

Abstract
Ischemic stroke remains a serious threat to human life. Generation of neuronal and vascular cells is an endogenous regenerative mechanism in the adult brain, which may contribute to tissue repair after stroke. However, the regenerative activity is typically insufficient for significant therapeutic effects after brain injuries. Pyruvate kinase isoform M2 (PKM2) is a key regulator for energy metabolism. PKM2 also has nonmetabolic roles involving regulations of gene expression, cell proliferation, and migration in cancer cells as well as noncancerous cells. In a focal ischemic stroke mouse model, recombinant PKM2 (rPKM2) administration (160 ng/kg, intranasal delivery) at 1 h after stroke showed the significant effect of a reduced infarct volume of more the 60%. Delayed treatment of rPKM2, however, lost the acute neuroprotective effect. We then tested a novel hypothesis that delayed treatment of PKM2 might show proregenerative effects for long-term functional recovery and this chronic action could be mediated by its downstream STAT3 signaling. rPKM2 (160 ng/kg) was delivered to the brain using noninvasive intranasal administration 24 h after the stroke and repeated every other day. Western blot analysis revealed that, 7 days after the stroke, the levels of PKM2 and phosphorylated STAT3 and the expression of angiogenic factors VEGF, Ang-1, and Tie-2 in the peri-infarct region were significantly increased in the rPKM2 treatment group compared with those of the stroke vehicle group. To label proliferating cells, 5-bromo-2′-deoxyuridine (BrdU, 50 mg/kg, i.p.) was injected every day starting 3 days after stroke. At 14 days after stroke, immunohistochemistry showed that rPKM2 increased cell homing of doublecortin (DCX)-positive neuroblasts to the ischemic cortex. In neural progenitor cell (NPC) cultures, rPKM2 (0.4–4 nM) increased the expression of integrin β1 and the activation/phosphorylation of focal adhesion kinase (FAK). A mediator role of FAK in PKM2-promoted cell migration was verified in FAK-knockout fibroblast cultures. In the peri-infarct region of the brain, increased numbers of Glut-1/BrdU and NeuN/BrdU double-positive cells indicated enhanced angiogenesis and neurogenesis, respectively, compared to stroke vehicle mice. Using Laser Doppler imaging, we observed better recovery of the local blood flow in the peri-infarct region of rPKM2-treated mice 14 days after stroke. Meanwhile, rPKM2 improved the sensorimotor functional recovery measured by the adhesive removal test. Inhibiting the STAT3 phosphorylation/activation by the STAT3 inhibitor, BP-1-102 (3 mg/kg/day, o.g.), abolished all beneficial effects of rPKM2 in the stroke mice. Taken together, this investigation provides the first evidence demonstrating that early treatment of rPKM2 shows an acute neuroprotective effect against ischemic brain damage, whereas delayed rPKM2 treatment promotes regenerative activities in the poststroke brain leading to better functional recovery. The underlying mechanism involves activation of the STAT3 and FAK signals in the poststroke brain.
Electronic supplementary material
The online version of this article (10.1007/s13311-018-0635-2) contains supplementary material, which is available to authorized users.
Key Words: Ischemic stroke, neuroprotection, pyruvate kinase isoform M2, STAT3, FAK, neuroblasts, proliferation, angiogenesis, neurogenesis, LCBF, sensorimotor function
Introduction
Stroke remains a leading cause of human death and long-term disability in the USA and around the world. To date, the effective treatments for stroke are still very limited. In addition to previous and current approaches of neuroprotective treatments, innovative therapeutics such as delayed treatments that can promote regenerative activities and improve functional recovery after stroke are the recent focus in stroke research [1, 2]. Pyruvate kinase M2 (PKM2) is one of the four pyruvate kinase isoforms (PKM1, PKM2, PKL, and PKR) expressed in mammalian cells [3]. Pyruvate kinases regulate the final rate-limiting step of glycolysis by catalyzing the transfer of a phosphate group from phosphoenolpyruvate to ADP to produce pyruvate and ATP [4]. Among the four isoforms, PKM1 and PKM2 are ubiquitously expressed in different types of cells and tissues [3]. PKM2 is highly expressed in proliferating cells such as cancer cells [3, 5]. Expression and activity of PKM2 are regulated at multiple levels, including gene expression, alternative splicing, and post-translational modification, and by metabolic intermediates and growth signaling pathways [6]. Hence, PKM2 is a unique multifaceted regulator that can improve cellular adaptation in their metabolic states to match physiological needs in different environments [7]. Some recent attention has been focused on PKM2 because of its critical roles in cellular metabolism and cell proliferation of cancer cells and immune cells [8]. PKM2 is thus indicated as a potential therapeutic target for cancer therapy [9, 10]. We hypothesized that the increased glycolytic capacity induced by PKM2 could be utilized to battle against hypoxia- and ischemia-induced brain injuries such as ischemic stroke. There is so far no report about the neuroprotective effect of PKM2.
In addition to regulating glycolysis, PKM2 has nonmetabolic functions such as transcriptional regulations of cell cycle progression [11–13]. In contrast to the mitochondrial respiratory reaction, energy regeneration by these pyruvate kinases is independent from oxygen supply and allows survival of the organs under hypoxic conditions [14]. PKM2 may also act as a coactivator of hypoxia-induced factor 1-alpha (HIF-1α); the latter behaves as a master transcription factor to regulate multiple signaling pathways in response to hypoxic insults [15]. Increased levels and activities of PKM2 are associated with enhanced motility and metastasis of tumor cells; the molecular mechanism involved in the cell migration is so far poorly understood.
We noticed that increased aerobic glycolysis and cell proliferation or migration are not unique to cancer and malignancy, but rather originate in normal biology and physiological development [16]. In physiological proliferation of neural progenitors or under hypoxia, PKM2 helps to reprogram energy metabolism to support growth and adaptation. Thus, metabolic transformation is a co-opting of developmental episodes integral to physiological growth [17]. Presently, these important metabolic and nonmetabolic roles of PKM2 were mainly identified in cancerous cells; the potential nonmetabolic function in normal cells or in the response to brain injuries such as ischemic stroke is unknown.
Because of its increased expression and significant changes in cellular metabolism, PKM2 has been identified as a biomarker for tissue damage such as acute kidney injury [18]. PKM2 can regulate gene transcription MEK5 by phosphorylating STAT3, which in turn activates numerous transcriptional factors to regulate cell proliferation, differentiation, migration, and survival [11, 12, 19]. PKM2 greatly promotes tumorous angiogenesis through enhancing the endothelial cell migration and extracellular matrix attachment [20]. PKM2 was recently found to be released by neutrophils at peripheral wound sites and to facilitate early wound healing by promoting angiogenesis [21]. Studies on functions of intracellular and extracellular PKM2 seem to suggest that PKM2 orchestrates tissue regeneration in response to various injuries and stresses by both adjusting cellular metabolism and promoting angiogenesis under stress conditions. This scenario well reflects the role of PKM2 in tissue damage repair. Whether PKM2 can be used as a therapeutic reagent to promote regeneration in neurological diseases is an intriguing but unexplored issue.
