

Abstract
We herein report a woman with chronic inflammatory demyelinating polyneuropathy (CIDP) in whom positivity for anti-neurofascin 155 antibodies was revealed 23 years after the onset of neuropathy. The patient initially reported numbness in the face at 50 years of age and subsequently manifested features compatible to typical CIDP. Steroid administration initiated at 54 years of age ameliorated her neuropathic symptoms. Although the nerve conduction indices at 59 years of age deteriorated, those at 68, 72, and 73 years of age showed a gradual recovery. The deterioration and subsequent restoration of compound muscle action potential amplitudes was the most dramatic, suggesting that a conduction block can be reversed earlier than other electrophysiological indices.
Keywords: chronic inflammatory demyelinating polyneuropathy, conduction block, neurofascin, paranode, prognosis, treatment
Introduction
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an immune-mediated neuropathy characterized by electrophysiological features suggestive of demyelination. Phagocytosis of myelin by macrophages that have infiltrated the endoneurial space has been thought to be the cause of demyelination, resulting in nerve conduction abnormalities in CIDP (1-5). However, recent studies have suggested the association of autoantibodies directed against paranodal junctional molecules in a subpopulation of patients (6-13). Among these autoantibodies, anti-neurofascin 155 antibodies induce the dissection of paranodes that results in nerve conduction abnormalities (14). This mechanism-inducing conduction abnormality is completely different from the classical macrophage-mediated mechanism of demyelination (14). Patients with anti-neurofascin 155 antibodies receive much attention from physicians because they tend to manifest unique clinical features. For example, patients with anti-neurofascin 155 antibodies tend to show distal limb involvement, sensory ataxia, tremor, and a poor response to an intravenous immunoglobulin treatment (8, 11-14). As these antibodies have only recently been recognized in CIDP patients, the long-term sequelae associated with this type of CIDP have not been clarified.
We herein report a patient with CIDP who was revealed to be positive for anti-neurofascin 155 antibodies 23 years after the onset of neuropathy. This case has not been included in other studies of anti-neurofascin 155 antibodies.
Case Report
A 73-year-old woman first noted numbness on the left side of her face at 50 years of age. Although the numbness spread to cover the whole face, it gradually subsided thereafter. Numbness and weakness in the lower limbs appeared three years later. As they gradually worsened, she visited our hospital and was hospitalized at 54 years of age. She had no remarkable personal or family history.
A neurological examination on admission revealed that she was alert and well-oriented. Mild paresthesia in the V2 and V3 branches of the left trigeminal nerve and moderate weakness of the bilateral sternocleidomastoideus and trapezius muscles were noted, while the other cranial nerves were intact. Moderate weakness was diffusely present bilaterally in both the upper and lower extremities. Muscle atrophy was not evident on inspection. Her grasp powers were 16/18 kg (right/left, respectively). A sensory deficit was noted in a distally accentuated globe-and-stocking pattern. Light touch and pain sensations were moderately impaired, and the vibration sensation was reduced severely in the distal portions of the lower limbs. No hand tremors were noted. The deep tendon reflexes were reduced in the upper extremities and absent entirely in the lower limbs. The plantar responses were flexor on both sides. A cerebrospinal fluid examination revealed an increased protein level and normal cell count (176 mg/dL; normal, 15-45 mg/dL). The findings of nerve conduction studies (NCSs), which were performed as described previously (15, 16), indicated demyelinating neuropathy (Table). The amplitudes of compound muscle action potentials (CMAPs) in the tibial nerve and sensory nerve action potentials in the median, ulnar, and sural nerves were markedly decreased or not elicited. Magnetic resonance imaging revealed no significant abnormalities in the brain or spinal cord. Thickening of the cauda equina was not evident.
Table.
