

Abstract
We previously discovered that phytonutrient genistein rapidly activates cAMP signaling in β-cells and improving islet mass in diabetic mice. However, the mechanism underlying these actions of genistein is still unclear. Here, we show that pharmacological or molecular inhibition of Gαs blocked genistein-stimulated adenylate cyclase activity in plasma membrane and intracellular cAMP production in INS1 cells and islets. Further, genistein stimulation of cAMP generation was abolished in islets exposed to a specific GPR30 inhibitor G15 or islets from GPR30 deficient (GPR30−/−) mice. In vivo, dietary provision of genistein (0.5 g/kg diet) significantly mitigated streptozotocin-induced hyperglycemia in male WT mice, which was associated with improved blood insulin levels and pancreatic islet mass and survival, whereas these effects were absent in Gpr30−/− mice. Genistein treatment promoted survival of INS1 cells and human islets chronically exposed to palmitate and high glucose. At molecular level, genistein activated CREB phosphorylation and subsequently induced Bcl-2 expression, and knockdown of CREB diminished the protective effect of genistein on β-cells induced by lipoglucotoxicity. Finally, deletion of GPR30 in β-cells or islets ablated genistein-induced CREB phosphorylation and its cytoprotective effect. These findings demonstrate that genistein is a survival factor for β-cells via GPR30-initiated, Gαs-mediated activation of CREB.
Keywords: genistein, cAMP, CREB, GPR30, apoptosis, islets, mice
1. Introduction
It is estimated that over 30.3 million or 9.4% of people in the US presently suffer from diabetes, and 57 million people have pre-diabetes [1]. While the availability of novel drugs, techniques, and surgical intervention has improved the survival rate of individuals with diabetes, the prevalence of diabetes is still rising in the U.S., with the number of people with diabetes projected to double by 2025 [2]. Type 2 diabetes (T2D) is a result of chronic insulin resistance and progressive loss of β-cell mass and function [3]. In experimental animals and humans, obesity is a leading pathogenic factor for developing insulin resistance, and insulin resistance will progress to T2D when β-cells are unable to secret adequate amount of insulin to compensate for decreased insulin sensitivity, which is largely due to insulin secretory dysfunction and significant loss of functional β-cells [4, 5]. Indeed, those individuals with T2D always manifest increased β-cell apoptosis and reduced β-cell mass [5–7]. Sustained hyperlipidemia and hyperglycemia in β-cells play an important role in contributing to β-cell apoptosis and dysfunction, thereby leading to the deterioration of glycemic control and the overt development of T2D [8, 9]. As such, the search for agents that promote β-cell survival may provide an effective strategy to prevent the onset of diabetes [10].
Genistein is a flavonoid compound present in legumes and Chinese herbal medicines Genista tinctoria Linn and Sophora subprostrala Chun. It is widely used as a dietary supplement in the U.S. for various presumed health benefits [11–13]. Genistein has been previously investigated for its potential beneficial effects on cancer treatment, cognitive function, and cardiovascular and skeletal health, with a primary focus on exploring its potential hypolipidemic, anti-oxidative and estrogenic effects [11–13]. While studies on whether genistein alone has an effect on diabetes are limited, available data showed that administration of genistein-containing soy products lowered plasma glucose in diabetic animals [14, 15] and in humans [16, 17]. Genistein has been reported to exhibit antioxidant activity [18, 19]. However, this effect of genistein is achieved only at concentrations ranging from 25-100 μM, which are well beyond those achievable through dietary ingestion of genistein supplement in both humans and rodents. Thus, the mechanism of genistein’s action as a potential anti-diabetic agent is still unknown. We recently observed for the first time that genistein at physiologically achievable concentrations (0.1-5 μM) activated cAMP signaling by inducing adenylate cyclase (AC) activity in β-cells and islets [20]. We further demonstrated that dietary intake of genistein preserved β-cell mass and mitigated diabetes in mice [21]. In the present study, we investigated whether genistein directly promotes β-cell survival and prevents diabetes by targeting the G-protein-coupled receptor (GPCR)-mediated signaling pathway.
2. Research design and Methods
2.1. Materials and Reagents
Cell culture medium, supplements, palmitate, fatty acid-free bovine serum albumin (BSA), H89, mellittin, and protease and phosphatase inhibitor cocktails were from Sigma (St. Louis, MO); genistein (purity≥99.0%) was from LC Laboratories (Woburn, MA); ICI 182,780 (ICI), 17β-estradiol (E2), and G15 were from Tocris Biosciences (Bristol, UK); insulin ELISA kits were obtained from Crystal Chemical Inc. (Chicago, IL); cell viability and caspase-3 assay kits, pRL luciferase control vector, and dual luciferase assay system were purchased from Promega (Madison, WI); cyclic AMP EIA kits were from Cayman Chemical Company (Ann Arbor, MI); terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kits were from Trevigen Inc. (Gaithersburg,MD); the active form of the caspase −3 antibody was from BD Biosciences (San Jose, CA); the rabbit polyclonal anti-insulin antibody was from Abcam (Cambridge, MA); antibodies for phospho-CREB (ser133), CREB, and β-actin were from Cell Signaling Technology (Beverly, MA); nitrocellulose membranes and protein assay kits were from Bio-Rad (Hercules, CA); CREB ShortCut® siRNA, scramble sequence of siRNA and transfection reagents were from New England Biolabs (Ipswich, MA); anti-GPR30, anti-Gαs, shGαs lentiviral particles, and GPR30 siRNA (rats) were from Santa Cruz Biotechnology (Santa Cruz, CA); a reporter plasmid containing a promoter region of the Bcl-2 gene linked to a firefly luciferase gene (Bcl-2-Luc) was a gift of Dr. Linda M. Boxer, Stanford University); a reporter plasmid containing multiple copies of a consensus cAMP responsive element (CRE)-binding sequence fused to a TATA-like promoter region upstream of the gene for firefly luciferase (CRE-luc) was from BD Clontech, CA; the plasmid transfection reagent was from Invitrogen, CA; the ImmPRESS anti-rabbit Ig (peroxidase) polymer detection kit, vector NovaRED peroxidase substrate, and vector SG peroxidase substrate were from Vector Laboratories (Burlingame, CA). Stock solutions of genistein and E2 at 20 mM dissolved in dimethyl sulfoxide (DMSO) were stored at –20 °C.
2.2. Cell and human islet culture
INS1 cells (provided by Dr. Pierre Maechler, University of Geneva Medical School) were cultured as previously described [22]. Human islets were supplied through the Integrated Islet Distribution Program (City of Hope Medical Center, Duarte, CA). The islet purity and viability was over 90%. The islets were maintained in CMRL-1066 medium containing 10% FBS. Palmitate was dissolved in medium containing 7% fatty acid-free BSA, sonicated, and sterile-filtered (final fatty acid/BSA molar ratio=6.6) [23]. For control experiments, palmitate-free BSA was prepared in the same way as stated above. For examining the effects of genistein on CREB phosphorylation, INS1 cells or islets were pre-incubated with Krebs Ringer Bicarbonate buffer for 20 min followed by treatment with genistein or vehicle for 30 min. In some experiments, INS1 cells or islets were preincubated with estrogen (E2) receptor (ER) antagonist ICI 182,780 (ICI; 10 μM), PKA inhibitor H89 (10 μM), Gαs inhibitor mellittin (M; 5 μM), GPR30 antagonist G15 (5 μM), or vehicle (DMSO) for 30 min before addition of genistein.
2.3. AC and intracellular cAMP measurements
For measuring AC activity, the plasma membranes from INS1 cells were isolated by differential centrifugation [24]. The membranes were pre-incubated with GDPβS (100 μM), Gαs antibody (1:200), non-immune IgG, or vehicle for 30 min at 25°C, followed by addition of genistein (2.5 μM), GLP-1 (20 nM), forskolin (5μM), or vehicle, for 10 min. AC activity was then measured by determining cAMP production as previously described [24]. For intracellular cAMP measurements, islets were treated with genistein (5 μM) for 20 min. Intracellular cAMP levels were measured by an EIA kit [20]. Data were normalized to the cellular protein concentration in the same samples.
2.4. Immunoblot analysis
Equal amounts of protein (50 μg) from cell extracts were subjected to immunoblot analysis as previously described [21]. Membranes were first probed with an antibody against Gαs, GPR30, Bcl-2, or phospho-CREB (p-CREB). The immunoreactive proteins were detected by chemiluminescence. Nitrocellulose membranes were then stripped and reprobed with antibodies against CREB or β-actin. The protein bands were digitally imaged for densitometric quantitation with Image Lab instrument(Bio-Rad). The expression of p-CREB and Bcl-2 was normalized to that of total CREB and β-actin, respectively, and expressed as a fold increase over the control.
