

Abstract
Background
Erlotinib is an epidermal growth factor receptor (EGFR)-targeting tyrosine kinase inhibitor approved for treatment of non-small cell lung cancer. The wide inter-individual pharmacokinetic (PK) variability of erlotinib may impact treatment outcome and/or toxicity. Recent in vivo studies reported a nonlinear uptake transport of erlotinib into the liver, suggesting carrier-mediated system(s) to mediate its hepatobiliary clearance. Erlotinib has been identified in vitro as a substrate of organic anion-transporting polypeptide (OATP) transporters which expression does not restrict to hepatocytes and may impact the tissue uptake of erlotinib in vivo.
Results
The impact of rifampicin (40 mg/kg), a potent OATP inhibitor, on the liver uptake and exposure to tissues of 11C-erlotinib was investigated in rats (4 animals per group) using positron emission tomography (PET) imaging. Tissue pharmacokinetics (PK) and corresponding exposure (area under the curve, AUC) were assessed in the liver, kidney cortex, abdominal aorta (blood pool) and the lungs. The plasma PK of parent 11C-erlotinib was also measured using arterial blood sampling to estimate the transfer rate constant (kuptake) of 11C-erlotinib from plasma into different tissues.
PET images unveiled the predominant distribution of 11C-erlotinib-associated radioactivity to the liver, which gradually moved to the intestine, thus highlighting hepatobiliary clearance. 11C-erlotinib also accumulated in the kidney cortex. Rifampicin did not impact AUCaorta but reduced kuptake, liver (p < 0.001), causing a significant 27.3% decrease in liver exposure (p < 0.001). Moreover, a significant decrease in kuptake, kidney with a concomitant decrease in AUCkidney (− 30.4%, p < 0.001) were observed. Rifampicin neither affected kuptake, lung nor AUClung.
Conclusions
Our results suggest that 11C-erlotinib is an in vivo substrate of rOATP transporters expressed in the liver and possibly of rifampicin-inhibitable transporter(s) in the kidneys. Decreased 11C-erlotinib uptake by elimination organs did not translate into changes in systemic exposure and exposure to the lungs, which are a target tissue for erlotinib therapy.
Keywords: Erlotinib, Solute carrier transporter, Solute carrier O, Organic anion-transporting polypeptide, Liver, Positron emission tomography, SLCO2B1, SLCO
Background
Erlotinib is a reversible inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase activity, and it is approved for treatment of advanced and metastatic non-small cell lung cancer (NSCLC) with mutated EGFR [1]. However, nearly 30% of patients who are carriers of the somatic EGFR mutation do not respond adequately to therapy with tyrosine kinase inhibitors [2]. NSCLC patients, receiving the therapeutic dose of 150 mg erlotinib once daily, showed a marked interpatient variability in trough plasma concentrations and area under the curve (AUC) of 64% and 51%, respectively [3]. Variability in the pharmacokinetics (PK) of erlotinib is therefore assumed to contribute to variability in therapeutic response [4]. Furthermore, a correlation of erlotinib plasma PK parameters and treatment efficacy and occurrence of side effects was found [5, 6].
The molecular determinants for this PK variability are not fully understood. ATP-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp, ABCB1) and breast cancer resistance protein (BCRP, ABCG2), were shown to play pivotal roles in restricting the intestinal absorption of erlotinib [7]. Erlotinib is mainly eliminated through hepatobiliary clearance and undergoes hepatic metabolism by cytochrome P450 (CYP) 3A4/3A5 and, to a lesser extent, by CYP1A1/2 [8]. Thus, CYP3A4 inducers and inhibitors are likely to affect erlotinib PK and may be responsible for drug-drug interactions (DDIs) [9, 10]. In addition to metabolism considerations, erlotinib is also a substrate of several transporters of importance for PK [11]. P-gp and BCRP are now recognized as major determinants for tissue distribution and clearance erlotinib [7, 12].
Recent positron emission tomography (PET) studies using carbon-11-labeled erlotinib provided novel information regarding the clearance of this drug. In baboons, injection of a high dose of unlabeled erlotinib decreased the plasma clearance of 11C-erlotinib, pointing to non-linear PK [13]. Similarly, a high dose of erlotinib in mice was associated with a decreased liver uptake of 11C-erlotinib, suggesting a saturable and possibly carrier-mediated transport mechanism for erlotinib at the basolateral membrane of hepatocytes [14]. This observation was recently confirmed in healthy human volunteers using a similar saturation approach [15].
The hepatic uptake of various xenobiotics is mediated by sinusoidal membrane transporters, mainly solute carrier (SLC) transporters, as a prerequisite for hepatobiliary elimination [16]. Three members of the organic anion-transporting polypeptide (OATP) family (SLCO gene), namely OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), and OATP2B1 (SLCO2B1), are expressed in hepatocytes and are considered of importance for PK [11]. Recent in vitro studies demonstrated that erlotinib is selectively transported by OATP2B1 among the other OATP transporters expressed in hepatocytes [15]. OATP2B1 expression is not restricted to the liver and may also play a role in the uptake of its substrates into other organs [17]. OATP2B1 function may thus account for variability in the distribution of erlotinib to the liver and to target/vulnerable tissues, with consequences for treatment efficacy and safety. OATP function was thus hypothesized to control the liver uptake of erlotinib with consequences for plasma and tissue kinetics in vivo.