Serial Nerve Conduction Studies.
| Nerve conduction (Right) |
The first examination | After 2 years | After 5 years | After 14 years | After 18 years | After 19 years | Controls* mean (SD) |
|---|---|---|---|---|---|---|---|
| Median nerve | |||||||
| MCV (m/s) | 25.0 | 36.0 | 23.0 | 27.0 | 31.0 | 32.0 | 57.6 (3.8) |
| DL (ms) | 9.1 | 9.8 | 7.4 | 8.6 | 6.9 | 7.1 | 3.4 (0.4) |
| Distal CMAP duration (ms)** | 6.7 | 5.6 | 5.1 | 5.5 | 5.7 | 6.3 | 4.7 (0.9) |
| CMAP (mV) | 12.8 | 5.7 | 0.9 | 4.3 | 6.3 | 8.3 | 8.2 (2.9) |
| FO (%) | 38.0 | ND | ND | ND | 43.8 | 43.8 | 67.6 (20.3) |
| F latency | 45.7 | ND | ND | ND | 50.0 | 43.6 | 22.3 (1.9) |
| SCV (m/s) | 16.0 | 19.8 | 56.3 (5.3) | ||||
| SNAP (μV) | 1.8 | NE | NE | NE | NE | 1.5 | 28.0 (11.5) |
| Ulnar nerve | |||||||
| MCV (m/s) | 26.0 | 30.0 | 29.0 | 32.0 | 35.0 | 35.0 | 58.0 (4.6) |
| DL (ms) | 7.6 | 6.6 | 6.4 | 6.1 | 5.6 | 5.3 | 2.6 (0.3) |
| Distal CMAP duration (ms)** | 7.0 | 5.9 | 5.2 | 5.7 | 6.1 | 6.7 | 5.1 (0.7) |
| CMAP (mV) | 7.5 | 4.1 | 2.4 | 3.6 | 4.8 | 4.0 | 7.4 (1.8) |
| SCV (m/s) | 21.0 | 22.8 | 54.5 (5.5) | ||||
| SNAP (μV) | NE | NE | NE | NE | 1.0 | 2.0 | 23.8 (10.3) |
| Tibial nerve | |||||||
| MCV (m/s) | 20.0 | 29.0 | 13.0 | 26.0 | 26.0 | 26.0 | 46.0 (3.8) |
| DL (ms) | 17.2 | 13.8 | 7.6 | 15.3 | 11.5 | 7.8 | 4.0 (0.6) |
| CMAP (mV) | 1.0 | 0.6 | 1.1 | 1.7 | 1.1 | 1.0 | 11.8 (3.5) |
| Sural nerve | |||||||
| SCV (m/s) | 19 | 49.2 (4.8) | |||||
| SNAP (μV) | NE | NE | 0.8 | NE | NE | NE | 16.8 (7.8) |
CMAP: compound muscle action potentials, DL: distal latency, MCV: motor nerve conduction velocity, ND: not determined, NE: not elicited, SCV: sensory nerve conduction velocity, SNAP: sensory nerve action potentials
*Control values were based on previously published reports (15, 16).
**Duration from onset to the first crossing of the baseline in CMAP was measured (15).
Under light microscopy, a sural nerve biopsy revealed preserved large and small myelinated fiber densities (2,786 and 4,789 fibers/mm2, respectively; control values (17), 3,129±462 and 5,118±429 [mean ± standard deviation] fibers/mm2). Marked endoneurial edema with a substantial increase in the subperineurial space was observed. Epieurial inflammatory cellular infiltration was not observed. Teased-fiber preparations showed little axonal degeneration (2.1%; control value, 1.8%±1.6%), with fibers showing slight widening of the node of Ranvier (i.e. segmental demyelination, 5.2%). Electron microscopy showed that unmyelinated fibers were preserved (26,479 fibers/mm2; control value, 30,655±2,731 fibers/mm2). An assessment of longitudinal sections revealed the detachment of the myelin terminal loops from the axolemma at the paranode, a finding consistent with a previous report on neurofascin 155 antibody-positive CIDP cases (Fig. 1) (14, 18, 19).
Figure 1.
