed on THY plates containing S. gordonii. (C) Survival of N2 [rol-6(su1006)], N2 [SKN-1B/C::GFP[rol-6(su1006)]] and N2 [SKN-1B/C S393A::GFP[rol-6(su1006)] worms on S. gordonii. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
Activation of SKN-1 requires the p38 MAPK pathway in response to H2O2 produced by the mitis group streptococci
Previous work has established SKN-1 activity is increased in the presence of E. faecalis and P. aeruginosa [32, 36]. The activity of SKN-1 can be determined by detecting the localization of this transcription factor into the intestinal nuclei using a transgenic line expressing SKN-1 fused to GFP. Using confocal microscopy, we observed greater levels of SKN-1B/C::GFP localization in the intestinal nuclei in worms on the WT and complementary strains of S. gordonii (Fig 4A and 4B). Significantly lower levels of SKN-1 localization was detected in worms exposed to the ΔspxB mutant. High levels of localization of SKN-1 was also observed when SKN-1B/C::GFP expressing worms were exposed to the clincial isolates (S6 Fig). These data further confirms SKN-1 is activated by H2O2 produced by this group of organisms. The activation of SKN-1 has been shown to regulated by p38 MAPK pathway in response to oxidative stresses and to pathogens. Using the SKN-1B/C::GFP transgenic worms, we knockdown components of the p38 MAPK pathway mainly nsy-1, sek-1, pmk-1 and skn-1 and determined the localization of SKN-1 on WT S. gordonii (Fig 4C and 4D). SKN-1 localization was significantly reduced in the sek-1, pmk-1 and skn-1 knockdown worms compared to the vector control treated worms. Partial reduction of SKN-1 localization was observed in nsy-1 knockdown on S. gordonii suggesting an additional pathway activates SKN-1 via the interaction with SEK-1. In addition, we determine the expression of gcs-1::gfp in nsy-1, sek-1, pmk-1 and skn-1 knockdown worms relative to the vector control treated worms (Fig 4E and 4F). Significant reduction in expression of gcs-1::gfp was observed in the sek-1, pmk-1 and skn-1 knockdown worms compared to the vector control. However, the gcs-1::gfp expression in nsy-1 knockdown worms was not comparable to the sek-1, pmk-1 and skn-1 knockdown worms, but significantly reduced relative to the vector control treated worms. To further determine if the p38 MAPK pathway is activated in response to H2O2 produced by the mitis group, we determined the level of phosphorylation of PMK-1 in nsy-1, sek-1, and pmk-1 mutant worms compared to N2 worms by western blot (Fig 4G). We observed no phosphorylation of PMK-1 in the sek-1 worms, whereas the level of phosphorylation in the nsy-1 mutant worms was significantly lower compared to the N2 worms on S. gordonii. pmk-1 mutant worms were used as a negative control. This data further indicates the p38 MAPK pathway is activated in response to the mitis group streptococci. However, the phosphorylation of PMK-1 is partially dependent on NSY-1 and further supports the SKN-1 localization data. A recent study showed the mitochondrial chaperone HSP-60 physically binds and stabilizes SEK-1, thereby activating PMK-1 and increasing immunity in response to P. aeruginosa [38]. Perhaps, HSP-60 interacts with SEK-1 to activate SKN-1 via PMK-1 in response to H2O2 produced by the mitis group streptococci. Future studies will be needed to address the activation of SKN-1 by HSP-60 via the p38 MAPK pathway.
Fig 4. SKN-1 activation is dependent on the p38 MAPK pathway in response to the pathogens.

