

Abstract
Normal cells acquire aggressive behavior by modifying signaling pathways. For instance, alteration of endocytosis profoundly impacts both proliferation and migration during tumorigenesis. Here we investigate the mechanisms that enable the endocytic machinery to coordinate these processes. We show that a membrane curvature-sensing protein, endophilin A3, promotes growth and migration of colon cancer cells through two competing mechanisms — an endocytosis pathway that is required for proliferation, and a GTPase regulatory pathway that controls cell motility. EndoA3 stimulates cell migration by binding the Rac GEF TIAM1 leading to activation of small GTPases. Competing interactions of EndoA3 with membrane versus TIAM1 modulate hyperproliferative and metastatic phenotypes. Disruption of EndoA3-membrane interactions stimulates TIAM1 and small GTPases in vitro, and further promotes prometastatic phenotypes in vivo. Together, these results uncover a coupling mechanism, by which EndoA3 promotes growth and migration of colon cancers, by linking membrane dynamics to GTPase regulation.
TOC Graphic
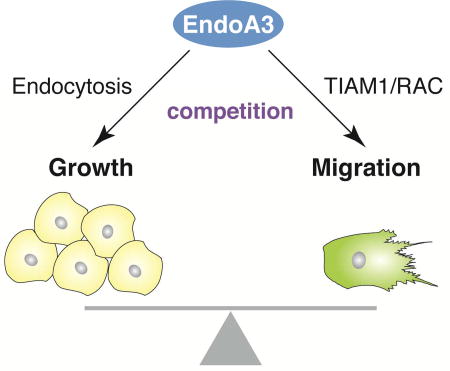
Poudel et al. show that Endophilin A3 (EndoA3) modulates proliferation and migration of colorectal cancer cells using two distinct pathways, i.e., EndoA3 facilitates cell proliferation by enhancing endocytosis, and it promotes migration by activating TIAM1 and Rac1. Modulation of the two pathways tunes tumor progression in vivo.
Introduction
Hyper-proliferation and migration are defining features of malignant tumor cells (Hanahan and Weinberg, 2000; Sporn, 1996). Coordination of migration and proliferation enables tumors to spread and to establish secondary areas of growth. It has been established that cancer cells have the ability to integrate complex signals from neighboring cells (e.g., stromal, immune, and vascular cells) for guiding their migration and proliferation. These findings highlight the existence of cellular pathways that couple external signals to the intrinsic properties of cancer cells. Among these pathways, endocytosis controls signaling, adhesion, and nutrient receptors on the cell surface, thereby playing a crucial role in cellular communication with the environment. While it is generally accepted that endocytosis is altered in cancer cells to enhance their proliferation and metastasis, the identity of endocytic proteins and their precise functions in cancer progression remain elusive (Lanzetii and Di Fiore, 2017; Mellman and Yarden, 2013; Mosesson et al., 2008).
The endophilin proteins facilitate endocytosis and support cellular signaling (Kjaerulff et al., 2011). These proteins harbor two functional domains, i.e., a N-terminal BAR (Bin–Amphiphysin–Rvs) domain that binds membranes in a curvature-dependent manner, and a C-terminal SH3 domain that mediates protein-protein interactions (Cestra et al., 1999; Farsad and Camilli, 2003). In living cells, endophilin promotes receptor and membrane internalization, thus serving as a key component for receptor-mediated signaling events (Boucrot et al., 2015; Petrelli et al., 2002; Renard et al., 2015; Soubeyran et al., 2002). Endophilin is known for its role in promoting endocytosis in neurons and other types of cells (Bai et al., 2010; Boucrot et al., 2015; Gad et al., 2000; Guichet et al., 2002; Milosevic et al., 2011; Renard et al., 2015; Rikhy et al., 2002; Ringstad et al., 1999; Schuske et al., 2003). However, the function of endophilin beyond endocytosis is less explored. Growing evidence suggests that endophilin has a key role in cancer biology (Kjaerulff et al., 2011). For example, mutations in endophilin have been linked to cancer progression (Ghosh et al., 2009; Giordani et al., 2002; Sinha et al., 2008). Moreover, endophilin expression levels and phosphorylation are altered in tumors (Aramaki et al., 2005; Bonner et al., 2003; Ghosh et al., 2009; Nguyen et al., 2007; Rikova et al., 2007; Sinha et al., 2008; Wu et al., 2005).
The endophilin A family includes three protein isoforms (EndoA1, EndoA2, and EndoA3) that are encoded by paralogous genes. All three isoforms have been linked to cancers. EndoA1 and EndoA2 are thought to be tumor suppressors because their expression is strongly reduced in metastatic tumors (Ghosh et al., 2009; Kjaerulff et al., 2011; Osterberg et al., 2009; Yam et al., 2004). However, subcutaneous injection of NIH3T3 cells that express EndoA2 leads to tumor formation in nude mice (Lua and Low, 2005). Subsequently, it was shown that the tyrosine-protein kinase Src phosphorylates the C-terminus SH3 domain of EndoA2 to promote degradation of the extracellular matrix (Wu et al., 2005), supporting a role of EndoA2 in promoting cancer metastasis. Among the three EndoA members, EndoA3 is the least studied isoform. Recently, EndoA3 was linked to cancer invasiveness (Delic et al., 2012; Li et al., 2016). In mouse xenografts expressing a constitutively active version of the tyrosine kinase receptor Ephrin, EndoA3 was identified as a ‘hub’ gene with altered expression levels in invasive colorectal tumors (Li et al., 2016). In addition, biochemical data suggest that the C-terminal SH3 domain of EndoA3 directly binds MTA1, a protein whose expression directly correlates with the metastatic ability of cancer cells (Aramaki et al., 2005). However, because MTA1 is primarily a nuclear protein and EndoA3 resides in the cytosol, the biological implications of EndoA3-MTA1 interactions are currently unclear.
Here, we show that EndoA3 promotes the progression of colon cancers through a mechanism involving two competing components. On one hand, EndoA3 facilitates cell proliferation by increasing endocytosis. On the other hand, EndoA3 stimulates cell migration by binding and activating the Rac GEF TIAM1. These results demonstrate a competitive mechanism for EndoA3-membrane and EndoA3-TIAM1 interactions to balance cancer growth and migration.
Results
EndoA3 is expressed in human and mouse colon cancers
Increased EndoA3 expression has been linked to poor outcomes in patients with advanced colon cancer (Jorissen et al., 2009). To examine the connection between EndoA3 expression and human colon cancer, we analyzed tissue microarrays from colon cancer patients using immunohistochemistry. Our results identified significant increases in EndoA3-positive areas in tumors (~29% of total area) compared to neighboring normal tissue (~9%) (Figure 1A-B). To quantify the expression level of EndoA3 on tumor stage, we employed the ‘H’ score (Metz et al., 2016), and found that ~20% of stage 1, ~52% of stage 2, and ~64% of stage 3 tumors have elevated EndoA3 levels (Figure 1C and Supplementary Figure 1B). These data indicate that more advanced tumors express EndoA3 at higher levels. To further examine the relationship between EndoA3 expression and colon cancer behavior, we performed western blotting using tumors derived from mouse models of intestinal cancers (Figure 1D). We examined mouse models that have intestinal neoplasms driven by mutated or deleted Apc, Tgfbr2 and Kras because these mutant genes can affect the initiation and progression of colon neoplasms (Amado et al., 2008; Markowitz et al., 1995; Morin et al., 1997; Sparks et al., 1998). These mouse models include mice with: 1) mutations in the Wnt pathway secondary to a truncation in one of the alleles of Apc (Apc1638/+) (ATT); 2) Pten and Tgfbr2 deletions combined in the intestinal epithelium (PPVcTT); 3) Apc truncation in combination with Tgfbr2 deletion in the intestinal epithelium (AVcTT); and 4) activated oncogenic KrasG13D and Tgfbr2 deletion (KVcTT) in the intestinal epithelium. We found that EndoA3 levels were highly elevated in these tumors (designated T) compared to neighboring intestinal mucosa (designated N) (Figure 1D).
Figure 1. EndoA3 is upregulated in aggressive colon cancers.

(A) Representative images for immunohistochemistry staining of human colon tissue microarrays (US Biomax). Images are shown for a tumor (grade 2, stage iii) (left) and normal mucosa (right) from the same patient. EndoA3 was visualized using rabbit polyclonal antibody against human EndoA3 (Abcam). Specificity of EndoA3 antibody for IHC was determined as described in Materials and Methods. In addition, control IgG showed no staining on human adrenal gland samples (Supplementary Figure 1A). (B) The average levels of EndoA3 in cancers and normal tissues from multiple patients were compared using an image deconvolution algorithm in ImageJ. The size of areas with positive EndoA3 staining was normalized to the size of total tissue area (Ruifrok and Johnston, 2001) (*** p<0.001, n = number of patient samples). (C) The ‘H’ score system (Metz et al., 2016) was used in stage analysis of EndoA3 expression. The percentage of tumors with higher H scores than normal neighboring tissues was plotted based on the tumor stage (stages 1-3). (D) Immunoblotting for EndoA3 in mice colon tumor sets (a-e). Normal mucosae (N) and adenocarcinoma tumors (T) were analyzed. EndoA3 levels were normalized to actin, and the fold changes were indicated below the EndoA3 blots (left and middle panels). Genotypes of the mouse models were listed at the bottom of the panel. EndoA3 expression levels in each sample set (a-e) that includes a tumor tissue and its neighboring normal mucosae were quantified using densitometry (3 sample sets were analyzed for each data point). Paired Student’s t-test was used for statistical analysis. ** p<0.01, * p<0.05. Fold changes of EndoA3 levels in tumors were plotted in the right panel. The dashed line indicates the expression level of EndoA3 in normal mucosae.
EndoA3 promotes endocytosis, growth, and migration in cancer cells
To determine the cellular consequence of increased expression of EndoA3, we generated DLD1 and HCT116 colon adenocarcinoma cell lines stably expressing mCherry-tagged EndoA3 (EndoA3-mCherry; designated EndoA3 DLD1 cells). First, we used the FM1-43 dye uptake assay to measure the levels of endocytosis in DLD1 cells (Betz et al., 1996; Meyers et al., 2003). Consistent with the known role of endophilin in promoting endocytosis, EndoA3-mCherry DLD1 cells exhibited increased levels of FM1-43 uptake after 5min dye incubation (Figure 2A). Next, we used automated assays to examine the proliferation and the migration of EndoA3 DLD1 and EndoA3 HCT116 cells. Significantly increased growth rates were observed in DLD1 cells (Figure 2B) and HCT116 cells (Supplementary Figure 2B left) that carry EndoA3-mCherry. These data indicate that EndoA3 further accelerates proliferation in fast growing DLD1 and HCT116 cell lines (Ahmed et al., 2013). To quantify cell migration, we used an automated 2D scratch wound assay where cell proliferation was blocked using reduced serum conditions (1%) and mitomycin. We found that DLD1 and HCT116 cells expressing EndoA3-mCherry migrated faster compared to DLD1 cells expressing empty vectors (Figure 2C, Supplementary Figure 2B right). To further test the link between EndoA3 expression levels and cell physiology, we reduced the endogenous EndoA3 expression in three different colorectal cancer cell lines (DLD1, HCT116, and SW480; Supplementary Figure 2C). Both growth and migration were attenuated in these cells upon siRNA-mediated EndoA3 knockdown (Figure 2D-E and Supplementary Figure 2D-E). By contrast, control siRNA with scrambled sequence had no effect. Together, these results from multiple cell lines demonstrate a new role of EndoA3 in promoting proliferation and migration in colon adenocarcinoma cells.
Figure 2. EndoA3 expression levels regulate endocytosis, growth, and migration of DLD1 cells.