In the present study, 11C-erlotinib PET imaging was performed in anesthetized rats in the absence and the presence of the broad spectrum OATP inhibitor rifampicin to understand whether rifampicin-inhibitable uptake transport may be responsible for the pharmacokinetic variability of erlotinib in patients.
Methods
Animals
Fourteen male Sprague-Dawley rats (Janvier, France) were included in the study (392 ± 31 g in weight at the time of experiment). Animals were housed in a temperature and humidity-controlled room with a 12-h light/dark cycle and with access to food and water ad libitum. Animal studies were conducted in accordance with the French legislation and European directives on the use of animals in research. The protocol has been accepted by a local ethics committee for animal use (protocol number A16/057).
Chemicals and radiochemicals
6-O-desmethyl erlotinib was purchased from Syncom (The Netherlands). Rifampicin (Rifadine®) was purchased from Sanofi-Aventis (France). 11C-erlotinib was synthesized by 11C-methylation of 6-O-desmethyl erlotinib as previously described [18]. The radiochemical purity of 11C-erlotinib was greater than 98% with a molar activity of 152 ± 65 GBq/μmol (decay-corrected to the end of bombardment).
PET imaging
PET imaging was performed using an Inveon® microPET system (Siemens, Germany). Anesthesia was induced and further maintained for the whole scan duration using 4% and 2% of isoflurane in O2, respectively. A tail vein was catheterized for radiotracer injection, and rats were then positioned on a heated bed with their abdominal region located at the center of the field of view. Dynamic PET acquisition (60 min) started at the time of 11C-erlotinib injection (injected amount 33 ± 7 MBq). 11C-erlotinib dynamic PET data were sorted into 24 frames with time durations of 3 × 0.5 min, 6 × 1 min, 5 × 2 min, 4 × 3 min, 2 × 4 min, 4 × 5 min, and 1 × 2.5 min. Dynamic images were reconstructed using the FORE+OSEM2D algorithm including normalization, attenuation, scatter and random corrections.
PET experiments were performed in the absence (control group, n = 4 animals) and in the presence (rifampicin group, n = 4 animals) of an intravenous (i.v.) pretreatment with rifampicin (40 mg/kg), given as a bolus at 5 min before radiotracer injection. Acute rifampicin administration was previously shown to inhibit OATP function in rat using different OATP-substrate probes [19–22].
Data analysis
PET data analysis was performed using PMOD software (version 3.8; PMOD Technologies, Zurich, Switzerland). For each animal, volumes of interest (VOIs) were manually drawn for the abdominal aorta (clearly observed at early time frames), liver, lungs, and kidney cortex. Corresponding time-activity curves (TACs) were generated. Radioactivity was corrected for injected dose and animal weight, and data were expressed in units of standardized uptake value (SUV). The highest concentration value of the time-activity curves was defined as SUVmax. Area under the TACs was estimated from the start to the end of acquisition for the abdominal aorta, the liver, the lungs, and the kidneys (AUCaorta, AUCliver, AUClungs, and AUCkidneys; SUV.min) using GraphPad Prism software (CA, USA).
Arterial input function
In parallel conditions and for each group, one additional animal was used to measure a metabolite-corrected arterial input function of 11C-erlotinib, obtained from arterial blood sampling, gamma counting, and HPLC analysis (injected amount 58 and 46 MBq for control and rifampicin condition, respectively). Twenty blood samples (50 μL) were collected at selected times from the femoral artery to describe the time course of total radioactivity in arterial plasma after radiotracer injection. Samples were centrifuged (3 min, 2054×g, 4 °C), and radioactivity in cell-free plasma (25 μL) was determined using a Cobra Quantum® (D5003, Perkin-Elmer) gamma counter. Additional samples (500 μL) were withdrawn at 5, 10, 15, 30, and 60 min after injection to determine the parent fraction of unmetabolized 11C-erlotinib in plasma using radio-HPLC analysis as previously described [13]. For each condition, the parent fraction of 11C-erlotinib versus time was fitted using a 1-exponential decay equation and applied to the total radioactivity kinetics to estimate a representative metabolite-corrected arterial input function of 11C-erlotinib. AUCplasma expressed as SUV.min represents the AUC of the metabolite-corrected arterial input function, obtained using arterial blood sampling from 0 to 60 min after radiotracer injection.
Determination of parent 11C-erlotinib in the liver
In parallel to the PET imaging experiments, the parent fraction of 11C-erlotinib was determined in the liver at 15 min after radiotracer injection. Two additional rats per condition received a 11C-erlotinib injection (injected amount 43 and 40 MBq for the control condition; 32 and 19 MBq for the rifampicin condition) under isoflurane anesthesia. After 15 min, animals were decapitated and liver fragments were removed and sonicated for extraction using acetonitrile. After centrifugation, the supernatant was counted for radioactivity and subjected to radio-HPLC analysis.
