

Abstract
Toxoplasma gondii is an important zoonotic pathogen infecting one-third of the world’s population and numerous animals, causing significant healthcare burden and socioeconomic problems. Vaccination is an efficient way to reduce global sero-prevalence, however, ideal vaccines are not yet available. We recently discovered that the Toxoplasma mutant lacking both lactate dehydrogenases LDH1 and LDH2 (Δldh) grew well in vitro but was unable to propagate in mice, making it a good live vaccine candidate. Here, we tested the protection efficacy of ME49 Δldh using a mouse model. Vaccinated mice were efficiently protected from the lethal challenge of a variety of wild-type strains, including type 1 strain RH, type 2 strain ME49, type 3 strain VEG, and a field isolate of Chinese 1. The protection efficacies of a single vaccination were nearly 100% for most cases and it worked well against the challenges of both tachyzoites and tissue cysts. Re-challenging parasites were unable to propagate in vaccinated mice, nor did they make tissue cysts. High levels of Toxoplasma-specific IgG were produced 30 days after immunization and stayed high during the whole tests (at least 125 days). However, passive immunization of naïve mice with sera from vaccinated mice did reduce parasite propagation, but the overall protection against parasite infections was rather limited. On the other hand, Δldh immunization evoked elevated levels of Th1 cytokines like INF-γ and IL-12, at early time points. In addition, splenocytes extracted from immunized mice were able to induce quick and robust INF-γ and other pro-inflammatory cytokine production upon T. gondii antigen stimulation. Together these results suggest that cellular immune responses are the main contributors to the protective immunity elicited by Δldh vaccination, and humoral immunity also contributes partially. We also generated uracil auxotrophic mutants in ME49 and compared their immune protection efficiencies to the Δldh mutants. The results showed that these two types of mutants have similar properties as live vaccine candidates. Taken together, these results suggest that mutants lacking LDH were severely attenuated in virulence but were able to induce strong anti-toxoplasma immune responses, therefore are good candidates for live vaccines.
Keywords: Toxoplasma, lactate dehydrogenase, live vaccine, toxoplasmosis, cellular immunity
Introduction
Toxoplasma gondii is an obligate intracellular parasite that infects all warm-blooded animals and humans (1). Generally, its infection in healthy people causes no or mild flu-like symptoms, thus most of the infections are not noticed. However, in susceptible pregnant women, T. gondii infection may have severe consequences such as abortion, neonatal death, congenital defects, and mental retardation of delivered babies (2, 3). In addition, it is also a high risk for individuals with compromised immune functions, such as AIDS and organ transplant patients (2). Due to the broad host range, a variety of agricultural important animals such as pigs and sheep are constantly challenged by T. gondii, causing substantial economic losses and public health problems (2, 4, 5). Folic acid metabolism inhibitors such as pyrimethamine and sulfadiazine are commonly used to treat toxoplasmosis but they do not work on chronic infections (6).
The control of Toxoplasma infection is rather difficult, one reason is that it has complex life cycle and multiple routes of transmission (7, 8). Cats are the definitive hosts of T. gondii and the oocysts shed by cats are thought to be a key source of human and animal infections (9). In addition, T. gondii can be transmitted between intermediate hosts through predation. Most of the Toxoplasma infection cases belong to chronic infection, where the parasites are encysted in muscles and central nerve system (called tissue cysts) of infected animals lifelong (7). Ingestion of raw or undercooked meat from such animals represents another important route of transmitting the parasites to humans and animals (2, 5). As mentioned above, encysted parasites at the chronic infection stage are resistant to most of the current therapeutics. Another challenge to the control of toxoplasmosis is the complex population structure of T. gondii strains. North America and Europe are dominated by three clonal strains (type I, II, and III), which display different acute virulence in mice (10, 11). However, in other parts of the world, the strains are much more diverse, particularly in South America (12, 13). A recent study demonstrated that genetically distinct strains may be able to superinfect the same host, indicating the lack of sufficient cross protection from immunization with one single strain (14). This study had important highlights for the design of Toxoplasma vaccines, particularly whole parasite-based vaccines.
Scientists have done tremendous amount of work to pursue an ideal vaccine against T. gondii. The first generation of vaccines contained killed parasites, or native antigens derived from soluble or secretory proteins of cultured tachyzoites. These vaccines only provided limited protection against further infections (15, 16). Then when the recombinant DNA technology became available, a variety of subunit vaccines were tried, mainly using surface or secretory proteins such as SAG1 and MIC2 (17–20). These recombinant protein or vector-based subunit vaccines were safer and easier to make than native antigen-based vaccines, however, they did not provide sufficient protection either [reviewed in Ref. (21, 22)]. The strategy that holds the most promise for a good Toxoplasma vaccine seems to be live attenuated vaccines. Currently there is one commercial vaccine (Toxovax®) available, which is derived from the S48 strain originally isolated from an aborted lamb and licensed for use to avoid congenital toxoplasmosis in ewes (23). The exact mechanisms of Toxovax® as a vaccine are not well understood, but thought to be linked to its inability to form cysts or oocysts to complete the life cycle (24). Tachyzoites can be cleared efficiently by hosts’ immunity, therefore mutants defective in cyst formation have the potential to be vaccines. Encouraged by the success of Toxovax® and to design safer live vaccines, scientists turned to genetically modified parasites. Among these, uracil auxotroph mutants defective in de novo UMP (uridine 5′-monophosphate) synthesis are promising (25–27). Mutants with inactivated CPSII or OMPDC grew well in vitro in the presence of extra uracil (25–27), but were unable to establish acute infection in animals, therefore were severely attenuated. These mutants were extensively studied in mice and displayed great potential to be good vaccines, but still need to be tested in other animals like pigs, sheep, and cats.
We recently discovered that T. gondii mutants with both lactate dehydrogenase genes deleted (Δldh) grew robustly in vitro but failed to propagate in vivo (28), very similar to the uracil auxotroph mutants. The reason for this growth difference is that, under in vivo conditions when oxygen is limited, lactate fermentation becomes a key energy supply pathway to support parasite replication. By contrast, oxidative phosphorylation alone provides enough energy for parasite reproduction in vitro when oxygen is rich. As such, Δldh mutant was greatly attenuated in vivo, even in immune-deficient animals (28). In this study, we set to analyze the protective immunity of the Δldh mutant as a vaccine using the mouse model. The results showed that vaccination of mice with tachyzoites of this mutant induced efficient protection against the challenge of a variety of strains.
Results
Tissue Cysts Derived From ME49 Δldh Were Severely Attenuated in Mice
Our previous study reported that the virulence of ME49 Δldh tachyzoites was significantly (>640-fold) reduced compared to that of the wild-type (WT) strain ME49, no mortality was detected even at the infection dose of 3.2 × 104 tachyzoites/mouse (28). However, we did observe that many of the Δldh tachyzoites infected mice formed brain cysts, although the amount was much less than that of WT parasites infected mice. To see whether the residual amount of cysts in Δldh infected mice have normal virulence, 50 cysts derived from WT or Δldh parasites were used to infect ICR mice by oral administration, and the survival of infected mice was monitored for 40 days. The results showed that 50 cysts of ME49 killed 80% of the mice. However, none of the Δldh cyst infected mice died (Figure 1), nor did they show any obvious clinical symptoms, suggesting that the virulence of the Δldh cysts was also significantly attenuated, just like the tachyzoites of this strain.
Figure 1.