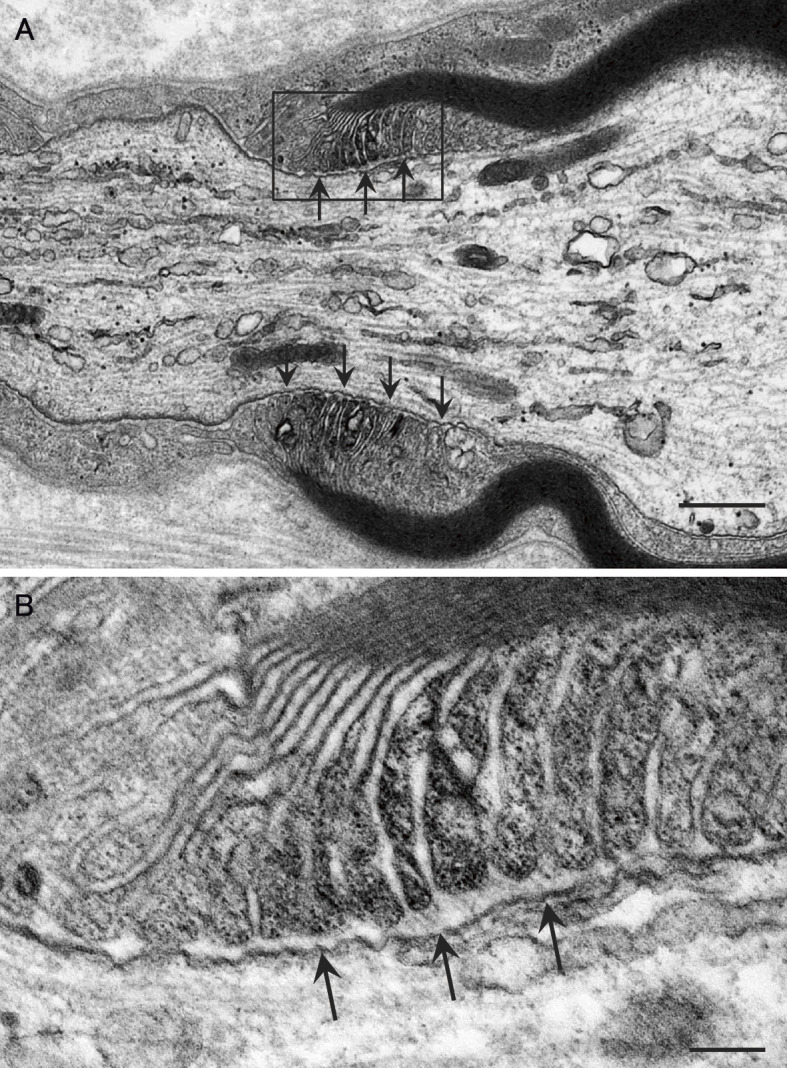
Electron microscopic findings of longitudinal sections of a sural nerve biopsy specimen. A clear space was frequently observed between the myelin terminal loops and the axolemma (arrows). A high-powered view of the region in the box in (A) is shown in (B). Uranyl acetate and lead citrate staining. Scale bars=0.5 µm (A) and 0.1 µm (B).
Based on the diagnosis of CIDP, steroid pulse treatment was administered intravenously (1,000 mg daily dose of methylprednisolone for 3 days); subsequently, 50 mg of prednisolone was administered orally for 1 month. The weakness and numbness gradually improved; therefore, the dosage of prednisolone was tapered by 5 mg every 2 weeks. She was discharged at 4 months after the initiation of treatment, at which time her grasp powers were 26/26 kg, and the cerebrospinal fluid protein level was 157 mg/dL. However, numbness in the face and limbs remained.
Five months after the discharge, the dosage of prednisolone was reduced to 15 mg/daily. Her grasp powers had increased to 30/34 kg, and she was able to run. Although she began to complain of mild hand tremors, the tapering of the prednisolone dosage continued because the numbness and weakness were not exacerbated. At 56 years of age, when she was taking a 5-mg daily dose of prednisolone, she complained of worsening of her sensory symptoms and difficulty walking. The prednisolone dose was increased to 10 mg daily. An NCS performed at that time revealed improvement in the motor conduction velocities despite a reduction in the CMAP amplitudes in the median and ulnar nerves. Muscle computed tomography (CT) performed at 57 years of age, as described previously (16), revealed no muscle atrophy of the arms, legs, or trunk.