Rate constant for the transfer of 11C-erlotinib from blood to tissues
Rate constants for the transfer of 11C-erlotinib from plasma to the liver, kidneys, and lungs (kuptake, liver, kuptake, kidney and kuptake, lung) were calculated using a previously described graphical method (integration plot analysis) and the following equation [21]:
where Ct, organ is the concentration of radioactivity in the investigated organ at time t, determined with PET. Ct, plasma is the concentration of parent 11C-erlotinib in the blood at time t, determined by arterial blood sampling, and AUC0 − t, plasma is the area under the blood concentration-time curve of 11C-erlotinib from time 0 to time t. kuptake is equal to the slope value of the regression line obtained by performing linear regression analysis of a plot of Ct, organ/Ct, plasma versus AUC0 − t, plasma/Ct, plasma. VE is the y-intercept of the integration plot, representing the initial distribution volume of 11C-erlotinib in the organ. Using this approach, linear uptake was observed from 0.25 to 6 min after tracer injection for the liver and from 1.25 to 6 min for the kidneys and lungs.
Statistical analysis
Data are reported as mean ± SD. Statistical analysis was performed using a two-way ANOVA with “treatment” and “organ” as factors. Statistical significance was set at p < 0.05.
Results
Figure 1 shows representative PET images centered on the abdominal region for a control and a rifampicin-pretreated rat. Radioactivity was primarily localized in the liver within the first minutes after 11C-erlotinib injection, and then it gradually moved to the intestine, thus dynamically highlighting the predominantly hepatobiliary elimination of radioactivity (Fig. 1). Radioactivity also accumulated in the kidneys, which were best visualized on late summation PET images. The lungs were visible in the very first frames only, probably due to their high vascular content (Fig. 1).
Fig. 1.
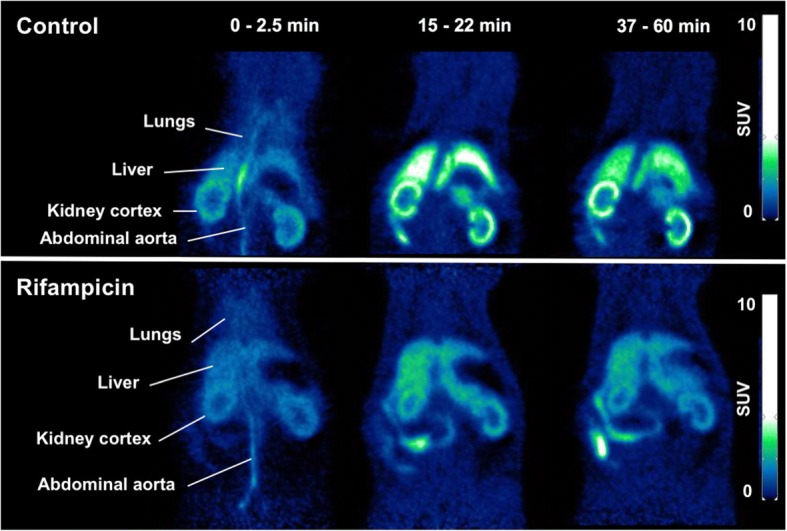
Representative PET images of the rat abdominal region after 11C-erlotinib injection in the control condition and after rifampicin pretreatment (40 mg/kg i.v; 5 min before 11C-erlotinib injection)
From time points > 3 min after injection, hepatic accumulation of radioactivity was lower in rifampicin-pretreated rats than in control animals (Fig. 1). SUVmax in the liver was significantly lower in rifampicin-treated animals (7.3 ± 1.2) as compared with control animals (9.9 ± 0.7, p < 0.05). Rifampicin pretreatment caused a significant ~ 27% decrease in AUCliver (p < 0.001; Fig. 2). In animals sacrificed at 15 min after injection, the percentage of unmetabolized 11C-erlotinib in the liver ranged from 96.4 to 100% in the control group and from 96.0 to 100% in the rifampicin-treated group, respectively. This shows a limited contribution of radiolabeled metabolites to the PET signal in the liver within the first 15 min of scanning. AUCliver from 0 to 15 min was significantly lower in rifampicin-treated animals (103.6 ± 15.2 SUV.min) as compared with the control animals (129.4 ± 10.1 SUV.min, p < 0.05).
Fig. 2.

Time-activity curves of 11C-erlotinib in the liver (a), kidneys (b), and lungs (c) and abdominal aorta (d) in the absence (control) and the presence of OATP inhibition with rifampicin (40 mg/kg i.v.). Corresponding exposure to tissues, expressed as area under the curve (AUC) in each organ, is reported in (e). Data are mean (n = 4) ± 1. S.D. ***, p < 0.001, n.s., non-significant
Radioactivity concentrations in the kidney cortex were also lower in the rifampicin-treated group, and the difference tended to increase with time (Fig. 2). AUCkidney was markedly reduced in rifampicin-treated animals (p < 0.001). The lungs showed a much lower exposure to radioactivity compared to the liver and the kidneys (Fig. 2). No significant differences in AUClung were observed between the two groups (Fig. 2).