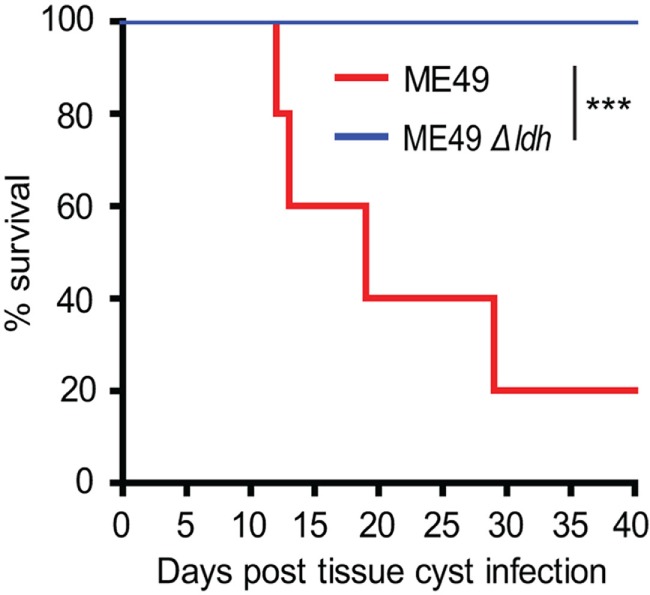
Virulence tests of Toxoplasma cysts in mice. ICR mice were infected with brain cysts of ME49 or ME49 Δldh (50 cysts/mouse, n = 5 mice for ME49 strain and n = 7 mice for ME49 Δldh strain) by oral administration and their survival was monitored for 40 days. ***p < 0.001, Gehan–Breslow–Wilcoxon test.
Δldh Vaccination Elicited Strong Protective Immunity Against Tachyzoites Infection
The Δldh mutant grew robustly in vitro but was unable to propagate in vivo, this unique property makes it a good vaccine candidate (28). To check this possibility and assess the immune protection offered by Δldh vaccination, ICR mice were first immunized with ME49 Δldh by intraperitoneal injection (104 tachyzoites/mouse). Thirty days later, they were challenged with 104 tachyzoites of the type 2 strain ME49, type 3 strain VEG, or Chinese 1 strain C7719. Subsequently survival of the mice was monitored for another 30 days. The mortality rates for non-immunized mice were 100% upon the infection of ME49 and C7719, whereas VEG caused 90% mortality. However, for Δldh-vaccinated mice, the survival rates were 100% upon the challenge of all three strains and no obvious symptoms were observed during the 30-day infection period (Figures 2A–C). These results indicate that Δldh vaccination is able to offer efficient protection against lethal parasite infections.
Figure 2.

Δldh parasite immunization protected mice from Toxoplasma gondii tachyzoites infection. ICR mice were pre-immunized with 104 tachyzoites of the ME49 Δldh mutant. (A–C) 30 days post-immunization, they were challenged with 104 tachyzoites of the ME49 (A), VEG (B), or C7719 (C) strains (10 mice for each strain) by intraperitoneal injection and their survival was monitored for another 30 days. (D–F) 75 days post-immunization, 104 tachyzoites of the RH Δhxgprt (D), ME49 (E), C7719 (F) strains were used to challenge the mice and their survival was monitored for another 30 days. (G,H) 125 days post-immunization, 104 tachyzoites of the ME49 (G), VEG (H) strains were used to challenge the mice and their survival was monitored for another 30 days. Non-immunized mice were included as control. ***p < 0.001, Gehan–Breslow–Wilcoxon tests.
The above results showed that 30 days post Δldh vaccination, mice were well protected. To see whether Δldh vaccination provides longer time protective immunity, mice were vaccinated with ME49 Δldh and 75 days later, they were challenged with type 1 strain RH Δhxgprt, type 2 strain ME49, or Chinese 1 strain C7719. The survival rates of immunized mice that were re-infected with ME49 or C7719 were 100% (Figures 2E,F), similar to the above infections 30 days post-vaccination. However, if challenged with the most virulent type 1 strain RH Δhxgprt, only 60% of the immunized mice survived (Figure 2D). As will be discussed below, the protection against RH infection was also 100% if the challenge occurred 20 days after immunization. To examine the efficiency of even longer time protection, ME49 Δldh vaccinated mice were challenged with WT parasites 125 days post-vaccination. The results showed that the immune protection against ME49 and VEG challenge was still 100% (Figures 2G,H). Together, these results suggest that ME49 Δldh immunization can provide both short- and long-term protection against strains with intermediate virulence, but the protection against strongly virulent strains is limited to a short time after vaccination and then decreased gradually.
Δldh Immunization Stimulated Protective Immunity Against Bradyzoites Infection
A significant portion of T. gondii infections were acquired through ingestion of tissue cysts that contain bradyzoites. In order to check whether Δldh vaccination provided protection against bradyzoites infection, ICR mice were first immunized with 104 tachyzoites of ME49 Δldh. Thirty days after immunization, they were orally challenged with 50 cysts of the ME49 strain. The results show that 100% of Δldh-immunized mice survived the cyst challenging, whereas all naïve mice died of this infection (Figure 3). These results suggest that Δldh strain also elicited protective immunity against bradyzoite infection.
Figure 3.
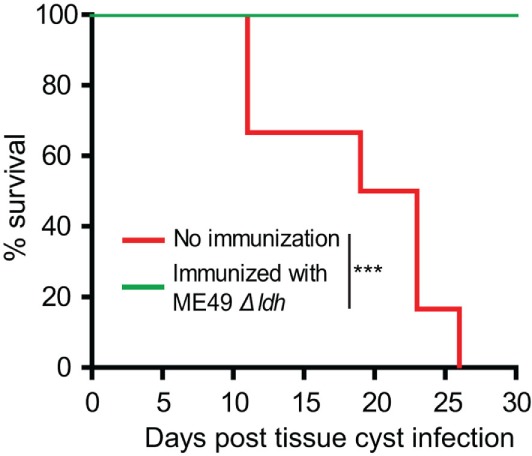
Δldh mutant vaccination protected mice from Toxoplasma gondii cysts infection. ICR mice were pre-immunized with ME49 Δldh as in Figure 2, 30 days post-immunization they were challenged with 50 fresh brain cysts of the ME49 strain and their survival was monitored for another 30 days (n = 10 mice for each strain). Non-immunized mice were included as control. ***p < 0.001, Gehan–Breslow–Wilcoxon test.
Δldh Immunization Blocked Cyst Formation From New Infections
One caveat of chemotherapies against acute toxoplasmosis is that the drugs may drive the parasites to slowly replicating bradyzoites to form tissue cysts, which lead to lifelong chronic infection. To examine whether the Δldh mutant vaccination could provide protective immunity against chronic T. gondii infection, the cyst forming competent but less virulent (compared with ME49) strain TgPIG-WH1 (genotyped as ToxoDB #3) was used to challenge the vaccinated ICR mice. As shown in Figure 4A, at the infection dose of 104 tachyzoites/mouse through intraperitoneal injection, TgPIG-WH1 killed only 10% of the non-immunized mice, consistent with its reduced virulence. Thirty days post TgPIG-WH1 challenge, survived mice were sacrificed and the numbers of tissue cysts in the brains were determined by DBA-FITC staining, and compared to that of vaccinated but not re-challenged, as well as non-immunized but TgPIG-WH1 infected mice. The results in Figure 4B showed that infection of naïve mice by TgPIG-WH1 resulted in about 500 cysts per brain on average. However, cyst number in Δldh immunized and subsequently TgPIG-WH1 infected mice was reduced to around 100, which was very similar to that of vaccinated but not re-challenged mice, suggesting that they were probably derived from vaccination but not TgPIG-WH1 infection. Together, these results indicate that Δldh immunization also provides protective immunity against chronic toxoplasmosis.
Figure 4.