(A) Representative images of the localization of SKN-1B/C::GFP in worms exposed to the WT, ΔspxB mutant and ΔspxB;spxB+ complement strains of S. gordonii. Close-ups are shown in the upper right-hand corner of each image. (B) The degree of nuclear localization of SKN-1B/C::GFP and the percentage of worms in each category fed on WT, ΔspxB mutant and ΔspxB;spxB+ complement strains of S. gordonii. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly lower levels of nuclear localization of SKN-1B/C::GFP was observed in the ΔspxB mutant (P<0.0001) compared to the WT and ΔspxB;spxB+ complement strains of S. gordonii. (C) Representative images of the localization of SKN-1B/C::GFP in nsy-1, sek-1, pmk-1, skn-1 knockdown and vector control treated worms on S. gordonii. Close-ups are shown in the upper right-hand corner of each image. (D) The degree of SKN-1B/C::GFP nuclear localization and the percentage of worms in each category fed on nsy-1, sek-1, pmk-1, skn-1 knockdown and vector control treated worms on S. gordonii. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly lower levels of nuclear localization of SKN-1B/C::GFP was observed in the nsy-1(P<0.01), sek-1 (P<0.001), pmk-1 (P<0.0001), skn-1 knockdown (P<0.0001) compared to the vector control treated worms on S. gordonii. (E) Representative images of gcs-1::gfp expression in nsy-1, sek-1, pmk-1, skn-1 knockdown and vector control treated worms on S. gordonii. (F) The level of gcs-1::gfp expression and the percentage of worms in each category fed on nsy-1, sek-1, pmk-1, skn-1 knockdown and vector control treated worms on S. gordonii. A total of more than 100 worms exposed to each strain was imaged and the experiment was repeated 3 times. Significantly lower levels of gcs-1::gfp expression was observed in the nsy-1(P<0.001), sek-1 (P<0.0001), pmk-1 (P<0.0001), skn-1 knockdown (P<0.0001) compared to the vector control treated worms on S. gordonii. (G) Western blot was used to analyze the level of phosphorylation of PMK-1 in N2, nsy-1, sek-1 and pmk-1 mutant worms exposed to S. gordonii for 2 hours. Phosphorylation of PMK-1 was partial reduced in nsy-1 mutant worms, while in the sek-1 mutant worms phospho-p38 was completely absent. pmk-1 mutant worms was used as a negative control and α-tubulin as a loading control. (H) Survival of N2 and nsy-1(ok593), sek-1(km4) and pmk-1(km25) IV L4 larvae when fed on THY plates containing S. gordonii. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
We next determined if the loss of nsy-1, sek-1 and pmk-1 reduced the survival of the worms relative to the WT strain when exposed to S. gordonii. A significant decrease in the survival of the nsy-1, sek-1 and pmk-1 mutant strains was observed compared to the N2 strain. The data shows the p38 MAPK pathway is required for the survival of C. elegans in the presence of S. gordonii.
IRE-1-TRAF (trf-1) is not required for the activation of SKN-1 in response to the mitis group
A recent study uncovered an unexpected function for the ER transmembrane protein IRE-1 in redox-regulated cytoplasmic signaling. On treatment with arsenite, IRE-1 and NSY-1 form a complex via the participation of TRAF-2 (trf-1) resulting in the activation of the p38 MAPK pathway [33]. In light of this discovery we investigated if IRE-1 and TRAF-2 (trf-1) are required for the activation of the p38 signaling in response to S. gordonii. We first determined the localization of SKN-1B/C::GFP in ire-1 and trf-1 knockdown worms relative to the vector treated worms on the pathogen (Fig 5A and 5B). The percentage of SKN-1 localization was similar in ire-1 and trf-1 knockdown worms compared to the vector control treated worms. We next determined the expression of gcs-1::gfp in ire-1 and trf-1 knockdown worms compared to the vector control treated worms on S. gordonii (Fig 5C and 5D). No significant difference in expression of gcs-1::gfp was observed between the knockdowns and the vector control treated worms. We further investigated if the levels of phosphorylation of PMK-1 is influenced by IRE-1 and TRAF-2 in the presence of S. gordonii (Fig 5E). No difference in the level of phosphorylation of PMK-1 by western blot was observed in the ire-1 mutant strain and the trf-1 knockdown strain compared to the vector control treated worms.
Fig 5. Activation of SKN-1 does not require IRE-1-TRAF (trf-1), when exposed to the S. gordonii.

