

Abstract
Background
Non-small cell lung cancer (NSCLC) is a life-threatening disease that has a poor prognosis and low survival rate. Cleavage factor Im 25 (CFIm25) is a RNA-binding protein that if down-regulated causes 3’UTR shortening and thus promotes the transcript stability of target genes. It is not clear whether CFIm25 and alternative polyadenylation (APA) play a role during cancer development. The purpose of this study is to explore the role of CFIm25 in lung cancer cell proliferation.
Methods
CFIm25 was knocked down in A549 cells. Western blots were carried out to determine the protein expression of CFIm25, insulin growth factor 1 receptor (IGF1R), CyclinD1 (CCND1) and TP53. Real-time qRT PCR was performed to determine the total transcript levels of CFIm25 targets and the normalized fold changes in their distal PAS (dPAS) usage. Immunofluorescence was carried out to check the expression of CFIm25, IGF1R and CCND1. Cell proliferation over time was determined using the WST-1 reagent.
Results
The transcript levels of CCND1 and GSK3β were significantly increased and the dPAS usage of several oncogenes (IGF1R, CCND1 and GSK3β) were decreased after CFIm25 knockdown. The protein level of IGF1R was increased, and we detected increased percentage of CCND1 positive cells and cell proliferation over time in CFIm25 knockdown cells. In addition, the mRNA and APA analysis of IGF1R using patient RNA-seq data from the Cancer Genome Atlas indicated that IGF1R is shortened in both lung adenocarcinoma and lung squamous cell carcinoma compared to normal controls.
Conclusions
Our findings suggest that CFIm25 plays an important role in lung cancer cell proliferation through regulating the APA of oncogenes, including IGF1R, and promoting their protein expression.
Keywords: Alternative polyadenylation, cancer, A549, IGF1R, CyclinDl, GSK3β, proliferation, apoptosis, P53
1. Introduction
The occurrence and development of lung cancer is a complicated process related to many signal pathways. Among the many classifications of lung cancers, non-small cell lung cancer (NSCLC) is the most common type that accounts for about 85% of all lung cancers [1]. Lung squamous cell carcinoma (LUSC, 30% of NSCLC) and lung adenocarcinoma (LUAD, 50% of NSCLC) are the two major subtypes of NSCLC. NSCLC has a poor prognosis, low 5-year survival rates (14% for stage IIIA and 5% for stage IIIB) and no effective preventive measures [2]. Lung cancer cells are highly proliferative by escaping cell cycle check points as well as programed cell death, a process that is normally mediated by mutations or changed expression of oncogenes, tumor-suppressor genes or microRNAs [3]. However, the underlying molecular mechanisms that account for altered oncogene expression and promote the abnormal proliferation of lung cancer cells are extremely complex and remain unknown.
Polyadenylation is an important RNA process for messenger RNA (mRNA) maturation. More than half of mammalian mRNA have more than one polyadenylation sites (PAS), thus usage of different PAS can result in transcripts with different sizes of 3’-untranslated regions (3’UTRs) or different transcripts, a process called alternative polyadenylation (APA) [4,5]. Transcripts with shorter 3’UTR are normally more stable due to the loss of binding sites for miRNAs and other regulatory proteins, and normally have higher protein output. APA was recently identified as a widespread mechanism controlling gene stability and expression, and the importance of APA has been emphasized in many cancers including glioblastoma tumor, hepatocellular carcinoma, prostate cancer and breast cancer [6,7,8]. APA can be regulated by multiple proteins involved in the polyadenylation machinery. Among these, cleavage factor Im 25 (CFIm25) was identified as the top regulator that suppresses the usage of proximal PAS, thus its depletion promotes 3’UTR shortening of target genes and normally enhances target gene stability and expression. CFIm25 down-regulation was also identified as a mechanism that promotes glioblastoma tumor growth [6]. However, it is not known whether CFIm25 also plays a role in lung cancer cell proliferation and lung cancer progress.