Colon adenocarcinoma DLD1 cells stably expressing EndoA3-mCherry were generated using retroviral transduction. (A) Increased EndoA3-mCherry expression (~3 fold over endogenous EndoA3, Supplementary Figure 2A) promotes endocytosis in DLD1 cells (blue) compared to DLD1 cells with empty vectors (gray) as measured by a FM1-43 dye intake assay (Betz et al., 1996; Meyers et al., 2003). Unpaired Student’s t-test was used for statistical analysis (*** p<0.001, n=3 independent replicates). (B) Automated cell growth assay shows that EndoA3 DLD cells (blue) proliferated faster compared to DLD1 cells carrying empty vectors (gray). (C) EndoA3-mCherry expression (blue) accelerates wound closure in a 2D scratch assay. Data from DLD1 cells with empty vectors are indicated in gray. (D-E) Knockdown of endogenous EndoA3 by siRNA reduces growth (D) and migration (E) of DLD1 cells. The error bars in panels B-E indicate SEM from 5 independent biological replicates. Two-way ANOVA tests were conducted to calculate statistical significance. *** indicates p<0.001 for the comparison between control and EndoA3 DLD1 cells.
EndoA3 alters cytoskeletal dynamics and invasiveness in DLD1 cells
To understand how EndoA3 increases cell growth and migration, we examined morphological changes in DLD1 cells that stably express EndoA3. We found that the number of long and thin actin-rich filopodia was significantly increased (~12-fold) in DLD1 cells expressing EndoA3 (Figure 3A), compared to cells expressing the empty vector. To quantify the filopodia dynamics, we stably expressed Lifeact-eGFP (Riedl et al., 2008) in DLD1 and EndoA3 DLD1 cells. We calculated the rates at which de novo actin protrusions extend and retract using an established kymograph analysis (Supplementary Figure 2F-G) (Sidani et al., 2007). We found that filopodia in EndoA3 DLD1 cells extend more rapidly than those in control DLD1 cells (EndoA3: 2.8 ± 0.2 µm/sec; control: 1.7 ± 0.1 µm/sec; Supplementary Figure 2G left). Similarly, the retraction rates were increased in EndoA3 cells (EndoA3: 2.0 ± 0.2 µm/sec; control: 1.2 ± 0.1 µm/sec; Supplementary Figure 2G middle). Finally, we found that the rate of de novo filopodia formation was higher in EndoA3 DLD1 cells (2.9 ± 0.3 per 10 min per cell) compared to control DLD1 cells (1.2 ± 0.1 per 10 min per cell) (Supplementary Figure 2G right). These data demonstrate that EndoA3 promotes the formation of filopodia. Interestingly, we found that lamellipodia were also enhanced in EndoA3 DLD cells. Using Lifeact-eGFP and live imaging analysis, we determined that about 61% EndoA3 DLD1 cells show lamellipodia within a 10-minute time frame (Figure 3B). By contrast, only ~26% control DLD1 cells had lamellipodia within the 10 minute time window. Together, these results show that EndoA3 has a significant and broad impact on actin-rich filopodia and lamellipodia structures.
Figure 3. Increases in EndoA3 expression alter cytoskeletal dynamics and invasiveness.

(A) Representative immunofluorescence images of EndoA3 DLD1 (left) and parental DLD1 cells (middle) stained with Alexa488-phalloidin (green). White arrows indicate examples of filopodia. Scale bar 2 µm. Fold changes of actin-rich protrusions in DLD1 cells that express either EndoA3 or empty vectors (compared to those in parental DLD1 cells) were plotted in the right panel. “n” = number of images quantified. (B) Representative images of enhanced lamellipodia in DLD1 cells that stably expressed EndoA3-mCherry and Lifeact-eGFP are shown in the left panels. The images were taken from a movie used for lamellipodia quantification. Percentage of cells that show lamellipodia in 10 minutes were plotted in the right panel. “n” = number of movies quantified. Unpaired Student’s t-test, *** p<0.001. (C) Representative images show increased invasiveness of EndoA3 DLD1 cells. 3D spheroids were embedded into mixed matrigel and collagen gels (1:1 volumetric ratio), and images were collected at day 7 post embedding. Control DLD1 cells with empty vectors (left), and EndoA3 DLD1 cells (middle). Dotted circles represent the initial spheroid area at day 1. Fold increase of volume (day 7/day 0) from five different spheroids were used for the statistical analysis (Student’s ttest, n= number of spheroids used for quantification, * p<0.05).
Because EndoA3 accelerates the 2D migration rates and increases filopodia and lamellipodia structures, we next examined whether EndoA3 DLD1 cells are more invasive using the 3D matrigel/collagen assay. Spheroids of EndoA3 DLD1 cells were cultured in mixed extracellular matrix of 50% collagen and 50% matrigel, which is permissive to both invasion and migration. We found that the volume of control DLD1 cells increased ~4.9 ± 0.8 fold in 7 days (Figure 3C left and right). By contrast, the volume of EndoA3 DLD1 cells expanded ~9.6 ± 1.2 fold in the same period (Figure 3C middle and right). These results support the notion that EndoA3 increases growth and motility of DLD1 cells.
EndoA3-membrane interactions play distinct roles in cell growth and migration
We next investigated the role of EndoA3-membrane interactions in EndoA3-induced phenotypes. We generated a cell line stably expressing a mutant version of EndoA3 denoted EndoA3ΔN (ΔN; a deletion of 20 amino acid residues that removes the N-terminal amphipathic helix), lacking membrane-binding activity (Supplementary Figure 3A-B). Fluorescence microscopy data showed that EndoA3WT resides in two populations – a membrane-bound pool (on plasma membrane and intracellular organelles) and a cytosolic pool (Supplementary Figure 3C left and 3D). These two pools of EndoA3WT appear to be exchangeable and dynamic (Supplementary Video 1 left). In contrast, EndoA3ΔN only exhibited a diffuse cytosolic pattern (Supplementary Figure 3C right, 3D, and Supplementary Video 1 right). Similarly, cancer-related mutations (K171N and R174L) (Cancer Genome Atlas Network, 2012; Cancer Genome Atlas Research Network, 2014) that disrupt the membrane-binding interface also shift EndoA3 localization to the cytosol (Supplementary Figure 3E). As expected, EndoA3ΔN failed to promote endocytosis (~1.1x, compared to parental DLD1 cell lines; Figure 4A) and slightly inhibited cell growth (~0.88x, compared to parental DLD1 cell lines; Figure 4B). These findings indicate a potential role of EndoA3-dependent receptor-mediated endocytosis in cell proliferation. To further examine the connection between cell growth and endocytosis, we measured growth rates in the presence of dynamin inhibitors (OctMAB and Dynole 34-2). Results show that these dynamin inhibitors strongly blocked the growth of DLD1 cells and EndoA3 DLD1 cells (Supplementary Figure 3F and data not shown), suggesting that endocytosis is required for EndoA3 to promote cell growth. Surprisingly, disruption of EndoA3-membrane interactions did not abolish filopodia and lamellipodia formation. Instead, EndoA3ΔN cells have more filopodia (ΔN: ~15-fold and WT: ~12-fold; Figure 4C left). We observed a higher number of de novo actin filopodia (ΔN: 4.9 ± 0.9 per 10 min per cell; WT: 2.9 ± 0.3 per 10 min per cell, Supplementary Figure 3G), and longer filopodia in EndoA3ΔN cells (ΔN: 3.7 ± 0.2 µm; WT: 2.6 ± 0.2 µm; control: 2.6 ± 0.3 µm; Supplementary Figure 3H). These data suggest that membrane interactions play an inhibitory role in the EndoA3-induced initiation and the stability of filopodia. In contrast to increased filopodia formation, fewer EndoA3ΔN cells have lamellipodia, when compared to EndoA3WT cells (ΔN: ~44%, and WT: ~62%; Figure 4C right). However, it is worth noting that lamellipodia formation in EndoA3ΔN cells still remains elevated when compared to control cells with empty vectors (~44% EndoA3ΔN cells, and ~26% control cells have lamellipodia; Figure 4C right). Together, these data demonstrate that membrane interactions are essential for EndoA3 to promote endocytosis and cell growth, but are not required for inducing membrane protrusions such as filopodia and lamellipodia.
Figure 4. EndoA3ΔN lacking the membrane-binding activity has increased activity in promoting migration.

The comparison of (A) endocytosis, and (B) growth among cells expressing EndoA3ΔN-mCherry (orange), EndoA3WT-mCherry (blue), and control vectors (gray). Error bars indicate SEM. (C) The number of filopodia protrusions per cell is shown in the left panel, and the fraction of cells with lamellipodia is shown in the right panel. “n” = number of images quantified. (D) Wound closure of EndoA3ΔN DLD1 cells was accelerated in the 2D migration assay. Error bars indicate SEM. (E) Xenotransplantation of human cancer cells into zebrafish hindbrain for assessing cell dissemination in vivo. (Upper left panel) A representative image of a zebrafish that was injected with DLD1 cells expressing EndoA3WT-mCherry into the fish embryo hindbrain. The image was taken 96 hours after injection. The white arrowhead indicates DLD1 cells that have disseminated from the injection site (indicated by the white arrow) in the zebrafish embryo hindbrain. (Lower left panel) An image showing the zoomed area with disseminated DLD1 cells. (Right panel) Percentage of fishes with disseminated DLD1 cells expressing empty vectors, EndoA3 WT-mCherry, and EndoA3ΔN-mCherry. “n”, the number of animals analyzed over 3 independent experiments. One-way ANOVA tests were used for multiple comparisons followed by unpaired Student’s t-test used for error analysis (* p<0.05, ** p<0.01, *** p<0.001).
To determine the impact of EndoA3ΔN on cancer cell motility, we examined cell migration using both in vitro and in vivo assays. First, we found that EndoA3ΔN cells migrated faster than cells carrying EndoA3WT in the automated 2D scratch assay (Figure 4D). Second, we examined the spontaneous metastasis of EndoA3WT and ΔN DLD1 cells using a human to fish xenotransplantation assay. We injected fluorescently labeled cells into the hindbrain of zebrafish embryos, and scored the fish at 96 hours post injection for disseminated cells outside of the hindbrain. Recently, the human to fish xenotransplantation method has been successfully used to quantify the migration of colon adenocarcinoma cells (Anderson et al., 2016). Transplanted DLD1 cells migrate in a solitary instead of collective manner in zebrafish (Anderson et al., 2016; Roh-Johnson et al., 2017) . Our results showed that compared to animals injected with cells expressing the control vector, fish carrying EndoA3 DLD1 cells showed a higher level (~24% compared to ~11%) of dissemination of cells from the injection site. DLD1 cells carrying membrane-binding defective EndoA3ΔN exhibited the highest level of migratory potential in vivo (Figure 4E; ΔN: ~33%). These data indicate that disruption of membrane interactions increases the ability of EndoA3 to stimulate cancer cell migration.
EndoA3 binds TIAM1
To understand how EndoA3 drives migration and invasion, we performed a yeast two-hybrid screen (data not shown) to identify proteins that bind endophilin. Our results showed that a potential binding partner of the endophilin is TIAM1 (T-cell Lymphoma Invasion and Metastasis 1). TIAM1 is an interesting candidate because it has a profound role in cancer cell migration and invasion (Guo et al., 2013; Huang et al., 2013; Malliri et al., 2002; Minard et al., 2004, 2006; Van Leeuwen et al., 1997; Xu et al., 2010). To confirm interaction between TIAM1 and EndoA3, we performed biochemical assays using both recombinant protein segments and full-length proteins. First, we carried out pull-down experiments using GST-tagged BAR domains of EndoA1, A2, and A3, respectively (Figure 5A). We found that EndoA3 BAR binds strongly to a TIAM1 fragment containing DH-PDZ domains (residues 841 to 1418 of mouse TIAM1; Figure 5A). By contrast, the EndoA2 BAR domain failed to bind the TIAM1 fragment, and the EndoA1 BAR domain showed a weak TIAM1 binding activity. To determine whether endogenous EndoA3 and TIAM1 form complexes in cells, we conducted co-immunoprecipitation experiments using extracts from colon cancer cell lines (DLD1, SW480, and HCT116). Using either EndoA3 or TIAM1 antibodies, we were able to detect EndoA3-TIAM1 complexes in immunoprecipitates (Figure 5B, Supplementary Figure 4A-B). These data demonstrate that TIAM1 is a bona fide binding partner of EndoA3.
Figure 5. EndoA3 binds and activates TIAM1.

