

Abstract
The dynamic interchange between monomeric globular actin (G-actin) and polymeric filamentous actin filaments (F-actin) is fundamental and essential to many cellular processes including cytokinesis and maintenance of genomic stability. Here we report that the long non-coding RNA LNC CRYBG3 directly binds G-actin to inhibit its polymerization and formation of contractile rings, resulting in M-Phase cell arrest. Knockdown of LNC CRYBG3 in tumor cells enhanced their malignant phenotypes. Nucleotide sequence 228–237 of the full-length LNC CRYBG3 and the ser14 domain of β-actin are essential for their interaction, and mutation of either of these sites abrogated binding of LNC CRYBG3 to G-actin. Binding of LNC CRYBG3 to G-actin blocked nuclear localization of MAL, which consequently kept serum response factor (SRF) away from the promoter region of several immediate early genes, including JUNB and Arp3, which are necessary for cellular proliferation, tumor growth, adhesion, movement, and metastasis. These findings reveal a novel lncRNA-actin-MAL-SRF pathway and highlight LNC CRYBG3 as a means to block cytokinesis and treat cancer by targeting the actin cytoskeleton.
INTRODUCTION
The dynamic actin cytoskeleton in eukaryotic cells plays multiple roles in regulating cellular morphology, motility, and vesicle trafficking by exerting mechanical forces that alter the shape of the plasma membrane (1,2). In vertebrates, three main groups of actin isoforms, α-, β-, and γ-actin, have been identified. The α-actin, mostly found in muscle, is a major constituent of the contractile apparatus. The β- and γ- actins coexist in most cell types as components of cytoskeleton and mediators of internal cell motility. It is widely recognized that the diverse range of structures formed by actin enable it to fulfill a number of specialized cellular functions including cell division, cell mobility, vesicle and organelle movement, embryogenesis, wound healing and tumor invasiveness, among others(3). Cell division is normally accomplished by separating a parent cell into two daughter cells through cytokinesis that involves a contractile ring composed of actin, myosin, and α-actinin (4). Actin is actively formed in the contractile ring with the participation of Arp3, formin Cdc12, profilin, and WASp, along with preformed microfilaments. Once the ring has been constructed, the structure is maintained by continual assembly and disassembly by the Arp2/3 complex and formins, which is key to one of the crucial steps of cytokinesis (5). In multicellular organisms where tissue specialization is critical for maintenance complex cellular function, e.g. cellular adhesion and motility in normal epithelial cells and cellular invasion and metastasis in cancer cells. These cellular processes require actin cytoskeleton as well as cadherins that act as extracellular elements in both normal and cancer cells (6) (7). As such, it is essential to clarify the regulatory process and molecular mechanisms underlying the remodeling of actin cytoskeleton.
The dynamic process of actin polymerization and depolymerization is regulated by a group of actin-binding proteins (ABPs) (8) including profilins (9), β-thymosins (10), Wiskott-Aldrich syndrome protein homology domain 2 (WH2)-containing proteins (10) (11), actin depolymerizing factor (ADF)/cofilins (collectively referred to cofilins) (12), twinfilins (13), cyclase-associated proteins (CAPs) (14) and Rho GTPases (15). Actin dynamics induce MAL nuclear accumulation and affect the activity of serum response factor (SRF), which activates numerous downstream genes (16). Furthermore, many mRNAs or non-coding RNAs (ncRNAs) can bind to actin (17) (18), but none of them is known to affect the structure of actin.
In recent years, long non-coding RNAs (lncRNAs), defined as non-coding RNA molecules greater than 200 nucleotides in length, have been shown to modulate cancer progression (19,20), as well as cellular proliferation (21) through targeting cytoskeleton structures. For example, disruption of TUG1 abolished the interaction of EZH2 with α-actin and accelerated depolymerization of F-actin (22). Here, we performed both in vitro and in vivo studies and found that a lncRNA, LNC CRYBG3, directly binds to globular actin (G-actin) to inhibit its polymerization and the formation of contractile rings. Consequently, cells are blocked in M phase, which results in a large number of binucleated cells and cell death.
MATERIALS AND METHODS
RNA quantitative real-time polymerase chain reaction (qRT-PCR), Immunofluorescence, Western blot and cell cycle assays were performed as mentioned previously (23). The details were seen as SI Materials and Methods.
Cell culture
A panel of lung cancer cell (A549, NCI-H1299, Calu-1, HCC827), immortalized human bronchial epithelial cell (Beas-2B) and immortalized Human embryonic lung fibroblast (MRC5) were used in this study. All cell lines were routinely cultured, maintained and used within 10–20 passages according to the requirements. All of these cell lines were purchase by the American Type Culture Collection. All the cell lines were authenticated using short tandem repeat (STR) profiling and LDH, G6PD, MDH,PEP-B isozyme detection by Cell Bank of Typical Culture Preservation Committee of Chinese Academy of Sciences. Cell lines were routinely tested mycoplasma free mycoplasma detection kit (Beyotime, Guangzhou, China). A549 and Calu-1is a K-ras mutant but EGFR wild type cell line. HCC827 is a K-ras wild type but EGFR mutant cell line. H1299 is K-ras and EGFR wild type NSCLC cell line.
Gene silencing, overexpression, and reporter plasmids
LNC CRYBG3 shRNA lenti-virus particles (Sangon, Shanghai, China) were transfected into cells and subsequently screened with medium contains 2 μg/mL puromycin (Invitrogen) and double-checked with RT-PCR. LNC CRYBG3 adenovirus particles (Sangon, Shanghai, China) and pcDNA 3.1-LNC CRYBG3 recombinant plasmid were used for LNC CRYBG3 overexpression. The SRF-lux reporter and MLV-lacZ reporter were used to detect SRF activity by luciferase assay. Luciferase assay were performed as described previously (24). A549 cells on a 12-well plate were co-transfected with 300 ng DNA. Luciferase activity was measured 48 h later using the Luciferase Reporter Assay System (Promega, WI, USA) with Multiscan Spectrum (BioTek Synergy 2, USA).