To understand the role of CFIm25 and APA in lung cancer cell proliferation, we used siRNA to knockdown CFIm25 expression in A549 cells, a well characterized lung adenocarcinoma cell line, and analyzed the expression of several proteins involved in cell proliferation and apoptosis. We observed increased A549 proliferation upon CFIm25 knockdown, possibly due to enhanced expression of IGF1R and CCND1. In addition, we confirmed that both IGF1R and CCND1 have 3’UTR shortening after CFIm25 depletion, and this may be sufficient to increase their protein expression. The 3’UTR shortening of IGF1R was also observed in lung cancer samples compared to normal tissues, suggesting APA is an important novel mechanism for increased IGF1R signaling in lung cancers. In summary, we have determined that CFIm25 down-regulation has a significant impact on lung cancer cell proliferation through APA.
2. Material and methods
2.1. Cell culture
Lung carcinoma from human cells (A549) were purchased from ATCC (Manassas, VA). A549 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) supplement with 10% fetal bovine serum (FBS) and antibiotics. Cells were cultured at 37°C in a humidified 5% carbon dioxide atmosphere.
2.2. Transfection
For siRNA transfection, cells were cultured in antibiotic-free media, transfected with 50 ng/ml miRNA mimic using Lipofectamine® RNAiMAX (Life technology, Grand Island, NY) on day 0 and day 2, and collected for Western Blot or real-time PCR analysis on day 4.
2.3. Western Blot
A549 cells were collected and lysed in RIPA lysis buffer (50mM Tris-HCl, pH 7.4; 150 mM NaCl; and 1% Nonidet P-40) containing a protease inhibitor cocktail (Thermo Fisher Scientific, Fair Lawn, NJ, USA). Equal amounts of protein were separated on SDS-PAGE and transferred to nitrocellular membranes. The membranes were then blocked with 5% (w/v) nonfat milk, washed with Tris-buffered saline-Tween-20 (TBST), and incubated with primary rabbit anti-CFIm25 (Proteintech, Chicago, IL), rabbit anti-IGFIR or mouse anti-β-Actin antibodies (Sigma-Aldrich, St. Louis, MO) overnight at 4°C, and then incubated with corresponding secondary antibodies conjugated to horseradish peroxidase (Cell Signaling, Danvers, MA) for 1 h at room temperature. Membranes were developed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Bio-Sciences, Pittsburgh, PA).
2.4. RNA purification and real-time quantitative PCR
A549 cells were lyzed using TRIzol (Life Technologies) and total RNA was isolated using RNeasy Mini Kit (Quagen, Valencia, CA). For real-time PCR analysis, RNA was treated with DNase and reverse-transcribed using iScript™ Reverse Transcription Supermix (Bio-Rad). Real-time PCR was performed under Lightcycler 96 (Roche, Indianapolis, IN) using primers listed in Table 1 and data were quantified using the comparative Ct method and presented as mean ratio to β-actin RNA. The percentage of dPAS usage was examined using a PCR-based method as described. Two pairs of primers were designed with one targeting the open reading frame to represent the total transcript level, and the other targeting sequences just before the dPAS to detect long transcripts that used the dPAS. Percentage of dPAS usage was calculated as ΔCT=CTdistal − CTtotal. Data were presented as fold changes normalized to control by calculating ΔΔCT=ΔCTaverage target − ΔCTaverage of control.
Table 1.
Primers used for real-time PCR
| Gene | Forward primer | Reverse Primer |
|---|---|---|
| Homoe_CFIm25 | TGAAGTTGAAGGACTAAAACGCT | ACCAGTTACCAATGCAATCGTC |
| Homo_GSK3β | CTGGTCCGAGGAGAACCCAATGTTTCG | CAGCCAACACACAGCCAGCAGACCATAC |
| Homo_GSK3β Long | GAGCTGAGCCCATGGTTGTGTGTAAC | GGTTCACTTCAGCAGGCAGGACAACTC |
| Homo_IGF1R | ATGCTGACCTCTGTTACCTCT | GGCTTATTCCCCACAATGTAGTT |
| Homo_IGF1R Long | CCGGTGAAAACACCTGTCTG | ATTTGCGGTGCATCCATTCC |
| Homo_CCND1 | CTGCCAGGAGCAGATCGAAG | AATGCTCCGGAGAGGAGGGAACT |
| Homo_CCND1 Long | ATCGAGAGGCCAAAGGCT | CGTCTTTTTGTCTTCTGCTGGA |
| Homo_ β-Actin | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
2.5. Immunofluorescence
A549 cells were fixed in 1% formaldehyde in PBS overnight. After washing with PBS, cells were blocked with Avidin/Biotin Blocking System (VectorLabs) and incubated in 5% normal goat serum for 1 hour. A549 cells were then incubated with primary antibodies against CFIm25 (1:200, proteintech), CCND1 and IGF1R (1:200, Cell signaling) overnight at 4°C, and with biotinylated anti-Rabbit antibodies (1:1000, VectorLabs) for 1hr at room temperature (RT). After washing, Vector® Red Substrate (VectorLab) was used for CFIm25/CCND1/IGF1R immunofluorescence dual-staining for 5 seconds at RT. Cells were finally mounted with DAPI (Life Technologies).
