

Abstract
Deregulation of cellular proteostasis due to the failure of the ubiquitin proteasome system to dispose of misfolded aggregation-prone proteins is a hallmark of various neurodegenerative diseases in humans. Microorganisms have evolved to survive massive protein misfolding and aggregation triggered by heat shock using their protein-unfolding ATPases (unfoldases) from the Hsp100 family. Because the Hsp100 chaperones are absent in homoeothermic mammals, we hypothesized that the vulnerability of mammalian neurons to misfolded proteins could be mitigated by expressing a xenogeneic unfoldase. To test this idea, we expressed proteasome-activating nucleotidase (PAN), a protein-unfolding ATPase from thermophilic Archaea, which is homologous to the 19S eukaryotic proteasome and similar to the Hsp100 family chaperones in rod photoreceptors of mice. We found that PAN had no obvious effect in healthy rods; however, it effectively counteracted protein-misfolding retinopathy in Gγ1 knock-out mice. We conclude that archaeal PAN can rescue a protein-misfolding neurodegenerative disease, likely by recognizing misfolded mammalian proteins.
SIGNIFICANCE STATEMENT This study demonstrates successful therapeutic application of an archaeal molecular chaperone in an animal model of neurodegenerative disease. Introducing the archaeal protein-unfolding ATPase proteasome-activating nucleotidase (PAN) into the retinal photoreceptors of mice protected these neurons from the cytotoxic effect of misfolded proteins. We propose that xenogeneic protein-unfolding chaperones could be equally effective against other types of neurodegenerative diseases of protein-misfolding etiology.
Keywords: chaperone, misfolding, neurodegeneration, photoreceptors, proteasome, proteostasis
Introduction
The folding algorithm for proteins is encoded in their primary structure and executed during synthesis by the ribosome (Balchin et al., 2016). This process is guided by small, negative free energy changes that occur when the residues of the emerging polypeptide begin to interact with one another and with the media. Certain mutations resulting in amino acid substitutions may significantly alter the folding algorithm to give rise to non-native or misfolded proteins. The cytotoxicity of misfolded proteins is not generally caused by their loss of function, but rather by their propensity to form oligomers and aggregates that impede the ubiquitin proteasome system (UPS), causing deregulation of cellular proteostasis in the long term (Gidalevitz et al., 2006, 2009; Kristiansen et al., 2007; Deriziotis et al., 2011; Guo et al., 2018; Thibaudeau et al., 2018). In this capacity, misfolded proteins are increasingly viewed as a common contributing factor for many neurodegenerative diseases (Dantuma and Bott, 2014; Hipp et al., 2014; Schmidt and Finley, 2014).
The ubiquitin proteasome, also called the 26S proteasome, maintains cellular proteostasis (Glickman and Ciechanover, 2002; Collins and Goldberg, 2017). The 26S proteasome consists of two subcomplexes: the 19S regulatory particle and the 20S catalytic particle. In eukaryotes, the 19S recognizes ubiquitin-tagged proteins and unfolds and injects them into the hollow-barrel shaped 20S catalytic particle. The 20S core cleaves proteins into peptides, which are then converted into amino acids and recycled. Unfortunately, in some neurodegenerative diseases, the activity of the 26S proteasome is impaired; recently, small toxic oligomers were even shown to directly inhibit the proteasome (Thibaudeau et al., 2018). One approach to counteract neurodegenerative disease is to decrease the amount of aggregated misfolded protein that can impede the UPS. This approach was effective in mouse models of Huntington's (Vacher et al., 2005), Alzheimer's disease and Parkinson's disease (Vashist et al., 2010), where expression of a protein-unfolding ATPase, Hsp104, originally from yeast, slowed the progression of neurodegeneration by reducing aggregate formation. To expand on this idea, we expressed a protein-unfolding ATPase from Archaea, called proteasome-activating nucleotidase (PAN), in a mouse model that displays neurodegenerative retinopathy of protein misfolding etiology.