(A) Representative images of the localization of SKN-1B/C::GFP in ire-1, trf-1 knockdown and vector control treated worms on S. gordonii for 2 hours. (B) The degree of SKN-1B/C::GFP nuclear localization and the percentage of worms in each category fed on ire-1, trf-1 knockdown and vector control treated worms on S. gordonii. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Similar levels of nuclear localization of SKN-1B/C::GFP was observed in the ire-1, trf-1 knockdown and vector control treated worms on S. gordonii. (C) Representative images of gcs-1::gfp expression in nsy-1, sek-1, pmk-1, skn-1 knockdown and vector control treated worms on S. gordonii. (D) The level of gcs-1::gfp expression and the percentage of worms in each category fed on ire-1, trf-1 knockdown and vector control treated worms on S. gordonii for 2 hours. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. No difference of gcs-1::gfp expression was observed in the ire-1, trf-1 knockdown worms compared to the vector control treated worms on S. gordonii. (E) Western blot was used to analyze the level of phosphorylation of PMK-1 in vector control treated worms, ire-1 mutant worms and trf-1 knockdown worms exposed to S. gordonii for 2 hours. No significant differences was observed in the level of phosphorylation of PMK-1 in the ire-1 mutant worms and trf-1 knockdown worms compared to the vector control treated worms. α-tubulin was used as a loading control. The experiment was performed three times.
Lastly, we determined the survival of the ire-1 mutant strain relative to the N2—strain and the trf-1 knockdown strain relative to the vector control treated worms on the pathogen (Fig 6A and 6B). A significant decrease in survival was observed in the ire-1 mutant strain compared to WT strain. However, no difference in survival was seen between the trf-1 knockdown and vector control treated worms in response to S. gordonii. Taken together the data suggests the phosphorylation of PMK-1 does not require the IRE-1 TRAF-2 complex, but is activated by a parallel pathway. Moreover, IRE-1 may be required for induction of the ER stress response when exposed to S. gordonii. Numerous studies have shown that oxidative stress induces ER stress and the pattern of UPR activation is dependent on the cell type [39–41]. The Unfolded protein response (UPR) may indeed play an important role in the protection of the worms in response to oxidative stress caused by the mitis group. Further investigation is required to elucidate the parallel pathway for p38 phosphorylation and the role of the UPR in response to the streptococcal H2O2.
Fig 6. IRE-1 is required for the survival of the worms on S. gordonii.

(A) Survival of N2 and ire-1(zc14) II; zcIs4 V L4 larvae when fed on THY plates containing S. gordonii. (B) Survival of vector control treated and trf-1 knockdown worms exposed to S. gordonii on THY plates. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
The activation of the p38 MAPK pathway in response to S. gordonii is not dependent on Ce-DUOX-1/BLI-3
The activation of SKN-1 in response to pathogens and arsenite has shown to be mediated by ROS released by the dual oxidase Ce-Duox1/BLI-3 [32, 33]. We examined if indeed H2O2 produced by Ce-Duox1/BLI-3 in response to S. gordonii contributes to the activation of the p38 MAPK pathway. To test this, we determine the levels of phosphorylation of PMK-1 by western blot in bli-3 knockdown and vector control treated worms exposed to S. gordonii WT, ΔspxB and E. coli OP50. No significant change in the level of phospho-p38 was observed between the bli-3 knockdown and vector control treated worms on WT S. gordonii (Fig 7A). Furthermore, we detected significantly lower levels of phospho-p38 in bli-3 knockdown and vector control treated worms fed on the ΔspxB mutant strain of S. gordonii or E. coli OP50. The data suggests the activation of the p38 MAPK pathway in response to S. gordonii is not dependent on Ce-Duox1/BLI-3. Furthermore, to confirm if Ce-Duox1/BLI-3 is required for the survival of the worms on S. gordonii, we determine the susceptibility of bli-3 knockdown and vector control treated L4 larvae. There was no significant difference in survival between bli-3 knockdown and the vector control treated worms on the pathogen (Fig 7B). Taken together, these findings suggest Ce-Duox1/BLI-3 is not activated to produce ROS in response to S. gordonii and hence does not contribute to the induction of p38 MAPK pathway.
Fig 7. Activation of the p38 MAPK pathway is not dependent on Ce-DUOX-1/BLI-3.