2.6. APA analysis
The RNA-seq BAM file of 503 LUSC and 51 normal samples, 515 LUAD and 59 normal samples were downloaded from the national cancer institute’s Genomic Dac Commons (GDC) (http://gdc.cancer.gov) The gene expressions were recalculated as reads per kilobase per million mapped reads (RPKM) across all samples. The expression data were then normalized using quartile normalization. To characterize the dynamic APA events of IGF1R in cancer and normal tissues, we used a well-established algorithm named Dynamic analysis of Alternative PolyAdenylation from RNA-seq (Dapars, https://github.com/ZhengXia/dapars) to identify the alternative proximal polyA sites and calculate the Percentage of Distal poly(A) Site Usage Index (PDUI) for IGF1R [9]. T-tests were then used to compare the PDUI of IGF1R in cancers and normal samples.
3. Results
3.1. CFIm25 knockdown promotes the IGF pathway
To understand the role of CFIm25 and APA in lung cancer development, we first silenced CFIm25 expression in A549 cells using CFIm25-specific siRNA. Western blot and immunofluorescence show that CFIm25 protein expression is successfully depleted in A549 cells transfected with CFIm25 siRNA (Fig. 1A and 1B). The insulin-like growth factor 1 receptor (IGF1R) has recently been identified as a potential therapeutic target in NSCLC [10]. IGF1R has increased expression in many NSCLC patients and is involved in promoting the tumor transformation, growth and survival of cancer cells. We found that pre-IGF1R expression is increased in A549 cells with CFIm25 KD. Similarly, immunofluorescence also showed an increased IGF1R expression in CFIm25 KD A549 cells (Fig. 1C). Taken together, our results indicate that KD of CFIm25 enhances IGF1R signaling in A549 cells.
Fig. 1.
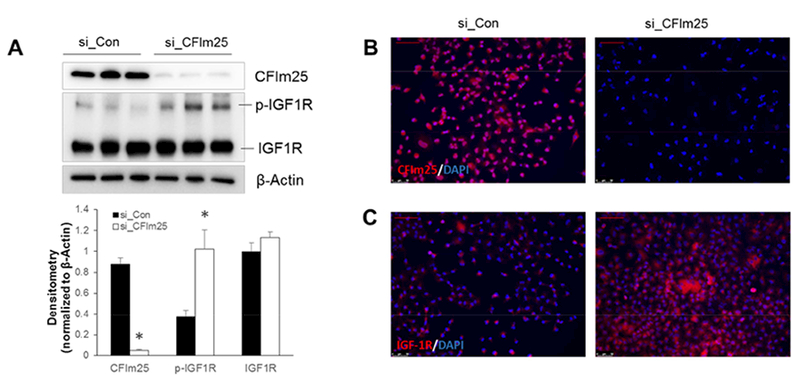
Knockdown of CFIm25 in A549 cells promotes IGF1R expression. A549 cells were transfected with Control (si_Con) or CFIm25 (si_CFIm25) siRNA on day 0 and day 1 and collected for analysis on day 4. (A) Western blots were carried out to determine the protein expression of CFIm25 and IGF1R. β-Actin was used as an internal control. (B and C) Immunofluorescence was carried out to check the expression of CFIm25 (B) and IGF1R (C). Scale bar=75μM. * P<0.05 vs si_Con.