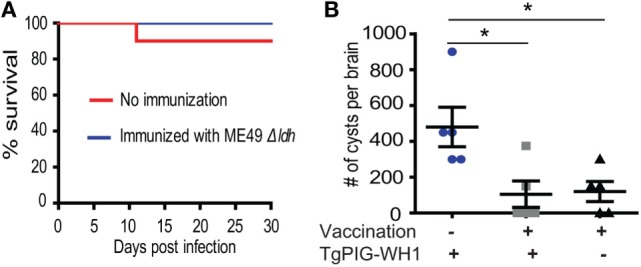
Δldh mutant immunization prevented cyst formation from further infections. (A) ICR mice were pre-immunized with ME49 Δldh as in Figure 2 and 30 days later challenged with 104 tachyzoites of TgPIG-WH1. Non-immunized mice were included as control and survival of mice were monitored for another 30 days (n = 10 mice for each strain). (B) Cyst loads in the brains of non-immunized but TgPIG-WH1 challenged mice, immunized and TgPIG-WH1 challenged mice, and Δldh immunized but not TgPIG-WH1 challenged mice [not shown in (A)]. The mice survived at day 30 in (A) were sacrificed, and the number of Toxoplasma cysts in the brain homogenate was determined by DBA-FITC staining and fluorescent microscopy. Data from the analysis of five mice in each group were graphed, *p < 0.05, Student’s t-test.
Δldh Vaccinated Mice Were Able to Clear Challenging Parasites Rapidly
The above results demonstrated that vaccinating mice with ME49 Δldh provided efficient protection against both acute and chronic toxoplasmosis. To further examine the fate of challenging parasites, the propagation dynamics of a luciferase expressing strain RH-Luc in vaccinated mice were determined by bioluminescent imaging. In naïve mice, RH-Luc infection resulted in rapid replication of the parasites. At the infection dose of 104 tachyzoites/mouse, luminescent signals were detectable just 1 day after infection and increased over 1,000-fold 5 days post-infection (Figures 5A,B). However, in the vaccinated mice, luminescent signals were not detectable at both time points, indicating little propagation and probably rapid clearance of the challenging RH-Luc (Figures 5A,B). It should be noted that in this experiment, RH-Luc infection was performed 30 days after Δldh immunization. We also followed the survival of these RH-Luc infected mice for 20 days, all naïve mice died within 8 days but all vaccinated mice survived with no symptoms (Figure 5C). These results suggest that, at least within a limited time period, Δldh immunization enables rapid clearance of the challenging parasites, which explains its efficient immune protection.
Figure 5.

Δldh parasite vaccination elicited rapid clearance of challenging parasites. (A) Naïve or pre-immunized mice were challenged with 104 tachyzoites of RH-Luc (five mice for each group). Parasite loads in mice 1 and 5 days post RH-Luc infection were analyzed by live animal imaging on the IVIS Spectra system. p/sec/cm2/sr: photons per second per cm2 per steradian. (B) Total bioluminescent signals of mice from (A) were calculated and graphed, **p < 0.01, Student’s t-test. (C) Survival curves of naïve or Δldh immunized mice infected with RH-Luc, ***p < 0.001, Gehan–Breslow–Wilcoxon test.
Δldh Vaccination Significantly Increased the Levels of Pro-Inflammatory Cytokines and T. gondii Specific IgG Antibodies
To estimate the potential mechanisms of immune protection offered by Δldh vaccination, we first examined the cytokine level changes upon vaccination. Sera samples obtained from vaccinated mice 30, 75, or 125 days post-vaccination were subject to ELISA analysis to check the levels of IFN-γ, IL-12, IL-10, and TNF-α. The IL-12–IFN-γ axis is key to activate cellular immune clearance of T. gondii. Their levels, as well as the levels of another pro-inflammatory cytokine TNF-α, increased significantly both 30 and 75 days post-vaccination, compared to control mice (Figures 6A–C). However, the levels at day 75 were lower than that at day 30, which was likely caused by the activation of anti-inflammatory response, as evidenced by high levels of IL-10 in vaccinated mice 30 days post-infection (Figure 6D). At 125 days post-infection, the levels of all cytokines were back to normal as in naïve mice (Figures 6A–D). We also measured the T. gondii specific IgG levels and found that ME49 Δldh vaccination induced high levels of IgG 30, 75, and 125 days post-immunization (Figure 6E). Although it looked like there was an increase in T. gondii specific IgG levels over time, the differences were not statistically significant (Figure 6E). The relatively stable levels of parasite-specific IgG were in contrast to the changes of cytokine levels. These results, along with the above-described short- and long-term protective immunity (Figure 2), suggest that Δldh immunization probably stimulated both humoral and cell-mediated immune responses to control secondary infections.
Figure 6.

Levels of selected cytokines and Toxoplasma specific IgG in sera of mice 30, 75, and 125 days after Δldh immunization. (A–D) Levels of indicated cytokines measured by ELISA. (E) Relative levels of Toxoplasma specific IgG, as determined by indirect ELISA. Sera from naïve mice were used as controls and three mice from each group were analyzed. *p < 0.05, **p < 0.01, ***p < 0.001, NS: not significant, Student’s t-test.
Passive Immunization With the Sera of Δldh-Vaccinated Mice Provided Partial Protection Against T. gondii Infection
Above results demonstrated that high levels of T. gondii specific IgG were produced in Δldh-vaccinated mice. To estimate the contribution of such antisera in restricting further parasite infection, naïve mice were infected with the WT strain ME49 through intraperitoneal injection. At day 0 and day 3 after infection, sera from the ME49 Δldh-vaccinated mice were collected 160 days post-immunization and administered into infected mice by tail vein injection. The protection of passive immunization was estimated in two ways, parasite burden and mice survival. One week post-infection, parasite burden in peritoneal fluid was determined by quantitative PCR. The results showed that passive immunization with the sera of Δldh-vaccinated mice did result in significantly lower level of parasite burden compared to immunization with sera of naïve mice (Figure 7A). However, when the survival of mice were followed, passively immunized mice only survived for two more days, compared with the negative immunization control (Figure 7B). Together these results suggested that the sera of Δldh-vaccinated mice are able to reduce parasite propagation to some degree but do not offer full protection against lethal infections.
Figure 7.

Passive immunization using the sera from Δldh-vaccinated mice provided partial protection against parasite infection. (A) ICR mice (n = 3 for each group) were injected with 104 tachyzoites of ME49 by intraperitoneal injection. At day 0 and day 3 post-infection, mice were treated with positive sera collected from the ME49 Δldh-vaccinated mice (160 days post-vaccination), or negative serum from naïve mice by tail intravenous injection. Parasite loads in peritoneal fluids 7 days post-infection were estimated by quantitative PCR. ***p < 0.0001, Student’s t-test. (B) Survival of passively immunized mice.
Robust Pro-Inflammatory Cytokine Production by Splenocytes of Δldh-Vaccinated Mice Upon T. gondii Antigen Stimulation
The above passive immunization results suggested that the contribution of humoral immunity to the protection of Δldh vaccination is limited. Next, we estimated cell-mediated immunity, as well as the antigen-specific memory responses by checking the antigen recall response in vaccinated mice. To do that, splenocytes from ME49 Δldh immunized mice were harvested 160 days post-vaccination, a time point when the cytokine levels in immunized mice was indistinguishable from that of naïve mice. These splenocytes were cultured in vitro and stimulated with total soluble Toxoplasma antigen (TSA) prepared from ME49 tachyzoites. Subsequently the supernatants from the cultures were collected to estimate the cytokine levels by ELISA. Compared with the no-stimulation or non-vaccinated splenocyte controls, TSA stimulated high levels of pro-inflammatory cytokines INF-γ and TNF-α (Figures 8A,B). Induction of IL-12 production was not as obvious (Figure 8C). Given that INF-γ is the key activator of cell-mediated immunity for T. gondii clearance, quick and robust INF-γ production from vaccinated splenocytes upon TSA stimulation suggests that efficient cellular immune responses would be activated during secondary infections.