Although archaeal PAN is homologous to the eukaryotic 19S regulatory particle, some of its features are similar to the protein-unfolding ATPases (unfoldases) of the Hsp100 family, Hsp104 and ClpB (Glover and Lindquist, 1998; Lee et al., 2003; Bösl et al., 2006). The Hsp100s are believed to have evolved to ensure the survival of poikilothermic unicellular organisms, which regularly experienced massive protein misfolding caused by temperature spikes. Because homoeothermic mammals do not usually experience temperature spikes and they have no known Hsp100s, their neurons may be poorly adapted to cope with aberrant proteins that are destabilized as a result of genetic mutations. Similar to the Hsp100 family, PAN can unfold proteins without ubiquitination or binding to the 20S proteasome, allowing PAN to function independently of the 20S core and to recognize substrates using a different identification system (Smith et al., 2006). For example, PAN unfolds stable β-sheet-rich GFP when an unstructured 9-residue ssrA degradation tag from bacteria is placed on GFP's C terminus (Smith et al., 2005). Therefore, PAN likely recognizes substrates by their exposed hydrophobic residues rather than if they have been conjugated with ubiquitin. Archaea do have ubiquitin-like proteins termed SAMPs that appear to target proteins for degradation (Maupin-Furlow, 2014); however, PAN, similar to the Hsp100 chaperones, can directly recognize misfolded proteins. This was shown in vitro, when PAN prevented aggregation of model proteins by acting as a molecular chaperone (Benaroudj and Goldberg, 2000). Based on PAN′s features that are distinct from the proteasome but are common to the protein-unfolding ATPases of the Hsp100 family, we hypothesized that PAN could recognize and unfold aberrant proteins that evade degradation by the UPS and that could potentially impair the 26S proteasome in mammals.
Materials and Methods
Animal models.
The coding sequence of PAN from Methanocaldococcus jannaschii was codon optimized for mammalian expression and commercially generated by GenScript. In addition, a Kozak sequence was introduced to the 5′ end and the sequences for a tandem FLAG-HA epitope tag (GGGDYKDDDKVKLYPYDVPDYA) were added to the 3′ end, followed by two stop codons. The final transgene included a 4.4 kb mouse rhodopsin promoter and a mouse protamine I polyadenylation sequence (Lem et al., 1991). The transgene was verified by sequencing and purified and injected into the pronuclei of zygotes from superovulated FVB females at the West Virginia University (WVU) Transgenic Animal Core Facility. Transgene integration was determined by PCR genotyping of tail DNA using forward primer CTG ATG CAG CTG CTC GCC GAA ATG and reverse primer ATC G TC CAT TGT GAC ATA ATC GCG CAG T. The colonies were established by crossing transgenic heterozygotes with wild-type partners of the 129-E background (Charles River Laboratories). All transgenic heterozygotes were from one transgenic founder with confirmed PAN protein expression in the retina. Gγ1 knock-out mice used in this study (Kolesnikov et al., 2011) were backcrossed into the 129-E background. All experiments involving mice were performed according to procedures approved by the Animal Care and Use Committee of WVU.
Tissue culture.
HEK 293 cells were maintained in DMEM: nutrient mixture F-12 (DMEM/F-12 from ATCC) complete medium at 37°C and supplemented with 10% (v/v) FBS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 2.5 mm l-glutamine, and 15 mm HEPES. For the transient expression of PANet, cells were plated in six-well plates and grown to at least 50% confluency. The following day, cells were transfected with 2 μg of total plasmid DNA using FuGENE 6 Transfection reagent (Roche and Promega) at a 6 (plasmid DNA):1 (FuGENE 6) ratio. Empty pTriEx 4 vector was used as a control. Cells were collected after 48 h of transfection, frozen on dry ice, and stored at −80°C.
Pull down of PANet.