Intranasal administration has been proven a noninvasive method with great clinical relevance to deliver protein, neuropeptides, and even cells into the brain by utilizing the olfactory neuronal distribution pathways in the cribriform plate, which bypasses the blood–brain barrier (BBB), and directly guides therapeutics from nose to brain tissues [22, 23]. In the present investigation, recombinant PKM2 (rPKM2) was delivered to the ischemic brain using the intranasal method for clinical feasibility. We tested the possibility that PKM2 downstream signals involving cell survival and regeneration such as STAT3 and other regenerative factors contributed to PKM2’s effects. The novel hypothesis that PKM2 could regulate key adhesion/migration factors such as the focal adhesion kinase (FAK) and therefore accelerate cell migration was examined in cultured FAK gene-modified cells and in the ischemic brain. Our data support that rPKM2 may be explored as an early stroke treatment for acute neuroprotection as well as a delayed stroke treatment to promote angiogenesis, neurogenesis, and functional recovery through activation of downstream intracellular signaling pathways.
Methods and Materials
Focal Ischemic Stroke Model of Mice
C57BL/6 (24–28 g, 9–13 weeks old) mice were housed at 21 to 22 °C room temperature with a 12-h light/dark cycle in the pathogen-free Laboratory Animal Center for Research at Emory University. Occlusions of the right middle cerebral artery (MCA) were performed according to previous procedures with modified artery occlusion procedures [24]. Briefly, animals were subjected to ketamine/xylazine (ketamine 80–100 mg/kg i.p., xylazine 10–12.5 mg/kg i.p.) anesthesia, and the right MCA supplying the sensorimotor cortex was permanently ligated by a 10-0 suture (Surgical Specialties CO., Reading, PA). The creation of the right sensorimotor cortex ischemia was completed by bilateral occlusion of the common carotid arteries (CCA) for 7 min followed by reperfusion. During surgery and recovery periods, body temperature was monitored and maintained at 37.0 ± 0.5 °C using a temperature-control unit and heating pads. All animal experiments and surgery procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University.
Drug Administration
Recombinant pyruvate kinase M2 (rPKM2) was produced by Dr. Zhi-Ren Liu’s group according to the published protocol [20, 21]. rPKM2 (160 ng/kg) was administered intranasally according to established procedures every other day starting 24 h after stroke until the animals were sacrificed [22]. To label proliferating cells, 5-bromo-2′-deoxyuridine (BrdU) (Sigma, St. Louis, MO) was administered to all animals (50 mg/kg/day, intraperitoneal injection) beginning on day 3 after stroke and continued once daily until sacrifice. The STAT3 inhibitor XVIII, BP-1-102 (3 mg/kg, Millipore, Billerica, MA) was administered orally once daily. Saline was used as vehicle control of drug treatments.
Immunohistochemistry
Fresh frozen coronal brain sections were cut at 10 μm thickness using a cryostat (Leica CM 9500, Leica Biosystems, Buffalo Grove, IL). Sections were dried on a slide warmer for 30 min, fixed with 10% buffered formalin for 10 min, permeabilized with 0.2% Triton-X 100 (in PBS) for 5 min, blocked for 1 h with 1% fish gel at room temperature, and then incubated with the primary antibodies Glut-1 (1:800; Millipore), NeuN (1:400; Millipore), collagen IV (Millipore), and integrin αvβ3 (Santa Cruz, Dallas, TX) overnight at 4 °C. Slides were incubated with anti-mouse, anti-rabbit, or anti-rat secondary antibodies for 2 h at room temperature. Vectashield mounting media for fluorescence (Vector Laboratory, Burlingame, CA) were used to coverslip slides in preparation for microscopy and image analysis. For BrdU (1:600, AbD serotec) staining, slides were incubated in 2 N HCl at 37 °C for 1 h, followed by 0.2% Trition X 100 incubation for 45 min. For DCX (1:50, Santa Cruz Biotechnology) staining, animals were transcardially perfused with warm saline and 4% paraformaldehyde. Pictures were taken by using fluorescence microscopy (BX61; Olympus, Tokyo, Japan) along the peri-infarct region defined morphologically as the region just outside the stroke core. Confocal images were taken on a confocal microscope (FV1000, Olympus) to confirm colocalization.
For systematic random sampling in design-based stereological cell counting, 6 coronal brain sections per mouse were selected, spaced 90 μm apart across the same region of interest in each animal. For multistage random sampling, 6 fields per brain section were randomly chosen in the peri-infarct/penumbra region of the brain. Sections from different animals represent the same area in the anterior to posterior direction. Vessel density was measured using ImageJ (NIH). Neuroblast migration distance was determined and quantification of neuroblast migration was performed as described before [25, 26]. Briefly, proliferating cells in the subventricular zone (SVZ) and their migration towards the ischemic cortex were captured in a series of 6 to 10 images, depending on the distance of migration, at × 10 or × 20 magnification with 6 sections analyzed per animal. Analyses were performed on coronal brain section images taken within the region of interest including the SVZ migration tract and the peri-infarct region. Cells colabeled with BrdU and DCX were counted as newly formed neural progenitors. Migration distance (μm) was evaluated from the SVZ to the furthest colabeled DCX- and BrdU-positive cell, determined by confocal imaging.
Western Blot Analysis
The brain tissue from the peri-infarct region was collected at 7 days and 14 days after stroke. The protein was extracted using lysis buffer containing protease inhibitor (1:100). Protein (30 μg) from each sample was loaded into a gradient gel and run at constant current until protein markers had adequately separated. They were transferred onto polyvinyl difluoride membranes that were then probed by using standard protocols. Primary antibodies PKM2 (1:1000; provided by Dr. Zhi-Ren Liu), phospho-Stat3 (Tyr705) (1:1000; Cell Signaling, Danvers, MA), Tie-2 (1:200; Santa Cruz), integrin β1 (1:1000, Cell Signaling), phospho-FAK (Tyr397) (1:1000, Millipore), and mouse β-actin antibody (1:6000; Sigma-Aldrich, St. Louis, MO) were applied overnight at 4 °C. Alkaline phosphatase-conjugated secondary antibodies were applied for 1 to 2 h at room temperature. Alkaline phosphatase-conjugated antibodies were developed by using nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate solution. The intensity of each band was measured and subtracted by the background using NIH ImageJ software. The expression ratio of each target protein was then normalized against β-actin.
Local Cerebral Blood Flow (LCBF) Measurement
Laser scanning imaging was used to measure LCBF as previously described [27] at 3 time points: immediately before MCA ligation, during the 7-min bilateral common carotid artery ligation, and 14 days after ischemia. Briefly, animals were anesthetized with an injection of 4% chloral hydrate solution and an incision was made to expose the skull above the territory of the right MCA. The laser was centered over the right coronal suture. Different from the conventional laser Doppler probe that measures a small point of blood flow, the scanner method measures a 2.4 × 2.4 mm2 area using the laser Doppler perfusion imaging system (PeriFlux System 5000-PF5010 LDPM unit, Perimed, Stockholm, Sweden). This scanning measurement largely avoids inaccurate or biased results caused by inconsistent locations of the laser probe. Data was analyzed using the LDPI Win 2 software (Perimed AB, Stockholm, Sweden).