3.2. CFIm25 depletion enhances cell proliferation and suppresses programed cell death.
Overexpression of IGF1R in cancer cells can promote abnormal cell proliferation [11,12,13,14]. To understand whether increased expression of IGF1R signaling in CFIm25 depleted cells promotes cell proliferation, we used the WST-1 assay to determine cell proliferation over time. Cell proliferation was significantly increased on day 2 and day 3 in CFIm25 KD cells compared to control siRNA transfected cells (p<0.05) (Fig. 2A). It’s been reported that signaling through IGF1R and insulin receptor substrate 1 (IRS-1) promotes cell proliferation by up-regulating Cyclin D1 (CCND1) [15]. CCND1 is an important protein which is normally overexpressed in cancer cells and is critical to the development and progression of cancer. Previously, CCND1 was also identified as an important CFIm25 target that accounts for increased HELA cell proliferation after CFIm25 KD. In CFIm25 KD A549 cells, we also observed an increased protein expression of CCND1 (Fig. 2B). Similarly, immunofluorescence also confirmed that the percentage of cyclinD1 positive cells was significantly increased in CFIm25 KD cells (p<0.05) (Fig. 2C–2D).
Fig. 2.
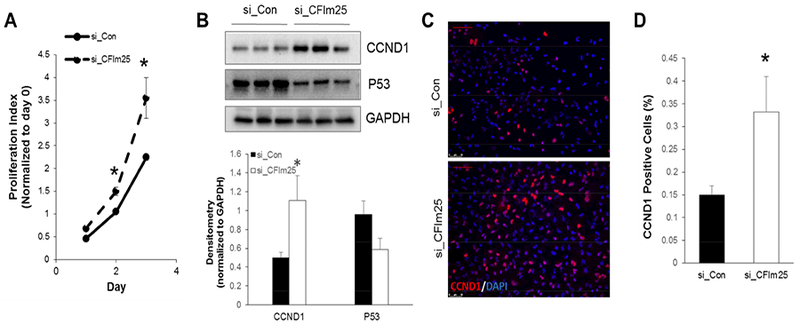
CFIm25 depletion enhances cell proliferation. (A) Cell proliferation over time was determined using the WST-1 reagent. n=5, * p<0.05 vs coresponding si_Con. (B) Western blots were carried out to determine the protein expression of CCND1 and P53. GAPDH was used as an internal control. (C) Immunofluorescence was carried out to examine the expression of CCND1 in A549 cells transfected with control or CFIm25 siRNA. (D) The percentage of CCND1 positive cells were counted and quantitated. N=3, * p<0.05 vs si_Con.
Suppression of the TP53 (P53) pathway is commonly seen in cancer cells, and it is one of the most important mechanisms for cancer cells to escape programmed cell death and become immortalized [16]. We detected a decreased P53 protein expression in CFIm25 KD cells (Fig. 2B), suggesting a down-regulation of apoptosis pathway in CFIm25 depleted cells that may partially contribute to increased cell proliferation. In summary, our studies indicate that CFIm25 depletion promotes lung cancer cell proliferation and CCND1 expression, and subsequently increases cell proliferation and suppresses apoptosis.
3.3. Knockdown of CFIm25 promotes the 3’UTR shortening of several oncogenes
Down-regulation of CFIm25 normally up-regulates targeted gene translation by promoting 3’UTR shortening. To find out whether IGF1R and CCND are directly targeted by CFIm25, we used a Realtime quantitive PCR (RT qPCR) based method to monitor the usage of dPAS usage by designing two primers for each gene, one targeting the end of the 3’UTR to represent the expression of the long version of transcript (Fig. 3A P2) and the other one targeting the translated region to represent the total transcript (Fig. 3A P1). We then calculated the percentage of the long version of the transcript and determined the log ratio of the percentage of CFIm25 KD cells to the percentage of control cells. The negative value indicates the target gene has 3’UTR shortening in CFIm25 KD cells. Using this method, we successfully detected 3’UTR shortening of Glycogen synthase kinase 3 beta (GSK3β), a known target of CFIm25, in CFIm25 KD cells (Fig. 3C). Interestingly, we found that both IGF1R and CCND1 have 3’UTR shortening in CFIm25 depleted cells (Fig. 3C), suggesting that both could be targeted by CFIm25. Among all of these three shortened genes, only the transcript level of CCND1 was increased (Fig. 1B). However, both IGF1R and CCND1 protein expression was increased, suggesting that the 3’ UTR shortening of IGF1R is sufficient to enhance its protein turn out. In summary, our data suggests that KD of CFIm25 promotes the 3’UTR shortening of IGF1R and CCND1, and that it is sufficient to promote protein expression and enhanced cancer cell proliferation.