Frozen retinas or HEK 293 cells transfected with either PANet or empty pTriEx 4 vector were homogenized in 50 mm Tris/HCl, pH 7.4, and 10% glycerol by short ultrasonic pulses. The homogenate with 2% IGEPAL CA-630 (56741; Sigma-Aldrich) was heated at 85°C for 15 min and cleared by centrifugation to remove insoluble parts and heat-precipitated proteins. The PANet complex that remained intact during heating was captured with anti-FLAG affinity gel (B23101; Biotool) for 1 h at room temperature. The anti-FLAG affinity gel was washed 2 times with 50 mm Tris/HCl, pH 7.4, 10% glycerol, 2% IGEPAL CA-630, and then 1 time with 50 mm Tris/HCl, pH 7.4, 10% glycerol. The captured PAN was eluted with 3% ammonium hydroxide solution and vacuum-dried. For Western blot analysis, lyophilized samples were reconstituted in SDS-PAGE sample buffer and analyzed using an Odyssey Infrared Imaging System (LI-COR Biosciences).
Protein-unfolding activity assay.
PANet and empty pTriEx 4 vector (negative control) were expressed in HEK 293 cells and purified using anti-FLAG affinity gel as described above. Wild-type PAN at a concentration of 20 nm was used as positive control and was purified as described previously (Smith et al., 2005). The protein-unfolding activity assay was adopted from that same study (Smith et al., 2005). In brief, substrate unfolding rate was measured by incubating 1 μm GFPssrA with or without PAN in a buffer containing 50 mm Tris, 5% glycerol, 20 mm MgCl2, 400 nm T20S (Thermoplasma acidophilum 20S) and 2 mm ATP at 37°C. PAN unfolds GFPssrA by recognizing the nine-residue ssrA degradation tag on the C terminus of GFP. T20S was added to degrade GFP that had been unfolded by PAN because GFP-ssrA will refold and regain fluorescence if it is not degraded or trapped in its unfolded state. The rate of GFPssrA fluorescence loss was measured on a BioTek synergy mx 96-well plate reader (λex: 485 nm, λem: 510 nm).
Retina.
PANet was isolated from 47 retinas of PANet (+) mice. For a negative control, equal number of PANet (−) retinas was used. The assay was conducted as described above except that it also contained 0.15 mg/ml (100 nm) of purified GroEL-D87K (in place of the T20S), which served as a trap preventing refolding of GFPssrA after unfolding by PANet (Weber-Ban et al., 1999).
20S proteasome gate-opening assay.
20S gate opening by PAN was measured using the internally quenched fluorogenic peptide substrate (LFP) in reaction buffer containing archaeal T20S (3 nm), wild-type PAN or PANet (200 nm), and MgCl2 (20 mm) (Smith et al., 2007). LFP was dissolved in DMSO and used at a final concentration of 10 μm either in the presence or absence of 10 μm ATPγS. LFP contains a fluorescent reporter (MCA) at the N terminus and a quenching group (DNP) at the C terminus. Upon cleavage of the peptide by the 20S proteasome, MCA is released and an increase in fluorescence can be observed at ex/em: 325/393. Rate of fluorescence increase (λex: 325, λem: 393) was measured every 20 s in a Biotek 96-well plate reader to determine activation (gate opening) by PAN.
Microscopy.
Enucleated eyes were sent to Excalibur Pathology for paraffin-embedding and hematoxylin and eosin staining of retinal cross-sections. The method for immunofluorescence confocal microscopy was described previously (Brooks et al., 2018).
ERG.
Mice were dark-adapted overnight before testing and all procedures were performed under dim red light. Mice were anesthetized by 1.5% isoflurane with 2.5 L/min oxygen delivered through a nose cone. The animal's pupils were dilated with a mixture of 1.25% phenylephrine hydrochloride and 0.5% tropicamide ophthalmic solution and visual responses were recorded simultaneously from both eyes using a Celeris rodent ERG system and Espion software (Diagnosis).
Experimental design and statistical analyses.
In all quantifications, the significance level was determined using the independent two-tailed Student's t test and the values are expressed as mean ± SEM. n = 1 represents one animal.