Adhesive Removal Test
The adhesive removal test measures sensorimotor function as previously described [28]. A small adhesive dot was placed on each forepaw, and the amount of time (seconds) needed to contact and remove the sticker from each forepaw was recorded. Recording stopped if the animal failed to contact the sticker within 2 min. Mice were trained 3 times before stroke surgery to ensure that they were able to remove the tape. The test was performed 3 times per mouse, and the average time was used in the analysis before stroke and 14 days after stroke.
Isolation and Culture of Mouse Neural Progenitor Cells (NPCs)
NPCs were isolated from C57/BL6 mice and cultured according to previously described protocol [29]. Briefly, the tissue from the subventricular zone (SVZ) of postnatal day 1 to 3 (P1–13) mice was isolated and dissociated to single-cell suspension. Cells were then plated in culture medium, containing DMEM and Ham’s F12 medium (DMEM/F12, 1:1) (Invitrogen, Carlsbad, CA) supplement with 2% B27 (Invitrogen), 20 ng/ml epidermal growth factor (EGF) (Invitrogen), and 20 ng/ml basic fibroblast growth factor (bFGF) (Invitrogen). 5 to 7 days after plating, neurospheres were mechanically split and plated in the same medium with a 1:3 ratio. Passage 2 to 4 NSCs were used in the present study.
Mouse Embryonic Fibroblast (MEF) Cell Cultures
FAK wild-type (FAK+/+) and FAK knockout (FAK−/−) MEF cell lines were generated in our lab [30]. Cells were cultured in DMEM medium (Invitrogen) supplemented with 10% FBS (Sigma).
Transwell Migration Assay
Neural progenitor cells were treated with rPKM2 (0.4–40 nM) in regular culture plates. The treated cells were resuspended in DMEM/F12 medium containing rPKM2 (0.4–40 nM) and then plated in transwell inserts with 8-μm pores (Corning, Durham, NC). The transwell inserts containing cell suspension were placed in 24-well plates with regular NPC culture medium and chemoattractant SDF-1 (200 ng/ml) in each well. 24 h after plating, the cells were fixed with 4% paraformaldehyde. The cells on the upper membrane surface of the inserts were removed using a cotton swab. The cells on the bottom membrane surface of the inserts were stained with Hoechst 33342 (Invitrogen) and Acti-stain 555 phalloidin (Cytoskeleton, Denver, CO). For migration assay performed on FAK+/+ or FAK−/− MEFs, a similar protocol was used as NPCs. However, treated MEFs were resuspended in DMEM medium without serum and plated in transwell inserts with 8-μm pores, which were placed in 24-well plates containing DMEM medium with 10% FBS. 2 h after plating, the number of MEFs on the bottom membrane of inserts was counted.
Statistical Analysis
Multiple comparisons were done using 1-way ANOVA followed by Tukey test or 2-way ANOVA followed by Bonferroni test. Changes were identified as significant if p was less than 0.05. Mean values were reported together with the standard error of the mean (SEM).
Results
Acute Neuroprotective Effect of PKM2 After Ischemic Stroke of the Mouse
A focal ischemic stroke model of the mouse caused the formation of infarction in the right sensorimotor cortex 3 days after the ischemic insult (Fig. 1A). Recombinant PKM2 (rPKM2, 160 ng/kg/day) was administered via intranasal delivery at 1 h or 24 h after stroke. The intranasal method is an established noninvasive brain delivery of drugs, neuropeptides, and even exogenous cells [22]. The acute treatment of 1-h delayed PKM2 markedly attenuated the infarct formation (~ 60% of reduction, Student t test, F = 11, p = 0.019; n = 6 per group), whereas the neuroprotective effect disappeared when PKM2 was given at 24 h after stroke (Fig. 1A).
Fig. 1.

Neuroprotective effect of rPKM2 and expression levels of PKM2 in the normal and stroke brain. Adult male mice were subjected to focal cerebral ischemia targeting the right sensorimotor cortex. The effect of rPKM2 intranasal administration (1 or 24 h after stroke) on brain damage was measured using 2,3,5-triphenyltetrazolium chloride (TTC) staining in brain sections 3 days after stroke. PKM2 and the downstream molecule STAT3 were measured using Western blotting. A Brain sections of TTC staining images in stroke control, stroke with 1-h-delayed rPKM2 (160 ng/kg, intranasal delivery), and stroke with 24-h-delayed rPKM2. White color represents the ischemic infarction. The bar graph on the right summarizes the infarction ratio in the 3 groups. With 1-h delay, rPKM2 showed significant neuroprotective effect of reducing the brain infarct formation. Asterisk, p < 0.05 versus stroke control, n = 6 per group. mean ± SEM. B Western blot analysis of PKM2 levels in the normal (sham) and stroke brains. A basal and significant expression of PKM2 protein was readily identified in the normal cortex. The bar graph on the right shows that after a focal ischemic insult, the PKM2 level was upregulated at 1 day after stroke. The PKM2 level gradually decreased thereafter and declined to a level significantly lower than 1 day after stroke as well as that in sham controls. 1-way ANOVA: asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus 1 day after stroke and sham, n = 5 per group. C PKM2 expression in the peri-infarct region 14 days after stroke with and without rPKM2 treatment (160 ng/kg per day, intranasal delivery). Student t test: asterisk, p < 0.05 versus sham. n = 5 in stroke group, n = 3 in stroke + PKM2 group. D The phosphorylation level of STAT3 expression in the peri-infarct region at 7 days after stroke. The pSTA3 level increased after stroke whereas the rPKM2 treatment further upregulated its expression. The STAT3 inhibitor BP-1-102 (BP) eliminated the effect of rPKM2. 2-way ANOVA: asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus stroke; delta, p < 0.05 versus stroke + PKM2. n = 4 in sham group, n = 5 in stroke, stroke + PKM2, and stroke + PKM2 + BP groups
Expression of PKM2 in the Normal and Stroke Brain of the Adult Mouse and Increased Phosphorylation of STAT3 by Intranasal Delivery of rPKM2
Although delayed rPKM2 treatment did not show a neuroprotective effect, we assume that its regenerative action might benefit tissue repair in the poststroke brain. To justify a chronic treatment with PKM2, we measured the expression level of PKM2 in the normal and stroke brain. PKM2 has been extensively investigated in cancer/tumor cells including neuroblastoma cells; its expression in normal brain tissues is obscure. We examined the PKM2 expression in the mouse sham control brain and possible changes after ischemic stroke. A basal level of PKM2 expression was clearly seen in the control cortex, whereas the PKM2 level was transiently increased in the peri-infarct region 1 day after focal cerebral ischemia. The PKM2 level then gradually declined to lower levels at 3 and 7 days after stroke (Fig. 1B). Based on this observation, exogenous rPKM2 was administered to prevent the decline of PKM2 in the poststroke brain, and we tested the hypothesis that the delayed administration of rPKM2 might show regenerative effects for poststroke recovery. Stroke mice received intranasally delivered rPKM2 (160 ng/kg/day) from 1 day after stroke. 7 days after stroke, the PKM2 level in the peri-infarct region of the ischemic cortex remained significantly higher than that in the stroke control (Fig. 1C).