Figure 8.

Cytokine production by splenocytes of ME49 Δldh-vaccinated mice after Toxoplasma antigen stimulation. ICR mice were vaccinated with 103 tachyzoites of ME49 Δldh and splenocytes were harvested 160 days post-vaccination. The in vitro cultured splenocytes were then treated with fetal bovine serum (control), total soluble Toxoplasma antigen (TSA) or Concanavalin A (conA, positive control) for 72 h. Subsequently the levels of IFN-γ (A), TNF-α (B) and IL-12 (C) in the culture supernatants were measured by ELISA. Splenocytes isolated from non-immunized naïve mice were included as controls. Three mice from each group were analyzed and each treatment condition was repeated three times, *p < 0.05, **p < 0.01, ***p < 0.0001, Student’s t-test.
Compare the Protection Efficiency of Δldh-Based Vaccines to That of Uracil Auxotrophic Mutants
Mutants defective in de novo pyrimidine biosynthesis are auxotrophic to uracil, therefore they are not replicating and avirulent in vivo (25–27). As such, those types of mutants (such as CPSII and OMPDC deficient strains) were considered great vaccine candidates. We sought to compare the protection efficiency of our Δldh-based vaccines to that based on uracil auxotrophic mutants. For this purpose, we first made an OMPDC deletion mutant in ME49, by CRISPR/Cas9-mediated gene replacement to replace OMPDC by loxP sites flanked DHFR* (Figure 9A). The resulted ME49 ompdc:DHFR* (Figure 9B) was then transiently transfected with a Cre recombinase expressing plasmid to remove the selection marker DHFR*, to obtain ME49 Δompdc (Figure 9C). We also generated a double knockout line (ME49 Δompdc Δldh1) by disrupting LDH1 in ME49 Δompdc (Figures 9D,E).
Figure 9.

Generation of OMPDC deletion mutants. (A) Schematic illustration of OMPDC replacement by the selection marker DHFR* and subsequent removal of DHFR* by Cre recombinase. (B) Diagnostic PCR on an ompdc:DHFR* clone. PCR1 and PCR2 check the integration of DHFR* at the 5′ and 3′ end of OMPDC, respectively, whereas PCR3 examines the presence of OMPDC sequence. (C) Diagnostic PCR confirming the removal of DHFR* by Cre recombinase. This PCR generated a 700 bp product from the endogenous DHFR locus, and a 250 bp product from DHFR* due to the absence of introns. Absence of the 250 bp product in ompdc:DHFR* treated with Cre indicates removal of DHFR*. (D) Diagram illustrating the deletion of LDH1 in Δompdc to make the double mutant Δompdc ldh1:DHFR*, by CRISPR/Cas9-mediated homologous gene replacement. (E) Diagnostic PCR on a Δompdc ldh1:DHFR* clone.
First, the virulence of these mutants were compared in mice. With the infection dose of 104 tachyzoites per mouse, ME49 killed all mice within 12 days. All mice infected with Δompdc, Δompdc Δldh1, or Δldh mutants were survived (Figure 10A), indicating significant virulence attenuation of these mutants. To further check the ability of these mutants to propagate in vivo, mice were infected with each mutant line (104 tachyzoites/mouse) and a week later, the parasite burden in peritoneal fluid was examined by qPCR. ME49 displayed robust replication and high parasite burden was detected in mice with this strain (Figure 10B). All three mutants showed minimal replication in mice, as the parasite burden in the corresponding mutants were very low, close to (Δldh) or below (Δompdc, Δompdc Δldh1) the reliable detection limit of qPCR (Figure 10B). It seemed like there were more parasites in Δldh-infected mice than Δompdc- and Δompdc Δldh1-infected ones, however, because of the sensitivity of the qPCR tests, it is hard to measure the exact parasite burden differences. Next, we compared the immune protection efficiencies of these mutants. Mice were first infected with each of these mutant lines, 30 days post-infection they were challenged with the WT strain ME49 and survival of the mice was monitored. All immunized mice survived the secondary infection (Figure 10C), which is consistent with previous results and indicates that these mutants offered similar protection efficiencies against further infections. Together, these results suggest that the Δldh mutant worked comparably well as the uracil auxotrophic mutants as a vaccine candidate.
Figure 10.

Comparison of Δldh mutants-based vaccines to that based on Δompdc auxotrophs. (A) Survival curves of mice infected with indicated strains. Tachyzoites of indicated strains were used to infect ICR mice (104 parasites/mouse, 10 mice for each strain) by intraperitoneal injection and the survival of mice were monitored for 30 days. ***p < 0.001, Gehan–Breslow–Wilcoxon test. (B) Replication of parasites in vivo. ICR mice were infected with 104 tachyzoites of indicated strains by intraperitoneal injection and parasite loads in peritoneal fluids 7 days post-infection were determined by quantitative PCR. ***p < 0.001, Student’s t-test. (C) Protection efficiency offered by vaccination of indicated Toxoplasma gondii mutants. ICR mice were immunized with indicated strains by intraperitoneal injection of 104 tachyzoites each. Thirty days post-immunization, they were challenged with 104 tachyzoites of the ME49 strain and survival of mice were monitored for another 30 days. Non-immunized mice were included as negative control. ***p < 0.001, Gehan–Breslow–Wilcoxon test.
Discussion
Decades of epidemiological and experimental studies have suggested that vaccination is probably the most effective way to control the ubiquitous pathogen T. gondii, and live attenuated vaccines hold the most promise among all vaccination strategies tried so far. Toxovax® derived from the incomplete strain S48 has been commercialized to reduce Toxoplasma caused abortion in sheep (23), and the uracil auxotroph mutants also look promising under experimental settings (25–27). However, these do not mean that current vaccines are ideal. In this study, we tested the possibility of using a lactate dehydrogenase null mutant (Δldh) as a live vaccine candidate to prevent animal toxoplasmosis, since our previous work has shown that this mutant was able to grow efficiently in vitro but unable to propagate in vivo (28). The results showed that, indeed ME49 Δldh immunization stimulated efficient protective immunity against the challenge of a variety of strains.
There is a long history for the search of good anti-toxoplasmic vaccines. Killed parasites, total antigens or secreted antigens extracted from cultured parasites, recombinant antigens in a variety of forms, and live attenuated parasites have all been tried over the last 60 years (16, 29–31). One important lesson from these studies is that live attenuated vaccines are the most efficient in stimulating protective immunity. The only commercially available Toxoplasma vaccine Toxovax® is a live vaccine derived from the S48 strain, which is used in breeding ewes to reduce incidences of dry ewes and abortion (23). Inability to complete its life cycle was thought to be why it can be used as a vaccine, since it can only grow as tachyzoites and survive in sheep for about 14 days before being cleared by sheep’s immune functions (32). However, this strain is still highly virulent in mice during acute infection. It may also revert to gain cysts or oocysts formation abilities. Because of these limitations, this vaccine is not widely used. Recently the de novo pyrimidine synthesis pathway was found to be a good target for vaccine design, as mutants (such as ΔcpsII and Δompdc) with defective de novo UMP biosynthesis activity were able to grow in vitro in the presence of uracil but unable to propagate in vivo (25–27). Both ΔcpsII and Δompdc mutants were reported to induce long-term protection against both acute and chronic toxoplasmosis. Although the uracil auxotroph mutants are promising, their protection is not always 100% (25–27). The Δldh mutant tested in this study is similar to the uracil auxotroph mutants as a vaccine candidate, due to its drastic growth differences between in vitro and in vivo conditions.