RNA pull down and immunoprecipitation analysis
LNC CRYBG3 and its antisense transcript were synthesized and constructed into 18T-pMD plasmid. LNC CRYBG3 RNA obtained with RNA in vitro transcription system containing biotin-labeled dUTP. Cells were harvested and re-suspended in 1 mL ice-cold RIP buffer containing RNase and protease inhibitors. The cell pellet was sheared and centrifuged at 15,000×g for 15 min at 4°C to clear the cell lysate. Folded RNA was mixed with cell lysate and incubated at room temperature for 2 h. 60 μL washed Streptavidin agarose beads were added to each binding reaction and further incubated at RT for 1 h. Beads were boiled in Laemmli loading buffer. Samples were performed SDS-PAGE and Western Blot assay. For RNA immunoprecipitation analysis, A549 cells were harvested by scraping cells in IP lysis buffer. β-actin anti-body (Abcam, NY, USA) was added to the cell extract and incubated overnight at 4°C. Streptavidin-coated magnetic beads were then added and incubated for 2h at 16°C before being resuspended in 1 mL TRIzol. The isolated RNA analyzed by qRT-PCR.
In vitro actin polymerization
In vitro actin polymerization was conducted with an Actin Polymerization Biochem Kit™ (Cytoskeleton, Inc. DENVER, U.S.A). General Actin Buffer with ATP was prepared as G-buffer. The pyrene actin was dissolved in G-buffer at 0.4 mg/mL (G-actin stock) and leave on ice for 1 h to depolymerize actin oligomers. Actin oligomers were centrifuged at 14,000 rpm at 4°C for 30 min. The supernatant was removed to 96 well plate 200 μL per well. G-buffer was added in wells as baseline controls and G-actin stock as positive controls. Actin Polymerization Buffer was added in the wells to start the reaction. Kinetic for over 120 cycles, 60 s interval time was set on Multiscan Spectrum (BioTek Synergy 2, U.S.A), fluorescence wavelengths was Ex. 360 nm and Em. 420 nm.
Animal experiment
1.6×106 A549-LV-NC cells or A549-LV- sh LNC CRYBG3 (sh1+sh2) cells were injected subcutaneously into the flanks of 5-week-old NOD/SCID mice (n=10). When the tumors volume reached 100 mm3, half of them were subjected to 1×107 LNC CRYBG3 adenovirus particles or equal amount of NC adenovirus particles. Tumor volumes were measured every two days for one month using a caliper. All tumor volume data were normalized to the ones obtained just before injection. All mice were maintained in the SPF Animal Laboratory, Soochow University. All animal studies were reviewed and approved by the Soochow University Institutional Animal Care and Use Committee.
Hematoxylin and eosin staining
Mouse tumor tissues were fixed with 4% formalin (V/V) and embedded with paraffin. Then tissues were spliced into 6 μm sections. Sections were deparaffinized with xylene and submerged into EDTA antigenic retrieval buffer for antigenic retrieval. At last, the sections were stained with hematoxylin-eosin. Digital images of organs were acquired by Nanozoomer (Hamamatsu Photonics).
Statistics
All experiments were independently repeated at least three times and all data were presented as the mean ± standard error. Student’s t-tests were used for statistic analysis. Probability (p) values less than 0.05 were considered to be statistically significant.
RESULTS
LNC CRYBG3 inhibits cytokinesis and resulting in cell death.
Using deep sequencing techniques, we identified a novel radiation- induced lncRNA, LNC CRYBG3, that is involved in the regulation of cell cycle progression. We synchronized cells at G0/G1 phase with thymidine and subsequently released them into S or G2/M phase (Fig. S1A). We found that the expression levels of LNC CRYBG3 correlated with the different cell cycle stages, the highest being in G0/G1 phase and the lowest in M phase (Fig. 1A). The low expression of LNC CRYBG3 in M phase was further confirmed with cells synchronized with nocodazole. At the same time, the F/G actin ratio was lowest in G0/G1 phase and the highest in G2/M and M phase (Fig. 1B & 1C). When LNC CRYBG3 was overexpressed in cells, the cell cycle was blocked in G2/M phase (Fig. 1D & S1B, S1C). Small hairpin RNAs used to knock down LNC CRYBG3 partially rescued the LNC CRYBG3- induced G2/M arrest (Fig. 1D). Using a live cell imaging system, we further confirmed the incomplete cytoplasmic division induced by LNC CRYBG3. As shown in Fig. 1E, A549 cells transfected with LNC CRYBG3 completed karyomitosis step but failed to undergo cytoplasmic division. Cell movement, deformation and proliferation were significantly reduced when A549 cells overexpressed LNC CRYBG3 (Supplementary video 1 and video 2). Fluorescent in situ hybridization (FISH) assay (Fig. S2) demonstrated that the overexpression of LNC CRYBG3 led to the production of binucleated cells, as the proportion of binucleated cells in all the A549, Calu-1, H1299, and MRC5 cells dramatically increased. The number of binucleated cells was alleviated by the knockdown of LNC CRYBG3 (Fig. 1F & 1G). These findings indicate that the overexpression of LNC CRYBG3 blocks cytokinesis. Overexpression of LNC CRYBG3 lead to obvious apoptosis (Fig. 1H). The proliferation curve showed that the proliferation of cells was significantly slowed down when cells were overexpressed with LNC CRYBG3 (Fig. 1I). Our results demonstrate that LNC CRYBG3 is a regulator of cell cycle process and its expression correlates with the cell death.
Figure 1. Overexpressed LNC CRYBG3 inhibits cytokinesis.