PKM2 can activate STAT3 and its transcription of a number of downstream genes in cancer cells [12]. In the ischemic brain 7 days after stroke, we investigated the effect of rPKM2 on the STAT3 signaling. Western blot analysis showed that, 7 days after stroke, animals that received the rPKM2 treatment had significantly higher levels of phosphorylated STAT3 (pSTAT3) (Fig. 1D). BP-1-102 is a STAT3 inhibitor that suppresses the phosphorylation of STAT3. In stroke animals that received rPKM2 and BP-1-102 (3 mg/kg/day, o.g.), the level of pSTAT3 in the peri-infarct region was significantly lower than that in animals that received rPKM2 treatment alone (Fig. 1D).
The effect of rPKM2 on STAT3 showed a dose-dependent manner. Reducing rPKM2 to 80 ng/kg did not cause a significant increase in p-STAT3 (Fig. 2A, B). Meanwhile, the rPKM2 treatment showed a dose-dependent upregulation on the angiogenic factors VEGF, Tie-2, and Ang-1 (Fig. 2). Based on above observations, the rPKM2 dosage of 160 ng/kg was selected to test in the following experiments.
Fig. 2.

Effects of rPKM2 treatment on the expression of angiogenic factors in the peri-infarct cortex. Western blot analysis was used to evaluate effects of rPKM2 treatment at 2 dosages (80 and 160 ng/kg, intranasal delivery from 1 day after stroke) on p-STAT3 and angiogenic factors in the peri-infarct cortex at 7 days after stroke. A–C The rPKM2 treatment shows dose-dependent increases in the expression of p-STAT3 and VEGF. At 160 ng/kg, rPKM2 significantly enhanced both factors. Note that 2 p-STAT3 bands were detected in this assay using the Phospho-Stat3 (Tyr705) antibody, which is consistent with the previous report (Cell Signaling; https://www.cellsignal.com/products/primary-antibodies/phospho-stat3-tyr705-d3a7-xp-rabbit-mab/9145). D–F rPKM2 at the higher dosage of 160 ng/kg significantly increased the expression of Tie-2 and Ang-1, whereas it also increased Tie-2 expression at a dosage of 80 ng/kg. n = 3 in sham control and n = 4 in rPKM2 groups. Asterisk, p < 0.05 versus sham controls
rPKM2 Treatment Promoted Angiogenesis After Ischemic Stroke
We tested the hypothesis that rPKM2 can promote angiogenesis in the ischemic brain through activation of STAT3 signaling. In the mouse focal ischemic stroke model, BrdU (50 mg/kg, i.p.) was injected to label the proliferating cells in the mouse. Immunohistochemical staining of the blood vessel marker Glut-1 and BrdU on brain sections was performed 14 days after stroke. The colabel of Glut-1 and BrdU indicated proliferating or growing vessels. In the peri-infarct region, there were more Glut-1/BrdU-colabeled cells 14 days after stroke compared with those in sham control animals, suggesting increased angiogenic activity after stroke. Stroke animals that received rPKM2 treatment showed an even greater number of Glut-1/BrdU-colabeled cells in the peri-infarct region compared to stroke vehicle controls (Fig. 3A–C). The STAT3 inhibitor BP-1-102 significantly reduced the number of Glut-1/BrdU-colabeled cells and Glut-1-positive vessels in the peri-infarct region compared with that of stroke animals that received rPKM2 (Fig. 3C). This data supported that the promoting effect of PKM2 on angiogenesis was largely mediated by the STAT3 signaling.
Fig. 3.

rPKM2 promoted angiogenesis after ischemic stroke. Immunohistochemical staining was applied to examine vascular markers in the peri-infarct cortical region. A Immunostaining images of the vascular endothelial cell marker Glut-1 (green) and the proliferating marker BrdU (red) in sham control, stroke, and experimental groups 14 days after stroke. Arrows point to representative BrdU and Glut-1-positive cells. B A confocal image shows the colabeling of Glut-1 (green) and BrdU (red), indication of proliferating endothelial cells. C Quantified data of Glut-1/BrdU-colabeled cells. rPKM2 treatment (stroke + PKM2) increased the number of proliferating vascular cells, indicating significantly enhanced angiogenesis in stroke animals. The STAT3 inhibitor BP-1-102 eliminated the effects of rPKM2. Mean ± SEM, n = 6 in sham group, n = 9 in stroke group, n = 6 in stroke animals that received rPKM2 treatment (stroke + PKM2) group, n = 6 in stroke animals received rPKM2 and STAT3 inhibitor BP-1-102 (stroke + PKM2 + BP) group. Asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus stroke; delta, p < 0.05 versus stroke + PKM2. D The immunostaining of integrin αvβ3 (red) as an angiogenic factor in the peri-infarct region at 14 days after stroke. E Costaining of integrin αvβ3 (red) and the extracellular matrix marker collagen IV. F The expression of integrin αvβ3 was quantified by the area fraction function using the NIH image J software. rPKM2 treatment significantly increased the level of integrin αvβ3 in the peri-infarct region, which was blocked by coapplied BP-1-102. n = 6 in each group; asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus stroke; delta, p < 0.05 versus stroke + PKM2
Since integrin αvβ3 is expressed on endothelial cells and plays an important responding role during the angiogenesis process [31–34], we examined the effect of rPKM2 treatment on integrin αvβ3 expression after stroke. Immunostaining imaging 14 days after stroke showed overlaid expression of integrin αvβ3 and collagen IV (Fig. 3E). The integrin αvβ3-positive area in stroke animals was lower in the peri-infarct region (2.77% of × 20 objective field) compared with that in animals in the sham group (3.95% of the field). rPKM2 treatment restored the level of integrin αvβ3 in stroke animals (3.71% per field). Inhibiting STAT3 with BP-1-102 blocked the effect of PKM2 (Fig. 3D, F).
rPKM2 Increased Migration Factors and Promoted NPC Migration In Vitro
In addition to its role in angiogenesis, STAT3 signaling has been shown to play a critical role in neurogenesis including neuroblast migration ad neuronal differentiation in the central nervous system (CNS) [35, 36]. The integrins and focal adhesive kinase (FAK) are key molecules in cell migration [30]. We examined the effect of rPKM2 on the expression of integrin β1 and phosphorylation/activation of FAK in neural progenitor cells (NPCs) isolated from the brain of C57/BL6 mouse P1–3 pups. Western blotting revealed that the level of integrin β1 in NPCs increased 24 h after 4 nM rPKM2 treatment. Moreover, phosphorylated FAK was increased by the rPKM2 treatment 48 h after adding rPKM2 (0.4 nM and 4 nM; Fig. 4A–C).
Fig. 4.