(A) EndoA3 shows robust binding with recombinant TIAM1. A scheme diagram of the GST pull-down assay is shown in the left panel. GST-tagged BAR domains of EndoA1, EndoA2, and EndoA3 (12 µg) were incubated with the recombinant TIAM1 fragment (20 µM, residues 841-1418) for 4 hours. Proteins bound to the GST beads were analyzed using SDS-PAGE and coomassie blue staining (right). “TIAM1 input” indicates 13% of the recombinant TIAM1 protein used in GST pull-down experiments. * indicates proteolytic fragments of the recombinant TIAM1. (B) Endogenous interactions between EndoA3 and TIAM1 in DLD1 cells. Co-immunoprecipitation experiments were performed using antibodies against EndoA3 (top) and TIAM1 (bottom). IgG was used as controls for immunoprecipitation experiments. Samples were analyzed using western blotting and enhanced chemiluminescence. (C) A scheme diagram showing that free phosphates (indicated as P) are released upon Rac1 GTP hydrolysis (left). The recombinant EndoA3 BAR domain (middle), but not the EndoA2 BAR domain (right), stimulates the TIAM1-dependent Rac1 GTPase activity in vitro. Free phosphates were detected using the malachite green absorbance. Unpaired Student’s t-tests were used for statistical analysis (n=5). *** p<0.001, ** p<0.01, and * p<0.05. (D) Expression of EndoA3 increases the levels of active Rac1 GTPase activity in DLD1 cells. Active Rac1 GTPases that bound to immobilized GST-tagged p21 activated kinase binding domain (PAK PBD) was detected using monoclonal anti-Rac1 antibody (Cytoskeleton Inc.) and enhanced chemiluminescence. Band intensities were quantified using densitometry (normalized to the EndoA2/tubulin ratio) (left). Increases of active RAC1 (fold) were quantified using data from three independent experiments (right). (E) Knockdown of TIAM1 (left) and the Rac1/Cdc42 inhibitor ML141 (right) blocked protrusion formation. Unpaired Student’s t-tests were used for 3 independent experiments (*** p<0.001).
EndoA3 stimulates TIAM1 mediated Rac1 activation
Because TIAM1 is a Rac-specific guanine nucleotide exchange factor (Worthylake et al., 2000), we examined whether EndoA3 affects TIAM1-dependent Rac1 activation. We monitored phosphate release upon GTP hydrolysis by Rac1 using a malachite green assay (Figure 5C) (Geladopoulos et al., 1991). Upon the addition of the EndoA3 BAR domain and recombinant TIAM1, release of free phosphates was accelerated, evidenced by increased malachite green absorbance (Figure 5C middle). As expected, the EndoA3 BAR domain did not stimulate Rac1 activation in the absence of TIAM1 (Supplementary Figure 4C). To test the specificity of EndoA3 in Rac1 activation, we examined the EndoA2 BAR domain that lacks the TIAM1-binding activity. The results showed that the EndoA2 BAR domain did not stimulate Rac1 activity in the presence of TIAM1 (Figure 5C right). To address whether EndoA3 increases the level of active Rac1 in cancer cells, we used the PBD domain of p21 activated kinase to pull down GTP bound Rac1 (active Rac1) as previously described (Bagrodia et al., 1998). We found higher levels of active Rac1 in EndoA3 cells, compared to cells expressing other endophilin isoforms (Figure 5D).
Because both TIAM1 and Rac GTPase are regulators of actin dynamics, we next examined whether EndoA3-induced filopodia rely on TIAM1 and Rac1 activation. First, siRNA down-regulation of TIAM1 led to a significant loss of protrusions (~ 6-fold less in cells transfected with TIAM1 siRNA compared to cells transfected with control siRNA; Figure 5E left). Second, a Rac1/Cdc42 inhibitor ML141 abolished the formation of filopodia (Figure 5E right), suggesting that activation of Rac1/Cdc42 is required. Third, siRNA down-regulation of either Rac1 or Cdc42 also blocked filopodia formation (Supplementary Figure 4D), indicating the involvement of a network of Rac GTPases. These results support the notion that TIAM1 and Rac GTPases activity are required for filopodia formation induced by EndoA3. To determine whether EndoA3 activates TIAM1 GEF activity, we determined the level of active TIAM1 using a nucleotide free Rac1 mutant. Previous studies have shown that the nucleotide free Rac1 mutant (G15A) binds specifically to active TIAM1 (García-Mata et al., 2006). Using this assay, we found that the Rac1 G15A (immobilized on beads) pulled down more TIAM1 from EndoA3 cells, compared with DLD1 cells with empty vectors (Supplementary Figure 4E). These data indicate that EndoA3 expression increases formation of active TIAM1-Rac1 complexes.
TIAM1-EndoA3 binding specificity
Our biochemical data show that EndoA3 exhibits specificity in binding TIAM1 and stimulating Rac1 activity. This is a surprising result given the high similarity among three EndoA isoforms (Kjaerulff et al., 2011). Thus, we further examined the specificity of EndoA3 in living cells using the protrusion and the 3D migration assays. We found that EndoA2 expression did not generate significant changes in the filopodia number (~2.5-fold; n.s., one-way ANOVA), while EndoA1 produced a slight but significant increase (~4 fold) (Supplementary Figure 4F). These findings show that EndoA1 and A2 lack the ability to robustly generate actin-rich filopodia. To determine whether the BAR domain of EndoA3 carries the specificity for enhancing invasiveness, we generated chimeric contrasts by swapping the SH3 and BAR domains of EndoA3 and EndoA2. Our results showed that the EndoA3 BAR_EndoA2 SH3 chimera increased invasiveness of DLD1 cells (Supplementary Figure 4G right), while the EndoA2 BAR_EndoA3 SH3 chimera did not (Supplementary Figure 4G left). These findings indicate that the specificity of EndoA3 in promoting invasiveness resides in the BAR domain.
TIAM1-EndoA3 and membrane-EndoA3 complexes are mutually exclusive
We next determined whether EndoA3 could simultaneously bind membranes and TIAM1. First, we performed in vitro cosedimentation experiments to examine interactions between membranes and EndoA3 BAR. As shown in Figure 6A, EndoA3 pellets with liposomes (70%PC, 24%PS, 1%PIP2, 5%NBD-PE) in the absence of TIAM1. When increasing amounts of TIAM1 were added, more EndoA3 BAR remained in the supernatant, suggesting the TIAM1 competes with membranes for EndoA3. Second, we used negative staining transmission electron microscopy to visualize membrane tubules induced by EndoA3 BAR (Figure 6B). Incubation of liposomes with EndoA3 BAR led to a significant amount of membrane tubulation. Addition of TIAM1 (20 µM) significantly decreased membrane tubules, confirming competition between TIAM1 and membranes for binding EndoA3 BAR. As a control, the presence of TIAM1 had no impact on the membrane tubulation activity of the EndoA2 BAR domain (Supplementary Figure 4H).
Figure 6. TIAM1 and membranes compete for EndoA3.

(A) Addition of recombinant TIAM1 fragments reduces membrane bound EndoA3 BAR. Cosedimentation assays were used to detect membrane-bound (pellet, P) and unbound (supernatant, S) fractions of EndoA3. Liposomes (1%PIP2, 24%PS, 70%PC, 5%NBD-PE, 0.5mM total lipids) and his6-tagged EndoA3 BAR domain (2 µM) were incubated with TIAM1 (0, 5, 10, and 20 µM, respectively) for 30 minutes. Samples were centrifuged at 75,000 rpm for 30 min. Supernatants and pellets were analyzed by western blotting and enhanced chemiluminescence. (B) Addition of TIAM1 reduces the tubulation activity of EndoA3. Liposomes (1%PIP2, 24%PS, 75%PC, 0.5mM total lipids) were incubated with his6-tagged EndoA3 BAR (1µM), with and without the recombinant TIAM1 fragment (20 µM), for 30 min at room temperature. Samples were analyzed using negative stain transmission electron microscopy. Representative images were shown at the left. The fraction of liposomes with membrane tubules were quantified and shown at the right. Unpaired Student’s t-tests were used for statistical analysis of 3 independent experiments. * p<0.05. (C) Membrane inhibits EndoA3-dependent, but not EndoA3ΔN-dependent, Rac1 activation. His6-tagged EndoA3 BAR (WT or ΔN; 8 µM) and TIAM1 fragments (0.5 µM) were incubated with Rac1 (2µM) in the absence and presence of liposomes (1%PIP2, 25%PS, 74%PC, 0.5mM total lipids). Fold activation of Rac1 GTPase activity was calculated using results from control experiments in the absence of his6-tagged EndoA3 BAR. Malachite green assay was used to quantify free phosphate groups released from GTP by Rac1. Student’s t-tests (** indicates p<0.01). (D) Anchoring EndoA3ΔN to the plasma membrane using an N-myristoylation tag reduces the levels of active TIAM1 in DLD1 cells. Lysates from DLD1 cells expressing EndoA3ΔN-GFP and Myr-EndoA3ΔN-GFP were analyzed. Active TIAM1 was detected using Rac1 G15A agarose beads. TIAM1 signal was normalized to tubulin levels. The ratio of active TIAM1 levels between EndoA3ΔN-GFP and Myr- EndoA3ΔN-GFP cells were indicated. (E) The ability of EndoA3ΔN to stimulate cell migration is significantly reduced when it is anchored to the plasma membrane. Migration (left) and growth (right) of DLD1 cells expressing either myr-EndoA3ΔN-GFP (green) or EndoA3ΔN-GFP (orange) was quantified using the automated assays described in Figure 2. Error bars indicate SEM. Statistical analysis was done using two-way ANOVA (** p<0.001). (F) EndoA3 localization was examined using fluorescence microscopy. EndoA3 cells were treated with DMSO (left), PTEN inhibitor 1 (bpV(HOpic), 200nM; middle), or PTEN inhibitor 2 (VO-OHpic, 200nM; right). (G) Ratio between membrane-bound and cytoplasmic EndoA3 was measured using ImageJ as previously described in Materials and Methods. “n” = the number of cells analyzed. ** p<0.01, one-way ANOVA. (H) Inhibition of PTEN accelerates the growth of EndoA3 DLD1 cells (4 independent experiments, * p<0.05). (I) Inhibition of PTEN reduces filopodia abundance in EndoA3 DLD1 cells (“n” = number of images analyzed). Three independent repeats. *** p<0.001, one-way ANOVA).
Next, we examined how membrane interactions affect the ability of EndoA3 to stimulate TIAM1 and Rac1 GTPase. We found that, upon the addition of phospholipid membranes (75%PC, 24%PS, 1%PIP2), EndoA3 failed to stimulate the Rac1 GTPase activity in the malachite green assay (Figure 6C). These data suggest that membranes play an inhibitory role in EndoA3-dependent Rac1 activation. We next tested whether disruption of EndoA3-membrane interactions could relieve the inhibitory effect of membranes. First, we found that deletion of N-terminal amphipathic helix does not disrupt EndoA3-TIAM1 interactions, as EndoA3ΔN binds the recombinant TIAM1 fragment to a similar extent as EndoA3WT (Supplementary Figure 5A), and remains active in stimulating TIAM1 in DLD1 cells (Supplementary Figure 5B). Indeed, the Rac1 activity was slightly enhanced in EndoA3ΔN DLD1 cells (Supplementary Figure 5C), even though the expression levels of EndoA3ΔN were lower than that of EndoA3WT (Supplementary Figure 5D). Because EndoA3ΔN does not bind membranes, we surmised that it may be able to escape the inhibitory regulation by membranes. Consistent with this idea, we found that the BAR domain of EndoA3ΔN remains active to stimulate Rac1 activity in the presence of membranes, as reported by the malachite green assay (Figure 6C). To further examine the inhibitory impact of membranes on EndoA3, we engineered a membraneanchored EndoA3ΔN by adding a myristoylation tag to the N-terminus of EndoA3 (myr-EndoA3ΔN; Figure 6D). Myr-EndoA3ΔN was expressed at similar levels as EndoA3ΔN in DLD1 cells (Supplementary Figure 5D). As expected, a large fraction of myr-EndoA3ΔN was associated with the plasma membrane (Supplementary Figure 5E). When compared to EndoA3ΔN DLD1 cells, myr-EndoA3ΔN cells have lower levels of active TIAM1 in pull-down experiments using the nucleotide free Rac1 mutant (Figure 6D), and reduced migration rates in the automated 2D migration assay (Figure 6E left). By contrast, levels of endocytosis and cell growth were indistinguishable between EndoA3ΔN and myr-EndoA3ΔN cells (Figure 6E right and Supplementary Figure 5F). These results demonstrate that targeting EndoA3 to membranes is sufficient to inhibit Rac1 activation and migration, suggesting a competitive link between endocytosis and cytoskeleton rearrangements.