An NCS performed at 59 years of age revealed a further reduction in the CMAP amplitudes in the median and ulnar nerves, particularly in the former (Fig. 2A). The reduction in the CMAP amplitudes in these nerves was not accompanied by a prolonged duration or polyphasia (i.e. temporal dispersion). At 60 years of age, she again complained of a worsening of numbness and gait disturbance; therefore, the dosage of prednisolone was increased to 20 mg daily and then tapered to 5 mg daily by 61 years of age. Although the patient complained of an unsteady gait, numbness in the lower limbs, and tremor in the upper limbs, her neuropathic symptoms have remained largely stable since then. Now, at 73 years of age, she has been taking a 5-mg daily dosage of prednisolone for 12 years.
Figure 2.
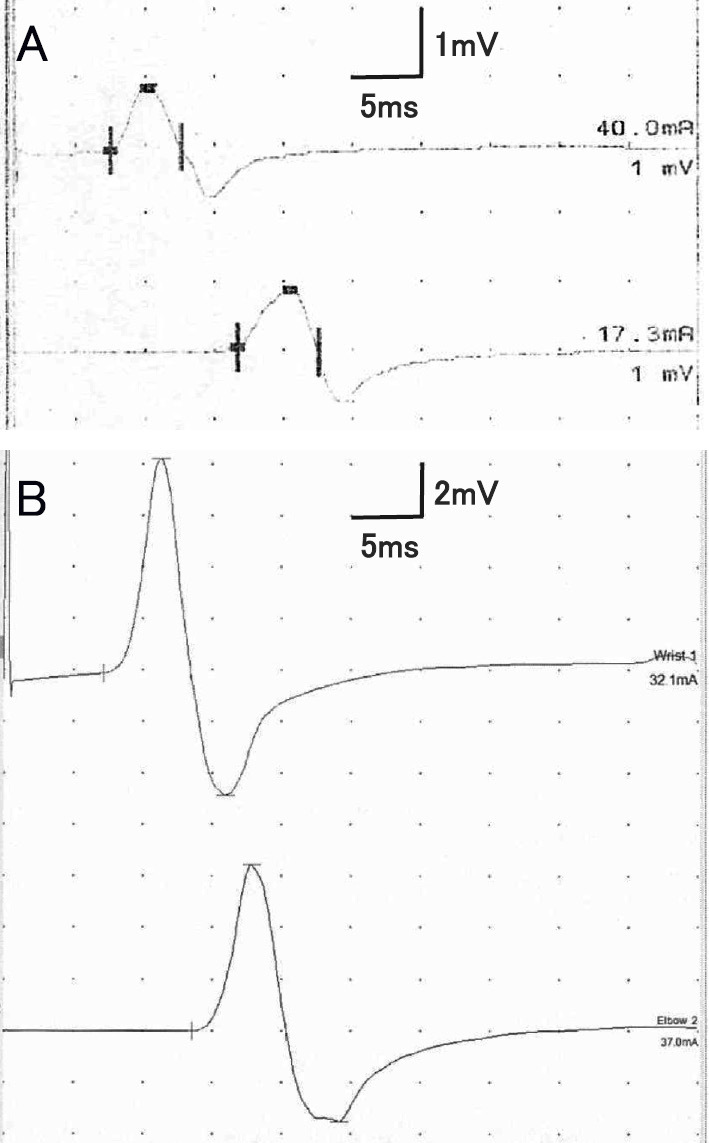
Compound muscle action potentials (CMAPs) in the median nerve. (A) A marked decrease in the CMAP amplitude unaccompanied by a prolonged duration and polyphasia (i.e. temporal dispersion) was observed at 59 years of age. (B) An improvement in the CMAP amplitude was observed at 73 years of age.
She reported an exacerbation of hand tremors at 67 years of age. However, serial NCSs performed at 68, 72, and 73 years of age (14, 18, and 19 years after the first examination, respectively) revealed a gradual improvement in the nerve conduction indices, particularly in the CMAP amplitudes in the median nerve (Fig. 2B). A serum examination using a cell-based assay (11) at 73 years of age revealed positivity for anti-neurofascin 155 antibodies (delta mean fluorescence intensity, 422.8). Muscle CT performed at that time revealed no muscle atrophy, which was similar to the results from 16 years earlier. A neurological examination at that time revealed mild weakness in the distal portions of the extremities. By contrast, her muscle strength was preserved in the proximal portions of the limbs, indicating a distribution compatible with a distal acquired demyelinating symmetric (DADS) form of CIDP (20). Her grasp power was 22/23 kg. Mild impairment of the light touch and pain sensations was noted in the distal portions of the lower limbs. The vibration sensation was mildly reduced in the hands and moderately reduced in the feet. Romberg's sign was mildly positive.