Fig. 3.
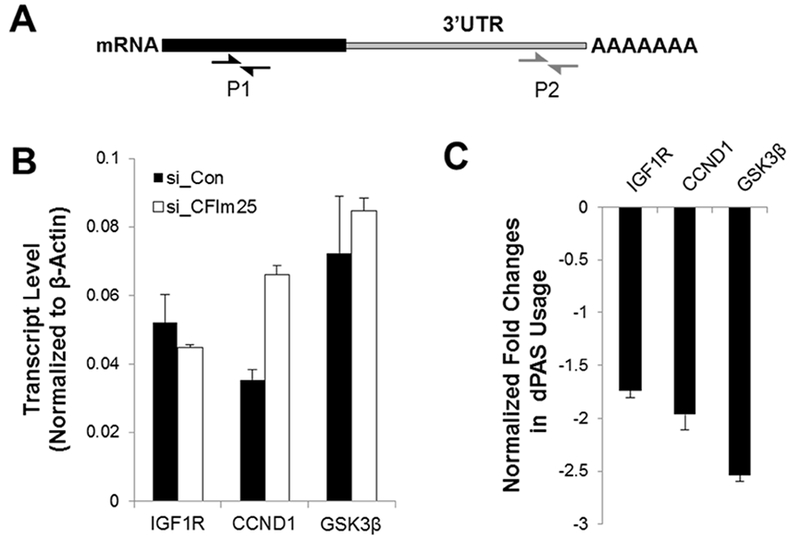
CFIm25 siRNA promotes the usage of proximal polyadenylation sites (pPAS). (A) Diagram showing the primers designed for the detection of total transcript (P1) and long variant of transcript (P2). (B and C) Real-time qRT PCR was carried out to determine the total transcript level of CFIm25 targets (IGF1R, CCND1 and GSK3β) (B) and the fold changes in the distal PAS (dPAS) usage (C).
3.4. The 3’UTR of IGF1R is shortened in NSCLC
The importance of IGF1R signaling in lung cancer has been intensively studied, however the mechanism that promote its protein expression in lung cancer is not well known. We have shown that CFIm25 depletion can promote the 3’UTR shortening of IGF1R, which is sufficient to increase its protein expression in A549 cells, suggesting that 3’UTR shortening of IGF1R is a potential mechanism to promote its protein expression in lung cancers. To understand whether the 3’UTR IGF1R is shortened in human lung cancers, we analyzed the APA of IGF1R using patient RNA-seq data from the Cancer Genome Atlas. Interestingly, we observed that the 3’UTR of IGF1R is significantly shortened in both LUAD and LUSC patients (Fig. 4C and 4D). The mRNA level of IGF1R has no difference in LUAD patients (Fig. 4A) and is only slightly increased in LUSC patients (Fig. 4B) compared to healthy donors. However, both LUAD and LUSC have been reported to have enhanced IGF1R signaling, suggesting that the 3’UTR shortening could be a new mechanism that mediates increased IGF1R protein expression in LUAD.
Fig. 4.