To further investigate the relationship between EndoA3-dependent endocytosis and cytoskeleton rearrangements, we examined EndoA3 localization and filopodia formation upon inhibition of the PIP3 phosphatase PTEN. It was recently demonstrated that PTEN inhibition promotes clathrin-mediated endocytosis by enhancing the recruitment of endophilin to membranes (Boucrot et al., 2015). Consistent with those data, we found that PTEN inhibitors (bPV(HOpic) and VO-OHpic) induced a dramatic shift of EndoA3 from the cytosol to the plasma membrane and intracellular vesicles in EndoA3WT DLD1 cells (Figure 6F-G). The growth of EndoA3WT DLD1 cells was accelerated upon PTEN inhibition (Figure 6H). In contrast, the localization of EndoA3ΔN and the growth of EndoA3ΔN DLD1 cells were not altered by PTEN inhibitors (Supplementary Figure 6), suggesting that EndoA3-mediated endocytosis is required for PTEN-regulated cell growth. Interestingly, PTEN inhibition significantly attenuated filopodia formation in EndoA3WT DLD1 cells (Figure 6I), supporting the notion that depletion of cytosolic EndoA3 limits the cytoskeleton rearrangements. Together, these results are in agreement with a membrane/cytosol competition for EndoA3 to balance endocytosis and cytoskeleton rearrangements in cancer cells.
EndoA3-membrane interactions modulate cancer cell proliferation and spread at secondary sites in mice
To examine whether the balance between EndoA3-membrane and EndoA3-TIAM1 interactions affects cancer progression in vivo, we examined the ability of cancer cells to colonize secondary sites (i.e., lungs) using an experimental metastasis procedure. We injected DLD1 cells with WT and ΔN EndoA3 into mouse tail veins and monitored their ability to colonize the lung. To determine the presence of metastatic clusters, we stained the lung sections using IHC (H&E, cytokeratin, and Ki-67, Supplementary Figure 7). To quantify the level of metastasis, we performed immunofluorescence analysis for the epithelial marker cytokeratin (8+18). An increased abundance of epithelial cell clusters (nodules) was found in lungs of mice injected with EndoA3ΔN DLD1 cells (ΔN: ~38 ± 10/mice), compared to mice with either EndoA3WT (WT: ~14 ± 1/mice) or control DLD1 cells (~8 ± 2/mice) (Figure 7A). These data are consistent with our findings that EndoA3ΔN is more potent than EndoA3WT in promoting tumor cell migration (Figure 4). Interestingly, the area of cytokeratin positive nodules from mice with EndoA3WT cells (0.028 ± 0.009 mm2/nodule) was significantly larger than mice with either EndoA3ΔN (0.008 ± 0.002 mm2/nodule) or control cells (0.002 ± 0.0005 mm2/nodule) (Figure 7B), suggesting that EndoA3WT cells proliferate more rapidly at secondary sites. To further examine the proliferative effect of EndoA3, we performed Ki-67 staining to assess tumor cell proliferation in lung lobes (Figure 7C). We found that Ki-67 positive areas were significantly larger in mice with EndoA3WT cells (0.013 ± 0.004 mm2/lobe) compared to control cells (0.0017 ± 0.0006 mm2/lobe), indicating a pro-proliferative function of EndoA3. Disruption of EndoA3-membrane interactions impaired the pro-proliferative activity, as mice carrying EndoA3ΔN cells had smaller Ki-67 positive areas compared to EndoA3WT cells (ΔN: 0.0046 ± 0.001 mm2/lobe, and WT: 0.013 ± 0.004 mm2/lobe). Taken together, these data suggest that membrane-bound and membrane-unbound EndoA3 promote different aspects of cancer progression, i.e., cytoplasmic EndoA3ΔN drives more DLD1 cells to migrate and colonize the lung, but membrane-bound EndoA3WT supports the proliferation of newly colonized tumor cells.
Figure 7. EndoA3 expression promotes tumor spread and proliferation at secondary sites in mice.

Experimental metastasis was examined in nude mice injected with DLD1 cells expressing vector control (gray), EndoA3-mCherry (blue), and EndoA3ΔN-mCherry (orange), respectively. (A) Number of metastatic nodules per mice in lungs upon tail vein injection of DLD1 cells. Metastatic nodules were visualized by immunofluorescence staining against Ck8/18. (B) Average area per nodule per mice in lung was measured. “n” indicates the number of animals. (C) The extent of cell proliferation was assessed using Ki-67 immunohistochemistry of pulmonary lobes. One-way ANOVA was used for multiple comparisons followed by unpaired Student's t-test was used for statistical analysis (*** p<0.001, ** p<0.01, * p<0.05).
Discussion
Growing evidence suggests that cancer cells hijack existing signaling pathways to drive mechanical changes and alter their interactions with the surrounding microenvironment (Lanzetii and Di Fiore, 2017; Lanzetti and Di Fiore, 2008; Mellman and Yarden, 2013; Mosesson et al., 2008). In this study, we uncover a pivotal role of EndoA3 in coordinating cytoskeletal rearrangement and endocytosis in cancer cells. Our results show that 1) EndoA3 promotes colon cancer metastasis by increasing both proliferation and migration, 2) EndoA3-mediated endocytosis is required for proliferation, but it is dispensable for cell migration, 3) EndoA3 stimulates Rac1 GTPase activity by direct interaction with the Rac1 GEF TIAM1, 4) EndoA3-dependent Rac1 activation determines membrane protrusion dynamics and subsequently controls cell migration, and 5) membranes inhibit EndoA3-TIAM1 interactions, revealing competition between EndoA3-mediated endocytosis and cell migration. Together, these findings highlight a role of EndoA3 in modulating small GTPase signaling. We propose that imbalanced activity of EndoA3 in cancer cells disrupts endocytosis and cell migration, which ultimately facilitates the metastasis of cancer cells. Below we discuss the implications of these results.
EndoA3 is a unique endocytic signature of colon cancer cells
Numerous studies have demonstrated that cancer cells modify endocytic pathways to gain pro-oncogenic phenotypes (Ben-Chetrit et al., 2015; Bendris et al., 2016; Eppinga et al., 2012; Frittoli et al., 2014; Hu et al., 2011; Razidlo et al., 2013; Rolland et al., 2014). It is generally thought that endocytosis promotes cancer cell proliferation and migration by internalization of cell surface receptors, adhesion molecules, and nutrients (Lanzetti and Di Fiore, 2008; Mosesson et al., 2008). For instance, endocytosis removes membrane receptors from cell surface, which deactivates signaling on the plasma membrane. Moreover, internalized receptors through endocytosis induce persistent signaling on intracellular organelles (e.g., endosomes) (Miaczynska et al., 2004), that could lead to altered spatial specificity and signal diversification in cancer cells. These changes broadly impact trafficking routes and signaling networks, and therefore represent an ‘endocytic signature’ of cancer cells (Lanzetii and Di Fiore, 2017). In this study, we show that EndoA3 is a unique component of the endocytic machinery that promotes oncogenic phenotypes in colon cancers through two distinct pathways. On one hand, EndoA3 accelerates the proliferation of colon cancer cells through enhanced endocytosis. On the other hand, it promotes cell migration by directly binding and activating Rac GEF TIAM1 and Rac GTPases in an endocytosis-independent manner. Interestingly, several proteins that act in the endophilin pathway also promote Rac signaling and are pro-metastatic. For example, the GTPase Dynamin 2 (Dyn2) activates Rac1 and invasive cellular migration of pancreatic tumor cells by binding and stabilizing the Rac1 GEF Vav1 (Razidlo et al., 2013). The Vav1-stablizing activity of Dyn2 is independent to the role of Dyn2 in endocytosis (Razidlo et al., 2013). Furthermore, the phosphoinositide phosphatase Synaptojanin 2 (SYNJ2) binds the active form of Rac1 (Malecz et al., 2000), and enhances invadopodia and lamellipodia formation in a Rac1-dependent manner (Chuang et al., 2004). Thus, the endophilin-dependent pathway is likely to represent a unique endocytic signature that engages both receptor internalization and small GTPase regulation.
Membrane interactions inhibit EndoA3-dependent TIAM1 activation
Small GTPases such as Cdc42, Rac1, and RhoA have profound impacts on cellular activities such as polarity, cytoskeletal dynamics, migration, and cell cycle (Etienne-Manneville and Hall, 2002; Hall, 1998; Raftopoulou and Hall, 2004; Schmitz et al., 2000). These GTPases behave as “molecular switches” that toggle between an inactive GDP-bound state and an active GTP bound state (Etienne-Manneville and Hall, 2002; Hall, 1998). Increasing evidence suggests that GTPase activity is tightly controlled by morphological changes of the plasma membrane (Frost et al., 2009; de Kreuk and Hordijk, 2012; Zhao et al., 2011). In particular, proteins that harbor curvature sensing BAR domains have emerged as key regulators of the small GTPase signaling. Several BAR proteins contain either scaffolding domains that bind active small GTPases, or GEF/GAP domains that facilitate the exchange and hydrolysis of GTP by GTPases (de Kreuk and Hordijk, 2012). Such a tandem configuration of BAR and GTPase scaffolding/modulatory domains enables the BAR proteins to localize GTPase signaling to the plasma membrane. In this study, we show that the BAR protein EndoA3 also has a dual role in membrane remodeling and GTPase regulation, i.e., it promotes endocytosis by EndoA3 BAR-membrane interactions, and it activates Rac GTPases through the binding between EndoA3 BAR and the Rac GEF TIAM1. However, the underlying mechanism for EndoA3 BAR to coordinate membrane and GTPase regulation is distinct from the proteins with tandem BAR and GTPase scaffolding/modulatory domains. In the case for EndoA3, membrane and TIAM1 interactions compete for the same BAR domain, resulting in a mutually exclusive control of EndoA3 endocytic versus GTPase signaling activities.
In support of the competition model, mutant EndoA3 (EndoA3ΔN) lacking membrane interactions exhibits increased ability to activate TIAM1 and Rac activity in biochemical assays. Physiologically, migration and metastasis of EndoA3ΔN cells were significantly enhanced both in vitro and in vivo. These findings uncover an unexpected role of EndoA3 in coordinating growth and migration of cancer cells, and therefore raise the possibility of dampening cancer aggressiveness by modulating the amount of cytosolic and membrane-bound EndoA3. That anchoring EndoA3ΔN on the plasma membrane by myristoylation inhibits migration without increasing endocytosis is consistent with the notion that modulating EndoA3 activity can influence neoplastic progression. More important, inhibition of the tumor suppressor PTEN shifts EndoA3 from the cytosol to membranes, which likely pushes cancer cell behavior towards proliferation rather than migration. Together, these data strongly suggest that EndoA3 connects two key processes of cancer progression – 1) assisting cancer cell proliferation through EndoA3-membrane dependent endocytosis, and 2) promoting cell migration by cytosolic EndoA3 activation of small GTPases.
Specific interactions between EndoA3 and TIAM1
Endophilin isoforms share a high-degree of similarity in both structural and biochemical features. For example, the primary sequence of the EndoA3 BAR domain is ~66% identical to the EndoA1 BAR and ~72% identical to the EndoA2 BAR3. Structurally, these BAR domains form similar crescent shaped dimers with positively charged residues distributed along the membrane-interacting concave face. Functionally, all three endophilin isoforms promote membrane internalization during endocytosis. However, despite high levels of similarity, endophilin isoforms are specialized for GTPase regulation. We find that the EndoA3 BAR domain binds robustly to TIAM1 and activates Rac1 activity in both biochemical and cell assays. By contrast, the EndoA2 BAR domain lacks detectable levels of TIAM1 interaction and fails to stimulate Rac1 GTPase activity. Interestingly, a recent study shows that the EndoA2 BAR domain strongly binds the Arf6 guanine nucleotide exchange factor EFA6 (Boulakirba et al., 2014) supporting the notion that endophilin isoforms have diverged in modulating specific GTPases. Indeed, we found that EndoA3 expression but not EndoA2 alters filopodia, lamellipodia, and cell migration in colon cancer cells. EndoA3-induced changes require the activation of Rac1, which may subsequently activate a network of GTPases to modulate cytoskeletal dynamics. Indeed, we found that EndoA3 expression led to CDC42-dependent filopodia formation (Supplementary Fig. 4D), even though CDC42 is not a direct target of TIAM1 (García-Mata et al., 2006). Together, these findings demonstrate that endophilin isoforms utilize specialized interactions with small GTPase modulators to control cellular dynamics.