Unexpectedly, rifampicin did not impact the image-derived blood kinetics. AUCaorta (80.4 ± 24.7 SUV.min) was not increased by rifampicin treatment (86.6 ± 23.8 SUV.min; p > 0.05). Radiotracer metabolism and binding to blood cells in either the presence or the absence of rifampicin may interfere with the estimation of the transfer of parent 11C-erlotinib from plasma to tissues. The plasma kinetics of parent 11C-erlotinib was therefore measured in one animal of each condition, from repeated arterial blood sampling, in order to provide a representative metabolite-corrected arterial input function and calculate kuptake in tissues. The percentage of parent 11C-erlotinib versus time was similar in the control and the rifampicin-pretreated rat (Fig. 3a). At 1 h after radiotracer injection, the percentage of unmetabolized 11C-erlotinib was 61% and 69% without and with rifampicin pretreatment, respectively. 11C-erlotinib plasma concentrations peaked rapidly after i.v. injection, followed by a rapid wash out (Fig. 3b). A plateau was reached after 15 min (SUV15 min = 0.34) with SUV60 min = 0.22 and 0.24 for the baseline and the rifampicin pretreated rats, respectively.
Fig. 3.

Metabolism and plasma kinetics of 11C-erlotinib in the absence (control) and the presence of rifampicin (40 mg/kg i.v.). Data were obtained using arterial blood sampling (femoral artery) in additional rats (1 animal for each condition). In a, the percentage of parent (unmetabolized) 11C-erlotinib versus time is displayed. Corresponding plasma kinetics of parent 11C-erlotinib (metabolite-corrected arterial input function) are shown in b
The lower liver and kidney exposures to radioactivity observed in the rifampicin-pretreated animals were consistent with significant decreases in kuptake, liver and kuptake, kidney (Fig. 4). There was no effect of rifampicin on kuptake, lung, which was consistent with the absence of an effect of rifampicin on AUClung (Fig. 2).
Fig. 4.

Estimation of the rate constant for the transfer of 11C-erlotinib from plasma into tissues. Integration plots of 11C-erlotinib uptake by the liver (a), kidneys (b), and lungs (c) are shown. Kuptake values are reported in d. Data are mean (n = 4) ± 1. S.D. *** p < 0.001; ** p < 0.01; n.s non-significant
Discussion
PET imaging is an appealing strategy to study the impact of OATP transporters on the tissue distribution of radiolabeled drugs [23]. In vitro studies using transfected cells showed that erlotinib is specifically transported by the human OATP2B1 [15]. Recent PET studies have demonstrated saturable liver uptake of 11C-erlotinib in humans [15]. To the best of our knowledge, the transport of erlotinib by the rodent rOATP2B1 has not been addressed but a similar saturation of the liver uptake of 11C-erlotinib was also observed in mice [14]. We therefore hypothesized that OATP inhibition with rifampicin may decrease the liver uptake of 11C-erlotinib with consequences for drug exposure to the lungs, as the therapeutic target tissue of erlotinib.
OATP transporters are recognized as the key determinants for the PK of many drugs [11]. They are expressed in the sinusoidal membrane of hepatocytes and were shown to control the uptake of their substrates from the blood into the liver. Conventional PK studies, based on the determination of drug concentrations in the plasma, have shown how alterations in hepatic OATP1B1 and OATP1B3 function may influence the plasma PK of diverse drugs. Genetic polymorphisms of the SLCO genes [24] as well as DDIs involving inhibitors of OATP transporters [25] have been deemed of clinical importance for PK variability. The importance of OATP2B1 in the liver is less understood, probably due to its overlapping substrate specificity with OATP1B1 and OATP1B3 [25]. Expression of OATP2B1 has been detected at many other blood-tissue interfaces than the sinusoidal membrane of hepatocytes, suggesting that OATP2B1 may control the target/vulnerable tissue exposure to its substrates in addition to its impact on hepatobiliary clearance [17].
Rifampicin is a prototypical OATP inhibitor used in clinical studies in drug development to assess the PK importance of OATP function [11, 26]. In vitro, rifampicin is a potent inhibitor of the human OATP1B1 (IC50 = 1.9 ± 0.16 μM) and OATP1B3 (IC50 = 6.4 ± 0.5 μM) and a weaker inhibitor of OATP2B1 (IC50 = 91.0 ± 14.6 μM) [27, 28]. Rifampicin was also shown to inhibit several rat OATPs including the liver-specific rOATP1B2 (Ki = 0.79 μM). Rifampicin also inhibits rOATP1A1 (Ki = 18 μM) and rOATP1A4 (Ki = 1.4–2.9 μM) [29–31]. The inhibition potency of rifampicin for other SLC transporters of PK importance has been studied in vitro and suggests a relatively good specificity for OATP transporters. Indeed, rifampicin inhibited neither organic cation transporter 1 (OCT1, SLC22A1, Ki > 100 μM) nor organic anion transporter 2 (OAT2, SLC22A7), which are both expressed in the sinusoidal hepatocyte membrane. Rifampicin most likely does not inhibit organic cation transporter 2 (OCT2, SLC22A2) and organic anion transporter 3 (OAT3, SLC22A8) function in vivo [32]. Plasma concentrations of rifampicin, when administered to rats at a dose of 40 mg/kg i.v., ranged from 100 to 300 μM, which exceeded the reported in vitro Ki values for inhibition of OATP transporters [20, 22]. The selected rifampicin dose was previously shown to inhibit the rOATP-mediated liver uptake of 11C-dehydropravastatin and 11C-rosuvastatin, as assessed with PET imaging in rats [20, 22]. In our study, we chose a simple i.v. bolus administration protocol, which did not require constant infusion during PET scanning.