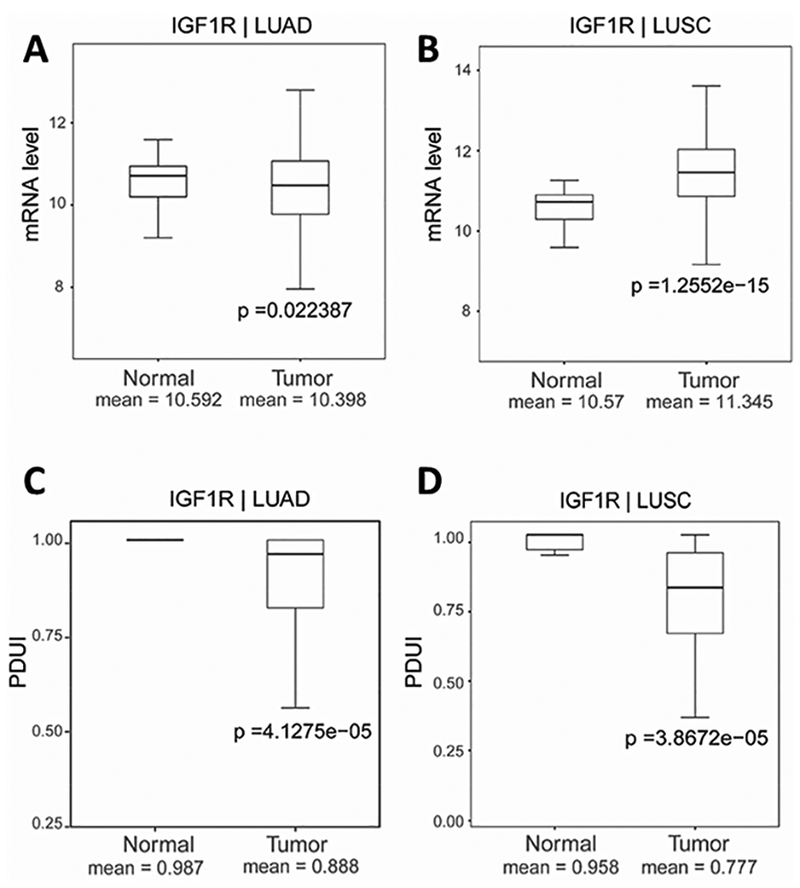
IGF1R has 3’UTR shortening in NSCLC samples. The transcript expression levels of IGF1R were determined using RNA-seq data from the Cancer Genome Altas in lung adenocarcinoma (LUAD) (A) and lung squamous cell carcinoma (LUSC) (B) compared to matched controls. The PDUI of IGF1R were calculated in LUAD (C) and LUSC (D) and compared to their controls to determine the usage of distal PAS.
4. Discussion
Non-small cell lung cancer (NSCLC) is the most common form of lung cancer that has no methods of prevention and no effective therapeutic treatment, and the mechanisms that promote uncontrolled cancer cell growth and division are not fully studied. In our study, we explored the role of CFIm25 in lung cancer cell proliferation. Using siRNA technology, we found that KD of CFIm25 promotes APA of several genes (IGF1R, CCND1and GSK3β) that play important roles in cancers, causing 3’UTR shortening of these genes and enhancing their protein expression (Fig. 5). We have also found that through up-regulation of these proteins, CFIm25 siRNA increases cancer cell proliferation and suppresses P53-mediated apoptosis.
Fig. 5.
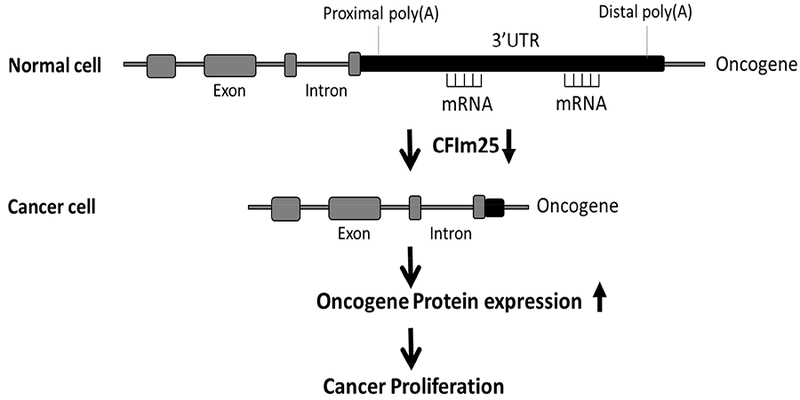
A model showing the effect of CFIm25 downregulation on cancer proliferation.