Antibodies.
PANet was detected using rat anti-HA antibody (11867423001; Roche) and rabbit anti-HA antibody (sc-805; Santa Cruz Biotechnology) for immunofluorescent confocal microscopy and Western blotting, respectively.
Results
Design of the PANet unfoldase
In Archaea, PAN unfolds substrates and facilitates their translocation into the cavity of the 20S core of the proteasome, where substrates undergo proteolysis (Zwickl et al., 1999). To prevent PAN from binding to the endogenous 20S complex, an HA-FLAG epitope tag (et) was placed on the C terminus of its subunit. This modification was expected to obstruct the HbYX sequence that is essential for binding between PAN and the 20S complex (Fig. 1A) (Smith et al., 2005). This construct termed PANet was first purified from HEK 293 cell culture and then its ability to unfold protein substrate and to couple to the 20S proteasome was compared with that of wild-type PAN. We found that PANet isolated from HEK 293 cells displayed significant protein-unfolding activity (Fig. 1B). This observation demonstrated that the PANet complex could be catalytically active in mammalian cells at mammalian body temperature. We also found that PANet did not stimulate the proteolytic activity of the 20S proteasome (Fig. 1C), demonstrating that this complex can only act as a protein-unfolding ATPase. Next, we targeted the expression of the PANet transgene to the rod photoreceptors of mice by using a 4.4 kb rhodopsin promoter (Lem et al., 1991). The expression of PANet was confirmed by Western blotting (Fig. 1D). Similar to PANet expressed in the cell culture, PANet isolated from the retina displayed robust protein-unfolding activity at 37°C, demonstrating that PANet from the retina is also functionally active (Fig. 1E). Next, we focused on characterizing PANet mice to determine whether PAN has an adverse effect on mouse rods.
Figure 1.

Design of the PANet unfoldase. A, Diagram illustrating the strategy for conversion of PAN to a protein-unfolding ATPase: a C-terminal epitope tag (et) obstructs the HbYX motif required for docking to the 20S proteasome. B, Protein unfolding activity of PANet isolated from HEK 293 cells compared with purified wild-type PAN. Data were normalized to control where an empty vector was used for transfection. C, ATP binding to PAN (as measured using the nonhydrolyzable ATP analog ATPγS) stimulates 20S gate opening in wild-type PAN (as expected), but not PANet. The stimulation of 20S activity (caused by PAN-induced 20S gate opening) was measured using saturating amounts of PAN and a 2 μm concentration of the fluorescent reporter peptide LFP with 20 mm MgCl2 and 2 mm ADP or 10 μm ATPγS (see Materials and Methods for details). D, Detection of PANet by Western blotting in anti-FLAG pull downs from retinal lysates. Specific band of 55 kDa (*) was visualized with antibody against HA. E, Protein-unfolding activity of PANet isolated from the retina compared with purified wild-type PAN. Data were normalized to control where transgene-negative mice were used.