An ideal live vaccine not only stimulates efficient protection against further infections but also leaves no parasites behind in order to prevent the risk of parasite dissemination through vaccination, particularly for meat producing animals. In the mouse model, Δldh vaccination induced strong protective immune responses against the challenge of a variety of strains, including type 1, 2, 3 strains and natural isolates belonging to ToxoDB #9 and ToxoDB #3. However, it should be noted that the protection to virulent type 1 strain RH-Luc is limited to short term, likely because our Δldh was made in the type 2 strain ME49. It was reported previously that vaccinated hosts can also be super-infected by wild strains if they contain virulent ROP5 and ROP18 alleles (14). This explains why ME49 Δldh vaccination only provided short-term protection against RH infection. It also suggests that using the endemic strains as starting material to design live vaccines is more likely to be successful. Nonetheless, inactivating LDH genes is still a feasible way of producing live attenuated vaccines. The other seemingly caveat of ME49 Δldh vaccination is that it still produced very small amount of tissue cysts, although we have shown that these cysts were severely attenuated and likely would not cause diseases in vaccinated animals or new hosts if transmitted to. Nevertheless, we have to admit that, ME49 Δldh still needs improvement in this regard, especially for meat producing animals due to the risk of transmitting the infection to humans. One strategy to improve, like we showed, is to combine Δldh with mutations that lead to defective de novo pyrimidine synthesis. The Δompdc Δldh1 mutant is as efficient as the Δldh mutant in terms of stimulating protective immunity against further infections. Yet, there was little parasite replication and no cyst formation in vivo after vaccinating animals with this strain (Figure 10B and data not shown), making it safer than the Δldh mutant. There is another advantage to combine multiple deletions in one strain for live vaccine design. If the vaccine were to be used in cats, combination of multiple mutations reduces the chance of reversing the vaccine strain back to the virulent parental strain through sexual reproduction, which can happen when the cats are infected with other strains during the vaccination period.
In efforts to assess the potential mechanism of protective immunity from Δldh vaccination, we found that it was a combination of humoral and cellular immune responses, with cellular immunity being the major contributor. Δldh administration stimulated high levels of IL-12 and IFN-γ, two pro-inflammatory cytokines known to be crucial for activation of cell-mediated clearance of T. gondii. The elevated levels of IL-12 and IFN-γ did stay for a while after vaccination and then gradually decreased. They were completely back to normal levels 125 days post-vaccination. At day 160 post Δldh immunization, splenocytes extracted from vaccinated mice were able to respond to T. gondii antigen quickly and produce high levels of IFN-γ and other pro-inflammatory cytokines, which could activate cellular immune responses and clear secondary infections efficiently. On the other hand, when testing the contribution of humoral immunity by passive immunization, it was found that sera from Δldh-vaccinated mice was able to reduce parasite propagation but the overall protection was rather limited, since the passively immunized mice only survived 2 days longer compared to controls (Figure 7B). Together these results suggested that cellular immunity, instead of humoral immunity is the main source of protection from Δldh vaccination, similar to the uracil auxotroph mutants-based vaccines (25, 27). In our cytokine measurement studies, we noticed that Δldh immunization induced relatively long duration of increased IL-12 and IFN-γ levels (Figures 6A–D). This was somewhat unexpected, but it may be caused by the unique propagation dynamics of the Δldh mutants in vivo. As was shown, after immunization Δldh parasites could form very low levels of cysts in mice, which suggested that some degree of parasite replication and differentiation must have occurred. The prolonged IL-12 and IFN-γ induction might be a consequence of these parasite activities, but further work is required to completely understand this observation. In addition, it is interesting to note that Δldh immunization provided full protection against the challenge of type 1 virulent strain in a relatively short term (30 days). If the challenge occurred too long (>75 days) after vaccination, the protection efficiency decreased significantly. This pattern correlated well with the IL-12 and IFN-γ level changes, which may indicate that the existing of an activated cell-mediated immunity is required for full control of virulent type 1 strains. Nonetheless, even after 75 days, vaccinated mice were still partially protected against RH Δhxgprt infection, and fully protected from the challenge of other strain types with intermediate virulence.
In conclusion, our study demonstrated that Δldh mutant can potentially be used as a live attenuated vaccine. Its administration stimulated a combination of humoral and cellular immune responses that protected animals from further infections of a variety of T. gondii strains. We also combined a uracil auxotroph conferring mutation with Δldh deletion to make the double knockout Δompdc Δldh1, which exhibited similar protection efficiency as the Δldh mutant but with further reduced in vivo replication and cyst formation, making it safer to use as vaccines. Although the work from mice models demonstrated the great potential of these mutants as vaccine candidates, further investigations are needed to check the safety of vaccination and efficiency of protection in large animals like pigs and sheep, as well as in cats to see the protection against oocyst shedding.
Materials and Methods
Mice and Parasite Strains
All 7-week-old ICR mice were purchased from the Hubei provincial center for disease control and prevention. All animals were maintained under standard conditions according to the regulations specified by Administration of Affairs Concerning Experimental Animals. Animal experiments were approved by the ethical committee of Huazhong Agricultural University (permit #: HZAUMO2016049).
Toxoplasma gondii type 1 strain RH Δhxgprt and its derivative RH-Luc that expresses a firefly luciferase, type 2 strain ME49, type 3 strain VEG, RFLP genotype ToxoDB #3 strain TgPIG-WH1, and ToxoDB #9 strain C7719 were used in this study, which were propagated with human foreskin fibroblast cells (purchased from ATCC, USA) and cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) (LifeTechnologies, Inc., Rockville, MD, USA). In addition, the Δompdc and Δompdc Δldh1 mutant strains were maintained in the presence of extra 250 µM uracil (25, 27) (LifeTechnologies, Inc., Rockville, MD, USA).
Virulence Test of Δldh Cysts
Fresh brain cysts derived from ME49 Δldh or ME49 were used to infect 7-week-old female ICR mice through oral administration (50 cysts/mouse). Mice survival was monitored for 40 days and blood sample were collected afterward. MIC3-based indirect ELISA was used to check the sera to confirm infection. Sero-negative mice were not included in analysis. Cumulative mortality was graphed as Kaplan–Meier survival plots and analyzed in Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Immune Protection Against Acute Infection Stimulated by T. gondii Mutants Vaccination
ICR mice were immunized with 104 tachyzoites of ME49 Δldh, ME49 Δompdc, or ME49 Δompdc Δldh1 through intraperitoneal injection. 30, 75, or 125 days post-immunization, mice were challenged with 104 tachyzoites of RH Δhxgprt, ME49, VEG, C7719, or TgPIG-WH1 by intraperitoneal injection (10 mice for each group), or 50 fresh cysts of ME49 by oral administration. Non-immunized naïve mice with the same challenging doses and routes were included as controls and subsequently these animals were monitored for another 30 days to check their clinical symptoms and survival. In addition, naïve or Δldh immunized mice were infected with 104 tachyzoites of the luciferase expressing RH-Luc strain by intraperitoneal injection (five mice/group). Propagation of RH-Luc in these mice was monitored 1 and 5 days post-infection, by bioluminescent imaging on the IVIS Spectrum imaging system (PerkinElmer, Inc., Boston, MA, USA). To do this, each mouse was injected with 200 µl of 15.4 mg/ml d-luciferin, anesthetized in a chamber containing 2% isoflurane and then imaged by IVIS Spectrum according to the manufacturer’s instructions, as well as previous descriptions (33). Data acquisition and analysis were performed using the Living Image 4 software (PerkinElmer, Inc., Boston, MA, USA).