CFIm25 is a broad proximal polyadenylation site (pPAS) suppressor, such that its down-regulation normally promotes the usage of pPAS and generates transcripts with truncated 3’UTRs. These shortened transcripts are normally more stable and have enhanced protein expression (Fig. 5). It has been reported that global 3’UTR shortening has been found in highly proliferating cells, suggesting that CFIm25 depletion may lead to cancer cell proliferation. Indeed, CFIm25 depletion has significantly enhanced cancer proliferation and increased glioblastoma tumor growth [6]. Our in vitro data showed significantly increased proliferation in CFIm25 KD cells, and recent findings from Dr. Leng’s lab using RNA sequencing data from the Cancer Genome Atlas revealed a global 3’UTR shortening in cancer cell lines. However, RNA-seq data from the Cancer Genome Atlas did not show any difference in CFIm25 transcript levels between lung cancers and normal donors (data not shown). Therefore, further studies are needed to understand whether CFIm25 protein expression is down-regulated in cancer samples.
IGF1R singling pathway activation has been studied for decades in various cancers and is critical for tumorigenesis [17]. The binding of IGF to its receptor IGF1R activates phosphoinositide 3-kinase (PI3K) and AKT through phosphorylation, and AKT subsequently regulates diverse cellular processes, including cell proliferation, anti-apoptosis, cell size, tissue invasion and angiogenesis [18]. Due to the important role of the IGF pathway in cancer, IGF1R has been identified as a potential therapeutic target in various cancer types. Although IGF signaling is enhanced in many cancers, the mechanism that enhances its signaling is not well known. In our study, we found KD of CFIm25 promoted its 3’UTR shortening. The shortened transcripts were sufficient to increase its protein turnover without increasing its transcript level, suggesting that 3’UTR shortening of IGF1R could be a mechanisms that promotes its protein expression in NSCLC. This mechanism was further confirmed by RNA-seq data from the Cancer Genome Atlas which showed a significant 3’UTR shortening of IGF1R in lung cancer samples compared to normal donors. Our results provide a novel mechanism for the up-regulation of IGF1R signaling in cancer at a RNA regulation level. However, the signaling that promotes 3’UTR shortening of IGF1R in lung cancers is not known. Recently, comprehensive APA characterization of cancer samples identified PABPN1 as a potential factor that regulates the APA profile across different cancer types [8]. Further studies are needed to understand whether PABPN1 is an upstream regulator that promotes the 3’UTR shortening of IGF1R in lung cancers.
Abrogated expression or mutation of proteins controlling the cell cycle process, including CCND1, is one of the major reasons by which cancer cells evade cell cycle checkpoints, thus accelerating their cell division rate [19]. Overexpression of CCND1 has been linked to the development of various cancers [20]. In addition, mutation or suppression of key genes that induce programmed cell death, including P53, is another cause of cancer development. In our study, we found that CFIm25 KD enhanced the expression of IGF1R and CCND1 through APA, subsequently enhancing A549 cell proliferation. The transcript of CCND1 can enhance IGF1R through the docking protein IRS-1, which is capable of activating CCND1 promoters [21], suggesting that increased IGF1R signaling in CFIm25 cancer cells could be a secondary mechanism that promotes CCND1 expression and subsequent cell proliferation. We also observed an inhibited expression of P53; however, P53 only has one known polyA site, so it may not be a direct target of CFIm25 and other indirect mechanisms might be involved in the regulation of P53 expression. Thus, these results identified APA of oncogenes as a potential novel mechanism for cancer development.
In summary, our studies are the first to reveal, to our knowledge, the role of CFIm25 and APA in lung cancer cell proliferation. Several important genes involved in cancer cell proliferation are identified to be regulated by APA to enhance their protein expression, subsequently promoting cell proliferation and inhibiting apoptosis. Our study provides a novel mechanism that promotes cancer proliferation and can be beneficial to the development of novel cancer drugs.