2.5. Transfection assays
INS1 cells were grown in a 24-well plate until 50% confluence. To determine whether genistein activates cAMP-regulated transcription, cells were co-transfected with 0.25 μg CRE-Luc reporter plasmid and 5 ng pRL reporter control vector per well using Lipofectamine 2000 transfection reagent according to the manufacturer’s protocol. To examine whether genistein enhances Bcl-2 transcription, cells were co-transfected with 0.5 μg Bcl-2-Luc vector and 1 ng pRL reporter plasmid per well. After transfection, cells were incubated with complete RPMI1640 medium for 24 h, and then treated with genistein or vehicle for 16 h for CRE-luc-transfected cells, or 24 h for Bcl-2-luc-transfected cells. Luciferase activity, normalized to pRL activity in the cell extracts was then determined with a dual luciferase reporter assay system.
For knockdown of CREB, INS1 cells were transfected with a heterogeneous mixture of target-specific CREB siRNA (50 nM) or corresponding amounts of scrambled siRNA using siRNA transfection reagents according to the manufacturers’ protocols. After 48 h of transfection, cells were incubated with or without genistein for 30 min for immunoblot analysis of CREB protein expression, or incubated with palmitate (0.5 mM) and high glucose (HG, 20 mM) in the presence or absence of genistein (5 μM) for 72 h followed by apoptosis assays. To delete GPR30, INS1 cells were transfected with 100 nM GPR30 siRNA or scrambled siRNA in serum- and antibiotic-free media for 16 h using Lipofectamine 2000 transfection reagent. Cells were then cultured in complete DMEM for 24 h, followed by treatment with 5 μM genistein or vehicle.
2.6. Cell apoptosis assays
INS1 cells or islets were cultured in complete RPMI-1640 medium with or without 0.5 mM palmitate and HG in the absence or presence of genistein (0.1, 1, and 10 μM) for 72 h, with genistein supplemented media replaced after 48 h. Cellular apoptosis was measured with a TUNEL-based quantitative apoptotic assay kit. For measuring cellular caspase-3 activity, cells or islets were cultured under the above glucolipotoxic condition with or without genistein for 48 h. The caspase-3 activity in the cell lysates was measured using an assay kit and normalized to the cellular protein concentration, measured with a Bio-Rad assay kit.
2.7. Animals and treatments
WT or Gpr30−/− mice (breeding colony provided by Dr. Deborah Clegg, UT Southwestern) were housed in a room maintained on a 12h light/dark cycle under constant temperature (22–25° C) with ad libitum access to food and water. Homozygosity was confirmed via quantitative RT-qPCR. When mice were 7-mo old, WT and Gpr30−/− male mice were divided into 4 groups with their initial body weight and blood glucose balanced among groups. The mice were then fed a phytoestrogen-free high-fat diet (HFD, 45% kcal fat) or HFD supplemented with 0.05% genistein. Body weight and food intake were recorded weekly. This genistein dosage was used (approximately a human intake of 25-200 mg/day) because it is within the range that humans can realistically consume through taking supplements [25]. After 4 wks of treatment, mice were injected intraperitoneally streptozotocin (STZ) dissolved in 0.1 mM cold sodium citrate buffer (pH 4.5) at 40 mg/kg daily for 3 consecutive days. Control mice were injected the same amount of vehicle. After this procedure, mice were remained on the same dietary treatment for 9 wks for the following metabolic measurements. Body weight and food intake were recorded weekly. The protocol of this study was approved by the Institutional Animal Care and Use Committee at Virginia Tech (protocol number: 11-176-HNFE).
2.8. Metabolic measurements
At the beginning of the experiment, fasting (12h) blood glucose was measured in tail vein blood sample using a glucometer (Kroger). Following STZ injection, blood glucose was measured weekly to assess the onset of hyperglycemia (non-fasting blood glucose >250 mg/dl) [26]. Plasma insulin concentrations were measured by ELISA (Crystal Chem, IL). Glucose tolerance and insulin sensitivity were assessed as previously described [21]. Fasting plasma total cholesterol and triacylglycerides were measured in triplicate using a Pointer 180 Analyzer (Pointe Scientific, Canton, MI) as described [27].
2.9. Immunohistochemistry and islet morphometry
At the end of the feeding study, mouse pancreata were collected and fixed as previously described [21]. A series of tissue sections (5-μm thickness) were prepared by AML Laboratory (Baltimore, MD), and immuno-stained to determine β-cell mass and apoptosis. The β-cell area was measured using images acquired from serial insulin-stained pancreatic sections (100 μm interval). The β-cell mass were calculated as previously described [21]. Apoptotic β-cells were detected by double labeling the sections with antibodies against activated caspase-3 and insulin using an ImmPRESS™ detection system as we previously described [28].
2.10. Statistical analysis
Data were analyzed with one-way ANOVA using SigmaPlot software. Treatment differences were subjected to Tukey’s test. A p-value < 0.05 was considered significant.
3. Results
3.1. Genistein activation of cAMP signaling is mediated via Gαs in β-cells
We recently found that genistein rapidly activates AC in β-cells [20]. AC activity is stimulated by G-protein α-subunit (Gαs), which is an important regulator of β-cell mass and function [29]. We found that genistein activation of AC in plasma membranes of β-cells was inhibited by GDPβS (Fig. 1A), which blocks the exchange of a GDP for a GTP and thereby inhibits the activation of G-proteins [30]. Further, pre-incubation of the plasma membranes from INS1 cells with anti-Gαs, which neutralizes and thereby blocks Gαs activation, ablated genistein activation of AC activity (Fig. 1B). Interestingly, genistein showed similar potency in inducing AC activity as glucagon-like-peptide-1 (GLP-1), a potent incretin that activates Gαs/AC signaling via its receptor β-cells. The effect of GDPβS or anti-Gαs on genistein-induced AC activation is specific because they did not influence forskolin-induced AC activity.
Fig. 1.
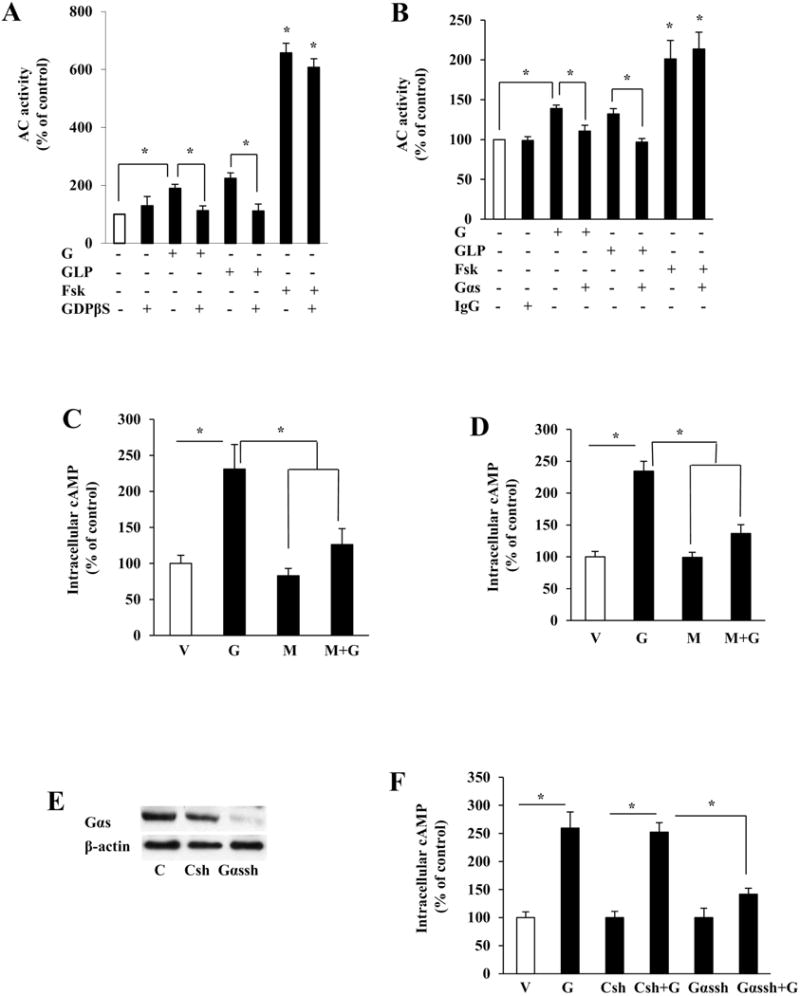
Genistein activated cAMP signaling via a Gαs-mediated mechanism in β-cells. (A) AC activity in INS1 cell plasma membranes preincubated with 100 μM GDPβS (A), Gαs antibody (Gαs, 1:200 (B), nonimmune IgG, or vehicle (V) for 30 min at 25°C, followed by stimulation with genistein (G, 2.5 μM), GLP-1 (GLP, 20 nM), forskolin (Fsk, 5μM) or vehicle for 10 min. Intracellular cAMP levels in INS1 cells (C) or human islets (D) pretreated with Gαs inhibitor mellittin (M, 5μM) for 30 min, followed by stimulation with genistein (G, 5μM) for 20 min. Islets were infected with lentiviral particles containing Gαs shRNA (Gαsh) or control shRNA (Csh) at MOI of 150. 48 h later, control and infected islets were either lysed for measuring Gαs and β-actin protein expression to determine the efficiency of knockdown (E), or incubated with genistein (G, 5μM) for 20 min to measure intracellular cAMP levels (F). Cyclic AMP levels were normalized to the protein contents, and expressed as percentage of the control value. Data are mean±SEM (n=4). *, p<0.05 vs. control.