Discussion
The initial symptom of CIDP in this patient seems to have been numbness in the face that appeared at 50 years of age. Although a transient improvement in that symptom was observed, exacerbation was noted as neuropathy in the extremities developed over the following three years. Facial sensory disturbance has been reported in some patients with anti-neurofascin 155 antibodies (11). A recent report demonstrated thickening of the cranial nerves in a patient manifesting facial sensory symptoms (21). In the present case, positivity for anti-neurofascin 155 antibodies was detected at 23 years after the onset of facial symptoms and 19 years after the initiation of steroid treatment. An antibody examination was not performed in the initial phase of neuropathy, but a sural nerve biopsy specimen obtained before the initiation of treatment showed a typical axo-glial detachment at the paranode, which has also been previously reported in anti-neurofascin 155 antibody-positive CIDP cases (14, 18, 19).
Previous studies of neurofascin 155 and contactin 1-deficient mice revealed the loss of transverse bands at the paranodal axo-glial junctions and slowing of the nerve conduction velocities (22, 23). These studies suggest that abnormalities of paranodal axo-glial junctional proteins may affect the tight connection between the myelin terminal loops and axolemma, inducing nerve conduction abnormalities. Therefore, the participation of anti-neurofascin 155 antibodies is strongly suggested in the pathogenesis of neuropathy, even in its early phase, in our patient. The lack of the blood-nerve barrier at the distal nerve terminals and nerve roots might enable anti-neurofascin 155 antibodies to access the paranodes at these sites. Marked abnormalities in the distal and F-wave latencies in our patient may support such a mechanism.
Some anti-neurofascin 155 antibody-positive CIDP patients are refractory to immunotherapies (24). In our patient, the nerve conduction indices improved after long-term steroid treatment, although neuropathic symptoms remained. The sequential changes in the CMAP amplitudes in the median and ulnar nerves, particularly in the former, were dramatic compared to the changes in the motor conduction velocities and distal latencies. Because the reduction in the CMAP amplitudes did not accompany a temporal dispersion in these nerves, a reversible conduction block similar to that in the acute motor axonal neuropathy form of Guillain-Barré syndrome may have occurred in our patient (25). Passive transfer of antibodies against paranodal junctional protein induced the conduction block (26). However, this model did not show a slowing of the nerve conduction indices, which is ubiquitously observed in patients with antibodies to paranodal junctional proteins, including anti-neurofascin 155 antibodies (11, 13, 14). Given the shorter duration of exposure to antibodies in animal models than in patients, this conduction block may represent a reversible functional deficit. As the duration of exposure to antibodies become longer, morphological abnormalities refractory to immunotherapies, such as paranodal dissection, as reported in patients with these antibodies (14), may occur at the paranode. This may result in additional abnormalities of conduction indices, such as slowing of nerve conduction velocities and distal latencies.
According to previous studies, the key clinical features suggesting that a patient with CIDP has anti-neurofascin 155 antibodies before the initiation of treatment are the features of DADS, sensory ataxia, and hand tremors (8, 11-14). On admission to our hospital at 54 years of age, our patient manifested symmetric, diffuse weakness in the upper and lower limbs, which is compatible with the definition of typical CIDP and not DADS (20). As hand tremors were not conspicuous in the initial phase of neuropathy, it was unlikely that the patient had anti-neurofascin 155 antibodies. However, as the weakness in the proximal portions of the upper limbs finally recovered, the patient exhibited features compatible with DADS. The tremors also became gradually evident with the increasing duration of neuropathy.
A previous study of 38 patients with CIDP found that approximately two-thirds of the patients did not require immunotherapies 5 years after the initiation of treatment (27). The present case suggests that the pathological condition caused by anti-neurofascin 155 antibodies persists for a long time and that long-term treatment in patients with these antibodies is required. However, the conduction block may be reversible to some extent. Further studies are needed to clarify the long-term outcomes of CIDP with anti-neurofascin 155 antibodies.