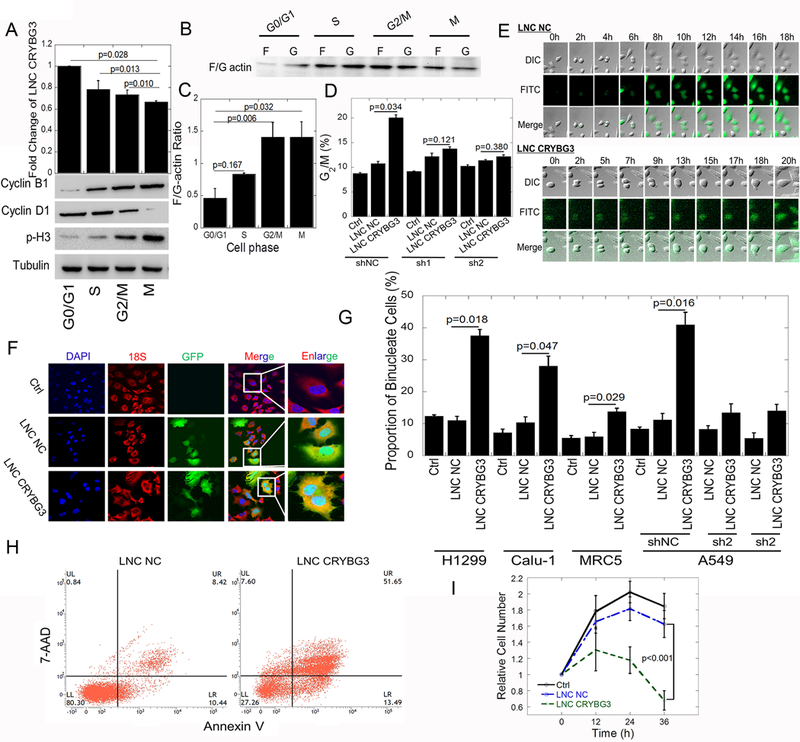
(A) LNC CRYBG3 expression in G0/G1, S, G2/M and M phase, detected by qRT-PCR (mean ± SE, n=3). Nocodazole and thymine were used for cell cycle synchronization. Prior to administration of 100 ng/mL nocodazole for 20h (M phase) or 2 mM thymine for 24h (G0/G1 phase) and release for 4h (S phase) and 15h (G2/M phase). The protein makers of different phase of cell cycle were confirmed by western blot. (B&C) F/G actin ratio in G0/G1, S, G2/M and M phase cells. (D) Cell cycle distribution after overexpression of LNC CRYBG3, assayed by flow cytometry. LNC NC, lncRNA Negative control. Sh1 or Sh2, LNC CRYBG3 short hairpin RNAs. (E) Confocal microscopy was used to observe cell mitosis after overexpressed with LNC NC or LNC CRYBG3. (F&G) The number of binucleated cells was counted. DAPI shows the nuclear; 18S rRNA probe shows cytoplasm; GFP shows the LNC CRYBG3 positive cells. Proportion of binucleated cells (mean ± SE, n=3) in Panel G. More than 1000 cells were counted in each group. (H) The apoptosis rate of cells when overexpressed with negative control or LNC CRYBG3. (I) Determination of cell proliferation curve when cells were overexpressed with negative control or LNC CRYBG3. Data represent the mean ± SE of three independent experiments.
LNC CRYBG3 suppresses tumor growth, as well as cell migration and invasion.
Since LNC CRYBG3 directly interacts with G-actin to inhibit the assembly of F-actin and arrests cells at the M phase, we hypothesized that LNC CRYBG3 may have an antitumor property. Using a set of tumor-bearing nude mice with subcutaneously inoculated human lung carcinoma A549 cells or A549 cells stably expressing LNC CRYBG3 shRNAs, we examined the role of LNC CRYBG3 in tumorigenesis. As shown in Fig. 2A-2B, A549 cells with knock down LNC CRYBG3 displayed markedly increased ability to form tumors compared to parental A549 cells. The tumors formed by A549 cells expressing sh LNC CRYBG3 grew more rapidly. Furthermore, when tumors of similar volume were overexpressed with LNC CRYBG3, tumor growth was significantly suppressed (Fig. 2B) and the tumor size and weight were significantly smaller than sham control (Fig. 2C-2E). In contrast, the expression of small hairpin RNAs removed the tumor suppression ability of LNC CRYBG3. Representative H&E staining of tumor tissues generated from transplanted LNC CRYBG3 knockdown A549 cell line showed a well differentiated histopathology compared with the sham control (Fig. 2F), indicating that LNC CRYBG3 induces tissue differentiation. Another consequence of the deconstructed actin cytoskeleton might be the limitation of cell movement. Therefore, we measured the metastasis and invasion of the in vivo and in vitro. As shown in Fig. 2G, after overexpression of LNC CRYBG3, the metastasis of A549 cells in the lungs was significantly reduced. As shown in Fig. 2H-2K, the mobility of A549 tumor cells was dramatically inhibited by LNC CRYBG3 regardless of the presence or absence of Matrigel and shRNAs partially released the inhibition. These results indicate a definitive role of LNC CRYBG3 in inhibiting tumor cell invasion and metastasis.
Figure 2. LNC CRYBG3 prevents tumor growth and reduces tumor cell metastasis and invasion.