Implications of EndoA3 mutations in cancers
In human cancers, several mutations occur within EndoA3 at residues that are critical for EndoA3- membrane interactions. For example, K171 and R174 are two conserved residues among EndoA isoforms. These positively charged residues support the electrostatic interactions that bring endophilin BAR domains to membranes with negatively charged headgroups (Gallop et al., 2006; Masuda et al., 2006). In colorectal cancers, the R174 residue is mutated to either Leu or Gln, which causes charge neutralization at the membrane-binding interface. Indeed, the R174(L/Q) mutation has been found also in lung cancer (Cancer Genome Atlas Network, 2012; Cancer Genome Atlas Research Network, 2014) and melanoma (Hodis et al., 2012), suggesting that EndoA3 mutations have a broader impact beyond colorectal cancers. Similarly, another charge neutralizing mutation K171N occurs in EndoA3 in colorectal and uterine cancers (Cancer Genome Atlas Network, 2012; Cancer Genome Atlas Research Network et al., 2013). Indeed, we found that both K171N and R174L increase the cytosolic pool of EndoA3. EndoA3 mutations are not limited to positively charged residues. The residue (S75) that is crucial for sensing and inducing membrane curvature is also altered in colorectal cancers (Cancer Genome Atlas Network, 2012; Matta et al., 2012). The S75 residue of all three EndoA isoforms can be phosphorylated by Leucine-rich repeat kinase 2 LRRK2 (Matta et al., 2012). In neurons, deregulation of the EndoA1 S75 phosphorylation accelerates neurodegeneration, likely due to changes in synaptic vesicle endocytosis and autophagy biogenesis (Matta et al., 2012; Soukup et al., 2016). In colorectal cancers, a S75L mutation occurs in EndoA3, but not EndoA1 and EndoA2. Such isoform specificity indicates that EndoA3 may have a stronger impact on colorectal cancers than its neuronal paralogues. These findings prompt us to speculate that mutations at the EndoA3- membrane interface alter the abundance of EndoA3-membrane and EndoA3-TIAM1 complexes, which could result in global changes in endocytosis, GTPase regulation, cytoskeleton rearrangement, and thus influence the progression and outcome of colorectal and perhaps other types of cancers.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jihong Bai (jbai@fredhutch.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish (Danio rerio)
Zebrafish (Danio rerio) were raised at the Fred Hutchinson Cancer Research Center, and animal care and experiments were approved by the Institutional Animal Care and Use Committee. All animals were maintained according to standard procedures [Westerfield M., The Zebrafish Book. 1993.] and staged as previously described (Kimmel et al. 1995).
Mouse (Mus musculus)
All mouse work was performed in accordance with Fred Hutchinson Cancer Center, IACUC #1195 and AAALAS guidelines. Crl:NU-Foxn1nu mice were purchased from Charles River laboratory.
METHODS DETAILS
Plasmids and Recombinant Protein Purification
Mouse cDNAs encoding EndoA1, A2, and A3 were amplified as previously described (Bai et al., 2010). DNA fragments encoding his6-SUMO was inserted into the NcoI and the BamHI sites of PET28A vector (Novagen, Madison, WA), which subsequently results in the plasmid BJP-A03. To construct plasmids for His6-SUMO-tagged recombinant proteins, DNA fragments encoding endophilin BAR domains were inserted into BJP-A03 between BamHI and NotI sites. His6-SUMO tagged fusion proteins were produced in the BL21(DE3) E. coli strain and were purified using a published protocol (Poudel et al., 2016). The his6-SUMO tag was removed from the recombinant proteins using the SUMO Protease (Thermo Fisher Scientific). To make plasmids for expressing GST-tagged recombinant proteins, DNA fragments encoding endophilin, Rac G15A, and mouse TIAM1 (amino acid residues 841-1418) were inserted into the pGEX-4T1 vector between BamHI and XhoI sites. These plasmids were transformed into Mach1 T1 E. coli cells, and GST-tagged recombinant proteins were purified using previously published protocol (Poudel et al., 2016). For retroviral transduction, cDNAs encoding full-length rat EndoA1, A2, and A3 were inserted into the pBABE vector (Cell Biolabs Inc) between BamHI and EcoR1 sites. For imaging actin dynamics, Lifeact-EGFP was inserted into the pBABE vector between BamHI and EcoRI sites. All mutations used in this study were generated using overlapping PCR amplification and the CloneEZ PCR cloning kit (Genscript). To generate chimera protein EndoA3 BAR_EndoA2 SH3, residues 1-243 from EndoA3 BAR domain was fused with residues 247-368 from the SH3 domain of EndoA2. To generate chimera protein EndoA2 BAR_EndoA3 SH3, residues 1-246 from EndoA2 BAR was fused with residues 247-347 from EndoA3 SH3.
Human Tissue Microarray Analysis
Analysis of human tissue microarray using immunohistochemistry (IHC) was performed by the IHC core facility at Fred Hutchinson Cancer Research Center (FHCRC) (Metz et al., 2016). Briefly, a colon cancer tissue microarray that consisted of 50 cases was obtained US Biomax inc (bc05118c). Patient data and prognostic status (age, sex, grade and TNM staging) were available through the US Biomax website. Inadequate sample and poor staining precluded an assessment of 8 cases from the microarray. The IHC core facility at FHCRC independently determined the specificity of EndoA3 antibody (abcam, 184008) for IHC through a multi-step procedure. First, a proper method for antigen retrieval was identified from several options to increase signal to noise ratio. Second, the EndoA3 antibody and a rabbit IgG were tested in brain and testis (positive controls) and skin (negative control). An in-house pathologist confirmed the antibody specificity. Third, human adrenal gland samples provided by US Biomax were used as negative controls for staining human tissue microarrays.
EndoA3 Detection from Mouse Tissues
All animal studies were performed according to the guidelines of the Fred Hutchinson Cancer Research Center. Normal mouse tissue and tumors were snap-frozen upon dissection, and total extracts were prepared in radioimmunoprecipitation assay buffer (pH7.6). Cells were lysed by sonication. The concentration of total protein in extracts was determined using the BCA assay (Thermo Fisher). Samples were diluted to contain 30µg total protein and were loaded onto SDS-PAGE gels. Samples were transferred onto nitrocellulose membranes. A rabbit polyclonal antibody against EndoA3 (Abcam 184008) was used to detect EndoA3. Endogenous EndoA3 was visualized using enhanced chemiluminescence and quantified using densitometry.
Cell Culture and Stable Cell Line Generation
Cells were cultured in 1x DMEM media (Life technologies) with 10% (vol/vol) fetal bovine serum and 100 U/ml penicillin/streptomycin. Cell lines that stably expressed genes of interest were generated by a retrovirus-based transduction method using pBABE vectors (Cell Biolabs). Briefly, plasmids containing genes of interest were inserted into the backbone of pBABE (hygro/puro) vectors. These plasmids were transiently introduced into a MLV based retroviral packaging cells (PT67, ATCC) using Lipofectamine 2000. After 36 hours, media was collected from the PT67 cells. Polybrene (5µM) (Sigma) was added along with fresh media at 1:1 ratio. The prepared viral titer was then subjected to target cells grown in 6 well plates. Upon introduction of virus, 6 well plates were subjected to centrifugation at 1500g for 30min. The same process was repeated at 48 hrs post transfection to improve efficiency of infection. After 72 hrs, fresh media was added and cells were allowed to grow for an additional 24 hrs, before introducing media that contained antibiotic for selection. Cells remained in selection media for 48 hrs before being transferred to growth media. Western blot analysis was carried out to confirm the expression of the gene of interest in selected cells. In cases where a fluorescent reporter was attached, fluorescence imaging was used to further confirm the expression of the gene of interest.
FM Dye Uptake Assay
FM dye uptake experiments were performed using established protocol with modifications (Dong et al., 2015; Hoopmann et al., 2012). Cells were cultured in glass bottom tissue culture plates (µ–Dish, Ibidi). Fresh media was added to the cells along with 10µM FM1-43 or FM4-64 dye (Thermo Fisher). Cells were incubated for 5 mins at 37°C to allow FM dye internalization. Cells were gently washed using 1x PBS containing 0.5mM EGTA and were incubated in 1x PBS containing 1 mM ADVASEP-7 (Sigma) for 5 min to remove excess dye. Images were taken using an Olympus FV1000 confocal microscope with a 60x/1.42 oil objective or a Zeiss spinning disk Axio Observer Z1 confocal microscope with a 40x water Objective. FM1-43 was excited with a 488 nm laser and FM4-64 was excited with a 560 nm laser. All images were analyzed using ImageJ. The number of total cells in each image was counted, along with the number of cells that contained FM1-43/FM4-64 fluorescence puncta, to calculate the percentage of cells with dye internalization. Student’s t-test was used for statistical analyses.
Automated Cell Growth and Migration Assays
For growth experiments, 5000 cells (counted using a hemocytometer) were plated into each well of clear bottom 96 well plate (Thermo Fisher). For migration experiments, 50- 70,000 cells were plated into each well of a Imagelock 96 well plate (Essen Bioscience). The amount of cells used in these studies was optimized to ensure the formation of smooth cell monolayers in 96 well plates. Prior to data collection, fresh media was added to the samples. For migration experiments, low serum media (1%) was used along with 5 µM mitomycin to limit proliferation to block proliferation. A wound maker device (Essen Bioscience) was used to make scratches. Data was collected automatically and analyzed using the Incucyte software. All graphs were plotted using Graphpad Prism.
Spheroid Cultures
Spheroid culture was performed as previously described (Timmins and Nielsen, 2007). Briefly, 500 cells were collected from a culture that was ~70% confluent. These cells were grown in suspension of hanging drops (500 µl medium) until spheroids were formed. Spheres were carefully re-suspended in 50% (vol/vol) mixture of collagen and matrigel. Extra care was applied to ensure that the spheroids did not contact plastic surface. The mixture was then covered with DMEM media to prevent spheroids from drying up. Growth and spread of the spheroid in the matrix was monitored by visual inspection using a Leica DMI3000B inverted microscope.
Fluorescence microscopy
Immunofluorescence was carried out using a published protocol (Conacci-Sorrell et al., 2014). Briefly, cells were cultured on glass coverslips or chamber slides, washed with PBS, and fixed with 4% paraformaldehyde for 30 mins. Next, cells were permeabilized using PBS buffer with 0.5% Triton X-100 for 5 min followed by treating with a blocking solution (Image IT FX, Invitrogen) for 30min. Cells were incubated with primary antibodies (1:100) for 1 hour. At the end of incubation, cells were washed 5x with 1x PBS and incubated with secondary antibodies for 1 hour. Samples were mounted to a microscope slide using ProLong Diamond Antifade Mountant (Thermo Fisher). All images were collected using an Olympus FV1000 microscope with a 60x/1.42 oil objective. To estimate the amount of membrane-bound and cytosolic mCherry-tagged EndoA3 variants, fluorescence images were collected using a Zeiss spinning disk Axio Observer Z1 confocal microscope with a 60x water objective, and were analyzed using ImageJ (Boucrot et al., 2015). Background fluorescence intensity was corrected for each image.
GST Pull-Down Assays
For EndoA3 binding experiments, GST-tagged rat EndoBAR proteins (12 µg), immobilized on glutathione beads (Genscript), were incubated with 20 µM TIAM1 fragment (His6-DH-PDZ, residues 845-1234) at 4°C for 4 hours in a HEPES buffer (50mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Triton X-100). Beads were washed three times using the HEPES Buffer. Samples were analyzed by SDS-PAGE and were stained by Coomassie Blue. For active Rac1 pulldown experiments were performed as previously described (García-Mata et al., 2006). Briefly, cells (freshly fed and ~50-60% confluent) were lysed using 1x RIPA buffer containing 2mM EDTA. Fresh lysates were prepared before the pulldown. Immobilized GST-PBD (PAK21) protein was incubated with the lysate (500 µg total proteins) for 2 hrs at 4°C. Samples were washed 3 times with 1xPBS buffer. Bound proteins were analyzed using SDS-PAGE gels. A similar protocol was used for assaying active TIAM1 except that GST-tagged Rac (G15A) was used (García-Mata et al., 2006).