To examine whether Gαs also mediates genistein-stimulated cAMP production in β-cells and islets, INS1 cells or human islets were pre-incubated in the absence or presence of the Gαs inhibitor, mellittin (5 μM), for 30 min [31], followed by addition of genistein (5μM) or vehicle for 20 min. Inhibition of Gαs with mellittin blocked genistein-stimulated cAMP production in INS1 cells (Fig. 1C) and human islet cells (Fig. 1D). We then utilized shRNA-expressing lentiviral particles to suppress Gαs expression in human islets, in order to further investigate whether Gαs mediates genistein-stimulated cAMP production in pancreatic islets. Consistently, infection of human islets with an Gαs-specific shRNA-expressing lentivirus ablated the expression of Gαs protein (Fig. 1E), and subsequently diminished genistein-triggered cAMP production (Fig. 1F), whereas the scrambled shRNA did not affect Gαs protein expression and genistein/cAMP signaling in the islets. Combined, these results provide evidence that Gas is essential in mediating genistein/cAMP signaling in β-cells.
3.2. Genistein acts through GPR30
Activation of AC could be linked to Gαs-coupled receptors [32] and genistein may bind to GPR30, a membrane-associated GPCR, in cancer cells [33]. We thus reasoned that genistein activation of cAMP signaling may be mediated via this receptor. We first confirmed that GPR30 protein is highly expressed in clonal rodent β-cells and human and mouse islets (Fig. 2A). Incubation of INS1 cells with G15, a specific GPR30 inhibitor, diminished genistein-stimulated cAMP production (Fig. 2B). Knockdown of Gpr30 with siRNA (Fig. 2C) greatly ablated genistein-stimulated cAMP production in INS1 cells, whereas scrambled siRNA had no such an effect (Fig. 2D). Furthermore, genistein stimulated cAMP generation in isolated WT mouse islets, but this genistein action was absent in Gpr30−/− islets (Fig. 2E). The deletion of GPR30 in islets from Gpr30−/− mice was confirmed by western blot analysis of GPR30 protein expression (Fig. 2F).
Fig. 2.
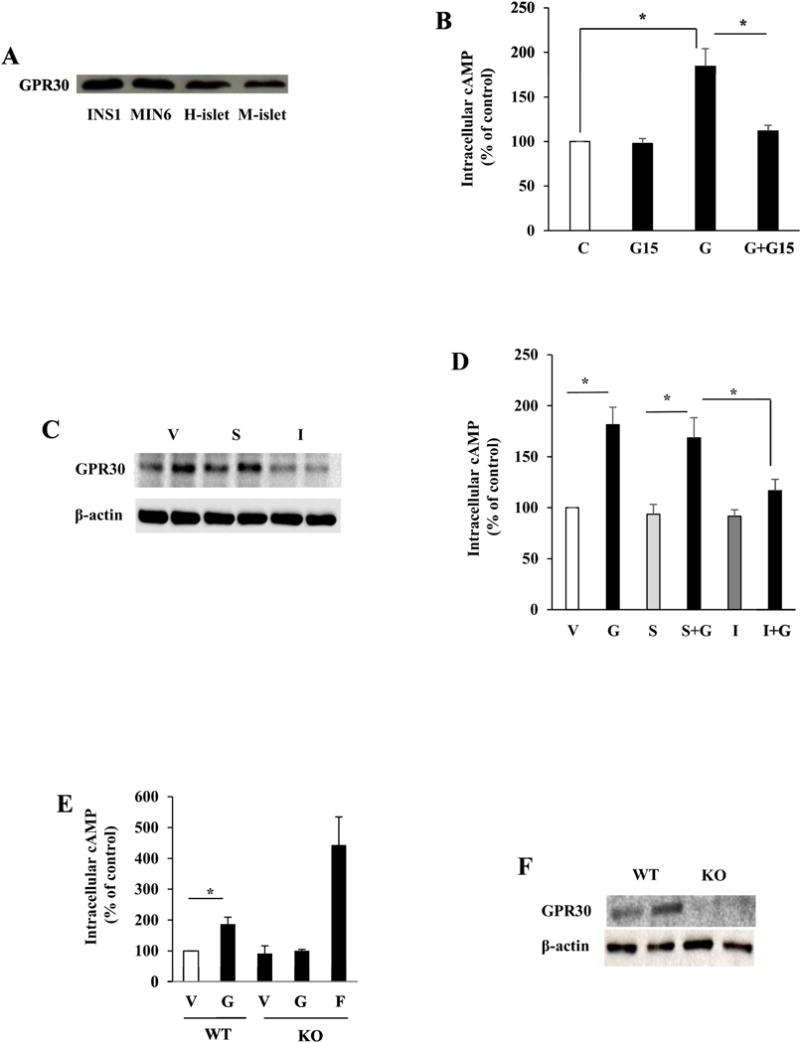
Genistein stimulation of cAMP production was mediated via GPR30 in β-cells. (A) protein expression of GPR30 in insulin-secreting cells and islets as determined by western blot (H=human, M=mouse). (B) Intracellular cAMP levels in INS1 cells preincubated with vehicle (V) or GPR30 antagonist G15 (5 μM) for 10 min before stimulation with genistein (G, 5 μM) for 20 min. (C) Knockdown of GPR30 protein expression in INS1 cells transfected with GPR30 siRNA (I) or scrambled siRNA (S). Images shown were from two independent transfection experiments. (D) Intracellular cAMP levels in GPR30 siRNA (I) or scrambled siRNA (S) transfected cells and exposed to 5 μM genistein or vehicle (V) for 20 min. (E) Intracellular cAMP levels in wild-type (WT) or GPR30−/− (KO) islets incubated with vehicle (V), genistein (G, 5 μM), or foskolin (F, 1 μM) for 20 min. (F) Western blot for GPR30 knockout in the islets. Cyclic AMP levels were normalized to the protein contents, and expressed as percentage of the control value. *, p<0.05 (n=3-4±SEM).
3.3. The anti-diabetic action of genistein in mice is mediated via GPR30
We next investigated whether GPR30 mediates the anti-diabetic effect of genistein in vivo. Before STZ injection, genistein had no effect on body weight gain, food intake or non-fasting blood glucose levels in both WT and Gpr30−/− mice (data not shown). As expected, STZ injection induced hyperglycemia in both WT and Gpr30−/− mice (Fig. 3A and B). We observed that WT diabetic mice fed a genistein supplemented HFD had significantly lower blood glucose levels as compared to HFD-fed WT diabetic mice (Fig. 3A), suggesting that genistein mitigated STZ-induced hyperglycemia. However, genistein failed to lower blood glucose levels in STZ-induced Gpr30−/− diabetic mice (Fig. 3B). Fasting blood glucose levels were not greatly increased by STZ injection or significantly modulated by genistein treatment (data not shown), indicating that STZ-induced elevation of blood glucose in our models is mainly due to a destruction of pancreatic β-cells, leading to deficiency of glucose-stimulated insulin secretion. Consistently, genistein significantly improved glucose tolerance (Fig. 3C and D) and postprandial circulating insulin levels (Fig.3E) in WT mice but not in Gpr30−/− mice (data not shown). Genistein had no effects on plasma lipid profiles (data not shown) and whole body insulin sensitivity in both WT (Fig. 3F) and Gpr30−/− (data not shown) mice.
Fig. 3.