The authors state that they have no Conflict of Interest (COI).
Financial Support
This work was supported by Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED, 16ek0109056h0003).
References
- 1.Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc 50: 621-637, 1975. [PubMed] [Google Scholar]
- 2.Prineas JW, McLeod JG. Chronic relapsing polyneuritis. J Neurol Sci 27: 427-458, 1976. [DOI] [PubMed] [Google Scholar]
- 3.Vital C, Vital A, Lagueny A, et al. . Chronic inflammatory demyelinating polyneuropathy: immunopathological and ultrastructural study of peripheral nerve biopsy in 42 cases. Ultrastruct Pathol 24: 363-369, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Vallat JM, Sommer C, Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol 9: 402-412, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Said G, Krarup C. Chronic inflammatory demyelinative polyneuropathy. Handb Clin Neurol 115: 403-413, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Ng JK, Malotka J, Kawakami N, et al. . Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 79: 2241-2248, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. . Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 73: 370-380, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. . Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 82: 879-886, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doppler K, Appeltshauser L, Wilhelmi K, et al. . Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry 86: 720-728, 2015. [DOI] [PubMed] [Google Scholar]
- 10.Miura Y, Devaux JJ, Fukami Y, et al. . Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 138: 1484-1491, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogata H, Yamasaki R, Hiwatashi A, et al. . Characterization of IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Ann Clin Transl Neurol 2: 960-971, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devaux JJ, Miura Y, Fukami Y, et al. . Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 86: 800-807, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadoya M, Kaida K, Koike H, et al. . IgG4 anti-neurofascin155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy: Clinical significance and diagnostic utility of a conventional assay. J Neuroimmunol 301: 16-22, 2016. [DOI] [PubMed] [Google Scholar]
- 14.Koike H, Kadoya M, Kaida KI, et al. . Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J Neurol Neurosurg Psychiatry 88: 465-473, 2017. [DOI] [PubMed] [Google Scholar]
- 15.Koike H, Hirayama M, Yamamoto M, et al. . Age associated axonal features in HNPP with 17p11.2 deletion in Japan. J Neurol Neurosurg Psychiatry 76: 1109-1114, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohyama K, Koike H, Katsuno M, et al. . Muscle atrophy in chronic inflammatory demyelinating polyneuropathy: a computed tomography assessment. Eur J Neurol 21: 1002-1010, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Koike H, Iijima M, Mori K, et al. . Neuropathic pain correlates with myelinated fibre loss and cytokine profile in POEMS syndrome. J Neurol Neurosurg Psychiatry 79: 1171-1179, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Vallat JM, Yuki N, Sekiguchi K, et al. . Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with anti-Neurofascin 155 antibodies. Neuromuscul Disord 27: 290-293, 2017. [DOI] [PubMed] [Google Scholar]
- 19.Kuwahara M, Suzuki H, Oka N, et al. . Electron microscopic abnormality and therapeutic efficacy in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin155 IgG4 antibody. Muscle Nerve 2017(Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 20.Joint Task Force of the EFNS and the PNS.. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society-First Revision. J Peripher Nerv Syst 15: 1-9, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Franques J, Chapon F, Devaux J, Mathis S. Cranial nerve hypertrophy in IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Neurology 88: e52, 2017. [DOI] [PubMed] [Google Scholar]
- 22.Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385-397, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res 87: 1773-1793, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Querol L, Rojas-García R, Diaz-Manera J, et al. . Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol Neuroimmunol Neuroinflamm 2: e149, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuwabara S, Yuki N, Koga M, et al. . IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barré syndrome. Ann Neurol 44: 202-208, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Manso C, Querol L, Mekaouche M, Illa I, Devaux JJ. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain 139: 1700-1712, 2016. [DOI] [PubMed] [Google Scholar]
- 27.Kuwabara S, Misawa S, Mori M, Tamura N, Kubota M, Hattori T. Long term prognosis of chronic inflammatory demyelinating polyneuropathy: a five year follow up of 38 cases. J Neurol Neurosurg Psychiatry 77: 66-70, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]