rPKM2 increased migration factors and migration of neural progenitor cells. In vitro assays were performed to examine the neural progenitor cells (NPCs) dissected from the SVZ region. A The protein level of integrin β1 and FAK activation was examined in NPC cultures using Western blotting after 24 and 48 h exposure to rPKM2 (0.4 and 4 nM). B Quantification of Western blot data showed that NPCs exposed to 24 h of rPKM2 (4 nM) expressed a higher level of integrin β1. C The level of FAK phosphorylation (p-FAK) increased in NPCs exposed to 48 h of rPKM2 (0.4 and 4 nM). 3 replicates in each group and experiments were repeated 3 times. Asterisk, p < 0.05 versus control group. D and E Immunochemistry staining showed that these NPCs expressed immature neuronal markers Nestin (green) and DCX (red). F Transwell migration assay was performed to detect the effect of rPKM2 on the migration of NPCs. NPCs were treated with rPKM2 at different concentrations for 48 h prior to the migration measurement. 24 h after plating in transwell inserts, NPCs of the bottom membrane of inserts were fixed and stained with nuclei marker Hoechst 33342 and cytoskeleton marker Acti-stain 555 phalloidin. rPKM2 (0.4–4 nM) was added in the inserts (upper chamber) and the chemoattractant factor SDF-1 was added to the bottom inset to attract cell migration. There were more NPCs migrated to the bottom membrane of the inserts with 0.4 and 4 nM rPKM2. n = 3 independent assays; asterisk, p < 0.05 versus control group
The effect of rPKM2 on cell migration was next tested in NPC cultures. Immunochemistry staining verified that these NPCs showed Nestin and doublecortin (DCX) expressions (Fig. 4D). In the transwell assay of directed migration in response to the chemoattractant SDF-1 placed on the bottom chamber, rPKM2 (0.4 nM and 4 nM) pretreatment for 48 h significantly increased the number of NPCs migrated to the bottom membrane of inserts (Fig. 4E, F).
The Mediating Role of FAK in rPKM2-Promoted Cell Migration
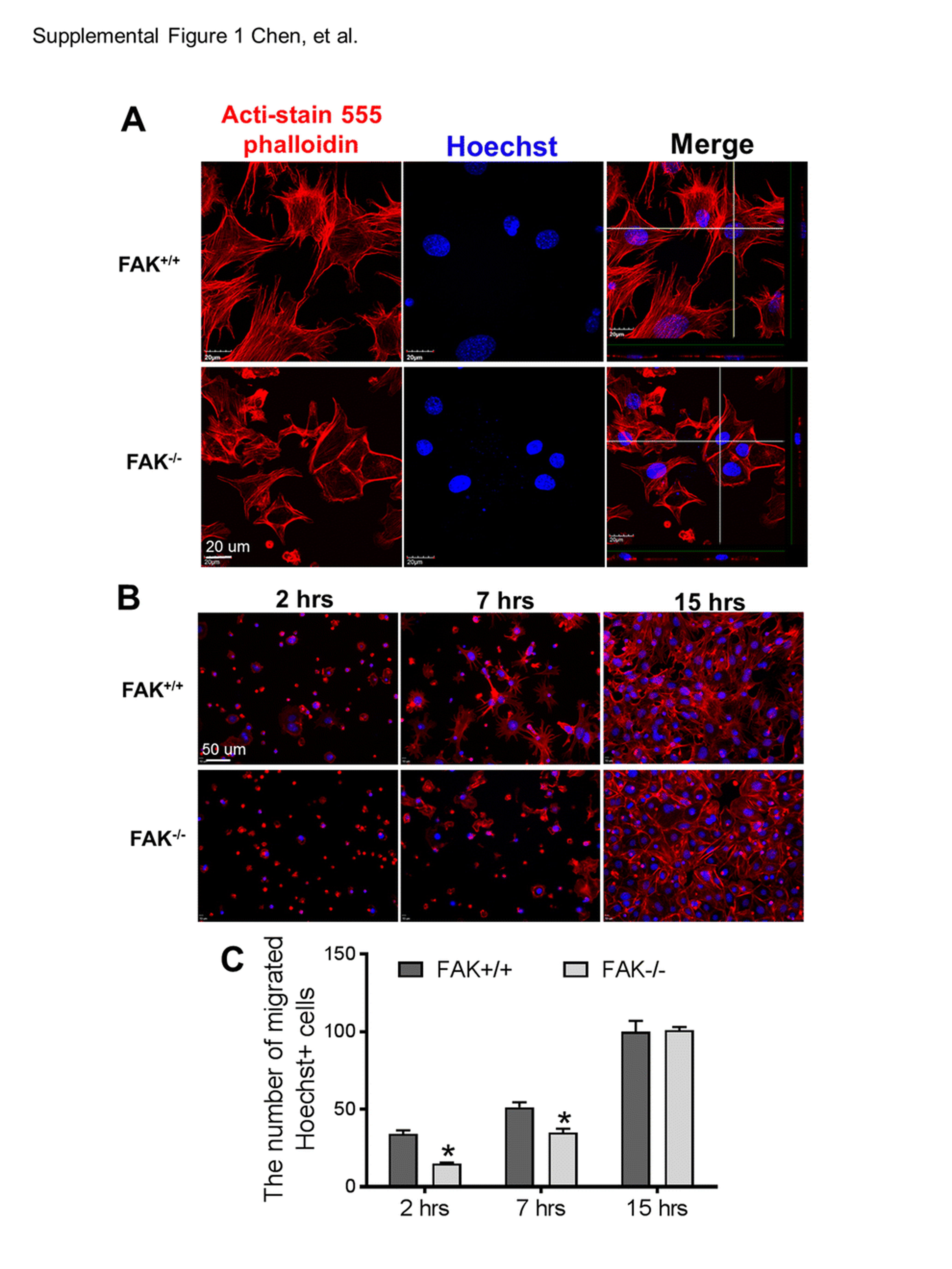
To further understand the molecular mechanism underlying the rPKM2 effect on cell migration, we performed the rPKM2 treatment in wild-type mouse embryonic fibroblast (FAK+/+ MEF) cultures and FAK knockout mouse embryonic fibroblast (FAK−/− MEFs) cultures. Immunocytochemistry of the cytoskeleton marker Acti-stain 555 phalloidin revealed that the cytoskeleton was degraded and migratory morphology including the formation of lamellipodia in FAK−/− MEFs was reduced (Suppl. Fig. 1A). Transwell migration assay showed that fewer FAK−/− MEFs migrated to the bottom membrane of inserts at 2 and 7 h after plating compared with FAK+/+ MEFs, although eventually no difference was seen at 15 h after plating (Suppl Fig. 1B, C). The rPKM2 exposure (4 nM or 40 nM) for 48 h prior to the transwell assay significantly increased the number of FAK+/+ MEFs migrating to the bottom membrane of inserts counted at 2 h after plating (Fig. 5A, B). This promoting effect of rPKM2, however, was not seen in FAK−/− MEFs (Fig. 5A, B). These results indicated that FAK plays an important mediator role in the effect of rPKM2 on cell migration.
Fig. 5.