Malachite Green Assay for GTPase Activity
A previously published protocol was modified to monitor malachite green absorbance upon Rac-GTP hydrolysis (DeVay et al., 2009). Rac1 (2 µM), TIAM1 (500 nM), and EndoA3 (8 µM), were mixed in PCR tubes. GTP (3.125 mM final concentration) was added at last. Samples (100 µl) were incubated at 37°C in a water bath. At each time point, 10 µl sample was taken out and was placed in a 96 well plate that was preloaded with 40 µl EDTA (200 mM) to stop the GDP-GTP exchange. At the end of experiments, 200µl of 0.98 mM malachite green was added to the wells and absorbance at 650 nm was recorded. Phosphate concentration was determined using a reference curve.
Liposome Preparation
Liposomes were prepared as previously described (Poudel et al., 2016). Briefly, lipid mixtures with desired composition were dried under compressed nitrogen for at least 3 hours. Dried lipids were lyophilized for 1.5 hours, and then were reconstituted into HEPES buffer. Small multilamellar liposomes were generated by sonication. For electron microscopy experiments, unilamellar liposomes were prepared by extrusion at least 15 times through polycarbonate membranes (1µm pores) using Mini extruders (Avanti Polar lipids).
Liposome Sedimentation Assay
Liposome sedimentation assay was carried out as previously described (Hui et al., 2006). Briefly, EndoA3 (2 µM), TIAM1 (5 µM), and liposomes (2 mM total lipids, 1%PIP2, 25%PS, 5%NBD-PE and 69%PC) were incubated at 4°C for 4 hours. Samples were centrifuged in a TA100.1 rotor (Beckman Coulter Inc.) at 75,000 rpm for 30 mins. Twenty microliters of the supernatant were collected, and pellets were re-suspended in 20µl SDS sample buffer. Samples were loaded onto SDS-PAGE gels and EndoA3 were visualized using Coomassie Blue.
Negative Staining and Transmission Electron Microscopy
Sample preparation for negative stain electron microscopy was carried out at the FHCRC electron microscopy facility as previously described (Poudel et al., 2016). Briefly, nickel grids were glow discharged for 40 sec, after which grids were incubated with samples. Samples were fixed using 0.5x Karnovsky’s fix at 2 minutes. Samples were then washed with one drop of 0.1 M cacodylate buffer, followed by a four-drop H2O wash. One drop of 1% uranyl acetate was touched on the sample. The grids were then carefully dragged through dry filter papers and put in a desiccator overnight to dry. Images were collected in a JEOL TEM 1400 transmission electron microscope, and measurements were done using ImageJ.
PTEN inhibition
PTEN inhibitors were obtained from Sigma Aldrich. Cells were treated for ~20 hours with DMSO, PTEN inhibitor 1 (bpV(HOpic, 200 nM), or PTEN inhibitor 2 (VO-OHpic, 200 nM) for imaging experiments. Media containing PTEN inhibitors were removed and replaced with Leibovitz medium. Images were collected using Zeiss AxioObserver Z1 spinning disc microscope using a 60x water objective. For growth measurements upon PTEN inhibition, cells were grown in 96-well imaging plates as described above. Growth media were replaced at t=0 to apply PTEN inhibitors. Plates were then loaded into IncuCyte Zoom and growth rates were monitored for 96 hours at 2-hour intervals.
Xenotransplantation Experiments
Nu/nu mice were commercially purchased from Charles River Laboratories, handled and caged under protocols approved by the animal facility at FHCRC (Institutional Animal Care and Use Committee, #1189). For tail vein injection experiments, cells were cultured as described above. Before injection, four million cells/200µl were aliquoted into tubes. Each aliquot was injected into a mouse tail vein using approved protocol. Veterinary technicians at FHCRC animal facility carried out all injections. Animals were then monitored for physical changes daily until euthanasia. Upon euthanization, lungs were fixed in formalin for 96 hours and were submitted to the immunohistochemistry core facility for sectioning and staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of Human Tissue Microarray staining
Digital images of IHC-stained TMA slides were obtained at 20x magnification (0.5 µm per pixel) using a whole-slide scanner (ScanScope AT, Aperio) with a 20x/0.75 Plan Apo objective lens (Olympus). Data from 15 different tumors and normal tissues were used to quantify the average IHC intensity of EndoA3 staining. Total tissue area and the percent of EndoA3 stained area were obtained using the color deconvolution plugin of ImageJ (Ruifrok and Johnston, 2001). For stage-based analysis, a published ‘H score’ methodology (Metz et al., 2016) was used to compare a tumor with its normal pair. Briefly, representative images were used as input parameters for recognition training in software (Genie, Aperio). Under technical supervision, the TMA samples were annotated using Aperio’s annotation software (ImageScope v12.2; Aperio). Only annotated images were used for analysis. The H-score for tumor grade analysis was calculated by a formula involving the positive percentages: score = 1.0 × (% weak) + 2.0 × (% medium) + 3.0 × (% strong).
Quantification of Human in Fish Xenotransplantation Assay
Transplantation of human DLD1 cells in zebrafish embryos was carried out as previously described (Anderson et al., 2016). Zebrafish embryos were dechorionated and treated with 0.2 mM phenylthiourea prior to injection to prevent melanization. DLD1 cells (~70% confluent) were collected and 25-50 cells were injected into hindbrain ventricle of wild type zebrafish embryos 48 hours post-fertilization (hpf). DLD1 cells were labelled with CellTracker Green (Invitrogen) prior to injection. Injected fish were incubated in 0.2 mM PTU at 31°C for 96 hours before scoring for metastasis. Metastasis was scored as the percentage of animals in which one or more cells had migrated from the primary injection site.
Quantification of Xenotransplantation Experiments in Mice
A metastasized nodule was scored when a cluster of five or more cells (determined by DAPI signals) shows cytokeratin 8/18 staining. A rabbit polyclonal antibody against Ki-67 was used to assess cell proliferation. Ki-67 area was quantified at regions where both Ki-67 and cytokeratin 8/18 immunostaining signals were observed. Area measurements and image collection were done using TissueFAXS Software (TissueGnostics) licensed to the imaging facility core at FHCRC.
Quantification of Spheroid Cultures
Radius of each sphere was measured using metamorph software. Volume of a sphere was used to calculate the volume of each sphere at t=0 and t=7 days. The ratio of volumes was then compared to calculate the fold change in volume.
Statistical Analysis
Statistical details for each experiment are presented in the figure and figure legends. Significance is defined as a p-value less than 0.05, as indicated by the statistical method reported for each experiment.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-SH3GL3 | Abcam | Cat#: ab184008 |
| Mouse monoclonal anti-SH3GL3 | Santa Cruz Biotech | Cat#: SC-376592 |
| Rabbit polyclonal anti-TIAM1 | Santa Cruz Biotech | Cat#: SC-872 |
| Mouse monoclonal anti-TIAM1 | Santa Cruz Biotech | Cat#: SC-393315 |
| Mouse monoclonal anti-Rac1 | Cytoskeleton Inc. | Cat#: ARC03 |
| Mouse monoclonal anti-FLAG | Sigma | Cat#: F-3165 |
| Rabbit polyclonal anti-His6 tag | Abcam | Cat#: ab9108 |
| Rabbit Polyclonal anti-Ki-67 | Abcam | Cat#: ab16667 |
| Mouse monoclonal anti-TROMA-1 | DSHB | Cat#: TROMA-1 |
| Rabbit polyclonal anti-dsRed | Takara | Cat#: 632496 |
| Mouse monoclonal anti-TIAM1 | FHCRC Shared Resource | 4E7b |
| Bacterial and Virus Strains | ||
| BL21 (DE3) Escherichia Coli | Thermo Fisher Scientific | Cat#: C601003 |
| Mach1 T1 Escherichia Coli | Thermo Fisher Scientific | Cat#: C8681201 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dako serum free block | Agilent | Cat#: X090930-2 |
| Acti-Stain 488 Phalloidin | Cytoskeleton Inc | Cat#: PHDG1-A |
| DAPI | Sigma | Cat#: D9542 |
| Malachite Green Oxalate Salt | Sigma | Cat#: M6880 |
| Prolong Diamond antifade reagent | Molecular Probes | Cat#: P36965 |
| Live Imaging Solution | Thermo Fisher Scientific | Cat#: A14291DJ |
| 0.25% Trypsin EDTA | Thermo Fisher Scientific | Cat#: 25200-056 |
| Opti-MEM Media | Thermo Fisher Scientific | Cat#: 31985062 |
| Leibovitz's L-15 Medium | Thermo Fisher Scientific | Cat#:11415064 |
| Collagen Type 1 | Thermo Fisher Scientific | Cat#: A1048301 |
| Matrigel | Corning | Cat#: 354234 |
| Fetal Bovine Serum | Omega Scientific | FB-01 |
| 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) | NOF America | Cat#: ME-6060 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | NOF America | Cat#: MC-6081 |
| 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS) | NOF America | Cat#: MS-8181LS |
| 1,2-dioleoyl-sn-glycero-3-phospho-(1'-myo-inositol-4',5'-bisphosphate) (PIP2) | Avanti Polar Lipids | Cat#:850155 |
| Recombinant EndoA1 BAR domain | Laboratory of Jihong Bai | BJP-B01 |
| Recombinant EndoA2 BAR domain | Laboratory of Jihong Bai | BJP-B595 |
| Recombinant EndoA3 BAR domain | Laboratory of Jihong Bai | BJP-B596 |
| Recombinant Full Length EndoA1 | Laboratory of Jihong Bai | BJP-B237 |
| Recombinant Full Length EndoA2 | Laboratory of Jihong Bai | BJP-R41 |
| Recombinant Full Length EndoA3 | Laboratory of Jihong Bai | BJP-R42 |
| Recombinant TIAM1 (His6-PDZ-DH) domain | This paper | BJP-674 |
| Recombinant PAK-PBD | This paper | BJP-A144 |
| Recombinant RAC G15A | This paper | BJP-R85 |
| DMEM | Thermo Fisher Scientific | Cat#: 11965092 |
| Critical Commercial Assays | ||
| Incucyte zoom system | Essen Bioscience | Cat#: 4647 |
| FM 1–43 Dye (N-(3-Triethylammoniumpropyl)-4-(4-(Dibutylamino) Styryl) Pyridinium Dibromide) | Thermo Fisher Scientific | Cat#: T3163 |
| FM 4–64 Dye (N-(3-Triethylammoniumpropyl)-4-(6-(4-(Diethylamino) Phenyl) Hexatrienyl) Pyridinium Dibromide) | Thermo Fisher Scientific | Cat#: T13320 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | Cat#: 13778030 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#: 11668027 |
| RAC/CDC 42 Inhibitor ML141 Inhibitor | Millipore Sigma | Cat#: 217708 |
| PTEN Inhibitor VO-OHpic | Sigma | Cat#: V8639 |
| PTEN Inhibitor bPV(HOpic) | Sigma | Cat#: SML0884 |
| Dynamin Inhibitors Toolbox | Abcam | Cat#: ab120468 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human colon adenocarcinoma cells: DLD1 | ATCC | Cat#: CCL-221 |
| Human colon adenocarcinoma cells: HCT116 | ATCC | Cat#: CCL-247 |
| Human colon adenocarcinoma cells: SW480 | ATCC | Cat#: CCL-228 |
| MLV-based retroviral packaging cell line: PT67 | ATCC | Cat#: CRL-12284 |
| Human Embryonic Kidney Cells | Sigma | Cat#: 85120602 |
| Experimental Models: Organisms/Strains | ||
| D. rerio: WT AB | Laboratory of Cecilia Moens | ZFIN: ZDB-GENO-960809-7 |
| M. musculus: NU/NU | Charles River | Crl:NU-Foxn1nu |
| Oligonucleotides | ||
| 3' for EndoA2 BAR in A2 BAR-A3SH3 chimera | ATGTTCACAGGCTTTGGCATGAACTCCCGCTTGGG | |
| 5' for EndoA2 BAR in A2 BAR-A3SH3 chimera | ATGCCAAAGCCTGTGAACA | |
| 3' for EndoA3 BAR in A3 BAR-A2 SH3 chimera | GGCTCCCGGGGCCGGGGCTTGAAcTCTCGCTTGGGG | |
| 5' for EndoA3 BAR in A3 BAR-A2 SH3 chimera | AAGCCCCGGCCCCGG | |
| 5' for Lifeact EGFP in pBABE Hygro | CTCCTTCTCTAGGCGCCGGCCGGTACCATGGGCGTGGCCGACCTGATCAAGAAGTTC | |
| 3' for Lifeact EGFP in pBABE Hygro | GGGTCGACCACTGTGCTGGCGAATTCTTACTTGTACAGCTCGTCCATGCCG | |
| 5' for EndoA3ΔN truncation | CTCTAGGCGCCGGCCGGATCCATGATAAGTGGTGCCGAAGGAACG | |
| 5' for K171N mutant of EndoA3 | CGCCTGGATTATGATTATAACAAGCGGCGGGTAGGTAAG | |
| 3' for K171N mutant of EndoA3 | CTTACCTACCCGCCGCTTGTTATAATCATAATCCAGGCG | |
| 5' for R174L of EndoA3 | TATGATTATAAAAAGCGGCTGGTAGGTAAGATCCCCGAG | |
| 3' for R174L of EndoA3 | CTCGGGGATCTTACCTACCAGCCGCTTTTTATAATCATA | |
| Recombinant DNA | ||
| Plasmid: mEndoA3-mCherry in pBABE Puro | Laboratory of Jihong Bai | BJP-R82.8 |
| Plasmid: mEndoA3ΔN-mCherry in pBABE Puro | Laboratory of Jihong Bai | BJP-R81.1 |
| Plasmid: Chimera mEndoA3 N-BAR::mEndoA2 SH3::3xFlag in pBABE PURO | Laboratory of Jihong Bai | BJP-R73 |
| Plasmid: Chimera mEndoA2 N-BAR::mEndoA3 SH3::3xFlag in pBABE PURO | Laboratory of Jihong Bai | BJP-R72 |
| Plasmid: mEndoA1::3xFlag in pBABE Puro | Laboratory of Jihong Bai | BJP-R55 |
| Plasmid: mEndoA2::3xFlag in pBABE Puro | Laboratory of Jihong Bai | BJP-R56 |
| Software and Algorithms | ||
| FIJI | NIH | https://fiji.sc |
| FlowJo | Tree Star | www.flowjo.com |
| Tissue Fax | Tissuegnostics | www.tissuegnostics.com/ |
| Zen | Zeiss | www.zeiss.com |
| Fluoview (FV10-ASW) | Olympus Corporation | http://www.olympusamerica.com/ |
| Metamorph Microscopy and Image Analysis Software | Molecular Devices | www.moleculardevices.com |
| Other | ||
| Human Tissue Microarray | US Biomax | Cat#: BC05118C |
Highlights.