Our PET data showed a predominant liver accumulation of radioactivity, which was consistent with hepatobiliary elimination of erlotinib [33]. A high hepatic accumulation of 11C-erlotinib has also been observed in mice and in humans [14, 15]. Rifampicin pretreatment significantly decreased the liver uptake and subsequent liver exposure to 11C-erlotinib. This suggested a role of sinusoidal rOATP transporters in controlling the hepatobiliary elimination of erlotinib. Radioactivity in the liver, analyzed at 15 min after injection, mainly consisted of unmetabolized 11C-erlotinib, regardless of the presence or absence of rOATP inhibition. This suggested that the quantification of kuptake, liver values was not influenced by radiotracer metabolism and accurately reflected the transfer constant of parent 11C-erlotinib from the blood into the liver.
Rifampicin is also a potent inhibitor of MRP2 (ABCC2) expressed at the canalicular side of hepatocytes [19]. Erlotinib is not a substrate of MRP2 [7]. Consistently, the slopes of 11C-erlotinib elimination from the liver were similar in the control and rifampicin-treated animals suggesting a predominant impact of rifampicin in reducing the basaloteral uptake of 11C-erlotinib by hepatocytes [19].
Control 11C-erlotinib PET images showed a relatively high uptake of the radioactivity in the kidney cortex. Unexpectedly, rifampicin also decreased the uptake of 11C-erlotinib into the kidneys, which led to a significant 30.4% decrease in kidney exposure. This effect is consistent with the saturable uptake of 11C-erlotinib by the kidneys reported in mice [14]. A similar phenomenon has been observed for the OATP substrate 11C-rosuvastatin, which kidney uptake was also decreased after rifampicin administration [20]. This suggested a role for rifampicin-inhibitable transporter(s) expressed in rat kidney epithelial cells in controlling the uptake and subsequent kidney exposure to erlotinib. The decrease in the kidney exposure was related to the decrease in kuptake, kidneys. This suggests a role for a basolateral blood-to-kidney transport rather than secretion or reabsorption process at the tubular level, as previously reported for 11C-rosuvastatin [20]. Rifampicin is an inhibitor of OAT1 and OAT2 but not OAT3 expressed at the basolateral membrane of kidney proximal tubules [20]. However, erlotinib is neither transported by OAT1 nor OAT2 but is a substrate of OAT3 [12]. Erlotinib is also a substrate of OCT2 which is not inhibited by rifampicin [12, 27]. Rifampicin is not an inhibitor of OATP4C1 [34] which importance for erlotinib transport is not known. Previous mice studies have shown that parent unmetabolized 11C-erlotinib could not be detected in urines [14]. It is therefore difficult to address whether changes in the 11C-erlotinib uptake by the kidneys translated into changes in the urinary clearance of the tracer. Species differences in the expression and function of membrane transporters may occur between rats and humans. Clinical experiments using 11C-erlotinib along with urine sampling would be useful to finally address the putative impact of these rifampicin-inhibitable transporter(s) in the uptake by the kidney tissue and whether it could drive any change in the renal elimination of erlotinib and its metabolites.
The lungs are the main target tissue for erlotinib therapy in patients. A decrease in the distribution of 11C-erlotinib to the major clearance organs liver and kidneys was expected to lead to an increase in plasma concentrations and a concomitant increase in lung exposure. Increase in plasma concentrations has been observed for 11C-rosuvastatin, as a consequence of decreased hepatic and renal uptake by rifampicin [20]. Interestingly, our results indicated that rifampicin decreased the distribution of erlotinib into the main elimination organs of the body with no or negligible consequences for blood and lung exposure. The lungs contain a high vascular fraction, and the concentration of radioactivity measured with PET in the lungs can therefore be expected to be influenced by the plasma kinetics of 11C-erlotinib, which did not differ between the control and rifampicin-treated rats.
The lack of an effect of rifampicin on systemic exposure suggested a limited role for rifampicin-inhibitable transporter(s) in controlling the total clearance of erlotinib. First, it can be hypothesized that a larger decrease in the uptake of 11C-erlotinib by the liver or kidneys would be necessary to detect changes into plasma PK. Indeed, saturation of the liver and kidney transport by unlabeled erlotinib led to significant changes in the plasma PK of 11C-erlotinib [14]. This suggests that substantial changes into the uptake of drugs by elimination organs may not directly translate into changes in plasma PK. This phenomenon could also be explained by compensatory mechanisms, such as a shift from hepatobiliary elimination to another (probably urinary) elimination pathway, which was previously reported for 11C-erlotinib in mice [14, 35]. It may also be hypothesized that efficient reabsorption of 11C-erlotinib at the enterohepatic level, which may not be inhibited by rifampicin, may counteract the decreased transport of 11C-erlotinib throughout hepatocytes. The impact of transporter function on the enterohepatic circulation of drugs is difficult to assess using PET. Nevertheless, such phenomenon would probably translate into changes in the early plasma kinetics [36], which was not observed in our experiments.
Rifampicin had no significant impact on kuptake, lung, suggesting that erlotinib uptake by the lungs is independent of rOATP transporter function. This is consistent with previous studies showing that rOATP1A1, rOATP1A4, and rOATP1B2, which can be inhibited with rifampicin, are not expressed in the lungs [37].