Knocking out FAK abolished the migratory effects of rPKM2 in mouse embryonic fibroblasts. The transwell migration assay was performed on cultured mouse embryonic fibroblasts (MEFs), taking the advantage of FAK gene manipulation in these cells. A The effect of rPKM2 on the migration of wild-type MEFs (FAK+/+) and FAK knockout MEFs (FAK−/−). Cells moved to the bottom membrane of inserts were stained with nuclei marker Hoechst 33342 (blue) and cytoskeleton marker Acti-stain 555 phalloidin (red). B The number of cells on the bottom membrane of inserts was increased by 48 h exposure to rPKM2 (4 nM and 40 nM) in wild-type MEFs (FAK+/+), whereas in FAK knockout MEF (FAK−/−) cultures there was no change in the number of cells on the bottom membrane of inserts by rPKM2 treatment. Asterisk, p < 0.05 versus control group
rPKM2 Promoted Neuroblast Migration in the Ischemic Brain
The regulatory effect of rPKM2 on neuroblast migration was then examined in the focal ischemic stroke mice. rPKM2 (160 ng/kg) was intranasally administered as described above. Immunochemistry staining of DCX and proliferation marker BrdU was performed on coronal brain sections at 14 days after stroke [37]. DCX/BrdU-colabeled cells were observed along the migrating track from the ipsilateral SVZ to the ischemic cortex (Fig. 6A). The distance of neuroblasts migrating towards the ischemic cortex was significantly longer in stroke animals that received rPKM2 treatment than those that received the vehicle control (Fig. 3B). There were a significantly greater number of DCX/BrdU-colabeled cells in stroke animals that received rPKM2 treatment than those that received the vehicle control (Fig. 6C, D).
Fig. 6.

The rPKM2 intranasal treatment enhanced neurogenesis and neuroblast migration after ischemic stroke in mice. The migration of neuroblasts from the SVZ towards the ischemic cortex was examined using immunohistochemical staining. A The neuroblast migration and regeneration along white matter between the ipsilateral SVZ (labeled as V in the image) to ischemic cortex was examined using the migrating neural progenitor marker DCX (green) and the proliferating cell marker BrdU (red) 14 days after stroke. B The migration distance was evaluated from the SVZ to the furthest colabeled DCX and BrdU-positive cell, determined by confocal imaging. rPKM2 treatment increased the average distance of neuroblast migrating towards the ischemic cortex. C and D Confocal images of DCX/BrdU-colabeled cells in the migration path 14 days after stroke. An increased number of DCX/BrdU cells were identified in stroke mice that received rPKM2 (D). Mean ± SEM, n = 5 in each group. Number sign, p < 0.05 versus stroke. E Immunochemistry staining of mature neuronal marker NeuN (green) and BrdU (red) was performed to evaluate neurogenesis 14 days after stroke. F and G The number of NeuN/BrdU-colabeled cells in confocal images of the peri-infarct region in each group was analyzed. The rPKM2 treatment significantly increased the number of BrdU-positive cells and the number of NeuN/BrdU-colabeled cells in the peri-infarct region 14 days after stroke, whereas STAT3 inhibitor BP-1-102 blocked the effect of rPKM2. Mean ± SEM, n = 6 in sham group, n = 6 in stroke group, n = 8 in stroke + PKM2 group, n = 5 in stroke + PKM2 + BP (BP-1-102) group. Asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus stroke; delta, p < 0.05 versus stroke + PKM2
rPKM2 Treatment Promoted Neuronal Differentiation in the Ischemic Brain
In poststroke neurogenesis, neuronal differentiation is a later but critical process. We examined newly formed neurons in the peri-infarct region. The mature neuronal marker NeuN and proliferation marker BrdU were costained at 14 days after stroke. More BrdU-positive cells and NeuN/BrdU-colabeled cells were found in stroke animals that received the rPKM2 treatment (Fig. 6E–G). Coapplied STAT3 phosphorylation inhibitor BP-1-102 (3 mg/kg) blocked the effect of rPKM2 on the neuronal differentiation in the ischemic brain (Fig. 6E–G).
rPKM2 Treatment Enhanced the LCBF and Behavioral Recovery After Ischemic Stroke
To examine whether improved neurogenesis by rPKM2 could result in functional improvements after stroke, we measured the local cerebral blood flow (LCBF) in the peri-infarct region using a laser Doppler scanner and the sensorimotor function using the adhesive removal test. At 14 days after stroke, the LCBF of stroke control animals showed 80% of the basal level measured before stroke. Stroke animals that received rPKM2 showed a significantly higher level of LCBF (94% of basal normal flow) (Fig. 7A, B). The STAT3 inhibitor BP-1-102 coapplied with rPKM2 blocked the effect of rPKM2 on LCBF (Fig. 7A, B).
Fig. 7.

rPKM2 improved LCBF restoration and attenuated the behavioral deficits after ischemic stroke. A The local cerebral blood flow (LCBF) in the peri-infarct region was measured 14 days after stroke using a laser Doppler image scanner. B The restoration of LCBF in each group was calculated by the ratio of the mean value of LCBF to the value before stroke. rPKM2 treatment promoted the LCBF restoration and STAT3 inhibitor BP-1-102 abolished the effect of rPKM2. Asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus stroke; delta, p < 0.05 versus stroke + PKM2. n = 7 in sham group, n = 13 in stroke group, n = 7 in stroke + PKM2 group, and n = 8 in stroke + PKM2 + BP (BP-1-102) group. C and D Adhesive removal test was used to assess sensorimotor function 3 and 14 days after stroke. The time needed for stroke animals to feel (time to contact) and remove the sticky dot from the left paw (time to remove) was recorded. The ischemic insult resulted in prolonged time for the stroke animals to remove the sticky dot 3 and 14 days after stroke. Fourteen days after stroke, animals that received rPKM2 treatment showed a significantly shorter time in feeling the sticky dot on their left paws, compared with stroke animals that received either vehicle control or a combination of rPKM2 and BP-1-102. 2-way ANOVA followed by Bonferroni test. Asterisk, p < 0.05 versus sham; number sign, p < 0.05 versus stroke; delta, p < 0.05 versus stroke + rPKM2. n = 5 in sham group; n = 8 in stroke group, stroke + PKM2 group, and stroke + PKM2 + BP (BP-1-102) group
In the adhesive removal test, the time spent for animals to feel the sticker (time to contact latency) and the time to remove the sticker (time to removal) at 3 and 14 days after stroke were examined. 3 days after stroke, the animals spent a significantly longer time to feel and remove the sticker on their affected left paws compared with the time needed before stroke (Fig. 7C, D). There were no differences in this functional deficit in animals that received the vehicle control, rPKM2, or rPKM2 plus BP-1-102 at this time point. At 14 days after stroke, however, stroke animals that received the rPKM2 treatment needed significantly shorter latency in feeling the sticker and shorter time in removing the sticker from their affected paws, suggesting that rPKM2 treatment improved the sensorimotor function of stroke animals (Fig. 7C, D). Consistent with earlier observations, the STAT3 inhibitor BP-1-102 attenuated the beneficial effects of rPKM2 (Fig. 7C, D).