Prevention of Cyst Formation by Δldh Immunization
ICR mice were first immunized with ME49 Δldh as above and then challenged with 104 tachyzoites of TgPIG-WH1. Subsequently the mice were monitored for another 30 days and seropositive mice were sacrificed to harvest the brains. Immunized but not TgPIG-WH1 challenged, and non-immunized but TgPIG-WH1 challenged mice were included as controls (10 mice per group). The number of Toxoplasma cysts in each mouse brain was determined by DBA-FITC staining, as previously described (34).
Cytokines and Toxoplasma-Specific IgG Level Measurements
Sera samples were collected from mice and stored at −20°C until analysis. The levels of IL12p70, INF-γ, IL-10, and TNF-α were measured using commercial ELISA kits following the manufacturer’s instructions (4A Biotech, Inc., Beijing, China). To analyze the levels of T. gondii specific IgG by ELISA, 96-well microtiter plates were coated with 100 μl/well 0.5 µg/ml soluble tachyzoite antigens diluted in PBS and incubated at 4°C overnight. The plates were then washed and blocked with 1% BSA. Sera samples were diluted 50-fold and added to each well to incubate for 60 min at 37°C. After extensive washing, the bound IgG antibodies were detected by HRP conjugated goat anti-mouse IgG secondary antibodies and tetramethylbenzidine as substrate. A minimum of three mice was included for each experiment and all sera samples were analyzed three times.
Cytokine Production by Splenocytes After T. gondii Antigen Stimulation
ICR mice were immunized with ME49 Δldh tachyzoites and seropositive animals were sacrificed 160 days after immunization. Subsequently the spleens were collected from the mice and homogenized in red cell lysis solution (Biosharp, Inc., Beijing, China) to obtain splenocyte suspensions. The recovered splenocytes were washed once in RPMI 1640 (LifeTechnologies, Inc., Rockville, MD, USA), resuspended and counted (35). Viability of the cells was determined by trypan blue staining. Then 3 × 105 viable splenocytes were plated in each well of 96-well plates and cultured in RPMI 1640 supplemented with 20% FBS (LifeTechnologies, Inc., Rockville, MD, USA) and 100 U/ml penicillin–streptomycin. Meanwhile the splenocytes were stimulated with total T. gondii soluble antigens (final concentration 50 µg/ml) isolated from tachyzoites of ME49 for 3 days. The supernatants from these cultures were harvested for cytokines level measurements, which was performed as above. T. gondii soluble antigens were prepared as the following: freshly egressed tachyzoites of ME49 were collected, purified by filtration, and lysed by sonication in a waterbath sonicator for 30 min. Subsequently the samples were spinned at 12,000 rcf for 10 min at 4°C and the supernatant was collected as TSA, which was subject to protein concentration determination by BCA Protein Assay Kit (Beyotime Biotechnology, Beijing, China) before use. The splenocytes were also stimulated with 5 µg/ml concanavalin A (LifeTechnologies, Inc., Rockville, MD, USA) or 20% FBS as positive and negative controls, respectively. The same experiments were also done with non-immunized naïve mice for further comparisons. Three naïve mice and three immunized mice were analyzed, and each supernatant sample was examined three times by ELISA for technical replication.
Passive Immunization With the Sera of Δldh-Vaccinated Mice
ICR mice (16 mice in total, two groups with 8 mice in each group) were infected with the WT strain ME49 through intraperitoneal injection (104 tachyzoites/mouse). At day 0 and day 3 after infection, sera from naïve mice or the ME49 Δldh-vaccinated mice 160 days post-immunization were collected and administered into infected mice by tail vein injection (100 μl/mouse). The protection of passive immunization was estimated in two ways. First, three mice from each group were sacrificed 7 days post-infection to measure the parasite burden in peritoneal fluids by quantitative PCR, as previously described (28). Second, the survival of the other five mice in each group was monitored daily for 30 days.
Generation of Δompdc and Δompdc Δldh1 Mutants
Both OMPDC and LDH1 deletions were made by CRISPR/Cas9-mediated homologous gene replacements, using the methods described previously (36, 37). All primers and plasmids used in the mutant constructions are listed in Tables 1 and 2, respectively. LDH1 and OMPDC specific CRISPR plasmids were generated by replacing the UPRT targeting guide RNA (gRNA) in pSAG1-Cas9-sgUPRT with corresponding gRNAs, using Q5 site-directed mutagenesis (36, 37). The plasmid pOMPDC:loxp-DHFR*-loxp was constructed by cloning the 5′- and 3′-homology arms (1,044 and 981 bps, respectively) of OMPDC, as well as the loxp-DHFR*-loxp (3,672 bps) amplified from pDONR-G265-loxP-DHFR*-loxp into pUC19 through Gibson assembly (New England Biolabs, Ipswich, MA, USA). It was used as the homologous template to replace OMPDC with the loxp-DHFR*-loxp. The plasmid pLDH1:loxp-DHFR*-loxp used to replace LDH1 with loxp-DHFR*-loxp was constructed in a similar way. All the plasmids were verified by DNA sequencing before use. To constructed the OMPDC knockout strain, ME49 was co-transfected with the OMPDC specific CRISPR plasmid and homologous template (OMPDC:loxp-DHFR*-loxp), selected with 1 µM pyrimethamine and 250 µM uracil (LifeTechnologies, Inc., Rockville, MD, USA), and single cloned by limiting dilution. The resulted ME49 ompdc:DHFR* was then transiently transfected with a Cre recombinase expressing plasmid (pmin-Cre-eGFP) (38) to remove the selection marker DHFR*, to obtain ME49 Δompdc. All clones were identified by diagnostic PCRs, as demonstrated in Figures 9A,D. The Δompdc Δldh1 double mutant were made by replacing LDH1 in ME49 Δompdc with loxp-DHFR*-loxp, using the same strategy as the OMPDC deletion.
Table 1.
Primers used in this study.