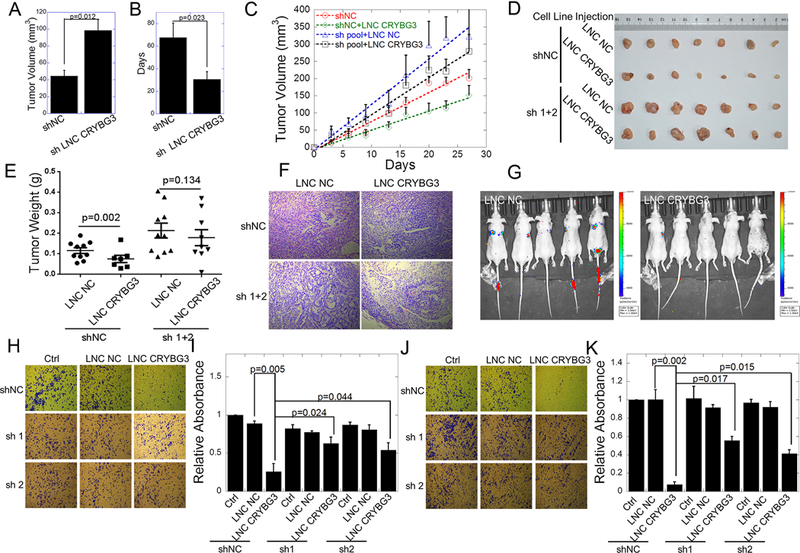
(A) The tumor volume was detected for each group on 30th day. (B) Calculate the theoretically time requirement for the tumor to grow to 100 mm3. The tumor formation frequency was calculated as described in methods (n = 10 for each group). (C) Xenografted tumor grows curve after overexpressed LNC CRYBG3 or negative control. The injection was begun on the time when xenografted tumor get 100 mm3 twice a week for 1 month (n=10 for each group). (D) The xenografed tumors obtained after 8 times injection (n=10 for each group). (E) The weight of xenografed tumors from different groups was compared (n=10 for each group). (F) Representative H&E staining of transplanted lung tumor tissues are shown. (G) In vivo cells metastasis experiment (n=5 for each group). (H&I) Up-regulation of LNC CRYBG3 reduced the metastatic potential of A549 cells. Images showing trans-well invasion of the A549 cells. The three images in the upper panel show invasion of A549 cells in control or overexpressed with negative control or LNC CRYBG3. The images in the bottom two panels show the short hairpin of LNC CRYBG3 RNA expressed cells overexpressed with LNC NC or LNC CRYBG3. (J&K) Up-regulation of LNC CRYBG3 influences the invasive property of A549 cells. Images showing matrigel invasion of the A549 cells. Data represent the mean ± SE of three independent biological experiments.
LNC CRYBG3 regulates cytoskeleton formation via interaction with G-actin.
Since LNC CRYBG3 overexpression blocks cytokinesis but not karyomitosis, it is possible that LNC CRYBG3 interferes with the formation of contractile ring. The results of immunofluorescent assay showed that LNC CRYBG3 overexpression caused contractile ring deficiency in metamitosis cells (Fig. 3A). Immunofluorescent assay also demonstrated that LNC CRYBG3 overexpression didn’t disturb the formation of tubulin but microfilament, i.e. F-actin (Fig. 3B & 3C). Western blot experiments showed that over express LNC CRYBG3 did not affect the expression of actin in cells (Fig. S3A-S3C). Since the assembly of F-actin by G-actin is a dynamic process and is the main component of contractile ring, we detected its formation with Western blot and quantified the decreased ratio of F-actin to G-actin in A549 cells overexpressed with LNC CRYBG3 (Fig. 3D & 3E). Using Jasplakinolide, which promotes F-actin assembly, we observed few actin filaments when A549 cells were overexpressed LNC CRYBG3 before the treatment with Jasplakinolide (Fig. 3F). However, if we treated cells with Jasplakinolide before overexpressing LNC CRYBG3, actin filaments were observed. To further confirm the role of LNC CRYBG3 in preventing G-actin polymerization into F-actin, small hairpin RNA was used to knock down LNC CRYBG3 expression. This resulted in promoting the actin polymerization and stable cytoskeletal structure (Fig. S3D). Frozen sections and immunofluorescent assay of tumors showed that F-actin content decreased when tumors were overexpressed with LNC CRYBG3 (Fig. 3G). These findings demonstrate that LNC CRYBG3 suppressed the formation of F-actin likely by inhibiting its assembly instead of promoting its disassembly.
Figure 3. LNC CRYBG3 regulates cytoskeleton formation.
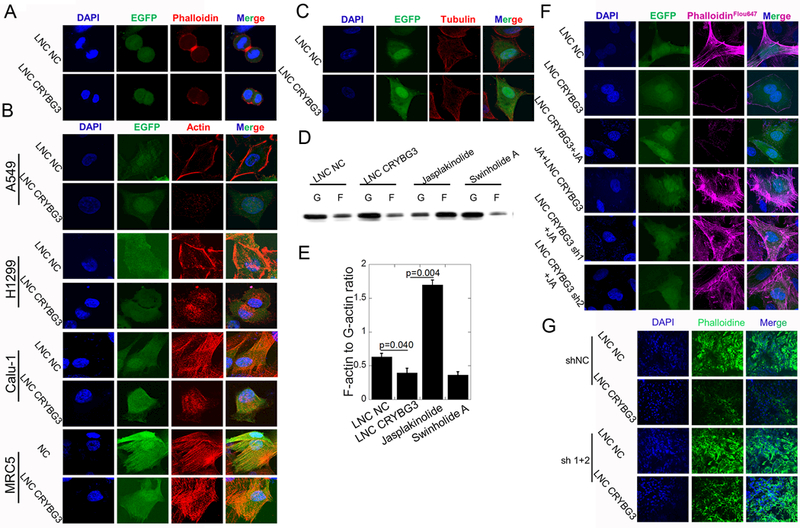
(A) Contractile ring dyeing in mitotic telophase. (B&C) Immunofluorescence analysis determined the microfilaments and microtubules morphology after cells overexpressed LNC CRYBG3. GFP shows the LNC CRYBG3 positive cells. (D&E) F-actin and G-actin protein levels in the A549 cells overexpressed with LNC CRYBG3 or negative control. (F) Immunofluorescence analysis determined the microfilaments morphology. (G) Frozen sections of tumors were stained with Alexa Fluor® 647 phalloidine to indicate the microfilaments and nuclear were stained with DAPI. Data represent the mean ± SE of three independent biological experiments.
LNC CRYBG3 directly binds to G-actin.