Imaging data obtained in the present animal study should be interpreted with respect to a clinical extrapolation. First, the expression, function, and tissue distribution of OATP transporters differ between rats and humans [38]. Second, the rifampicin dose used in this study was considerably higher than the rifampicin dose used to test OATP-mediated DDIs in humans (10 mg/kg). Third, we used in the present study a microdose of erlotinib, on a relatively short time, which may not reflect the PK behavior of a pharmacological dose of erlotinib. In fact, a previous 11C-erlotinib PET study in healthy volunteers suggested that OATP2B1-mediated liver uptake of erlotinib was negligible at pharmacological doses, due to saturation of transport activity [15]. Nevertheless, our results demonstrate that discrepancies may exist between the PK of drugs in elimination organs, their systemic PK, and subsequent exposure to target tissues. Our data suggest that OATP-mediated DDIs, with consequences for liver and kidney exposure, may not necessarily translate into changes in systemic PK. Previous studies have demonstrated the feasibility of 11C-erlotinib PET imaging in healthy volunteers [15], which may be combined with rifampicin administration [39]. A clinical translation of our results is thus possible to validate 11C-erlotinib as a PET probe for imaging of OATP2B1 function and to assess the impact of OATP inhibition on the liver distribution and PK of erlotinib in humans.
Passive diffusion is often assumed to be the main mechanism for erlotinib to enter cancer cells and reach the EGFR kinase domains. However, it is not known whether TKIs such as erlotinib can freely cross membranes by passive diffusion or whether SLC transporters play a role in the tumor uptake [40]. Such phenomenon may control drug delivery to the tumor and account for therapeutic efficiency, with limited impact on the plasma PK. 11C-erlotinib PET imaging, performed with and without transporter inhibitors such as rifampicin, could be useful to address the impact of membrane transporters in controlling the uptake of erlotinib by tumor cells in vivo [41, 42].
Conclusion
In this study, the impact of rifampicin on erlotinib uptake by the liver and kidneys was assessed using 11C-erlotinib PET imaging in rats. Unexpectedly, despite significant decreases in liver and kidney exposure, rifampicin did not affect systemic exposure and subsequent exposure to the lungs as the main therapeutic target tissue of erlotinib. The consequences for erlotinib PK of these newly reported rifampicin-inhibitable transports, possibly OATP, should be addressed in humans using 11C-erlotinib PET imaging [39].
Acknowledgements
We thank Maud Goislard, Thierry Lekieffre, and Christine Coulon for technical assistance.
Funding
Sylvain Auvity received a public grant overseen by the French National Research Agency (ANR) as part of the « Investissement d’Avenir » program, through the “Lidex-PIM” project funded by the IDEX Paris-Saclay, ANR-11-IDEX-0003-02. This work was performed on a platform of France Life Imaging network partly funded by the grant “ANR-11-INBS-0006.”
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABC
ATP-binding cassette
- AUC
Area under the curve
- CYP
Cytochrome P-450
- EGFR
Epidermal growth factor receptor
- NSCLC
Non-small-cell lung cancer
- OAT
Organic anion transporter
- OATP
Organic anion-transporting polypeptide
- PET
Positron emission tomography
- P-gp
P-glycoprotein
- PK
Pharmacokinetics
- SLC
Solute carrier
- SUV
Standardized uptake value
Authors’ contributions
DA, SA, and SG performed the rat studies. FC performed the radiochemical production. DA, SA, and SM analyzed and interpreted the PET data. NT, SA, MB, and OL designed the study. DA and NT wrote the manuscript. SA, MB, and OL critically revised the manuscript. All authors read and approved the final manuscript.
Ethics approval
Animal studies were conducted in accordance with the French legislation and European directives on the use of animals in research. The protocol has been accepted by a local ethics committee for animal use (protocol number A16/057).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dorra Amor, Email: dorra.amor@rns.tn.
Sébastien Goutal, Email: sebastien.goutal@cea.fr.
Solène Marie, Email: solene.marie@aphp.fr.
Fabien Caillé, Email: fabien.caille@cea.fr.
Martin Bauer, Email: martin.m.bauer@meduniwien.ac.at.
Oliver Langer, Email: oliver.langer@ait.ac.at.
Sylvain Auvity, Email: sylvain.auvity@aphp.fr.
Nicolas Tournier, Phone: +33 1 69 86 77 12, Email: nicolas.tournier@cea.fr.