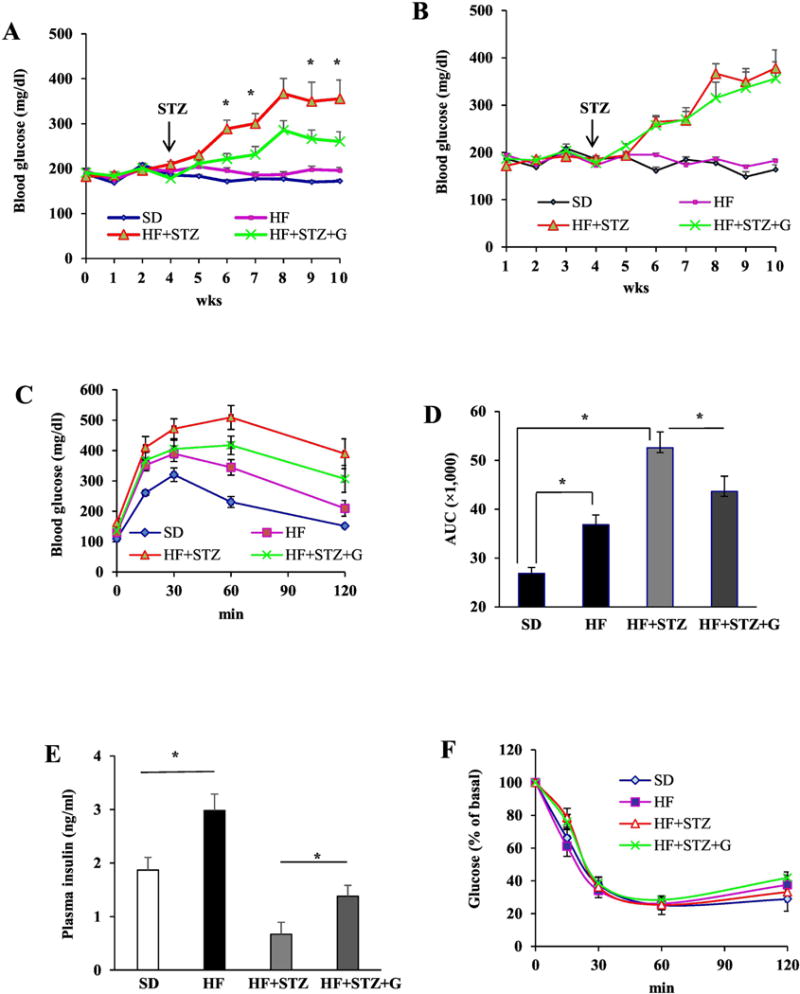
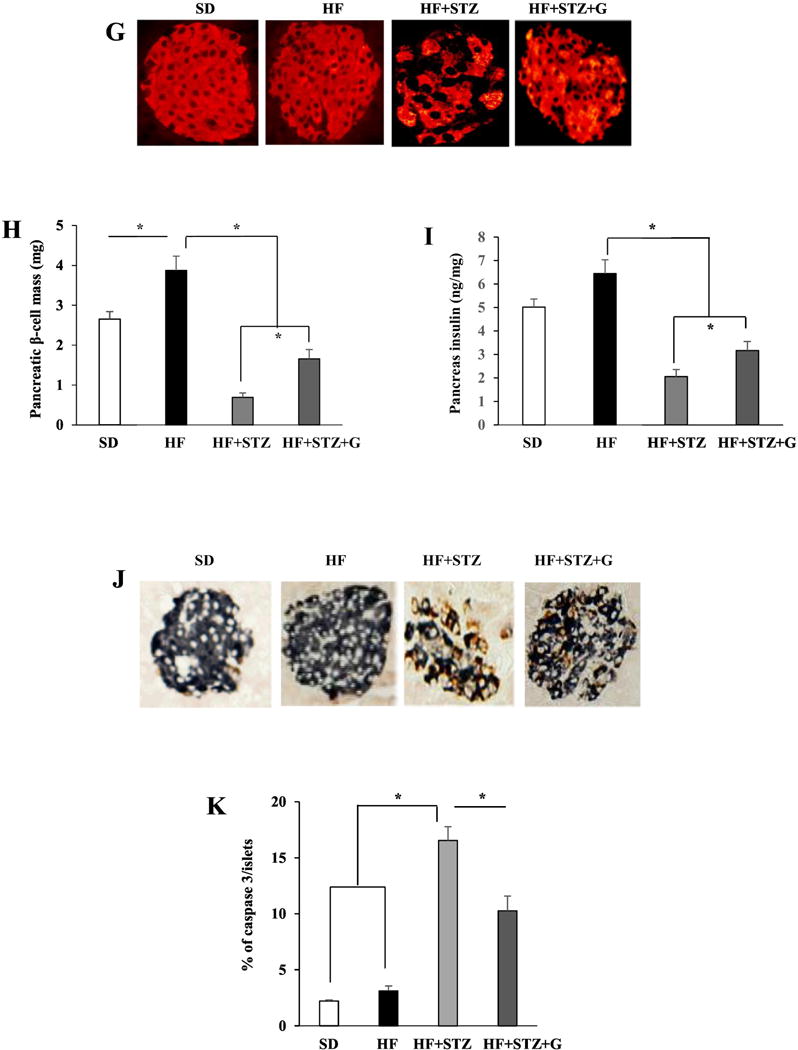
The anti-diabetic effect of genistein in streptozotocin (STZ)-induced obese diabetic mice was GPR30-dependent. WT or GPR30−/− male mice were fed a standard cow diet (SD), a high fat diet (HF), or HF supplemented with 0.05% genistein (HF+G) for 4 wks. Mild hyperglycemia in HF-fed mice was then induced by injection of STZ. Non-fasting blood glucose levels in WT (A) and GPR30−/− (B) mice were measured at the indicated time points. Data of intraperitoneal glucose tolerance test (C) and area under the curve (AUC) calculated using the trapezoidal rule (D) in WT mice. (E) Non-fasting plasma insulin levels in WT mice. (F) Insulin tolerance test in WT mice at 5 wks after the first STZ injection. (G) A set of representative micrographs of pancreatic sections stained with insulin antibody. (H) The β-cell mass determined as described in “Materials and Methods”. (I) Pancreas insulin contents measured by ELISA. (J) Representative activated caspase-3 (red) and insulin (blue) double staining of pancreatic sections from WT mice. (K) Quantification of caspase-3-positive β-cells expressed as percentage of total insulin-stained cells. Four pancreatic sections from each mouse with 6 mice per group were evaluated. Data are shown as means ± SE or mean±SEM (n= 6 mice/group). *, P<0.05.
Next, we examined whether genistein treatment preserved β-cell mass in WT diabetic mice. STZ administration severely decreased β-cell mass and disrupted the islet architecture. However, genistein-treated diabetic mice had significantly more islet β-cell mass and improved islet structure (Fig. 3G and H) concomitant with greater insulin content in the pancreas (Fig. 3I) as compared to control diabetic mice. Consistently, genistein reduced islet β-cell apoptosis, as determined by double immunolabeling of insulin and the activated caspase-3 (Fig. 3J and K), a key protease involved in the terminal steps of cell apoptosis. These results suggest that genistein improvement of hyperglycemia in STZ-induced obese diabetic mice is primarily due to GPR30-dependent protection of islet function.
3.4. Genistein prevents apoptosis of INS1 cells and islets exposed to glucolipotoxicity
To determine whether genistein directly promotes survival of β-cells, we first evaluated whether genistein protects INS1 cells exposed to excessive amounts of saturated fatty acids and glucose (glucolipotoxicity), which are believed to cause β-cell apoptosis and impair its function, thereby contributing to the pathogenesis of T2D [23]. Incubation of INS1 cells with palmitate (0.5 mM) and HG for 72 h significantly increased apoptosis. However, co-treatment with genistein dose-dependently prevented cell apoptosis induced by chronic glucolipotoxicity (Fig. 4A), with 1 μM genistein exerting a significant effect. In parallel to decreased apoptosis, the cellular caspase-3 activity was markedly increased in INS1 cells under glucolipotoxicity, which was significantly reduced by genistein treatment (Fig. 4B). In consistent with this cytoprotective effect in INS1 cells, genistein treatment also promoted survival of mouse (Fig. 4C and D) and human (Fig. 4E and 4F) islets chronically exposed to palmitate (0.5 mM) and HG (20 mM). To test whether genistein modulates anti-apoptotic Bcl-2 expression, which could be a mechanism through which genistein promotes islet cell survival, we incubated INS1 cells with genistein for 24 h, and found that genistein greatly induced Bcl-2 protein expression (Fig. 4G), which was accompanied with increased Bcl-2 promoter activity (Fig. 4H) as determined by a reporter gene assay, suggesting that genistein may regulate Bcl-2 expression at the transcriptional level in β-cells. Collectively, these results demonstrate that genistein directly promotes islet β-cell survival.