Supplementary Material
Acknowledgements
This project was sponsored by the National Natural Science Foundation of China granted to X. L. (No. 81470501, 81770440); and National Institutes of Health (NIH) grants to M.R.B. (HL70952); and the Cancer Prevention & Research Institute of Texas granted to L.H. (RR150085)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- [1].Rotow J, Bivona TG, Understanding and targeting resistance mechanisms in NSCLC, Nat Rev Cancer 17 (2017) 637–658. [DOI] [PubMed] [Google Scholar]
- [2].Cancer Stat Facts: Lung and Bronchus Cancer, National Cancer Institute Archived from the original on 4 March 2016. Retrieved 5 March 2016.
- [3].Perlikos F, Harrington KJ, Syrigos KN, Key molecular mechanisms in lung cancer invasion and metastasis: a comprehensive review, Crit Rev Oncol Hematol 87 (2013) 1–11. [DOI] [PubMed] [Google Scholar]
- [4].Mayr C, Bartel DP, Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells, Cell 138 (2009) 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tian B, Manley JL, Alternative polyadenylation of mRNA precursors, Nat Rev Mol Cell Biol 18 (2017) 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ, CFIm25 links alternative polyadenylation to glioblastoma tumour suppression, Nature 510 (2014) 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Morris AR, Bos A, Diosdado B, Rooijers K, Elkon R, Bolijn AS, Carvalho B, Meijer GA, Agami R, Alternative cleavage and polyadenylation during colorectal cancer development, Clin Cancer Res 18 (2012) 5256–5266. [DOI] [PubMed] [Google Scholar]
- [8].Xiang Y, Ye Y, Lou Y, Yang Y, Cai C, Zhang Z, Mills T, Chen NY, Kim Y, Muge Ozguc F, Diao L, Karmouty-Quintana H, Xia Y, Kellems RE, Chen Z, Blackburn MR, Yoo SH, Shyu AB, Mills GB, Han L, Comprehensive Characterization of Alternative Polyadenylation in Human Cancer, J Natl Cancer Inst (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, Li W, Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types, Nat Commun 5 (2014) 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fidler MJ, Shersher DD, Borgia JA, Bonomi P, Targeting the insulin-like growth factor receptor pathway in lung cancer: problems and pitfalls, Ther Adv Med Oncol 4 (2012) 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dupont J, Le Roith D, Insulin-like growth factor 1 and oestradiol promote cell proliferation of MCF-7 breast cancer cells: new insights into their synergistic effects, Mol Pathol 54 (2001) 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Heidegger I, Ofer P, Doppler W, Rotter V, Klocker H, Massoner P, Diverse functions of IGF/insulin signaling in malignant and noncancerous prostate cells: proliferation in cancer cells and differentiation in noncancerous cells, Endocrinology 153 (2012) 4633–4643. [DOI] [PubMed] [Google Scholar]
- [13].Werner H, For debate: the pathophysiological significance of IGF-I receptor overexpression: new insights, Pediatr Endocrinol Rev 7 (2009) 2–5. [PubMed] [Google Scholar]
- [14].Pollak MN, Schernhammer ES, Hankinson SE, Insulin-like growth factors and neoplasia, Nat Rev Cancer 4 (2004) 505–518. [DOI] [PubMed] [Google Scholar]
- [15].Salisbury TB, Tomblin JK, Insulin/Insulin-like growth factors in cancer: new roles for the aryl hydrocarbon receptor, tumor resistance mechanisms, and new blocking strategies, Front Endocrinol (Lausanne) 6 (2015) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Meek DW, Tumour suppression by p53: a role for the DNA damage response?, Nat Rev Cancer 9 (2009) 714–723. [DOI] [PubMed] [Google Scholar]
- [17].Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L, Luu HH, Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance, Genes Dis 2 (2015) 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Altomare DA, Testa JR, Perturbations of the AKT signaling pathway in human cancer, Oncogene 24 (2005) 7455–7464. [DOI] [PubMed] [Google Scholar]
- [19].Alao JP, The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention, Mol Cancer 6 (2007) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hunter T, Pines J, Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age, Cell 79 (1994) 573–582. [DOI] [PubMed] [Google Scholar]
- [21].Wu A, Chen J, Baserga R, Nuclear insulin receptor substrate-1 activates promoters of cell cycle progression genes, Oncogene 27 (2008) 397–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.