References
- 1.Rusch V, Mendelsohn J, Dmitrovsky E. The epidermal growth factor receptor and its ligands as therapeutic targets in human tumors. Cytokine Growth Factor Rev. 1996;7:133–141. doi: 10.1016/1359-6101(96)00016-0. [DOI] [PubMed] [Google Scholar]
- 2.Mayo C, Bertran-Alamillo J, Molina-Vila MÁ, Giménez-Capitán A, Costa C, Rosell R. Pharmacogenetics of EGFR in lung cancer: perspectives and clinical applications. Pharmacogenomics. 2012;13:789–802. doi: 10.2217/pgs.12.54. [DOI] [PubMed] [Google Scholar]
- 3.Hidalgo M, Bloedow D. Pharmacokinetics and pharmacodynamics: maximizing the clinical potential of Erlotinib (Tarceva) Semin Oncol. 2003;30:25–33. doi: 10.1016/S0093-7754(03)70012-8. [DOI] [PubMed] [Google Scholar]
- 4.Tan AR, Yang X, Hewitt SM, Berman A, Lepper ER, Sparreboom A, et al. Evaluation of biologic end points and pharmacokinetics in patients with metastatic breast cancer after treatment with erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor. J Clin Oncol. 2004;22:3080–90. [DOI] [PubMed]
- 5.Gao B, Yeap S, Clements A, Balakrishnar B, Wong M, Gurney H. Evidence for therapeutic drug monitoring of targeted anticancer therapies. J Clin Oncol. 2012;30:4017–25. [DOI] [PubMed]
- 6.Terada T, Noda S, Inui K. Management of dose variability and side effects for individualized cancer pharmacotherapy with tyrosine kinase inhibitors. Pharmacol Ther. 2015;152:125–134. doi: 10.1016/j.pharmthera.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MAJ, Beijnen JH, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1−/−/Mdr1a/1b−/− (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7:2280–2287. doi: 10.1158/1535-7163.MCT-07-2250. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–7. [DOI] [PubMed]
- 9.Rakhit A, Pantze MP, Fettner S, Jones HM, Charoin J-E, Riek M, et al. The effects of CYP3A4 inhibition on erlotinib pharmacokinetics: computer-based simulation (SimCYP) predicts in vivo metabolic inhibition. Eur J Clin Pharmacol. 2008;64:31–41. doi: 10.1007/s00228-007-0396-z. [DOI] [PubMed] [Google Scholar]
- 10.Hamilton M, Wolf JL, Drolet DW, Fettner SH, Rakhit AK, Witt K, et al. The effect of rifampicin, a prototypical CYP3A4 inducer, on erlotinib pharmacokinetics in healthy subjects. Cancer Chemother Pharmacol. 2014;73:613–621. doi: 10.1007/s00280-014-2390-3. [DOI] [PubMed] [Google Scholar]
- 11.Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmeliegy MA, Carcaboso AM, Tagen M, Bai F, Stewart CF. Role of ATP-binding cassette and solute carrier transporters in erlotinib CNS penetration and intracellular accumulation. Clin Cancer Res. 2011;17:89–99. [DOI] [PMC free article] [PubMed]
- 13.Tournier N, Goutal S, Auvity S, Traxl A, Mairinger S, Wanek T, et al. Strategies to inhibit ABCB1- and ABCG2-mediated efflux transport of erlotinib at the blood-brain barrier: a PET study on nonhuman primates. J Nucl Med. 2017;58:117–22. [DOI] [PubMed]
- 14.Traxl A, Wanek T, Mairinger S, Stanek J, Filip T, Sauberer M, et al. Breast cancer resistance protein and P-glycoprotein influence in vivo disposition of 11C-erlotinib. J Nucl Med. 2015;56:1930–6. [DOI] [PubMed]
- 15.Bauer M, Matsuda A, Wulkersdorfer B, Philippe C, Traxl A, Özvegy-Laczka C, et al. Influence of OATPs on hepatic disposition of erlotinib measured with positron emission tomography. Clin Pharmacol Ther. 2018;104:139-147. [DOI] [PMC free article] [PubMed]
- 16.Giacomini KM, Huang S-M. Transporters in drug development and clinical pharmacology. Clin Pharmacol Ther. 2013;94:3–9. doi: 10.1038/clpt.2013.86. [DOI] [PubMed] [Google Scholar]
- 17.Hagenbuch B, Stieger B. The SLCO (former SLC21) superfamily of transporters. Mol Asp Med. 2013;34:396–412. doi: 10.1016/j.mam.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bahce I, Smit EF, Lubberink M, van der Veldt AAM, Yaqub M, Windhorst AD, et al. Development of [(11)C] erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:183–193. doi: 10.1158/1078-0432.CCR-12-0289. [DOI] [PubMed] [Google Scholar]
- 19.Neyt S, Huisman MT, Vanhove C, De Man H, Vliegen M, Moerman L, et al. In vivo visualization and quantification of (disturbed) Oatp-mediated hepatic uptake and Mrp2-mediated biliary excretion of 99mTcmebrofenin in mice. J Nucl Med. 2013;54:624–30. [DOI] [PubMed]
- 20.He J, Yu Y, Prasad B, Link J, Miyaoka RS, Chen X, et al. PET imaging of Oatp-mediated hepatobiliary transport of [(11)C] rosuvastatin in the rat. Mol Pharm. 2014;11:2745–2754. doi: 10.1021/mp500027c. [DOI] [PubMed] [Google Scholar]
- 21.Takashima T, Hashizume Y, Katayama Y, Murai M, Wada Y, Maeda K, et al. The involvement of organic anion transporting polypeptide in the hepatic uptake of telmisartan in rats: PET studies with [11C]telmisartan. Mol Pharm. 2011;8:1789–1798. doi: 10.1021/mp200160t. [DOI] [PubMed] [Google Scholar]