Bioinformatic assay showed that G-actin is one of the candidate targets of LNC CRYBG3. Therefore, we performed RNA pull-down and RNA immunoprecipitation assay and found that there was an interaction between β-actin and LNC CRYBG3 (Fig. 4A & 4B). Using a cell-free system, we monitored the dynamics of the interaction of LNC CRYBG3 and actin. As shown in Fig. 4C, LNC CRYBG3 inhibited the assembly of G-actin. Based on the predicted secondary structure of LNC CRYBG3, we synthesized three truncated fragments of LNC CRYBG3 (Fig. 4D) and conducted the same assay. None of the three individual clusters could inhibit the assembly of G-actin (Fig. 4E). We hypothesized that the length of the lncRNA was crucial for its functions. We then introduced mutations into the sequence of LNC CRYBG3 to obtain structural alterations in each cluster (Fig. 4F & 4G). With the same assay, we found that the structural cluster at loci 228–237 of its nucleotide sequence is crucial for LNC CRYBG3 to maintain its functions in inhibiting the assembly of G-actin (Fig. 4H). Finally, we designed different mutations to the functional domains of actin. Immunofluorescent assay found that when the 14th Serine of β-actin was mutated to cysteine, the LNC CRYBG3 lost its ability to inhibit F-actin formation. Thus, Ser14 of β-actin is critical for its binding to LNC CRYBG3 (Fig. 4I). These results demonstrate the direct interaction of LNC CRYBG3 with β-actin.
Figure 4. LNC CRYBG3 directly interacts with G-actin.

(A) RNA pull-down analysis determined the Actin protein-LNC CRYBG3 interaction in A549 cells using 5’-biotin-linked RNAs. (B) Histogram of LNC CRYBG33 enrichment after RNA immunoprecipitation assays (RIP). Actin antibodies were used. (C) In vitro polymerisation of pyrene-labelled actin (0.1 μg/μL) was monitored using time-based fluorimetry. Additional components were: 20 μg/μL LNC CRYBG3 in vitro transcription product. Pyrene-labelled actin was monitored using time-based fluorimetry. (D) Diagram showing the truncated LNC CRYBG3. (E) In vitro polymerisation of pyrene-labelled actin treated with truncated LNC CRYBG3. (F&G) Diagram showing the mutant of LNC CRYBG3. (H) In vitro polymerisation of pyrene-labelled actin treated with mutant LNC CRYBG3. (I) Immunofluorescence analysis determined the microfilaments morphology after A549 cells were transfected with actin mutant plasmid and then overexpress LNC CRYBG3 or negative control. Data represent the mean ± SE of three independent biological experiments.
Binding of LNC CRYBG3 with G-actin blocks MAL nuclear accumulation and inhibits transcriptional activity of SRF.
MAL is known as a co-activator of SRF whose transcriptional activity responds to G-actin. Since LNC CRYBG3 inhibits the G-actin polymerization, we further tested the effects of LNC CRYBG3 on MAL subcellular localization and SRF activity. As shown in Fig. 5A, in negative control group, MAL evenly distributed in both cytoplasm and nucleus. LNC CRYBG3 overexpression inhibited nuclear accumulation of endogenous MAL, whereas Jasplakinolide efficiently induced nuclear accumulation of MAL. Swinholide A, which promotes F-actin depolymerization, induced accumulation of MAL in cytoplasm instead of nucleus. Luciferase assay showed that Jasplakinolide could enhance the activity of SRF, whereas Swinholide A and LNC CRYBG3 could reduce the activity of SRF (Fig. 5B). Swinholide A and LNC CRYBG3 reduced the expression of JUNB and Arp3 (Fig. 5C). The transplanted tumors were harvested and their RNAs were extracted for gene expression assay as above. The expressions of c-Fos and MLH1 were not significantly altered (p≤ 0.20 and p≤0.055, respectively when compare with parental cells) whereas those for JUNB and Arp3 were down-regulated significantly (p≤0.0005 and p≤0.0001, respectively in Fig. 5D). Taken together, these results suggest that the translocation of MAL in nucleus and the activation of SRF can be blocked by overexpression of LNC CRYBG3.
Figure 5. LNC CRYBG3 induces MAL nuclear accumulation and affects the activity of SRF.

(A) Immunofluorescence analysis determined MAL and SRF localization in cells. (B) Luciferase assay showed the SRF activity. (C) Genes expression shown by qRT-PCR. (D) Genes expression in xenografted tumors shown by qRT-PCR. Data represent the mean ± SE of three independent biological experiments.
DISCUSSION
In the present study, we demonstrated the function of a novel lncRNA, LNC CRYBG3, in regulating the formation of contractile ring by directly binding to G-actin and inhibiting the assembly of F-actin. We also found that the nucleotide sequence 228–237 LNC CRYBG3 and the ser14 of β-actin are essential to their direct binding. LNC CRYBG3 overexpression induced actin dynamic alteration and resulted in the accumulation of MAL in the cytoplasm and the inactivation of SRF necessary for the transcription of MLH1 and Arp3 genes under both in vivo and in vitro conditions. Overexpression of LNC CRYBG3 led to pathologic collapse of actin cytoskeleton and inhibited tumor growth, migration and invasion, providing a therapeutic application of LNC CRYBG3 for tumor treatment. The fragmentation of the microfilament caused by LNC CRYBG3 directly leads to the inability of the contractile ring formation. Eventually the cell failed to complete the cytoplasmic division and formed binuclear cells, which lead to cell death (Fig. 6). These conclusions prove that LNC CRYBG3 functioning as a new tumor suppressor gene. We overexpressed oncogenes, i.e. K-ras G12V and EGFR T790M/L858R in primary human cells and also found that LNC CRYBG3 content decreased significantly (Fig. S4A & S4B) .
Figure 6. Model for LNC CRYBG3-actin-MAL-SRF pathway.