Discussion
The present study reveals a novel link between PKM2 and STAT3/FAK signaling pathways that play critical roles in cell regeneration and migration both in vitro and in vivo. We evaluated the therapeutic benefits of a delayed treatment of recombinant PKM2 in promoting the tissue regeneration and functional recovery after ischemic stroke. In a focal ischemic stroke mouse model, we demonstrate that rPKM2 can be administered to the ischemic brain through the nasal route, and this treatment promotes neuroblast migration and increases endogenous angiogenesis and neurogenesis in the ischemic brain. The treatment enhanced local blood flow and long-term functional recovery after ischemic stroke. We show that phosphorylation/activation of STAT3 plays a critical role in the therapeutic benefits of rPKM2. We also identified that FAK activation is essential in mediating the rPKM2 effect on regeneration-related cell migration. The role of PKM2 as a protein kinase in regulating cell proliferation and migration has been explored in recent years [11, 12]. The dimeric form of PKM2 in the nucleus can directly phosphorylate STAT3 and lead to activation of transcription of STAT3 and downstream genes to promote cell proliferation [12]. STAT3 has been shown to upregulate angiogenic factors and promote angiogenesis in cancer cells [38]. The present study demonstrates increased phosphorylation of STAT3 and FAK in the poststroke brain, which thereafter promotes the angiogenesis and neurogenesis in the peri-infarct region after stroke. This was further confirmed by using the selective STAT3 inhibitor BP-1-102 as well as the FAK−/− cells, which abolished the effects of rPKM2. Together, these results provide novel evidence for targeting PKM2 as a therapeutic candidate and the underlying molecular mechanism in the treatment of stroke.
PKM2, the M2 isoform of pyruvate kinases, plays an important role in aerobic glycolysis and is a mediator of the Warburg effect in tumors. It was thus popularly believed that tumor cells switch expression of PKM from normal tissue-expressed PKM1 to tumor-specific PKM2 via an alternative splicing mechanism. This view was challenged by our data and other recent reports demonstrating that PKM2 is expressed in normal cells and tissues. Increasing evidence supports that PKM2 is a major PKM isoform expressed in a variety of normal tissues including the brain. For example, a recent study using mass spectrometry to precisely determine PKM1 and PKM2 protein levels provided clear evidence that PKM2 instead of PKM1 is found in normal differentiated tissues; therefore, there is no isoform shift from PKM1 towards PKM2 in most of the tumors [3]. The concept that PKM2 is a major pyruvate kinase in normal cells and tissues and is significantly regulated under stress or pathological conditions makes this metabolic regulatory gene an important target for disease therapy as shown in the stroke model in this investigation. Western blot data of the brain cortex reveal that PKM2 protein exists in the normal brain. An ischemic insult upregulates the PKM2 expression. This increased PKM2 level may occur in brain cells or be due to infiltrated immune cells. Identification of the upregulation mechanism of PKM2 in the brain and related specific cell type/s will be important in future studies.
As a glycolytic enzyme, PKM2 generally locates in the cytoplasm. PKM2 can translocate into the nucleus upon stresses such as mitogenic and oncogenic stimulation [39, 40]. In the nuclei, PKM2 acts as a transcriptional coactivator and a protein kinase that phosphorylates histones, highlighting the crucial role of PKM2 in the epigenetic regulation of gene transcription that is important for the G1–S phase transition and the Warburg effects leading to a high glycolysis state and increased cell proliferation [41]. This characteristic feature of PKM2 has been extensively demonstrated for its role in tumorigenesis [4]. On the other hand, the possibility that PKM2-induced glycolysis may be utilized by normal cells for survival and regeneration under ischemic and injurious conditions has not been investigated. Indeed, emerging evidence suggests that neurogenesis can be regulated by metabolic states such as increased glycolysis during development [42].
Phosphoinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) activation can increase PKM2 expression through hypoxia-inducible factor 1α (HIF1α)-regulated transcription of the PKM gene [43, 44]. Peroxisome proliferator-activated receptor γ (PPARγ), a nuclear hormone receptor, can also specifically and transcriptionally regulate PKM2 expression. This regulatory signaling pathway apparently links the PKM2 regulation with hypoxic and ischemic conditions. FAK is a key mediator in cell migration [45]. Its upstream signaling protein integrins have been shown to mediate the migration process [45]. Our data showed that PKM2 increased the expression of integrin β1 and phosphorylated FAK in cultured NPCs and promoted the migration of NPCs. To further elucidate the role of FAK in the effects of PKM2 on migration, a migration assay was performed in wild-type mouse embryonic fibroblasts and FAK knockout MEFs. These experiments provide a compelling evidence for the mediating role of FAK activation in the rPKM2 effect of promoting the directed migration initiated by the chemoattractant factor SDF-1.
Taken together, results in the present study suggest a therapeutic potential of PKM2 for stroke treatment. The mechanism of its effect may involve activating the STAT3 pathway which in turn promotes tissue repair and functional recovery via enhancing neuroblast migration, neurogenesis, and angiogenesis after stroke.
Electronic Supplementary Material
(PDF 1225 kb)
{kind=link}
Knocking out FAK slowed down the migration of MEFs. A. Immunocytochemistry staining of cytoskeleton marker Acti-stain 555 phalloidin (red) and nuclei marker Hoechst 33,342 (blue) showed the change of cytoskeleton expression in FAK−/− MEFs. B. Transwell migration assay was performed in FAK+/+ MEFs and FAK−/− MEFs. At 2 h, 7 h and 15 h after plating, the cells on the bottom membrane of inserts were stained with Acti-stain 555 phalloidin (red) and Hoechst 33,342 (blue). C. Cell counting data showed that there were significantly less FAK−/− MEFs migrating to the bottom membrane of inserts 2 h and 7 h after plating, however, there were no differences between FAK+/+ MEFs and FAK−/− MEFs 15 h after plating. *p < 0.05 vs. FAK+/+ group. (GIF 238 kb)
Acknowledgments
This work was partly supported by NIH grants NS085568 (LW/SPY), NS091585 (LW), and NS073378 (SPY) and VA Merit Award RX000666, RX001473 (SPY).
Authors’ Contributions
DC contributed to experimental design, performed many experiments, and participated in manuscript formation; LW contributed to concept development, experimental design, immunohistochemical staining and imaging, and data analysis, and participated in manuscript formation; ZL contributed to concept development, data analysis, and manuscript editing; JY and LPL contributed to concept development, data analysis, and experimental design; ZZW and XG contributed to animal surgery and data collection and analysis; SPY contributed to concept development, experimental design, data analysis, grant supports, and the writing/revising of the manuscript.
Compliance with Ethical Standards
All animal experiments and surgery procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
The original version of this article was updated to correct a misspelling of Li-Ping Liu’s name.
Change history
6/26/2018
The original version of this article was updated to correct the misspelling of Li-Ping Liu’s name.