- 22.Shingaki T, Takashima T, Ijuin R, Zhang X, Onoue T, Katayama Y, et al. Evaluation of Oatp and Mrp2 activities in hepatobiliary excretion using newly developed positron emission tomography tracer [11C] dehydropravastatin in rats. J Pharmacol Exp Ther. 2013;347:193–202. doi: 10.1124/jpet.113.206425. [DOI] [PubMed] [Google Scholar]
- 23.Marie S, Cisternino S, Buvat I, Declèves X, Tournier N. Imaging probes and modalities for the study of solute carrier O (SLCO)-transport function in vivo. J Pharm Sci. 2017;106:2335–2344. doi: 10.1016/j.xphs.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 24.Giacomini KM, Balimane PV, Cho SK, Eadon M, Edeki T, Hillgren KM, et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther. 2013;94:23–26. doi: 10.1038/clpt.2013.12. [DOI] [PubMed] [Google Scholar]
- 25.König J, Müller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–966. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 26.Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther. 2009;85:78–85. doi: 10.1038/clpt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parvez MM, Jung JA, Shin HJ, Kim DH, Shin J-G. Characterization of 22 antituberculosis drugs for inhibitory interaction potential on organic anionic transporter polypeptide (OATP)-mediated uptake. Antimicrob Agents Chemother. 2016;60:3096–3105. doi: 10.1128/AAC.02765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55:4740–4763. doi: 10.1021/jm300212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fattinger K, Cattori V, Hagenbuch B, Meier PJ, Stieger B. Rifamycin SV and rifampicin exhibit differential inhibition of the hepatic rat organic anion transporting polypeptides, Oatp1 and Oatp2. Hepatol Baltim Md. 2000;32:82–86. doi: 10.1053/jhep.2000.8539. [DOI] [PubMed] [Google Scholar]
- 30.Lau YY, Okochi H, Huang Y, Benet LZ. Pharmacokinetics of atorvastatin and its hydroxy metabolites in rats and the effects of concomitant rifampicin single doses: relevance of first-pass effect from hepatic uptake transporters, and intestinal and hepatic metabolism. Drug Metab Dispos. 2006;34:1175–81. [DOI] [PubMed]
- 31.Shitara Y, Sugiyama D, Kusuhara H, Kato Y, Abe T, Meier PJ, et al. Comparative inhibitory effects of different compounds on rat oatpl (slc21a1)- and Oatp2 (Slc21a5)-mediated transport. Pharm Res. 2002;19:147–153. doi: 10.1023/A:1014264614637. [DOI] [PubMed] [Google Scholar]
- 32.Parvez MM, Kaisar N, Shin HJ, Jung JA, Shin J-G. Inhibitory interaction potential of 22 antituberculosis drugs on organic anion and cation transporters of the SLC22A family. Antimicrob Agents Chemother. 2016;60:6558–6567. doi: 10.1128/AAC.01151-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling J, Johnson KA, Miao Z, Rakhit A, Pantze MP, Hamilton M, et al. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 2006;34:420–6. [DOI] [PubMed]
- 34.Sato T, Mishima E, Mano N, Abe T, Yamaguchi H. Potential drug interactions mediated by renal organic anion transporter OATP4C1. J Pharmacol Exp Ther. 2017;362:271–277. doi: 10.1124/jpet.117.241703. [DOI] [PubMed] [Google Scholar]
- 35.Traxl A, Komposch K, Glitzner E, Wanek T, Mairinger S, Langer O, et al. Hepatocyte-specific deletion of EGFR in mice reduces hepatic Abcg2 transport activity measured by [(11)C] erlotinib and positron emission tomography. Drug Metab Dispos Biol Fate Chem. 2017;45:1093–1100. doi: 10.1124/dmd.117.077081. [DOI] [PubMed] [Google Scholar]
- 36.Abbiati RA, Manca D. Enterohepatic circulation effect in physiologically based pharmacokinetic models: the sorafenib case. Ind Eng Chem Res. 2017;56:3156–3166. doi: 10.1021/acs.iecr.6b03686. [DOI] [Google Scholar]
- 37.Cattori V, van Montfoort JE, Stieger B, Landmann L, Meijer DK, Winterhalter KH, et al. Localization of organic anion transporting polypeptide 4 (Oatp4) in rat liver and comparison of its substrate specificity with Oatp1, Oatp2 and Oatp3. Pflugers Arch. 2001;443:188–195. doi: 10.1007/s004240100697. [DOI] [PubMed] [Google Scholar]
- 38.Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–1287. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takashima T, Kitamura S, Wada Y, Tanaka M, Shigihara Y, Ishii H, et al. PET imaging-based evaluation of hepatobiliary transport in humans with (15R)-11C-TIC-Me. J Nucl Med. 2012;53:741–8. [DOI] [PubMed]
- 40.Neul C, Schaeffeler E, Sparreboom A, Laufer S, Schwab M, Nies AT. Impact of membrane drug transporters on resistance to small-molecule tyrosine kinase inhibitors. Trends Pharmacol Sci. 2016;37:904–932. doi: 10.1016/j.tips.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Mann A, Semenenko I, Meir M, Eyal S. Molecular imaging of membrane transporters’ activity in cancer: a picture is worth a thousand tubes. AAPS J. 2015;17:788–801. doi: 10.1208/s12248-015-9752-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tournier N, Stieger B, Langer O. Imaging techniques to study drug transporter function in vivo. Pharmacol Ther. doi: 10.1016/j.pharmthera.2018.04.006. [Epub ahead of print]. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.