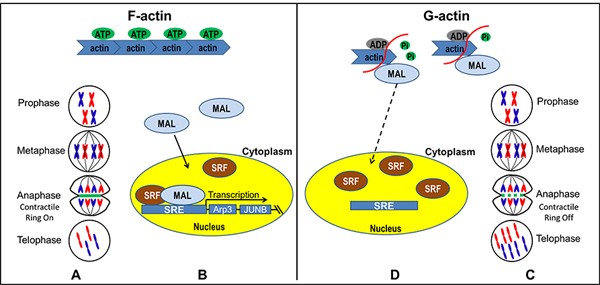
(A&D) As one of the most important components of contractile ring, the fragmentation of the microfilament caused by LNC CRYBG3 directly leads to the inability of the contractile ring formation. Eventually the cell failed to complete the cytoplasmic division and formed binuclear cells which leads cell death. (C) In LNC CRYBG3 overexpressed cells, MAL is predominantly cytoplasmic where it is sequestered by actin monomers. (B) In control group, an accumulation of F-actin and a commensurate decrease in the level of G-actin. As a consequence, MAL is no longer sequestered and is free to translocate to the nucleus where it associates with SRF and activates SRE-mediated gene expression.
It is well known that the cell cycle of a normal cell is precisely regulated by cyclins and cell division cycle kinases (25). Non-coding RNAs are important cell cycle modulators and levels of miR-138, miR-195, and miR-206 have been predicted to promote CCND1 expression, which regulates the progression from G1 phase to S phase of the cell cycle (26) (27). There is evidence that long non-coding RNA ALT1 controls the cell cycle progression via ACE2 and Cyclin D1 pathway (28). In the present study, we report that the lncRNA CRYBG3 regulates cell division and its expression is dynamically associated with cell cycle progression, being lowest in M phase and highest in G1 phase. Abnormal expression of LNC CRYBG3 results in cell cycle blockage in M phase due to the failure in contractile ring formation and absence of cytokinesis. These findings shed new light on the regulatory network in cell cycle process.
Actin cytoskeleton is one of the basic but essential subcellular organs in a cell and performs multiple functions in diverse cellular processes. As a target for drug design, cytoskeleton is closely related to the clinical treatment of cancer. In order to examine the causative role of cytoskeleton collapse in cancer development and progression, a number of studies have confirmed that the cytoskeleton is involved in both tumor initiation and development (29) (30). As one of the three main kinds of cytoskeletons, microfilaments play an important role in the growth, adhesion, migration, and metastasis of tumor cells, therefore, it can rationally be used as an anti-tumor target. Latrunculins A and Latrunculins B (macrolide compounds), inhibitors of the G-actin polymerization, can effectively reduce the formation of microfilaments so that they possess anti-cancer capabilities and inhibit cell metastasis and migration (31). In addition, marine macrolide toxins of the trisoxazole family, Mycalolide B, Kabiramide, and Jaspisamide A, target on actin with high affinity and specificity and thus exhibit potent filament-severing and monomer-sequestering activities that consequently result in cell death and suppression of cancer cell migration (32,33). Besides, some non-coding RNAs have also been reported to influence microfilaments functions. For instance, miR-30c, a biomarker of human breast cancer, regulates cell invasion by targeting the cytoskeleton genes encoding Twinfilin 1 (TWF1) and Vimentin (VIM) which regulate F-actin formation (30) and epithelial-to-mesenchymal transition (EMT). MicroRNA-375 regulates surfactant secretion via the reorganization of cytoskeleton (34). As such, it is likely that the regulation of actin dynamics and cell motility occurs at multiple levels. Here, we revealed that LNC CRYBG3 binds to β-actin Ser14 and inhibits G-actin polymerization, demonstrating for the first time that LNC CRYBG3 can inhibit tumor growth, metastasis, and invasion by using both in vitro and in vivo experiment models.
The binding modes of lncRNAs with target proteins are diverse. Guo et al. investigated the region of OLA1P2 responsible for the binding activity with phosphorylated STAT3. They designed various fragments or mutants of OLA1P2 and found that only the “T-G-T” ribonucleotides in the tail of OLA1P2 were significantly recovered by the phosphorylated STAT3 (Tyr705) protein (35). In our study, none of the three truncated fragments could inhibit the assembly of F-actin, but the 228–237 nucleotide mutant inhibited the G-actin polymerization. Thus, the spatial structures of the LNC CRYBG3 are crucial for its function. Meanwhile, Ser14 appears to be crutial for the binding of LNC CRYBG3 with G-actin. It is reported that mutant actin such as the S14C could increase the stability of F-actin (36). The introduction of S14C mutation into human β-actin results in the sustained conformation of F-actin by suppressing phosphate release (36) (37). The mechanism of this suppression remains unclear. In ATP-actin, Ser14 is involved in hydrogen bonding to the ATP gamma-phosphate (38). In contrast, in free ADP-actin, Ser14 adopts its alternative and preferred rotamer to interact with the nucleotide beta-phosphate (39). Cysteine has both a lower hydrogen bonding capacity and the opposite rotamer preference to serine (40). Therefore, it is likely that the Ser14 of β-actin is involved in the structural reorganization of β-actin after ATP hydrolysis. Since ser14 is one of the ATP binding sites on G-actin protein, the binding of LNC CRYBG3 with ser14 may block this ATP binding site, which leads to the fact that ADP-actin cannot be activated into ATP-actin. This may be main reason that G-actin can not polymerize and form F-actin filaments.