| Primer | Sequence | Use |
|---|---|---|
| gRNA-LDH1-Fw | 5′-GCCGGTCTGACCAAGGTGCCGTTTTAGAGCTAGAAATAGC-3′ | To construct the LDH1 specific CRISPR plasmid |
| gRNA-OMPDC-Fw | 5′-GAGCTTGACTCCACCCTCACGTTTTAGAGCTAGAAATAGC-3′ | To construct the OMPDC specific CRISPR plasmid |
| gRNA-R | 5′-AACTTGACATCCCCATTTAC-3′ | To construct gene specific CRISPR plasmids |
| pUC19-Fw | 5′-GGCGTAATCATGGTCATAGC-3′ | Amplification of pUC19 for Gibson assembly |
| pUC19-Rv | 5′-CTCGAATTCACTGGCCGTCG-3′ | Amplification of pUC19 for Gibson assembly |
| loxp-DHFR*-loxp-Fw | 5′-CAACCCGCGCAGAAGACATC-3′ | Amplification of loxp-DHFR*-loxp for Gibson assembly |
| loxp-DHFR*-loxp-Rv | 5′-GGACACGCTGAACTTGTGGC-3′ | Amplification of loxp-DHFR*-loxp for Gibson assembly |
| U5ompdc-Fw | 5′-CGACGGCCAGTGAATTCGAGGTACGTTTGCGATTGTGAGC-3′ | Amplification of 5′-homology of OMPDC for pOMPDC:loxp-DHFR*-loxp construction |
| U5ompdc-Rv | 5′-GATGTCTTCTGCGCGGGTTGCAGTTCTTTGGAATTGTCACGG-3′ | Amplification of 5′-homology of OMPDC for pOMPDC:loxp-DHFR*-loxp construction |
| U3ompdc-Fw | 5′-GCCACAAGTTCAGCGTGTCCCGTGCTGAAAGCGAAACACTATC-3′ | Amplification of 3′-homology of OMPDC for pOMPDC:loxp-DHFR*-loxp construction |
| U3ompdc-Rv | 5′-GCTATGACCATGATTACGCCGTCAGTTTCTCTGTGCGAGTTG-3′ | Amplification of 3′-homology of OMPDC for pOMPDC:loxp-DHFR*-loxp construction |
| U5ldh1-Rv | 5′-GATGTCTTCTGCGCGGGTTGTTCTCCTCTGCACAAGTGCG-3′ | Amplification of PUC19:LDH1-5UTR-3UTR for pLDH1: loxp-DHFR*-loxp construction |
| U3ldh1-Fw | 5′-GCCACAAGTTCAGCGTGTCCTGGCAAAACAGGAGCGGAAT-3′ | Amplification of PUC19:LDH1-5UTR-3UTR for pLDH1: loxp-DHFR*-loxp construction |
| 5′-UpU5OMPDC | 5′-GACAGTTATGGCCCGTCTTC-3′ | PCR1 of Δompdc:DHFR* |
| 3′-InDHFR*-Fw | 5′-CGTGACCACGCCAAAGTAG-3′ | PCR1 of Δompdc:DHFR* |
| 5′-InDHFR*-Rv | 5′-GCACTTGCAGGATGAATTCC-3′ | PCR2 of Δompdc:DHFR* |
| 3′-DnU3OMPDC | 5′-GAGCATTGTGCATTTGCGTC-3′ | PCR2 of Δompdc:DHFR* |
| 5′-UpgRNA OMPDC | 5′-CGTCAGCTTCGTTAGACCAG-3′ | PCR3 of Δompdc:DHFR* |
| 3′-DngRNA OMPDC | 5′-CAGCTGTACTATGAACGGGTG-3′ | PCR3 of Δompdc:DHFR* |
| 5′-InDHFR | 5′-GTCCGAGTCTGTGCTTCAC-3′ | To identify the deletion of loxP-DHFR* |
| 3′-InDHFR | 5′-CCTGCGCAGCAGTTGATTTG-3′ | To identify the deletion of loxP-DHFR* |
| 5′-InU5ldh1 | 5′-CTGTCCAGCGTAGCAATCAC-3′ | PCR1 of Δompdc Δldh1 |
| 3′-In DHFR* | 5′-ACCACGCCAAAGTAGAAAGG-3′ | PCR1 of Δompdc Δldh1 |
| 5′-InDHFR* | 5′-TGCTGGACTGTTGCTGTCTG-3′ | PCR2 of Δompdc Δldh1 |
| 3′-InU3ldh1 | 5′-GTGGCCGACCTTTTCAGAGC-3′ | PCR2 of Δompdc Δldh1 |
| 5′-UpgRNA ldh1 | 5′-GCTAGACGCGGAGAGTTGTG-3′ | PCR3 of Δompdc Δldh1 |
| 3′-DngRNA ldh1 | 5′-AGCTGCTTCTCCGTGACTAC-3′ | PCR3 of Δompdc Δldh1 |
| RT-tubulin-F | 5′-CACTGGTACACGGGTGAAGGT-3′ | β-tubulin-based qPCR |
| RT-tubulin-R | 5′-ATTCTCCCTCTTCCTCTGCG-3′ | β-tubulin-based qPCR |
Table 2.
Plasmids used in this study.
| Plasmid name | Use | Source |
|---|---|---|
| pSAG1-Cas9-sgUPRT | Template for gene-specific CRISPR plasmid construction | Addgene plasmid #54467 |
| pSAG1-Cas9-sgLDH1 | LDH1-specific CRISPR plasmid | (28) |
| pSAG1-Cas9-sgOMPDC | OMPDC-specific CRISPR plasmid | This work |
| pOMPDC: loxp-DHFR*-loxp | To replace OMPDC with loxp-DHFR*-loxp | This work |
| pLDH1: loxp-DHFR*-loxp | To replace LDH1 with loxp-DHFR*-loxp | This work |
| pLDH1:DHFR | Template for PUC19:LDH1-5UTR-3UTR amplification | (28) |
| pDONR-G265-loxP-DHFR*-loxp | Template for loxp-DHFR*-loxp amplification | This work |
| pmin-Cre-eGFP | To remove the selection marker DHFR* | (38) |
Statistical Analysis
Statistical comparisons were performed in Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA) using Student’s t-tests, Gehan–Breslow–Wilcoxon test or one-way ANOVA with Bonferroni posttests as indicated in figure legends.
Ethics Statement
All animals were maintained under standard conditions according to the regulations specified by Administration of Affairs Concerning Experimental Animals. Animal experiments were approved by the ethical committee of Huazhong Agricultural University (permit #: HZAUMO2016049).
Author Contributions
BS, NX, and TZ conceived and designed the experiments; NX, TZ, XL, SY, PZ, and JY performed the experiments; BS, NX, TZ, YZ, and JZ analyzed the data; BS and NX wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Drs. Min Cui, Min Hu, Rui Fang, and Lan He from the college of veterinary medicine, Huazhong Agricultural University for their thoughtful suggestions during this study.
Footnotes
Funding. This work was supported the National Key Research and Development Program of China (Grant No. 2017YFD0501304), the National Natural Science Foundation of China (Grant No. 31572508), and the Natural Science Foundation of Hubei Province (Project 2017CFA020). The funders had no role in the study design, data collection and analysis, preparation of the manuscript, or decision to submit the work for publication.
References
- 1.Elmore SA, Jones JL, Conrad PA, Patton S, Lindsay DS, Dubey JP. Toxoplasma gondii: epidemiology, feline clinical aspects, and prevention. Trends Parasitol (2010) 26(4):190–6. 10.1016/j.pt.2010.01.009 [DOI] [PubMed] [Google Scholar]
- 2.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet (2004) 363(9425):1965–76. 10.1016/S0140-6736(04)16412-X [DOI] [PubMed] [Google Scholar]
- 3.Petersen E, Pollak A, Reiter-Owona I. Recent trends in research on congenital toxoplasmosis. Int J Parasitol (2001) 31(2):115–44. 10.1016/S0020-7519(00)00140-5 [DOI] [PubMed] [Google Scholar]
- 4.Dubey JP. Toxoplasmosis in pigs – the last 20 years. Vet Parasitol (2009) 164(2–4):89–103. 10.1016/j.vetpar.2009.05.018 [DOI] [PubMed] [Google Scholar]
- 5.Dubey JP, Hill DE, Jones JL, Hightower AW, Kirkland E, Roberts JM, et al. Prevalence of viable Toxoplasma gondii in beef, chicken, and pork from retail meat stores in the United States: risk assessment to consumers. J Parasitol (2005) 91(5):1082–93. 10.1645/GE-683.1 [DOI] [PubMed] [Google Scholar]
- 6.Alday PH, Doggett JS. Drugs in development for toxoplasmosis: advances, challenges, and current status. Drug Des Devel Ther (2017) 11:273–93. 10.2147/DDDT.S60973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubey JP. Advances in the life cycle of Toxoplasma gondii. Int J Parasitol (1998) 28(7):1019–24. 10.1016/S0020-7519(98)00023-X [DOI] [PubMed] [Google Scholar]
- 8.Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol (2012) 10(11):766–78. 10.1038/nrmicro2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.VanWormer E, Fritz H, Shapiro K, Mazet JA, Conrad PA. Molecules to modeling: Toxoplasma gondii oocysts at the human-animal-environment interface. Comp Immunol Microbiol Infect Dis (2013) 36(3):217–31. 10.1016/j.cimid.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis (1995) 172(6):1561–6. 10.1093/infdis/172.6.1561 [DOI] [PubMed] [Google Scholar]