Fig. 4.
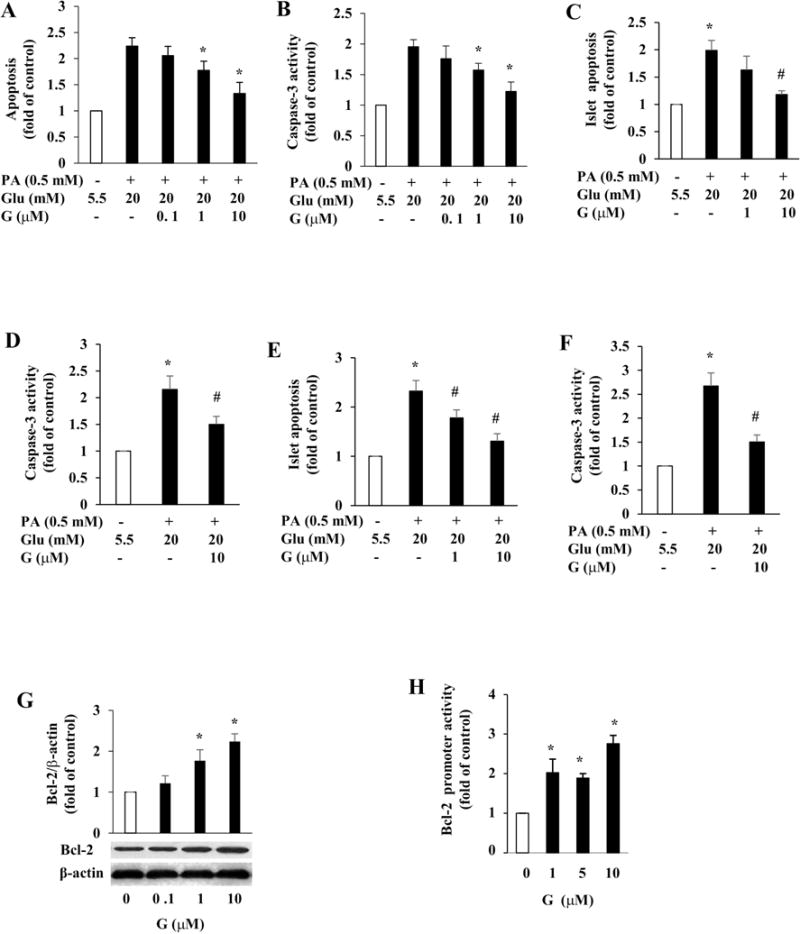
Genistein protected INS1 cells and islets from apoptosis. INS1 cells (A, B), mouse (C, D), or human (E, F) islets were pretreated with or without genistein (G) at indicated concentrations for 12 h, followed by addition of 0.5 mM palmitate (PA) and 20 mM glucose (Glu). Cellular apoptosis (A, C, E) and caspase-3 activity (B, D, F) were measured after 72 h and 48 h of treatment, respectively. Values were expressed as fold of the control. (G) INS1 cells were treated with genistein (G) or vehicle for 48 h. Cellular Bcl-2 protein was detected by western blot and normalized to β-actin content. (H) INS1cells were co-transfected with Bcl-2 promoter and control plasmids, followed by addition of genistein for 24 h. Luciferase activity in the cell lysates was measured. *, p<0.05 vs. control; #, p<0.05 vs. Glu and PA-alone treated islets (n=4±SEM).
3.5. The anti-apoptotic effect of genistein is independent of estrogen receptor (ER)
As genistein may have weak estrogenic effects on some tissues, and previous studies showed that E2 can promote β-cell survival [34, 35], we examined whether genistein promotion of islet survival was mediated via ERs. Genistein or E2 alone reduced human islet cell apoptosis (Fig. 5A). However, incubation of human islets with ICI182,780, a specific antagonist of ERα and ERβ, reduced the cytoprotective effect of E2 by 70%, but had no effect on genistein-mediated action. Moreover, daidzein, an analogue of genistein that is essentially inactive as an ER ligand [36], also prevented β-cell apoptosis (Fig.5B). These results indicate that genistein actions in β-cells are likely independent of ERα- and ERβ-mediated signaling machinery.
Fig. 5.
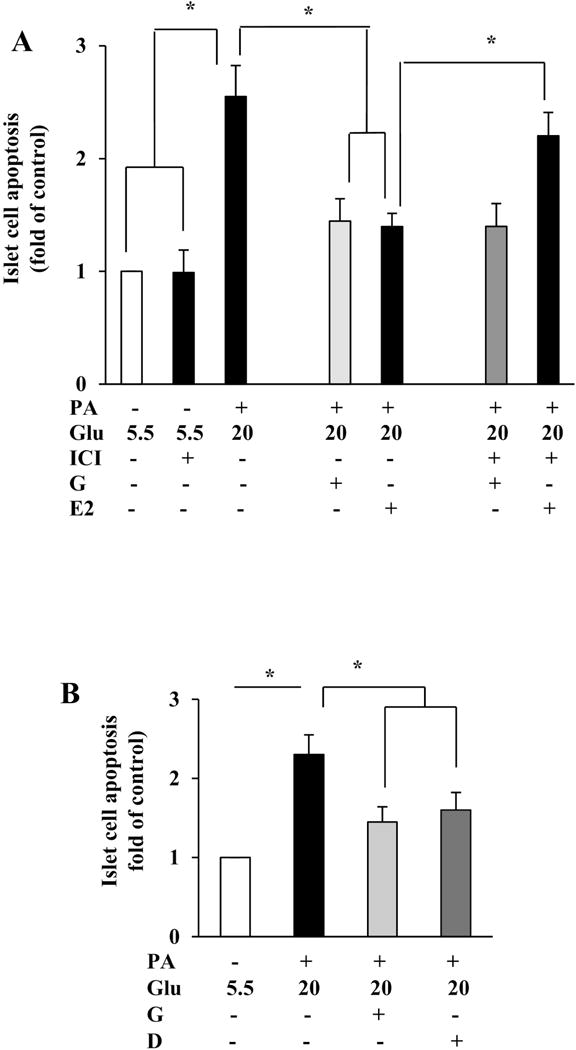
The anti-apoptotic effect of genistein was independent of ERα and ERβ. (A) Co-incubation of human islets with ER inhibitor ICI 182,780 (ICI, 10 μM) blocked the effect of 17β-estradiol (E2; 0.1μM), but no that of genistein (Gen, 1 μM), on high glucose (Glu, 20 mM) and palmitate (PA, 0.5 mM) induced islet apoptosis. (B) Genistein (G, 5 μM) and its non-estrogenic analog daidzein (D, 5 μM) protected human islets from high glucose (Glu; 20 mM) and palmitate (PA; 0.5 mM) induced apoptosis. *, p<0.05 (n= 4±SEM).
3.6. The cytoprotective effect of genistein in β-cells depends on GPR30-mediated activation of CREB
To investigate the underlying mechanism through which genistein promotes β-cell survival, we first determined whether genistein treatment activates CREB, an important β-cell survival factor [37]. Exposure of INS1 cells (Fig. 6A) and human islets (Fig. 6B) to genistein for 30 min activated CREB, a pattern that is consistent with its effects on cAMP production and cell survival. Pretreatment of cells with PKA inhibitor H89 abolished genistein-induced CREB phosphorylation (Fig. 6C), suggesting that PKA mediates the genistein-stimulated phosphorylation of CREB. We then tested whether the activation of CREB by genistein is sufficient to affect transcription of cAMP-regulated genes, which include Bcl-2. Genistein at as low as 1 μM concentration significantly stimulated CRE-mediated luciferase activity in INS1 cells, with 10 μM dose inducing a more than 1.5-fold increase in transcription over the control (Fig. 6D). Next, we used RNA interference to examine whether genistein protection of β-cell apoptosis is mediated via activation of CREB. Transfection of β-cells with CREB-specific siRNA greatly reduced CREB protein expression, whereas the scrambled siRNA did not affect CREB expression (Fig. 6E). Accordingly, disruption of CREB expression with siRNA diminished genistein-induced CREB phosphorylation (Fig. 6F), Bcl-2 protein expression (Fig. 6G), and its cytoprotective effect (Fig. 6H) in INS1 cells, whereas transfection with a scrambled siRNA had no effect on genistein-mediated actions. To further investigate whether genistein activation of CREB is mediated via GPR30, we transfected INS1 cells with siRNA of GPR30 or its scrambled control. Knockdown of GPR30 mRNA, which greatly reduced its protein expression (Fig. 2C), ablated genistein-stimulated CREB phosphorylation without affecting total CREB expression in INS1 cells (Fig. 6I). Similarly, genistein stimulated CREB phosphorylation (Fig. 6J) and protected against apoptosis (Fig. 6K) of WT islets exposed to palmitate and HG, but these effects were absent in Gpr30−/− islets. Collectively, these data for the first time demonstrated that genistein prevents β-cells from apoptosis via GPR30-mediated activation of CREB.