EndoA3 promotes proliferation and migration of cells using competitive mechanisms.
EndoA3-mediated endocytosis is required for proliferation, but not for cell migration.
EndoA3 stimulates Rac1 GTPase activity by directly binding the Rac1 GEF TIAM1.
EndoA3-Rac1 activation increases membrane protrusion dynamics and cell migration.
Acknowledgments
We thank Yan Liu, Ming Yu, and Fred Hutchinson Cancer Research Center Shared Resources for technical assistance. We thank Susan Parkhurst for comments on the manuscript. We thank Suzanne Hoppins and the Hoppins lab at University of Washington for advice on the Malachite Green Assay. This work was supported by grants from the National Institutes of Health (R01-NS085214 to 46 JB, R01-CA057138 to RE, K99-CA190836 to MR, CPRIT 50C0579903, Welch 52c1049001 to MCS, and the FHCRC/UW Cancer Consortium Cancer Center Support Grant P30-CA015704). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. KP is supported by a Dual Mentor Fellowship from an NIH Interdisciplinary Training in Cancer Research Training Grant (T32-CA080416), and an American Heart Association Postdoctoral Fellowship (14POST18230006).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
KP, JB, RE, MR, and MC designed the project. KP, TH, AS carried out biochemical experiments and data analysis. KP conducted all cell culture experiments. KP, MR, CM designed fish xenotransplantation, MR and KP performed fish xenotransplantation experiments, and SA conducted the analysis. SA performed western blot analysis of EndoA3 in tumors. HM and WG helped with mice experiments. KP, JB, RE, and MR interpreted data and co-wrote the manuscript. WG, MC, HM, CM provided critical feedback on the manuscript.
Declaration of Interests
No competing interests.
References
- Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2:e71. doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-Type KRAS Is Required for Panitumumab Efficacy in Patients With Metastatic Colorectal Cancer. J. Clin. Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- Anderson S, Poudel KR, Roh-Johnson M, Brabletz T, Yu M, Borenstein-Auerbach N, Grady WN, Bai J, Moens CB, Eisenman RN, et al. MYC-nick promotes cell migration by inducing fascin expression and Cdc42 activation. Proc. Natl. Acad. Sci. 2016;113:E5481–E5490. doi: 10.1073/pnas.1610994113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramaki Y, Ogawa K, Toh Y, Ito T, Akimitsu N, Hamamoto H, Sekimizu K, Matsusue K, Kono A, Iguchi H, et al. Direct interaction between metastasis-associated protein 1 and endophilin 3. FEBS Lett. 2005;579:3731–3736. doi: 10.1016/j.febslet.2005.05.069. [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Taylor SJ, Jordon KA, Aelst LV, Cerione RA. A Novel Regulator of p21-activated Kinases. J. Biol. Chem. 1998;273:23633–23636. doi: 10.1074/jbc.273.37.23633. [DOI] [PubMed] [Google Scholar]
- Bai J, Hu Z, Dittman JS, Pym ECG, Kaplan JM. Endophilin Functions as a Membrane-Bending Molecule and Is Delivered to Endocytic Zones by Exocytosis. Cell. 2010;143:430–441. doi: 10.1016/j.cell.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Chetrit N, Chetrit D, Russell R, Körner C, Mancini M, Abdul-Hai A, Itkin T, Carvalho S, Cohen-Dvashi H, Koestler WJ, et al. Synaptojanin 2 is a druggable mediator of metastasis and the gene is overexpressed and amplified in breast cancer. Sci. Signal. 2015;8:ra7. doi: 10.1126/scisignal.2005537. [DOI] [PubMed] [Google Scholar]
- Bendris N, Stearns CJS, Reis CR, Rodriguez-Canales J, Liu H, Witkiewicz AW, Schmid SL. Sorting nexin 9 negatively regulates invadopodia formation and function in cancer cells. J. Cell Sci. 2016;129:2804–2816. doi: 10.1242/jcs.188045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Curr. Opin. Neurobiol. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Bonner AE, Lemon WJ, Devereux TR, Lubet RA, You M. Molecular profiling of mouse lung tumors: association with tumor progression, lung development, and human lung adenocarcinomas. Oncogene. 2003;23:1166–1176. doi: 10.1038/sj.onc.1207234. [DOI] [PubMed] [Google Scholar]
- Boucrot E, Ferreira APA, Almeida-Souza L, Debard S, Vallis Y, Howard G, Bertot L, Sauvonnet N, McMahon HT. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature. 2015;517:460–465. doi: 10.1038/nature14067. [DOI] [PubMed] [Google Scholar]
- Boulakirba S, Macia E, Partisani M, Lacas-Gervais S, Brau F, Luton F, Franco M. Arf6 exchange factor EFA6 and endophilin directly interact at the plasma membrane to control clathrin-mediated endocytosis. Proc. Natl. Acad. Sci. 2014;111:9473–9478. doi: 10.1073/pnas.1401186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdisso JE, González Á, Arregui CO. PTP1B promotes focal complex maturation, lamellar persistence and directional migration. J Cell Sci. 2013;126:1820–1831. doi: 10.1242/jcs.118828. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestra G, Castagnoli L, Dente L, Minenkova O, Petrelli A, Migone N, Hoffmüller U, Schneider-Mergener J, Cesareni G. The SH3 Domains of Endophilin and Amphiphysin Bind to the Proline-rich Region of Synaptojanin 1 at Distinct Sites That Display an Unconventional Binding Specificity. J. Biol. Chem. 1999;274:32001–32007. doi: 10.1074/jbc.274.45.32001. [DOI] [PubMed] [Google Scholar]
- Chuang Y, Tran NL, Rusk N, Nakada M, Berens ME, Symons M. Role of Synaptojanin 2 in Glioma Cell Migration and Invasion. Cancer Res. 2004;64:8271–8275. doi: 10.1158/0008-5472.CAN-04-2097. [DOI] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Ngouenet C, Anderson S, Brabletz T, Eisenman RN. Stress-induced cleavage of Myc promotes cancer cell survival. Genes Dev. 2014;28:689–707. doi: 10.1101/gad.231894.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delic S, Lottmann N, Jetschke K, Reifenberger G, Riemenschneider MJ. Identification and functional validation of CDH11, PCSK6 and SH3GL3 as novel glioma invasion-associated candidate genes. Neuropathol. Appl. Neurobiol. 2012;38:201–212. doi: 10.1111/j.1365-2990.2011.01207.x. [DOI] [PubMed] [Google Scholar]
- DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Gou Y, Li Y, Liu Y, Bai J. Synaptojanin cooperates in vivo with endophilin through an unexpected mechanism. eLife. 2015:e05660. doi: 10.7554/eLife.05660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle AD, Kutys ML, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Micro-environmental control of cell migration - myosin IIA is required for efficient migration in fibrillar environments through control of cell adhesion dynamics. J Cell Sci. 2012;125:2244–2256. doi: 10.1242/jcs.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinga RD, Krueger EW, Weller SG, Zhang L, Cao H, McNiven MA. Increased Expression of the Large GTPase Dynamin 2 Potentiates Metastatic Migration and Invasion of Pancreatic Ductal Carcinoma. Oncogene. 2012;31:1228–1241. doi: 10.1038/onc.2011.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Farsad K, Camilli PD. Mechanisms of membrane deformation. Curr. Opin. Cell Biol. 2003;15:372–381. doi: 10.1016/s0955-0674(03)00073-5. [DOI] [PubMed] [Google Scholar]
- Frittoli E, Palamidessi A, Marighetti P, Confalonieri S, Bianchi F, Malinverno C, Mazzarol G, Viale G, Martin-Padura I, Garré M, et al. A RAB5/RAB4 recycling circuitry induces a proteolytic invasive program and promotes tumor dissemination. J Cell Biol. 2014;206:307–328. doi: 10.1083/jcb.201403127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A, Unger VM, De Camilli P. The BAR Domain Superfamily: Membrane-Molding Macromolecules. Cell. 2009;137:191–196. doi: 10.1016/j.cell.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H, Ringstad N, Löw P, Kjaerulff O, Gustafsson J, Wenk M, Di Paolo G, Nemoto Y, Crun J, Ellisman MH, et al. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron. 2000;27:301–312. doi: 10.1016/s0896-6273(00)00038-6. [DOI] [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJG, Evans PR, Langen R, McMahon HT. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of Activated GAPs and GEFs in Cell Lysates. In: B-M, editor. Enzymology. Academic Press; 2006. pp. 425–437. [DOI] [PubMed] [Google Scholar]
- Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. A malachite green colorimetric assay for protein phosphatase activity. Anal. Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Ghosh S, Maiti GP, Sabbir MG, Alam N, Sikdar N, Roy B, Roychoudhury S, Panda CK. SH3GL2 and CDKN2A/2B loci are independently altered in early dysplastic lesions of head and neck: correlation with HPV infection and tobacco habit. J. Pathol. 2009;217:408–419. doi: 10.1002/path.2464. [DOI] [PubMed] [Google Scholar]
- Giordani L, Iolascon A, Servedio V, Mazzocco K, Longo L, Tonini GP. Two regions of deletion in 9p22~p24 in neuroblastoma are frequently observed in favorable tumors. Cancer Genet. Cytogenet. 2002;135:42–47. doi: 10.1016/s0165-4608(01)00640-9. [DOI] [PubMed] [Google Scholar]
- Guichet A, Wucherpfennig T, Dudu V, Etter S, Wilsch-Bräuniger M, Hellwig A, González-Gaitán M, Huttner WB, Schmidt AA. Essential role of endophilin A in synaptic vesicle budding at the Drosophila neuromuscular junction. EMBO J. 2002;21:1661–1672. doi: 10.1093/emboj/21.7.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wang M, Jiang J, Xie C, Peng F, Li X, Tian R, Qin R. Balanced Tiam1-Rac1 and RhoA Drives Proliferation and Invasion of Pancreatic Cancer Cells. Mol. Cancer Res. 2013;11:230–239. doi: 10.1158/1541-7786.MCR-12-0632. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann P, Rizzoli SO, Betz WJ. Imaging Synaptic Vesicle Recycling by Staining and Destaining Vesicles with FM Dyes. Cold Spring Harb. Protoc. 2012;2012 doi: 10.1101/pdb.prot067603. pdb.prot067603. [DOI] [PubMed] [Google Scholar]
- Hu J, Mukhopadhyay A, Craig AWB. Transducer of Cdc42-dependent Actin Assembly Promotes Epidermal Growth Factor-induced Cell Motility and Invasiveness. J. Biol. Chem. 2011;286:2261–2272. doi: 10.1074/jbc.M110.157974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ye X, Guan J, Chen B, Li Q, Zheng X, Liu L, Wang S, Ding Y, Ding Y, et al. Tiam1 is associated with hepatocellular carcinoma metastasis. Int. J. Cancer. 2013;132:90–100. doi: 10.1002/ijc.27627. [DOI] [PubMed] [Google Scholar]
- Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, Kerr D, Aaltonen LA, Arango D, Kruhøffer M, et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009;15:7642–7651. doi: 10.1158/1078-0432.CCR-09-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaerulff O, Brodin L, Jung A. The Structure and Function of Endophilin Proteins. Cell Biochem. Biophys. 2011;60:137–154. doi: 10.1007/s12013-010-9137-5. [DOI] [PubMed] [Google Scholar]
- de Kreuk B-J, Hordijk PL. Control of Rho GTPase function by BAR-domains. Small GTPases. 2012;3:45–52. doi: 10.4161/sgtp.18960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetii L, Di Fiore PP. Behind the Scenes: Endo/Exocytosis in the Acquistion of Metastatic Traits. Cancer Res. 2017;77:1813–1817. doi: 10.1158/0008-5472.CAN-16-3403. [DOI] [PubMed] [Google Scholar]
- Lanzetti L, Di Fiore PP. Endocytosis and Cancer: an “Insider” Network with Dangerous Liaisons. Traffic. 2008;9:2011–2021. doi: 10.1111/j.1600-0854.2008.00816.x. [DOI] [PubMed] [Google Scholar]
- Li M, Yang C, Liu X, Yuan L, Zhang F, Wang M, Miao D, Gu X, Jiang S, Cui B, et al. EphA3 promotes malignant transformation of colorectal epithelial cells by upregulating oncogenic pathways. Cancer Lett. 2016;383:195–203. doi: 10.1016/j.canlet.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Lua BL, Low BC. Activation of EGF receptor endocytosis and ERK1/2 signaling by BPGAP1 requires direct interaction with EEN/endophilin II and a functional RhoGAP domain. J. Cell Sci. 2005;118:2707–2721. doi: 10.1242/jcs.02383. [DOI] [PubMed] [Google Scholar]
- Malecz N, McCabe PC, Spaargaren C, Qiu R-G, Chuang Y, Symons M. Synaptojanin 2, a novel Rac1 effector that regulates clathrin-mediated endocytosis. Curr. Biol. 2000;10:1383–1386. doi: 10.1016/s0960-9822(00)00778-8. [DOI] [PubMed] [Google Scholar]
- Malliri A, Van Der Kammen RA, Clark K, Van Der Valk M, Michiels F, Collard JG. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417:867–871. doi: 10.1038/nature00848. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, Mochizuki N. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 2006;25:2889–2897. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock P-J, Morais VA, Vilain S, Haddad D, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Mellman I, Yarden Y. Endocytosis and Cancer. Cold Spring Harb. Perspect. Biol. 2013;5:a016949. doi: 10.1101/cshperspect.a016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz HE, Kargl J, Busch SE, Kim K-H, Kurland BF, Abberbock SR, Randolph-Habecker J, Knoblaugh SE, Kolls JK, White MF, et al. Insulin receptor substrate-1 deficiency drives a proinflammatory phenotype in KRAS mutant lung adenocarcinoma. Proc. Natl. Acad. Sci. 2016;113:8795–8800. doi: 10.1073/pnas.1601989113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JR, MacDonald RB, Duggan A, Lenzi D, Standaert DG, Corwin JT, Corey DP. Lighting up the Senses: FM1-43 Loading of Sensory Cells through Nonselective Ion Channels. J. Neurosci. 2003;23:4054–4065. doi: 10.1523/JNEUROSCI.23-10-04054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr. Opin. Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O’Toole E, Ferguson S, Cremona O, et al. Recruitment of Endophilin to Clathrin-Coated Pit Necks Is Required for Efficient Vesicle Uncoating after Fission. Neuron. 2011;72:587–601. doi: 10.1016/j.neuron.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minard ME, Kim L-S, Price JE, Gallick GE. The Role of the Guanine Nucleotide Exchange Factor Tiam1 in Cellular Migration, Invasion, Adhesion and Tumor Progression. Breast Cancer Res. Treat. 2004;84:21–32. doi: 10.1023/B:BREA.0000018421.31632.e6. [DOI] [PubMed] [Google Scholar]
- Minard ME, Ellis LM, Gallick GE. Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin. Exp. Metastasis. 2006;23:301–313. doi: 10.1007/s10585-006-9040-z. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-Catenin-Tcf Signaling in Colon Cancer by Mutations in β-Catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat. Rev. Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- Nguyen ST, Hasegawa S, Tsuda H, Tomioka H, Ushijima M, Noda M, Omura K, Miki Y. Identification of a predictive gene expression signature of cervical lymph node metastasis in oral squamous cell carcinoma. Cancer Sci. 2007;98:740–746. doi: 10.1111/j.1349-7006.2007.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterberg L, Levan K, Partheen K, Delle U, Olsson B, Sundfeldt K, Horvath G. Potential predictive markers of chemotherapy resistance in stage III ovarian serous carcinomas. BMC Cancer. 2009;9:368. doi: 10.1186/1471-2407-9-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. The endophilin–CIN85–Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. 2002;416:187–190. doi: 10.1038/416187a. [DOI] [PubMed] [Google Scholar]
- Poudel KR, Dong Y, Yu H, Su A, Ho T, Liu Y, Schulten K, Bai J. A time-course of orchestrated endophilin action in sensing, bending, and stabilizing curved membranes. Mol. Biol. Cell mbc. 2016 doi: 10.1091/mbc.E16-04-0264. E16-04-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA. Dynamin 2 Potentiates Invasive Migration of Pancreatic Tumor Cells through Stabilization of the Rac1 GEF Vav1. Dev. Cell. 2013;24:573–585. doi: 10.1016/j.devcel.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard H-F, Simunovic M, Lemière J, Boucrot E, Garcia-Castillo MD, Arumugam S, Chambon V, Lamaze C, Wunder C, Kenworthy AK, et al. Endophilin-A2 functions in membrane scission in clathrin-independent endocytosis. Nature. 2015;517:493–496. doi: 10.1038/nature14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. Lifeact: a versatile marker to visualize F-actin. Nat. Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikhy R, Kumar V, Mittal R, Krishnan KS. Endophilin is critically required for synapse formation and function in Drosophila melanogaster. J. Neurosci. Off. J. Soc. Neurosci. 2002;22:7478–7484. doi: 10.1523/JNEUROSCI.22-17-07478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Ringstad N, Gad H, Löw P, Di Paolo G, Brodin L, Shupliakov O, De Camilli P. Endophilin/SH3p4 Is Required for the Transition from Early to Late Stages in Clathrin-Mediated Synaptic Vesicle Endocytosis. Neuron. 1999;24:143–154. doi: 10.1016/s0896-6273(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Roh-Johnson M, Shah AN, Stonick JA, Poudel KR, Kargl J, Yang GH, di Martino J, Hernandez RE, Gast CE, Zarour LR, et al. Macrophage-Dependent Cytoplasmic Transfer during Melanoma Invasion In Vivo. Dev. Cell. 2017;43:549–562.e6. doi: 10.1016/j.devcel.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland Y, Marighetti P, Malinverno C, Confalonieri S, Luise C, Ducano N, Palamidessi A, Bisi S, Kajiho H, Troglio F, et al. The CDC42-Interacting Protein 4 Controls Epithelial Cell Cohesion and Tumor Dissemination. Dev. Cell. 2014;30:553–568. doi: 10.1016/j.devcel.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001;23:291–299. [PubMed] [Google Scholar]
- Schmitz AA, Govek E-E, Böttner B, Van Aelst L. Rho GTPases: signaling, migration, and invasion. Exp. Cell Res. 2000;261:1–12. doi: 10.1006/excr.2000.5049. [DOI] [PubMed] [Google Scholar]
- Schuske KR, Richmond JE, Matthies DS, Davis WS, Runz S, Rube DA, van der Bliek AM, Jorgensen EM. Endophilin Is Required for Synaptic Vesicle Endocytosis by Localizing Synaptojanin. Neuron. 2003;40:749–762. doi: 10.1016/s0896-6273(03)00667-6. [DOI] [PubMed] [Google Scholar]
- Sidani M, Wessels D, Mouneimne G, Ghosh M, Goswami S, Sarmiento C, Wang W, Kuhl S, El-Sibai M, Backer JM, et al. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J. Cell Biol. 2007;179:777–791. doi: 10.1083/jcb.200707009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Chunder N, Mukherjee N, Alam N, Roy A, Roychoudhury S, Panda CK. Frequent deletion and methylation in SH3GL2 and CDKN2A loci are associated with early-and late-onset breast carcinoma. Ann. Surg. Oncol. 2008;15:1070–1080. doi: 10.1245/s10434-007-9790-0. [DOI] [PubMed] [Google Scholar]
- Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002;416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- Soukup S-F, Kuenen S, Vanhauwaert R, Manetsberger J, Hernández-Díaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, et al. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron. 2016;92:829–844. doi: 10.1016/j.neuron.2016.09.037. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational Analysis of the APC/β-Catenin/Tcf Pathway in Colorectal Cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- Sporn MB. The war on cancer. The Lancet. 1996;347:1377–1381. doi: 10.1016/s0140-6736(96)91015-6. [DOI] [PubMed] [Google Scholar]
- Timmins NE, Nielsen LK. Generation of Multicellular Tumor Spheroids by the Hanging-Drop Method. SpringerLink. 2007:141–151. doi: 10.1007/978-1-59745-443-8_8. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen FN, Kain HE, Van Der Kammen RA, Michiels F, Kranenburg OW, Collard JG. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 1997;139:797–807. doi: 10.1083/jcb.139.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake DK, Rossman KL, Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000;408:682–688. doi: 10.1038/35047014. [DOI] [PubMed] [Google Scholar]
- Wu X, Gan B, Yoo Y, Guan J-L. FAK-Mediated Src Phosphorylation of Endophilin A2 Inhibits Endocytosis of MT1-MMP and Promotes ECM Degradation. Dev. Cell. 2005;9:185–196. doi: 10.1016/j.devcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Xu K, Rajagopal S, Klebba I, Dong S, Ji Y, Liu J, Kuperwasser C, Garlick JA, Naber SP, Buchsbaum RJ. The role of fibroblast Tiam1 in tumor cell invasion and metastasis. Oncogene. 2010;29:6533–6542. doi: 10.1038/onc.2010.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam JWP, Jin D-Y, So CW, Chan LC. Identification and characterization of EBP, a novel EEN binding protein that inhibits Ras signaling and is recruited into the nucleus by the MLL-EEN fusion protein. Blood. 2004;103:1445–1453. doi: 10.1182/blood-2003-07-2452. [DOI] [PubMed] [Google Scholar]
- Zhao H, Pykäläinen A, Lappalainen P. I-BAR domain proteins: linking actin and plasma membrane dynamics. Curr. Opin. Cell Biol. 2011;23:14–21. doi: 10.1016/j.ceb.2010.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.