The serum response element (SRE) is found within the promoter region of the so-called “immediate early genes” whose expression is rapidly induced by serum. A complex of two transcription factors, SRF and TCF (ternary complex factor), binds directly to SRE to transcript downstream genes, such as c-Fos (41), MLH1 (42), JUNB (43), Arp3 (7). Substantial evidence supports a role for Rho signaling in the activation of SRF as a transcriptional regulator at the SRE (44). SRF activity responds to G-actin. MAL is a coactivator of SRF (16). Cytochalasin D and swinholide A, which do not promote actin polymerization, induce non-nuclear accumulation of expressed MAL. Jasplakinolide, which promotes the assembly of F-actin and activates SRF, efficiently induces the nuclear accumulation of MAL. G-actin, likely acting as a cofactor, binds with MAL and thereby occludes nuclear import signals or creates a nuclear export signal (16) to block the transportation MAL into the nucleus. Theoretically, in normal conditions or at the cell cycle phases of relatively low expression of LNC CRYBG3, such as M phase, G-actin polymerization leads to the accumulation of MAL in nucleus, subsequently activate SRF to transcript downstream genes including JUNB and Arps, and consequently promote cell proliferation, tumor growth, adhesion, movement, and metastasis (Fig. 6). However, at M phase or in case of abnormally overexpression of LNC CRYBG3, LNC CRYBG3 binds with G-actin and MAL, and consequently restrain the accumulation of MAL in nucleus, which results in the inactivation of SRF and its downstream genes. Consequently, the cells cannot form contractile ring to accomplish cell division and are blocked in M phase. These cells will become binucleated and ultimately die as shown in Figure 1H. Our findings might inspire a new strategy for tumor treatment.
In summary, LNC CRYBG3 regulates cell cycle process and its overexpression inhibits the assembly of microfilament by directly binding with G-actin and blocks cells in M phase. This aspect may be further exploited as a possible mean in cancer treatment based not only on its ability to induce incomplete cell division and cell death but also its suppression of tumor invasion and metastasis. Ultimately, its clinical efficiency will depend on how best to deliver the lncRNA into cancer cells efficiently and precisely to avoid side effects on normal tissues. Furthermore, in considering the rational selection of patients in targeting LNC CRYBG3 for treatment, the determination of LNC CRYBG3 expression status in patients may be necessary.
Supplementary Material
ACKNOWLEDGEMENTS
We thank G. Posern and Stefano Piccolo for advice and gift of reagents of SRF-lux reporter used for detecting SRF activity; Primal de Lanerolle for YFP Beta-Actin S14C and K268R; We sincere thanks Chuanyuan Li and Weinmin Gong for their valuable discussion and proof reading of the manuscript. This work was supported by the China National Natural Science Foundation awards (No. 81602794, 11335011, 81673151, 11405235); China Postdoctoral Science Foundation Project (2016M591904, 2017T100399); Natural Science Foundations of Jiangsu Province, China (BK20160334); Jiangsu Postdoctoral Science Foundation (1701176B) and the U.S. National Institutes of Health grant R01-ES 011804–10.
H. Pei, W. Hu and Z. Guo performed most of the experiments and data analyses. H. Chen, J. Ma and W. Mao helped the flow cytometry experiments. B. Li, A. Wang, J. Wan, J. Zhang, and J. Nie performed some of animal experiments. H. Pei and T.K. Hei drafted the manuscript. T.K. Hei and G. Zhou revised the manuscript, conceived the study and designed the experiments.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
REFERENCES
- 1.Pollard TD, Weihing RR. Actin and myosin and cell movement. CRC critical reviews in biochemistry 1974;2:1–65. [DOI] [PubMed] [Google Scholar]
- 2.Doherty GJ, McMahon HT. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Ann Rev Biophys 2008;37:65–95. [DOI] [PubMed] [Google Scholar]
- 3.Vindin H, Gunning P. Cytoskeletal tropomyosins: choreographers of actin filament functional diversity. J Muscle Res Cell M 2013;34:261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujiwara K, Porter ME, Pollard TD. Alpha-actinin localization in the cleavage furrow during cytokinesis. J Cell Biol 1978;79:268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelham RJ, Chang F. Actin dynamics in the contractile ring during cytokinesis in fission yeast. Nature 2002;419:82–6. [DOI] [PubMed] [Google Scholar]
- 6.Adams CL, Nelson WJ, Smith SJ. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J Cell Biol 1996;135:1899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kazazian K, Go C, Wu H, Brashavitskaya O, Xu R, Dennis JW, et al. Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res 2017;77:434–47. [DOI] [PubMed] [Google Scholar]
- 8.Makkonen M, Bertling E, Chebotareva NA, Baum J, Lappalainen P. Mammalian and Malaria Parasite Cyclase-associated Proteins Catalyze Nucleotide Exchange on G-actin through a Conserved Mechanism. J Biol Chem 2013;288:984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard TD, Cooper JA. Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry 1984;23:6631–41. [DOI] [PubMed] [Google Scholar]
- 10.Husson C, Cantrelle FX, Roblin P, Didry D, Le KHD, Perez J, et al. Multifunctionality of the beta-thymosin/WH2 module: G-actin sequestration, actin filament growth, nucleation, and severing. Ann Ny Acad Sci 2010;1194:44–52. [DOI] [PubMed] [Google Scholar]
- 11.Didry D, Cantrelle FX, Husson C, Roblin P, Moorthy AME, Perez J, et al. How a single residue in individual beta-thymosin/WH2 domains controls their functions in actin assembly. Embo J 2012;31:1000–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paavilainen VO, Merckel MC, Falck S, Ojala PJ, Pohl E, Wilmanns M, et al. Structural conservation between the actin monomer-binding sites of twinfilin and actin-depolymerizing factor (ADF)/Cofilin. J Biol Chem 2002;277:43089–95. [DOI] [PubMed] [Google Scholar]
- 13.Poukkula M, Kremneva E, Serlachius M, Lappalainen P. Actin-depolymerizing factor homology domain: a conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton 2011;68:471–90. [DOI] [PubMed] [Google Scholar]