Fig. 6.
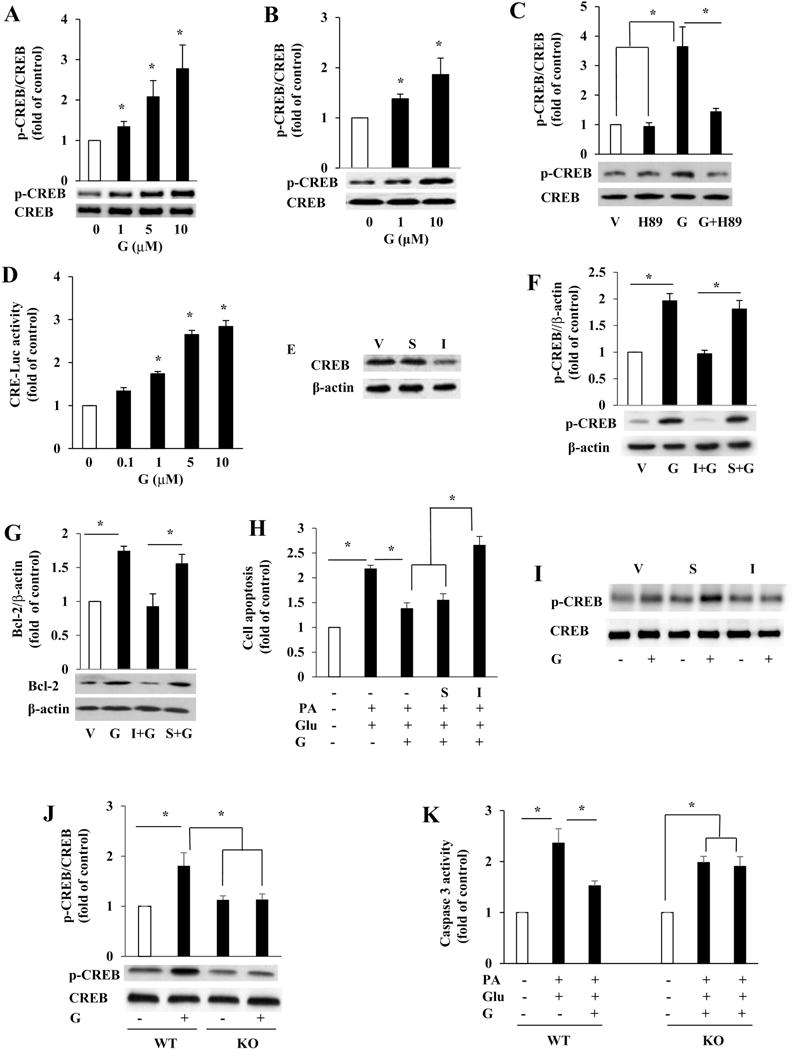
The cytoprotecitve effect of genistein on β-cells was dependent of GPR30-mediated activation of CREB. Western blot analyses of phosphorylated (p-CREB) and total CREB in INS1 cells (A) or human islets (B) incubated with various concentrations of genistein or vehicle for 30 min. (C) Total CREB and p-CREB protein levels in INS1 cells pretreated with PKA inhibitor H89 (10 μM) or vehicle (V) for 30 min, followed by addition of genistein (G, 5 μM) for 30 min. (D) INS1 cells transfected with reporter plasmid pCRE-Luc were incubated with vehicle or genistein for 24 h. Luciferase activity was measured and normalized to the control plasmids in the cell extracts. (E) INS1 cells were transfected with vehicle (V), scrambled siRNA (S), or siRNA directed against CREB (I) for 48 h. The efficiency of knockdown in transfected cells was verified by immunoblot analysis of CREB protein levels. (F) Total and p-CREB levels in control or transfected INS1 cells treated with genistein (G, 5 μM) or vehicle (V) for 30 min. (G) Bcl-2 protein levels in control or transfected INS1cells treated with genistein (G, 5 μM) or vehicle (V) for 48 h. (H) Quantification of apoptosis in control or transfected INS1 cells following treatment with 0.5 mM palmitate (PA) and high glucose (Glu, 20 mM) or vehicle in the presence or absence of 5 μM genistein (G) for 72 h. I. INS1 cells were transfected with scrambled control siRNA (S) or GPR30 siRNA (I). 48 h later, genistein (5 μM) effect on p-CREB and total CREB protein contents in cells were measured by western blot. (J) total CREB and p-CREB levels in WT and GPR30−/− (KO) islets isolated from male mice and treated with or without genistein (G, 5 μM) for 30 min. (K) Cellular caspase-3 activity in WT and KO islets incubated with palmitate (PA, 0.5 mM) plus 20 mM glucose (Glu) in the presence or absence of genistein (G, 5 μM) for 24 h. Values are mean ± SEM (n=4). *, p< 0.05.
4. Discussion
It has been shown that administration of phytonutrient genistein lowered plasma glucose in diabetic animals [14, 15] and in humans [16, 17] without affecting insulin sensitivity and fat metabolism. However, the molecular mechanism of this genistein action is still unclear. In the present study, we provide evidence for the first time that genistein directly promotes β-cell survival via the GPR30/Gα/AC/cAMP/PKA/CREB signaling pathway. Notably, this finding is physiologically relevant, as the effective doses of genistein (1-10 μM) overlap with those achievable by dietary intake of genistein supplement, which can achieve the total plasma genistein levels in the range of 1-7 μM in rodents and humans [13]. Consistent with these ex vivo findings, we further demonstrated that oral administration of genistein at a dose that humans can take as dietary supplement [25]. significantly mitigated hyperglycemia and preserved pancreatic β-cell mass in HFD and STZ-induced obese diabetic mice, an effect that was abolished in GPR30−/− mice. The significance of our findings is that they establish a mechanistic basis for the physiological actions of genistein. As the loss of functional β-cell mass plays a central role in the deterioration of blood glucose control in both T1D and T2D [5], the search for agents that promote β-cell survival and thereby preserve functional β-cell mass may provide an effective strategy to prevent the onset of diabetes. In this regard, genistein may be a safe and low-cost novel natural compound that can be used to prevent or ameliorate diabetes.
We recently discovered that genistein is a potent activator of cAMP signaling in islet β-cells [20]. However, the underlying mechanism of this genistein effect is unclear. In the present study, we performed a series of complementary experiments with the results indicating that Gαs activation is the upstream of the genistein elicited cAMP signaling in β-cells. As genistein activates AC via Gαs in plasma membranes of β-cells, and cell membrane-associated G-proteins are typically activated by GPCRs, we speculated that this genistein action is transmitted via a membrane-bound receptor, which subsequently activates intracellular signaling in β-cells. Genistein is known to have an affinity for ERα and ERβ, although it is unclear whether the physiological effects of genistein observed in vivo are mediated by these classical ERs. Recent in vitro studies provide evidence that plasma membrane-associated GPR30 and ER36α, a 36-kDa variant of ERα, are novel membrane ERs that mediate the membrane-initiated E2 signaling in cancer cells [38, 39]. We chose to study GPR30 as we showed that it is highly expressed in islets and β-cells consistent with recent observations [35, 40], whereas ER36α is not detectable. Incubation of β-cells with G15, a selective GPR30 antagonist that do not interfere with the classical nuclear ER signaling [41], abolished genistein-stimulated cAMP production. In accordance with this result, knockdown of GPR30 with siRNA in β-cells or deletion of Gpr30 in mouse islets greatly reduced genistein-stimulated intracellular cAMP production. These data for the first time, as to our knowledge, demonstrated that genistein may act on the cell membrane, eliciting a GPR30-mediated action that leads to the activation of cAMP signaling in β-cells.