(A) Western blot was used to analyze the level of phosphorylation of PMK-1 in bli-3 knockdown and vector control treated worms exposed to S. gordonii WT, ΔspxB and E. coli OP50 for 2 hours. No significant difference in phospho-p38 levels was observed in bli-3 knockdown worms relative to vector control treated worms. Phosphorylation of PMK-1 was absent in vector control treated and bli-3 knockdowns worms on the S. gordonii ΔspxB mutant strain and E. coli OP50. α-tubulin was used as a loading control. The experiment was repeated 3 times (B) Survival of vector control treated and bli-3 knockdown worms exposed to S. gordonii on THY plates. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
Conclusion
In this study, we demonstrate the oxidative stress response transcription factor SKN-1 is required for the survival of the worms when exposed to the mitis group streptococci. Furthermore, we demonstrate H2O2 produced by this group of bacteria activates SKN-1 via the p38 MAPK pathway. However, the activation of p38 signaling is not via the IRE-1-NSY-1 complex and is not dependent on Ce-Duox1/BLI-3, suggesting that a parallel mechanism senses H2O2 produced by the mitis group and activates p38 via NSY-1. In addition, our data suggests an NSY-1 independent mechanism activates the p38 pathway through SEK-1. Even though IRE-1 does not initiate the p38 signaling, our data indicates this kinase is required for the survival of the worms in the presence of the mitis group. Defining these mechanisms will be important areas of future research. Using C. elegans as a model host, we are able to determine the significance of H2O2 produced by the mitis group of streptococci on cellular mechanisms in context of a whole organism.
Supporting information
(DOCX)
(XLSX)
Survival of N2 L4 larvae exposed to S. gordonii, S. oralis, S. mitis, S. salivarius S. mutans and E. coli OP50 on THY plates in the presence of 1000U of catalase. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
(PDF)
Survival of N2 L4 larvae exposed to S. gordonii, S. oralis, S. mitis, S. salivarius, S. mutans and E. coli OP50 on THY plates supplemented with 100U of superoxide dismutase. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
(PDF)
Survival of N2 L4 larvae exposed to S. oralis (VGS#3), S. oralis (VGS#4), S. mitis (VGS#10), S. mitis (VGS#13) and E. coli OP50 on THY plates. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
(PDF)
qRT-PCR analysis of Phase II genes gst-4 and gcs-1 induced in worms fed for 2 hours on S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50. Experiments were performed with three separate replicates; each replicate was measured in duplicate and standardized to the control gene act-1. Error bars represent the standard error of the mean (SEM), P<0.001.
(PDF)
Representative images of gcs-1::gfp expression in worms exposed to S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50.for 2 hours. The level of gcs-1::gfp expression and the percentage of worms in each category fed on S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly high levels of gcs-1::gfp expression in worms was observed on S. oralis (VGS#3) and S. mitis (VGS#10) (P<0.0001) compared to E. coli OP50.
(PDF)
Representative images of the localization of SKN-1B/C::GFP in worms exposed to t S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50.for 2 hours. The degree of nuclear localization of SKN-1B/C::GFP and the percentage of worms in each category fed on S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly high levels of nuclear localization of SKN-1B/C::GFP were observed in worms on S. oralis (VGS#3) and S. mitis (VGS#10) strains (P<0.0001) compared to E. coli OP50.