References
- 1.Wei L, Wei ZZ, Jiang MQ, Mohamad O, Yu SP. Stem cell transplantation therapy for multifaceted therapeutic benefits after stroke. Prog Neurobiol. 2017;157:49–78. doi: 10.1016/j.pneurobio.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahota P, Savitz SI. Investigational therapies for ischemic stroke: neuroprotection and neurorecovery. Neurotherapeutics. 2011;8:434–51. doi: 10.1007/s13311-011-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bluemlein K, Grüning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011;2:393–400. doi: 10.18632/oncotarget.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang W, Lu Z. Pyruvate kinase M2 at a glance. J Cell Sci. 2015;128:1655–1660. doi: 10.1242/jcs.166629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desai S, Ding M, Wang B, et al. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget. 2014;5:8202–8210. doi: 10.18632/oncotarget.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao X, Wang H, Yang JJ, et al. Reciprocal regulation of protein kinase and pyruvate kinase activities of pyruvate kinase M2 by growth signals. J Biol Chem. 2013;288:15971–15979. doi: 10.1074/jbc.M112.448753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu VM, Vander Heiden MG. The role of pyruvate kinase M2 in cancer metabolism. Brain Pathol. 2015;25:781–783. doi: 10.1111/bpa.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alves-Filho JC, Palsson-McDermott EM. Pyruvate kinase M2: a potential target for regulating inflammation. Front Immunol. 2016;7:145. doi: 10.3389/fimmu.2016.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta V, Wellen KE, Mazurek S, Bamezai RN. Pyruvate kinase M2: regulatory circuits and potential for therapeutic intervention. Curr Pharm Des. 2014;20:2595–2606. doi: 10.2174/13816128113199990484. [DOI] [PubMed] [Google Scholar]
- 11.Dong G, Mao Q, Xia W, et al. PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett. 2016;11:1980–1986. doi: 10.3892/ol.2016.4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Heiden MG, Locasale JW, Swanson KD, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D, Fiske BP, Birsoy K, et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Liu H, Riker AI, et al. Emerging metabolic targets in cancer therapy. Front Biosci (Landmark Ed) 2011;16:1844–1860. doi: 10.2741/3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 17.Tech K, Deshmukh M, Gershon TR. Adaptations of energy metabolism during cerebellar neurogenesis are co-opted in medulloblastoma. Cancer Lett. 2015;356:268–272. doi: 10.1016/j.canlet.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheon JH, Kim SY, Son JY, et al. Pyruvate kinase M2: a novel biomarker for the early detection of acute kidney injury. Toxicol Res. 2016;32:47–56. doi: 10.5487/TR.2016.32.1.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Lee MM, Leung MM, et al. Regulator of G protein signaling 20 enhances cancer cell aggregation, migration, invasion and adhesion. Cell Signal. 2016;28:1663–1672. doi: 10.1016/j.cellsig.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Zhang Y, Qiao J, Yang JJ, Liu ZR. Pyruvate kinase M2 in blood circulation facilitates tumor growth by promoting angiogenesis. J Biol Chem. 2014;289:25812–25821. doi: 10.1074/jbc.M114.576934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Li L, Liu Y, Liu ZR. PKM2 released by neutrophils at wound site facilitates early wound healing by promoting angiogenesis. Wound Repair Regen. 2016;24:328–336. doi: 10.1111/wrr.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Lee J, Gu X, Wei L, Yu SP, Intranasal delivery of apelin-13 is neuroprotective and promotes angiogenesis after ischemic stroke in mice. ASN Neuro, 2015. 7. [DOI] [PMC free article] [PubMed]
- 23.Wermeling DP. Intranasal delivery of antiepileptic medications for treatment of seizures. Neurotherapeutics. 2009;6:352–358. doi: 10.1016/j.nurt.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei L, Rovainen CM, Woolsey TA. Ministrokes in rat barrel cortex. Stroke. 1995;26:1459–1462. doi: 10.1161/01.STR.26.8.1459. [DOI] [PubMed] [Google Scholar]
- 25.Espinera AR, Ogle ME, Gu X, Wei L. Citalopram enhances neurovascular regeneration and sensorimotor functional recovery after ischemic stroke in mice. Neurosci. 2013;247:1–11. doi: 10.1016/j.neuroscience.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang LL, Chen D, Lee J, et al. Mobilization of endogenous bone marrow derived endothelial progenitor cells and therapeutic potential of parathyroid hormone after ischemic stroke in mice. PLoS One. 2014;9:e87284. doi: 10.1371/journal.pone.0087284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Lu Z, Keogh CL, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043–1054. doi: 10.1038/sj.jcbfm.9600417. [DOI] [PubMed] [Google Scholar]
- 28.Bouet V, Boulouard M, Toutain J, et al. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 2009;4:1560–1564. doi: 10.1038/nprot.2009.125. [DOI] [PubMed] [Google Scholar]
- 29.Gu H, Yu SP, Gutekunst CA, Gross RE, Wei L. Inhibition of the Rho signaling pathway improves neurite outgrowth and neuronal differentiation of mouse neural stem cells. Int J Physiol Pathophysiol Pharmacol. 2013;5:11–20. [PMC free article] [PubMed] [Google Scholar]
- 30.Wei JF, Wei L, Zhou X, et al. Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J Cell Physiol. 2008;217:544–557. doi: 10.1002/jcp.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S, Harris M, Varner JA. Regulation of integrin alpha vbeta 3-mediated endothelial cell migration and angiogenesis by integrin alpha5beta1 and protein kinase A. J Biol Chem. 2000;275:33920–33928. doi: 10.1074/jbc.M003668200. [DOI] [PubMed] [Google Scholar]
- 33.Lakshmikanthan S, Sobczak M, Chun C, et al. Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin alphavbeta(3) Blood. 2011;118:2015–2026. doi: 10.1182/blood-2011-04-349282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Somanath PR, Malinin NL, Byzova TV. Cooperation between integrin alphavbeta3 and VEGFR2 in angiogenesis. Angiogenesis. 2009;12:177–185. doi: 10.1007/s10456-009-9141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaeferhoff K, Michalakis S, Tanimoto N, et al. Induction of STAT3-related genes in fast degenerating cone photoreceptors of cpfl1 mice. Cell Mol Life Sci. 2010;67:3173–3186. doi: 10.1007/s00018-010-0376-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira L, Font-Nieves M, Van den Haute C, et al. IL-10 regulates adult neurogenesis by modulating ERK and STAT3 activity. Front Cell Neurosci. 2015;9:57. doi: 10.3389/fncel.2015.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Couillard-Despres S, Winner B, Schaubeck S, et al. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- 38.Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 39.Lv L, Xu YP, Zhao D, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang W, Zheng Y, Xia Y, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, Li X, Yang W, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 2014;53:75–87. doi: 10.1016/j.molcel.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caravas J, Wildman DE. A genetic perspective on glucose consumption in the cerebral cortex during human development. Diabetes Obes Metab. 2014;16:21–5. doi: 10.1111/dom.12333. [DOI] [PubMed] [Google Scholar]
- 43.Iqbal MA, Siddiqui FA, Gupta V, et al. Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol Cancer. 2013;12:72. doi: 10.1186/1476-4598-12-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Q, Chen X, Ma J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsen M, Tremblay ML, Yamada KM. Phosphatases in cell-matrix adhesion and migration. Nat Rev Mol Cell Biol. 2003;4:700–711. doi: 10.1038/nrm1199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1225 kb)
Knocking out FAK slowed down the migration of MEFs. A. Immunocytochemistry staining of cytoskeleton marker Acti-stain 555 phalloidin (red) and nuclei marker Hoechst 33,342 (blue) showed the change of cytoskeleton expression in FAK−/− MEFs. B. Transwell migration assay was performed in FAK+/+ MEFs and FAK−/− MEFs. At 2 h, 7 h and 15 h after plating, the cells on the bottom membrane of inserts were stained with Acti-stain 555 phalloidin (red) and Hoechst 33,342 (blue). C. Cell counting data showed that there were significantly less FAK−/− MEFs migrating to the bottom membrane of inserts 2 h and 7 h after plating, however, there were no differences between FAK+/+ MEFs and FAK−/− MEFs 15 h after plating. *p < 0.05 vs. FAK+/+ group. (GIF 238 kb)