Intriguingly, GPR30 is not only expressed in the plasma membrane, but also in the endoplasmic reticulum and the Golgi complex [42–48], suggesting that GPR30 is an atypical GPCR. Thus, it is possible that GPR30 is activated intracellularly by genistein and then translocates to the cell membranes to initiate cellular signaling [45, 46]. While it is still unclear how GPR30 exactly interacts with the receptor ligands, which is largely due to the challenge in purifying active protein from the membrane, it appears that GPR30 can be activated by structurally diverse molecules such as E2, G1, and ER antagonist tamoxifen and fulvestrant [49], suggesting that it has large plasticity in accepting ligands. Interestingly, our unpublished data show that a large group of structurally related flavonoids also potently activates cAMP-mediated signaling. It may thus be speculated that GPR30 is a common target for flavonoids, confirmation of which could provide novel insights regarding the physiological roles of many flavonoids that are ubiquitous in the human diet.
We further investigated whether GPR30 mediates the antidiabetic action of genistein using WT and Gpr30−/− mice. In that regard, we used STZ to induce diabetes by directly causing the destruction of β-cells. We show that oral administration of genistein (50 mg/kg/d) significantly ameliorated hyperglycemia and glucose intolerance in WT obese diabetic mice, but these protective effects of genistein were completely absent in Gpr30−/− mice. The antidiabetic effect of genistein observed in this study might be relevant to humans, because this dose of genistein used in the present study (equivalent to the human intake of 75-100 mg/day) is within the dose range typically consumed by humans [50]. In addition, we chose to examine genistein effect in older adult mice (7 mo), because T2D usually occurs at middle and older age in humans. The diabetic mouse models used in this study were generated by HFD feeding and 3 mild doses of STZ administration that did not cause diabetes in chow-fed mice as we previously determined [51]. This mouse model shares the metabolic characteristics of human T2D with peripheral insulin resistance and reduced β-cell mass and function. In the present study, we observed that genistein had no effect on fasting blood glucose, which is primarily derived from hepatic glucose production [52], suggesting that gluconeogenesis was not altered by genistein. In line with our recent finding [21], genistein didn’t alter body weight gain, food intake, fat mass, plasma lipid profile, or insulin sensitivity, indicating that the blood glucose-lowering effect of genistein is not a secondary action thereby genistein modulated these variables. In contrast, genistein treatment significantly improved nonfasting plasma insulin levels in WT diabetic mice. These results suggest that the improvements in glycemic control by genistein treatment is primarily due to the improved pancreatic β-cell function, given that STZ induces diabetes by directly causing β-cell destruction and insulin deficiency. Consistently, dietary supplementation of genistein to WT mice partially prevented β-cell apoptosis and preserved islet mass concomitant with higher pancreas insulin content.
The present study showed that genistein directly promotes β-cell and islet survival. While genistein can bind to the ERs, and it was reported that E2 can protect islets against stimuli-induced apoptosis via the ER-dependent mechanisms [35], the protective effect of genistein against glucolipotoxicity-induced apoptosis in β-cells was not dependent on the ER-mediated pathway. First, ICI, a highly specific ER inhibitor, blocked E2 effect but did not affect the cytoprotective effect of genistein in β-cells. In addition, daidzein, an analogue of genistein that is essentially inactive as an ER ligand [36], also prevented β-cell apoptosis. Moreover, genistein primarily binds to ERβ but not ERα [36], and a recent study indicates that E2 acts primarily through ERα to protect β-cells from apoptosis [35]. Indeed, ERα expression level in the islets is 10-fold higher than that of ERβ [40]. These results further imply that genistein may prefer to activate GPR30 in β-cells, while E2 uses ERα to promote cell survival when all three receptors are present.
We further revealed that genistein prevents islets from apoptosis via GPR30-mediated activation of cAMP/PKA/CREB signaling. Our data showed that genistein strongly induced PKA-dependent, rapid phosphorylation of CREB in INS1 cells and islets, which was associated with the induction of the expression of the antiapoptotic Bcl-2 protein. Further, we demonstrated that Bcl-2 is a downstream effector of CREB in the genistein-triggered signaling cascade and cell survival, because knockdown of CREB diminished genistein-induced Bcl-2 protein expression and islet cell survival, indicating that CREB-mediated upregulation of Bcl-2 protein expression plays a major role in genistein promotion of β-cell survival. It is conceivable that genistein regulates Bcl-2 at the transcriptional levels, given that the cAMP responsive element (CRE) site present within Bcl-2 promoter plays a major role in inducing its expression [53]. Indeed, we found that genistein stimulated both cAMP-regulated gene expression and Bcl-2 promoter activity in β-cells. Finally, knockdown or deletion of GPR30 abolished genistein-stimulated CREB phosphorylation in β-cells, supporting that genistein acts on GPR30 to initiate the antiapoptotic signaling in β-cells.
In summary, we provide both in vitro and in vivo evidence for the first time that genistein stimulates cAMP signaling via a GPR30-mediated mechanism in β-cells, leading to improved islet survival. Loss of functional β-cell mass is the key for the deterioration of glycemic control in both T1D and T2D. In this context, genistein could be a naturally occurring agent that can be used as an alternative or complementary treatment for diabetes.
Highlights.
Phytonutrient genistein activates cAMP signaling via GPR30-mediated activation of Gαs in pancreatic beta-cells.
Genistein ameliorates hyperglycemia in diabetic mice via GPR30.
Genistein directly prevents islets from glucolipotoxicity-induced apoptosis via GPR30-mediated activation of CREB.
Acknowledgments
The work was supported by grants from National Center for Complementary and Integrated Health of National Institutes of Health (1R01AT007077 to D. Liu). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have no Conflict of Interest.
Author contributions
J. L., A.W., Y. W., and D. L. generated animal models and designed experiments; J.L., A. W., W. Z., D. L., Y. W., H. S., Z. J., H. A., Z. C., E. G., and B. X. performed research; J. L., A.W., and D. L. analyzed the data; J. L., A. W., D. L., E. G., and Z. C. wrote the paper.
References
- 1.AD A. Total Prevalence of Diabetes & Pre-diabetes. 2017 http://wwwdiabetesorg/diabetes-statistics/prevalencejsp.
- 2.King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care. 1998;21:1414–31. doi: 10.2337/diacare.21.9.1414. [DOI] [PubMed] [Google Scholar]
- 3.Stoffers DA. The development of beta-cell mass: recent progress and potential role of GLP-1. Horm Metab Res. 2004;36:811–21. doi: 10.1055/s-2004-826168. [DOI] [PubMed] [Google Scholar]
- 4.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006;27:356–70. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- 5.Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535–41. doi: 10.1210/jc.2004-0150. [DOI] [PubMed] [Google Scholar]
- 6.Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 7.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 8.Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–42. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 9.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–24. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 10.Rolin B, Larsen MO, Gotfredsen CF, Deacon CF, Carr RD, Wilken M, et al. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. American journal of physiology Endocrinology and metabolism. 2002;283:E745–52. doi: 10.1152/ajpendo.00030.2002. [DOI] [PubMed] [Google Scholar]
- 11.Erdman JW., Jr AHA Science Advisory: Soy protein and cardiovascular disease: A statement for healthcare professionals from the Nutrition Committee of the AHA. Circulation. 2000;102:2555–9. doi: 10.1161/01.cir.102.20.2555. [DOI] [PubMed] [Google Scholar]
- 12.Sacks FM, Lichtenstein A, Van Horn L, Harris W, Kris-Etherton P, Winston M. Soy protein, isoflavones, and cardiovascular health: an American Heart Association Science Advisory for professionals from the Nutrition Committee. Circulation. 2006;113:1034–44. doi: 10.1161/CIRCULATIONAHA.106.171052. [DOI] [PubMed] [Google Scholar]
- 13.Si H, Liu D. Genistein, a soy phytoestrogen, upregulates the expression of human endothelial nitric oxide synthase and lowers blood pressure in spontaneously hypertensive rats. The Journal of nutrition. 2008;138:297–304. doi: 10.1093/jn/138.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali AA, Velasquez MT, Hansen CT, Mohamed AI, Bhathena SJ. Modulation of carbohydrate metabolism and peptide hormones by soybean isoflavones and probiotics in obesity and diabetes. J Nutr Biochem. 2005 doi: 10.1016/j.jnutbio.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Mezei O, Banz WJ, Steger RW, Peluso MR, Winters TA, Shay N. Soy isoflavones exert antidiabetic and hypolipidemic effects through the PPAR pathways in obese Zucker rats and murine RAW 264.7 cells. The Journal of nutrition. 2003;133:1238–43. doi: 10.1093/jn/133.5.1238. [DOI] [PubMed] [Google Scholar]