(PDF)
Acknowledgments
We thank Dr. Bing-Yan Wang and Dr. Gena Tribble (The University of Texas, School of Dentistry) for providing the streptococcal strains. We also thank Dr. Richard Lamont (University of Louisville, School of Dentistry) for providing the ΔspxB mutant and the ΔspxB;spxB+ complement strains of S. gordonii. We are grateful to Dr. Samuel Shelburne (MD Anderson Cancer Center) for providing clinical strains of S. oralis and S. mitis and Dr. Keith Blackwell for the C. elegans strains. Final we thank Dr. Danielle Garsin and her lab (The University of Texas, McGovern Medical School) for providing reagents and worm strains to conduct the study. Some worm strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by University of Texas Health Science Center at Houston, School of Dentistry startup funds awarded to R. van der Hoeven (PI). Some worm strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. 10.1038/nature11234 ; PubMed Central PMCID: PMC3564958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Douglas CW, Heath J, Hampton KK, Preston FE. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol. 1993;39(3):179–82. 10.1099/00222615-39-3-179 . [DOI] [PubMed] [Google Scholar]
- 3.Dyson C, Barnes RA, Harrison GA. Infective endocarditis: an epidemiological review of 128 episodes. J Infect. 1999;38(2):87–93. . [DOI] [PubMed] [Google Scholar]
- 4.Mitchell J. Streptococcus mitis: walking the line between commensalism and pathogenesis. Mol Oral Microbiol. 2011;26(2):89–98. 10.1111/j.2041-1014.2010.00601.x . [DOI] [PubMed] [Google Scholar]
- 5.van der Meer JT, van Vianen W, Hu E, van Leeuwen WB, Valkenburg HA, Thompson J, et al. Distribution, antibiotic susceptibility and tolerance of bacterial isolates in culture-positive cases of endocarditis in The Netherlands. Eur J Clin Microbiol Infect Dis. 1991;10(9):728–34. . [DOI] [PubMed] [Google Scholar]
- 6.Shelburne SA 3rd, Chaftari AM, Jamal M, Al Wohoush I, Jiang Y, Abughazaleh S, et al. Identification and characterization of catheter-related bloodstream infections due to viridans group streptococci in patients with cancer. American journal of infection control. 2014;42(10):1127–9. 10.1016/j.ajic.2014.06.012 . [DOI] [PubMed] [Google Scholar]
- 7.Shelburne SA, Sahasrabhojane P, Saldana M, Yao H, Su X, Horstmann N, et al. Streptococcus mitis strains causing severe clinical disease in cancer patients. Emerging infectious diseases. 2014;20(5):762–71. 10.3201/eid2005.130953 ; PubMed Central PMCID: PMC4012796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahasrabhojane P, Galloway-Pena J, Velazquez L, Saldana M, Horstmann N, Tarrand J, et al. Species-level assessment of the molecular basis of fluoroquinolone resistance among viridans group streptococci causing bacteraemia in cancer patients. International journal of antimicrobial agents. 2014;43(6):558–62. 10.1016/j.ijantimicag.2014.01.031 . [DOI] [PubMed] [Google Scholar]
- 9.Zhu L, Kreth J. The role of hydrogen peroxide in environmental adaptation of oral microbial communities. Oxidative medicine and cellular longevity. 2012;2012:717843 10.1155/2012/717843 ; PubMed Central PMCID: PMC3405655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng X, Redanz S, Cullin N, Zhou X, Xu X, Joshi V, et al. Plasticity of the Pyruvate Node Modulates Hydrogen Peroxide Production and Acid Tolerance in Multiple Oral Streptococci. Appl Environ Microbiol. 2018;84(2). 10.1128/AEM.01697-17 ; PubMed Central PMCID: PMCPMC5752870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kreth J, Zhang Y, Herzberg MC. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J Bacteriol. 2008;190(13):4632–40. 10.1128/JB.00276-08 ; PubMed Central PMCID: PMCPMC2446780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreth J, Vu H, Zhang Y, Herzberg MC. Characterization of hydrogen peroxide-induced DNA release by Streptococcus sanguinis and Streptococcus gordonii. J Bacteriol. 2009;191(20):6281–91. 10.1128/JB.00906-09 ; PubMed Central PMCID: PMCPMC2753043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okahashi N, Nakata M, Sumitomo T, Terao Y, Kawabata S. Hydrogen peroxide produced by oral Streptococci induces macrophage cell death. PloS one. 2013;8(5):e62563 10.1371/journal.pone.0062563 ; PubMed Central PMCID: PMC3643943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okahashi N, Okinaga T, Sakurai A, Terao Y, Nakata M, Nakashima K, et al. Streptococcus sanguinis induces foam cell formation and cell death of macrophages in association with production of reactive oxygen species. FEMS microbiology letters. 2011;323(2):164–70. 10.1111/j.1574-6968.2011.02375.x . [DOI] [PubMed] [Google Scholar]