- 14.Dodatko T, Fedorov AA, Grynberg M, Patskovsky Y, Rozwarski DA, Jaroszewski L, et al. Crystal structure of the actin binding domain of the cyclase-associated protein. Biochemistry 2004;43:10628–41. [DOI] [PubMed] [Google Scholar]
- 15.Alberts AS, Geneste O, Treisman R. Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell 1998;92(4):475–87. [DOI] [PubMed] [Google Scholar]
- 16.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003;113(3):329–42. [DOI] [PubMed] [Google Scholar]
- 17.Zhang S, Buder K, Burkhardt C, Schlott B, Gorlach M, Grosse F. Nuclear DNA helicase II/RNA helicase A binds to filamentous actin. J Biol Chem 2002;277:843–53. [DOI] [PubMed] [Google Scholar]
- 18.Doukhanine E, Gavino C, Haines JD, Almazan G, Richard S. The QKI-6 RNA binding protein regulates actin-interacting protein-1 mRNA stability during oligodendrocyte differentiation. Mol Biol Cell 2010;21:3029–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun TT, He J, Liang Q, Ren LL, Yan TT, Yu TC, et al. LncRNA GClnc1 Promotes Gastric Carcinogenesis and May Act as a Modular Scaffold of WDR5 and KAT2A Complexes to Specify the Histone Modification Pattern. Cancer Discov 2016;6(7):784–801. [DOI] [PubMed] [Google Scholar]
- 20.Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, et al. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Molr Biol 2013;20(7):908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013;500(7464):598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen R, Kong P, Zhang F, Shu YN, Nie X, Dong LH, et al. EZH2-mediated alpha-actin methylation needs lncRNA TUG1, and promotes the cortex cytoskeleton formation in VSMCs. Gene 2017;616 :52–57. [DOI] [PubMed] [Google Scholar]
- 23.Pei H, Zhang J, Nie J, Ding N, Hu W, Hua J, et al. RAC2-P38 MAPK-dependent NADPH oxidase activity is associated with the resistance of quiescent cells to ionizing radiation. Cell cycle 2017;16:113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu W, Xu S, Yao B, Hong M, Wu X, Pei H, et al. MiR-663 inhibits radiation-induced bystander effects by targeting TGFB1 in a feedback mode. RNA Bio 2014;11(9):1189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi X, Ma M, Lin S. Cell Cycle-Dependent Expression Dynamics of G1/S Specific Cyclin, Cellulose Synthase and Cellulase in the Dinoflagellate Prorocentrum donghaiense. Front Microbiol 2017;8:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai CK, Zhao GY, Tian LY, Liu L, Yan K, Ma YL, et al. miR-15a and miR-16–1 downregulate CCND1 and induce apoptosis and cell cycle arrest in osteosarcoma. Oncol Rep 2012;28(5):1764–70. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Lv XB, Wang XP, Sang Y, Xu S, Hu K, et al. MiR-138 suppressed nasopharyngeal carcinoma growth and tumorigenesis by targeting the CCND1 oncogene. Cell Cycle 2012;11(13):2495–506. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Wang R, Ma JY, Wang M, Cui J, Wu WB, et al. A Human Long Non-Coding RNA ALT1 Controls the Cell Cycle of Vascular Endothelial Cells Via ACE2 and Cyclin D1 Pathway. Cell Physiol Bioche 2017;43(3):1152–67. [DOI] [PubMed] [Google Scholar]
- 29.Chen QY, Xu W, Jiao DM, Wu LJ, Song J, Yan J, et al. Silence of ezrin modifies migration and actin cytoskeleton rearrangements and enhances chemosensitivity of lung cancer cells in vitro. Mol Cell Biochem 2013;377:207–18. [DOI] [PubMed] [Google Scholar]
- 30.Bockhorn J, Yee K, Chang YF, Prat A, Huo D, Nwachukwu C, et al. MicroRNA-30c targets cytoskeleton genes involved in breast cancer cell invasion. Breast Cancer Res Treat 2013;137:373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morton WM, Ayscough KR, McLaughlin PJ. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Bio 2000;2(6):376–8. [DOI] [PubMed] [Google Scholar]
- 32.Klenchin VA, Allingham JS, King R, Tanaka J, Marriott G, Rayment I. Trisoxazole macrolide toxins mimic the binding of actin-capping proteins to actin. Nat Struct Biol 2003;10(12):1058–63. [DOI] [PubMed] [Google Scholar]
- 33.Spector I, Braet F, Shochet NR, Bubb MR. New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Micros Res Techniq 1999;47:18–37. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Mishra A, Chintagari NR, Gou D, Liu L. Micro-RNA-375 inhibits lung surfactant secretion by altering cytoskeleton reorganization. IUBMB life 2010;62:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo H, Liu J, Ben Q, Qu Y, Li M, Wang Y, et al. The aspirin-induced long non-coding RNA OLA1P2 blocks phosphorylated STAT3 homodimer formation. Genome Biol 2016;17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posern G, Sotiropoulos A, Treisman R. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol Biol Cell 2002;13(12):4167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belmont LD, Orlova A, Drubin DG, Egelman EH. A change in actin conformation associated with filament instability after Pi release. P Natl Acad Sci USA 1999;96:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U. The structure of crystalline profilin-beta-actin. Nature 1993;365:810–6. [DOI] [PubMed] [Google Scholar]
- 39.Otterbein LR, Graceffa P, Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science 2001;293:708–11. [DOI] [PubMed] [Google Scholar]
- 40.Ponder JW, Richards FM. Tertiary Templates for Proteins - Use of Packing Criteria in the Enumeration of Allowed Sequences for Different Structural Classes. J Mol Biol 1987;193:775–91. [DOI] [PubMed] [Google Scholar]
- 41.Kataki A, Sotirianakos S, Memos N, Karayiannis M, Messaris E, Leandros E, et al. P53 and C-FOS overexpression in patients with thyroid cancer: an immunohistochemical study. Neoplasma 2003;50(1):26–30. [PubMed] [Google Scholar]
- 42.Wilczak W, Rashed S, Hube-Magg C, Kluth M, Simon R, Buscheck F, et al. Up-regulation of mismatch repair genes MSH6, PMS2 and MLH1 parallels development of genetic instability and is linked to tumor aggressiveness and early PSA recurrence in prostate cancer. Carcinogenesis 2017;38(1):19–27. [DOI] [PubMed] [Google Scholar]
- 43.Selvaraj A, Prywes R. Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol Biol 2004;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 1995;81(7):1159–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.