- 16.Atteritano M, Marini H, Minutoli L, Polito F, Bitto A, Altavilla D, et al. Effects of the phytoestrogen genistein on some predictors of cardiovascular risk in osteopenic, postmenopausal women: a two-year randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2007;92:3068–75. doi: 10.1210/jc.2006-2295. [DOI] [PubMed] [Google Scholar]
- 17.Villa P, Costantini B, Suriano R, Perri C, Macri F, Ricciardi L, et al. The differential effect of the phytoestrogen genistein on cardiovascular risk factors in postmenopausal women: relationship with the metabolic status. J Clin Endocrinol Metab. 2009;94:552–8. doi: 10.1210/jc.2008-0735. [DOI] [PubMed] [Google Scholar]
- 18.Wei H, Wei L, Frenkel K, Bowen R, Barnes S. Inhibition of tumor promoter-induced hydrogen peroxide formation in vitro and in vivo by genistein. Nutrition & Cancer. 1993;20:1–12. doi: 10.1080/01635589309514265. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz-Larrea MB, Mohan AR, Paganga G, Miller NJ, Bolwell GP, Rice-Evans CA. Antioxidant activity of phytoestrogenic isoflavones. Free Radical Research. 1997;26:63–70. doi: 10.3109/10715769709097785. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, Zhen W, Yang Z, Carter JD, Si H, Reynolds KA. Genistein acutely stimulates insulin secretion in pancreatic beta-cells through a cAMP-dependent protein kinase pathway. Diabetes. 2006;55:1043–50. doi: 10.2337/diabetes.55.04.06.db05-1089. [DOI] [PubMed] [Google Scholar]
- 21.Fu Z, Zhang W, Zhen W, Lum H, Nadler J, Bassaganya-Riera J, et al. Genistein induces pancreatic beta-cell proliferation through activation of multiple signaling pathways and prevents insulin-deficient diabetes in mice. Endocrinology. 2010;151:3026–37. doi: 10.1210/en.2009-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–78. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 23.Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003;52:726–33. doi: 10.2337/diabetes.52.3.726. [DOI] [PubMed] [Google Scholar]
- 24.Liu D, Jiang H, Grange RW. Genistein activates the 3′,5′-cyclic adenosine monophosphate signaling pathway in vascular endothelial cells and protects endothelial barrier function. Endocrinology. 2005;146:1312–20. doi: 10.1210/en.2004-1221. [DOI] [PubMed] [Google Scholar]
- 25.Adams MR, Golden DL, Williams JK, Franke AA, Register TC, Kaplan JR. Soy protein containing isoflavones reduces the size of atherosclerotic plaques without affecting coronary artery reactivity in adult male monkeys. The Journal of nutrition. 2005;135:2852–6. doi: 10.1093/jn/135.12.2852. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Chen M, Fialkow LB, Ellett JD, Wu R, Nadler JL. The novel anti-inflammatory compound, lisofylline, prevents diabetes in multiple low-dose streptozotocin-treated mice. Pancreas. 2003;26:e99–104. doi: 10.1097/00006676-200305000-00021. [DOI] [PubMed] [Google Scholar]
- 27.Jia X, Chen Y, Zidichouski J, Zhang J, Sun C, Wang Y. Co-administration of berberine and plant stanols synergistically reduces plasma cholesterol in rats. Atherosclerosis. 2008;201:101–7. doi: 10.1016/j.atherosclerosis.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Fu Y, Luo J, Jia Z, Zhen W, Zhou K, Gilbert E, et al. Baicalein Protects against Type 2 Diabetes via Promoting Islet beta-Cell Function in Obese Diabetic Mice. Int J Endocrinol. 2014;2014:846742. doi: 10.1155/2014/846742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie T, Chen M, Zhang QH, Ma Z, Weinstein LS. Beta cell-specific deficiency of the stimulatory G protein alpha-subunit Gsalpha leads to reduced beta cell mass and insulin-deficient diabetes. Proc Natl Acad Sci U S A. 2007;104:19601–6. doi: 10.1073/pnas.0704796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parhami F, Fang ZT, Yang B, Fogelman AM, Berliner JA. Stimulation of Gs and inhibition of Gi protein functions by minimally oxidized LDL. Arterioscler Thromb Vasc Biol. 1995;15:2019–24. doi: 10.1161/01.atv.15.11.2019. [DOI] [PubMed] [Google Scholar]
- 31.Alenghat FJ, Tytell JD, Thodeti CK, Derrien A, Ingber DE. Mechanical control of cAMP signaling through integrins is mediated by the heterotrimeric Galphas protein. J Cell Biochem. 2009;106:529–38. doi: 10.1002/jcb.22001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghahremani MH, Cheng P, Lembo PM, Albert PR. Distinct roles for Galphai2, Galphai3, and Gbeta gamma in modulation offorskolin- or Gs-mediated cAMP accumulation and calcium mobilization by dopamine D2S receptors. Journal of Biological Chemistry. 1999;274:9238–45. doi: 10.1074/jbc.274.14.9238. [DOI] [PubMed] [Google Scholar]
- 33.Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006;102:175–9. doi: 10.1016/j.jsbmb.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 34.Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, et al. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103:9232–7. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, et al. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes. 2009;58:2292–302. doi: 10.2337/db09-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–63. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 37.Granata R, Settanni F, Gallo D, Trovato L, Biancone L, Cantaluppi V, et al. Obestatin promotes survival of pancreatic beta-cells and human islets and induces expression of genes involved in the regulation of beta-cell mass and function. Diabetes. 2008;57:967–79. doi: 10.2337/db07-1104. [DOI] [PubMed] [Google Scholar]
- 38.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–32. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 39.Tong JS, Zhang QH, Wang ZB, Li S, Yang CR, Fu XQ, et al. ER-alpha36, a novel variant of ER-alpha, mediates estrogen-stimulated proliferation of endometrial carcinoma cells via the PKCdelta/ERK pathway. PLoS One. 2010;5:e15408. doi: 10.1371/journal.pone.0015408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balhuizen A, Kumar R, Amisten S, Lundquist I, Salehi A. Activation of G protein-coupled receptor 30 modulates hormone secretion and counteracts cytokine-induced apoptosis in pancreatic islets of female mice. Mol Cell Endocrinol. 2010;320:16–24. doi: 10.1016/j.mce.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 41.Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5:421–7. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakrabarti S, Davidge ST. G-protein coupled receptor 30 (GPR30): a novel regulator of endothelial inflammation. PLoS One. 2012;7:e52357. doi: 10.1371/journal.pone.0052357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng SB, Graeber CT, Quinn JA, Filardo EJ. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids. 2011;76:892–6. doi: 10.1016/j.steroids.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 44.Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149:4846–56. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]
- 45.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, et al. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. 2007;2:536–44. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 46.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 47.Chevalier N, Vega A, Bouskine A, Siddeek B, Michiels JF, Chevallier D, et al. GPR30, the non-classical membrane G protein related estrogen receptor, is overexpressed in human seminoma and promotes seminoma cell proliferation. PLoS One. 2012;7:e34672. doi: 10.1371/journal.pone.0034672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang C, Prossnitz ER, Roy SK. G protein-coupled receptor 30 expression is required for estrogen stimulation of primordial follicle formation in the hamster ovary. Endocrinology. 2008;149:4452–61. doi: 10.1210/en.2008-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendez-Luna D, Bello M, Correa-Basurto J. Understanding the molecular basis of agonist/antagonist mechanism of GPER1/GPR30 through structural and energetic analyses. J Steroid Biochem Mol Biol. 2016;158:104–16. doi: 10.1016/j.jsbmb.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Teede HJ, Dalais FS, Kotsopoulos D, Liang YL, Davis S, McGrath BP. Dietary soy has both beneficial and potentially adverse cardiovascular effects: a placebo-controlled study in men and postmenopausal women. J Clin Endocrinol Metab. 2001;86:3053–60. doi: 10.1210/jcem.86.7.7645. [DOI] [PubMed] [Google Scholar]
- 51.Alkhalidy H, Moore W, Zhang Y, McMillan R, Wang A, Ali M, et al. Small Molecule Kaempferol Promotes Insulin Sensitivity and Preserved Pancreatic beta -Cell Mass in Middle-Aged Obese Diabetic Mice. J Diabetes Res. 2015;2015:532984. doi: 10.1155/2015/532984. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Consoli A, Nurjhan N, Capani F, Gerich J. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes. 1989;38:550–7. doi: 10.2337/diab.38.5.550. [DOI] [PubMed] [Google Scholar]
- 53.Pugazhenthi S, Miller E, Sable C, Young P, Heidenreich KA, Boxer LM, et al. Insulin-like growth factor-I induces bcl-2 promoter through the transcription factor cAMP-response element-binding protein. J Biol Chem. 1999;274:27529–35. doi: 10.1074/jbc.274.39.27529. [DOI] [PubMed] [Google Scholar]