- 15.Okahashi N, Sumitomo T, Nakata M, Sakurai A, Kuwata H, Kawabata S. Hydrogen peroxide contributes to the epithelial cell death induced by the oral mitis group of streptococci. PloS one. 2014;9(1):e88136 10.1371/journal.pone.0088136 ; PubMed Central PMCID: PMC3909332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okahashi N, Nakata M, Kuwata H, Kawabata S. Streptococcus oralis Induces Lysosomal Impairment of Macrophages via Bacterial Hydrogen Peroxide. Infection and immunity. 2016;84(7):2042–50. 10.1128/IAI.00134-16 ; PubMed Central PMCID: PMCPMC4936348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loose M, Hudel M, Zimmer KP, Garcia E, Hammerschmidt S, Lucas R, et al. Pneumococcal hydrogen peroxide-induced stress signaling regulates inflammatory genes. The Journal of infectious diseases. 2015;211(2):306–16. 10.1093/infdis/jiu428 ; PubMed Central PMCID: PMC4334831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuen GJ, Ausubel FM. Enterococcus infection biology: lessons from invertebrate host models. Journal of microbiology. 2014;52(3):200–10. 10.1007/s12275-014-4011-6 ; PubMed Central PMCID: PMC4556283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nature reviews Immunology. 2010;10(1):47–58. 10.1038/nri2689 ; PubMed Central PMCID: PMC2965059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolm M, Jansen WT, Schnabel R, Chhatwal GS. Hydrogen peroxide-mediated killing of Caenorhabditis elegans: a common feature of different streptococcal species. Infection and immunity. 2004;72(2):1192–4. 10.1128/IAI.72.2.1192-1194.2004 ; PubMed Central PMCID: PMC321644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE, et al. A simple model host for identifying Gram-positive virulence factors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10892–7. 10.1073/pnas.191378698 ; PubMed Central PMCID: PMCPMC58570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen WT, Bolm M, Balling R, Chhatwal GS, Schnabel R. Hydrogen peroxide-mediated killing of Caenorhabditis elegans by Streptococcus pyogenes. Infection and immunity. 2002;70(9):5202–7. 10.1128/IAI.70.9.5202-5207.2002 ; PubMed Central PMCID: PMC128270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, Isik M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free radical biology & medicine. 2015;88(Pt B):290–301. 10.1016/j.freeradbiomed.2015.06.008 ; PubMed Central PMCID: PMCPMC4809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free radical biology & medicine. 2015;88(Pt B):93–100. 10.1016/j.freeradbiomed.2015.06.006 . [DOI] [PubMed] [Google Scholar]
- 25.An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes & development. 2003;17(15):1882–93. 10.1101/gad.1107803 ; PubMed Central PMCID: PMC196237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16(2):123–40. 10.1111/j.1365-2443.2010.01473.x . [DOI] [PubMed] [Google Scholar]
- 27.An JH, Vranas K, Lucke M, Inoue H, Hisamoto N, Matsumoto K, et al. Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(45):16275–80. 10.1073/pnas.0508105102 ; PubMed Central PMCID: PMC1283458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glover-Cutter KM, Lin S, Blackwell TK. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS genetics. 2013;9(9):e1003701 10.1371/journal.pgen.1003701 ; PubMed Central PMCID: PMC3772064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kell A, Ventura N, Kahn N, Johnson TE. Activation of SKN-1 by novel kinases in Caenorhabditis elegans. Free radical biology & medicine. 2007;43(11):1560–6. 10.1016/j.freeradbiomed.2007.08.025 ; PubMed Central PMCID: PMCPMC2212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Robida-Stubbs S, Tullet JM, Rual JF, Vidal M, Blackwell TK. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS genetics. 2010;6(8). 10.1371/journal.pgen.1001048 ; PubMed Central PMCID: PMC2916858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, et al. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes & development. 2005;19(19):2278–83. 10.1101/gad.1324805 ; PubMed Central PMCID: PMC1240035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoeven R, McCallum KC, Cruz MR, Garsin DA. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS pathogens. 2011;7(12):e1002453 10.1371/journal.ppat.1002453 ; PubMed Central PMCID: PMC3245310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hourihan JM, Moronetti Mazzeo LE, Fernandez-Cardenas LP, Blackwell TK. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Molecular cell. 2016;63(4):553–66. 10.1016/j.molcel.2016.07.019 ; PubMed Central PMCID: PMCPMC4996358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hope IA, Hames BD, editor. C. elegans A Practical Approach. Oxford: Oxford University Press; 1999. [Google Scholar]