- 11.Khan A, Dubey JP, Su C, Ajioka JW, Rosenthal BM, Sibley LD. Genetic analyses of atypical Toxoplasma gondii strains reveal a fourth clonal lineage in North America. Int J Parasitol (2011) 41(6):645–55. 10.1016/j.ijpara.2011.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorenzi H, Khan A, Behnke MS, Namasivayam S, Swapna LS, Hadjithomas M, et al. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat Commun (2016) 7:10147. 10.1038/ncomms10147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behnke MS, Khan A, Lauron EJ, Jimah JR, Wang Q, Tolia NH, et al. Rhoptry proteins ROP5 and ROP18 are major murine virulence factors in genetically divergent South American strains of Toxoplasma gondii. PLoS Genet (2015) 11(8):e1005434. 10.1371/journal.pgen.1005434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen KD, Camejo A, Melo MB, Cordeiro C, Julien L, Grotenbreg GM, et al. Toxoplasma gondii superinfection and virulence during secondary infection correlate with the exact ROP5/ROP18 allelic combination. MBio (2015) 6(2):e02280. 10.1128/mBio.02280-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ezz Eldin HM, Kamel HH, Badawy AF, Shash LS. A comparative study between excretory/secretory and autoclaved vaccines against RH strain of Toxoplasma gondii in murine models. J Parasit Dis (2015) 39(3):526–35. 10.1007/s12639-013-0390-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waldeland H, Frenkel JK. Live and killed vaccines against toxoplasmosis in mice. J Parasitol (1983) 69(1):60–5. 10.2307/3281275 [DOI] [PubMed] [Google Scholar]
- 17.Darcy F, Maes P, Gras-Masse H, Auriault C, Bossus M, Deslee D, et al. Protection of mice and nude rats against toxoplasmosis by a multiple antigenic peptide construction derived from Toxoplasma gondii P30 antigen. J Immunol (1992) 149(11):3636–41. [PubMed] [Google Scholar]
- 18.Petersen E, Nielsen HV, Christiansen L, Spenter J. Immunization with E. coli produced recombinant T. gondii SAG1 with alum as adjuvant protect mice against lethal infection with Toxoplasma gondii. Vaccine (1998) 16(13):1283–9. 10.1016/S0264-410X(98)00039-5 [DOI] [PubMed] [Google Scholar]
- 19.Dautu G, Munyaka B, Carmen G, Zhang G, Omata Y, Xuenan X, et al. Toxoplasma gondii: DNA vaccination with genes encoding antigens MIC2, M2AP, AMA1 and BAG1 and evaluation of their immunogenic potential. Exp Parasitol (2007) 116(3):273–82. 10.1016/j.exppara.2007.01.017 [DOI] [PubMed] [Google Scholar]
- 20.Beghetto E, Nielsen HV, Del Porto P, Buffolano W, Guglietta S, Felici F, et al. A combination of antigenic regions of Toxoplasma gondii microneme proteins induces protective immunity against oral infection with parasite cysts. J Infect Dis (2005) 191(4):637–45. 10.1086/427660 [DOI] [PubMed] [Google Scholar]
- 21.Pan M, Lyu C, Zhao J, Shen B. Sixty years (1957–2017) of research on toxoplasmosis in China-an overview. Front Microbiol (2017) 8:1825. 10.3389/fmicb.2017.01825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang NZ, Chen J, Wang M, Petersen E, Zhu XQ. Vaccines against Toxoplasma gondii: new developments and perspectives. Expert Rev Vaccines (2013) 12(11):1287–99. 10.1586/14760584.2013.844652 [DOI] [PubMed] [Google Scholar]
- 23.Buxton D, Innes EA. A commercial vaccine for ovine toxoplasmosis. Parasitology (1995) 110(Suppl):S11–6. 10.1017/S003118200000144X [DOI] [PubMed] [Google Scholar]
- 24.Buxton D, Thomson K, Maley S, Wright S, Bos HJ. Vaccination of sheep with a live incomplete strain (S48) of Toxoplasma gondii and their immunity to challenge when pregnant. Vet Rec (1991) 129(5):89–93. 10.1136/vr.129.5.89 [DOI] [PubMed] [Google Scholar]
- 25.Fox BA, Bzik DJ. Avirulent uracil auxotrophs based on disruption of orotidine-5’-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect Immun (2010) 78(9):3744–52. 10.1128/IAI.00287-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature (2002) 415(6874):926–9. 10.1038/415926a [DOI] [PubMed] [Google Scholar]
- 27.Fox BA, Bzik DJ. Nonreplicating, cyst-defective type II Toxoplasma gondii vaccine strains stimulate protective immunity against acute and chronic infection. Infect Immun (2015) 83(5):2148–55. 10.1128/IAI.02756-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia N, Yang J, Ye S, Zhang L, Zhou Y, Zhao J, et al. Functional analysis of Toxoplasma lactate dehydrogenases suggests critical roles of lactate fermentation for parasite growth in vivo. Cell Microbiol (2018) 20(1). 10.1111/cmi.12794 [DOI] [PubMed] [Google Scholar]
- 29.Cutchins EC, Warren J. Immunity patterns in the guinea pig following Toxoplasma infection and vaccination with killed Toxoplasma. Am J Trop Med Hyg (1956) 5(2):197–209. 10.4269/ajtmh.1956.5.197 [DOI] [PubMed] [Google Scholar]
- 30.Krahenbuhl JL, Ruskin J, Remington JS. The use of killed vaccines in immunization against an intracellular parasite: Toxoplasma gondii. J Immunol (1972) 108(2):425–31. [PubMed] [Google Scholar]
- 31.Zhang NZ, Wang M, Xu Y, Petersen E, Zhu XQ. Recent advances in developing vaccines against Toxoplasma gondii: an update. Expert Rev Vaccines (2015) 14(12):1609–21. 10.1586/14760584.2015.1098539 [DOI] [PubMed] [Google Scholar]
- 32.Innes EA, Panton WR, Thomson KM, Maley S, Buxton D. Kinetics of interferon gamma production in vivo during infection with the S48 vaccine strain of Toxoplasma gondii. J Comp Pathol (1995) 113(1):89–94. 10.1016/S0021-9975(05)80074-X [DOI] [PubMed] [Google Scholar]
- 33.Saeij JP, Boyle JP, Grigg ME, Arrizabalaga G, Boothroyd JC. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect Immun (2005) 73(2):695–702. 10.1128/IAI.73.2.695-702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huskinson-Mark J, Araujo FG, Remington JS. Evaluation of the effect of drugs on the cyst form of Toxoplasma gondii. J Infect Dis (1991) 164(1):170–1. 10.1093/infdis/164.1.170 [DOI] [PubMed] [Google Scholar]
- 35.Vercammen M, Scorza T, Huygen K, De Braekeleer J, Diet R, Jacobs D, et al. DNA vaccination with genes encoding Toxoplasma gondii antigens GRA1, GRA7, and ROP2 induces partially protective immunity against lethal challenge in mice. Infect Immun (2000) 68(1):38–45. 10.1128/IAI.68.1.38-45.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen B, Brown KM, Lee TD, Sibley LD. Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio (2014) 5(3):e1114–1114. 10.1128/mBio.01114-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen B, Brown K, Long S, Sibley LD. Development of CRISPR/Cas9 for efficient genome editing in Toxoplasma gondii. Methods Mol Biol (2017) 1498:79–103. 10.1007/978-1-4939-6472-7_6 [DOI] [PubMed] [Google Scholar]
- 38.Heaslip AT, Nishi M, Stein B, Hu K. The motility of a human parasite, Toxoplasma gondii, is regulated by a novel lysine methyltransferase. PLoS Pathog (2011) 7(9):e1002201. 10.1371/journal.ppat.1002201 [DOI] [PMC free article] [PubMed] [Google Scholar]