- 35.Zhu L, Xu Y, Ferretti JJ, Kreth J. Probing oral microbial functionality—expression of spxB in plaque samples. PloS one. 2014;9(1):e86685 10.1371/journal.pone.0086685 ; PubMed Central PMCID: PMCPMC3906080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papp D, Csermely P, Soti C. A role for SKN-1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans. PLoS pathogens. 2012;8(4):e1002673 10.1371/journal.ppat.1002673 ; PubMed Central PMCID: PMCPMC3343120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132(6):1025–38. 10.1016/j.cell.2008.01.030 ; PubMed Central PMCID: PMCPMC2367249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong DE, Lee D, Hwang SY, Lee Y, Lee JE, Seo M, et al. Mitochondrial chaperone HSP-60 regulates anti-bacterial immunity via p38 MAP kinase signaling. The EMBO journal. 2017;36(8):1046–65. 10.15252/embj.201694781 ; PubMed Central PMCID: PMCPMC5391144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He S, Yaung J, Kim YH, Barron E, Ryan SJ, Hinton DR. Endoplasmic reticulum stress induced by oxidative stress in retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. 2008;246(5):677–83. 10.1007/s00417-008-0770-2 . [DOI] [PubMed] [Google Scholar]
- 40.Pallepati P, Averill-Bates DA. Activation of ER stress and apoptosis by hydrogen peroxide in HeLa cells: protective role of mild heat preconditioning at 40 degrees C. Biochim Biophys Acta. 2011;1813(12):1987–99. 10.1016/j.bbamcr.2011.07.021 . [DOI] [PubMed] [Google Scholar]
- 41.Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. The Journal of biological chemistry. 2005;280(40):33917–25. 10.1074/jbc.M505818200 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(XLSX)
Survival of N2 L4 larvae exposed to S. gordonii, S. oralis, S. mitis, S. salivarius S. mutans and E. coli OP50 on THY plates in the presence of 1000U of catalase. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
(PDF)
Survival of N2 L4 larvae exposed to S. gordonii, S. oralis, S. mitis, S. salivarius, S. mutans and E. coli OP50 on THY plates supplemented with 100U of superoxide dismutase. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
(PDF)
Survival of N2 L4 larvae exposed to S. oralis (VGS#3), S. oralis (VGS#4), S. mitis (VGS#10), S. mitis (VGS#13) and E. coli OP50 on THY plates. The data are representative of experiments repeated two or more times with an n = 60–90 worms for each condition. Kaplan-Meier log rank analysis was used to compare survival curves and to calculate the median survival. P-values <0.05 were considered to be statistically significant.
(PDF)
qRT-PCR analysis of Phase II genes gst-4 and gcs-1 induced in worms fed for 2 hours on S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50. Experiments were performed with three separate replicates; each replicate was measured in duplicate and standardized to the control gene act-1. Error bars represent the standard error of the mean (SEM), P<0.001.
(PDF)
Representative images of gcs-1::gfp expression in worms exposed to S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50.for 2 hours. The level of gcs-1::gfp expression and the percentage of worms in each category fed on S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly high levels of gcs-1::gfp expression in worms was observed on S. oralis (VGS#3) and S. mitis (VGS#10) (P<0.0001) compared to E. coli OP50.
(PDF)
Representative images of the localization of SKN-1B/C::GFP in worms exposed to t S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50.for 2 hours. The degree of nuclear localization of SKN-1B/C::GFP and the percentage of worms in each category fed on S. oralis (VGS#3), S. mitis (VGS#10) and E. coli OP50. A total of more than 100 worms exposed to each strain were imaged and the experiment was repeated 3 times. Significantly high levels of nuclear localization of SKN-1B/C::GFP were observed in worms on S. oralis (VGS#3) and S. mitis (VGS#10) strains (P<0.0001) compared to E. coli OP50.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.