Characterization of PANet mice
PANet mice were healthy and viable. The gross ocular morphology of 1-year-old PANet (+) mice was similar to the morphology of their PANet (−) littermates (Fig. 2A). The total number of rod photoreceptors in PANet (+) retinas also remained normal based on the thickness of the outer nuclear layer of the retina, which contains rod nuclei (Fig. 2B). Examining the expression of PANet in the retina by immunofluorescent microscopy revealed that PANet was present in all subcellular compartments of rod photoreceptor, except for the rod outer segments (Fig. 2C). This exclusion is likely due to the size of the PAN complex because large complexes cannot access the outer segment. The expression of PANet (+) transgene in the retina lasted until at least 1 year of age. To determine the percentage of rods that express transgene, we visualized PANet-expressing rods in flat-mounted retinal preparations (Fig. 2D). We found that at least 40% of rods expressed PANet in the areas with the highest degree of mosaicism shown in Figure 2D, whereas 70% or more of rods expressed PANet in the areas with the least amount of mosaicism (data not shown). Therefore, according to our most conservative assessment at least 50% of rods expressed the transgene. In the assay, shown in Figure 1E, we had to capture PANet from 47 retinas to match the activity of 5.6 × 1010 wild-type PAN complexes used as a positive control. Assuming that PANet and wild-type PAN have the same specific activity and that there are 6.4 × 106 rods in a mouse retina (Carter-Dawson and LaVail, 1979; Jeon et al., 1998), half of which express the transgene, we could estimate that each rod cell expressed ∼370 PANet complexes. Last, we analyzed visual responses of mice by ERG using the amplitude of the a-wave as a quantitative readout for the status of phototransduction. We found that visual responses of PANet(−) and PANet(+) siblings were statistically indistinguishable across a wide range of stimulating flashes (Fig. 2E), which indicated that the levels of phototransduction proteins in PANet-expressing rods remained unaltered. Therefore, we found no evidence that PANet may compromise viability or function of rod photoreceptors by unfolding their native proteins.
Figure 2.

Normal retinal morphology and rod visual function of 1-year-old PANet mice. Mice of the indicated genotype were analyzed at 1 year of age. A, Ocular cross-section stained with hematoxylin and eosin. B, Central retina morphology. The indicated retinal layers are as follows: OS, outer segments layer; IS, inner segments layer; ONL, outer nuclear layer; OPL, outer plexiform layer; and INL, inner nuclear later. Diagram illustrates the location of rod cells within the retina. C, Protein localization was determined in frozen retinal cross-sections by immunofluorescent confocal microscopy. The immunostaining of PANet (red) and the DIC image with cell nuclei stained with DAPI (blue) are shown. D, PANet (red) was visualized in flat-mounted retinas by immunofluorescent confocal microscopy using antibody against HA. E, Visual responses of rods were analyzed by ERG and the maximum amplitude of elicited a-wave was plotted as a function of flash intensity. Error bars indicate SEM (n = 4). Each dataset was fitted with a simple rectangular hyperbola with two parameters.
PANet protects rod photoreceptors against misfolded proteins
Next, we tested the efficiency of PANet to protect rod photoreceptors against misfolded proteins in Gγ1 knock-out mice (Kolesnikov et al., 2011). In this model, the misfolding of a major rod protein, the Gβ1 subunit of transducin, was shown to overload the UPS (Lobanova et al., 2013). The ensuing deregulation of proteostasis leads to complete degeneration of Gγ1-null rods (Lobanova et al., 2008; Kolesnikov et al., 2011). To determine whether PANet can protect rods of Gγ1-null mice, PANet mice were backcrossed on Gγ1-null background. Then, PANet(−) and PANet(+) littermates (PANet−/−; Gγ1−/−, and PANet+/−; Gγ1−/−, respectively) were raised under identical environmental conditions and comparatively analyzed. By the age of 7 months, PANet (−) mice on Gγ1-null background had lost virtually all of their rods, as evident from disappearance of the entire outer nuclear layer of their retinas. However, in PANet (+) mice, more than a half of total rod population, presumably expressing the transgene (Fig. 2D), had survived (Fig. 3A–C). Consistent with this observation, PANet (−) mice did not elicit any visual responses when analyzed by ERG, whereas their PANet (+) littermates continued to generate robust a-waves (Fig. 3D,E). Moreover, the amplitudes of the a-wave in 7-month-old PANet+/−; Gγ1−/− mice and 1-month-old Gγ1−/− mice were mostly identical across a wide range of stimulating flashes, except for the two brightest flashes of 4 and 10 cd s m−2 (Fig. 3D), when the reduction in the a-wave was directly proportional to the number of surviving rods (Fig. 3C). These data demonstrate that PANet can vastly improve the survival rate of Gγ1-null rod photoreceptors. Furthermore, PANet may be able to completely rescue Gγ1 knock-out mice if expression of the PANet transgene was uniform with no mosaicism.
Figure 3.
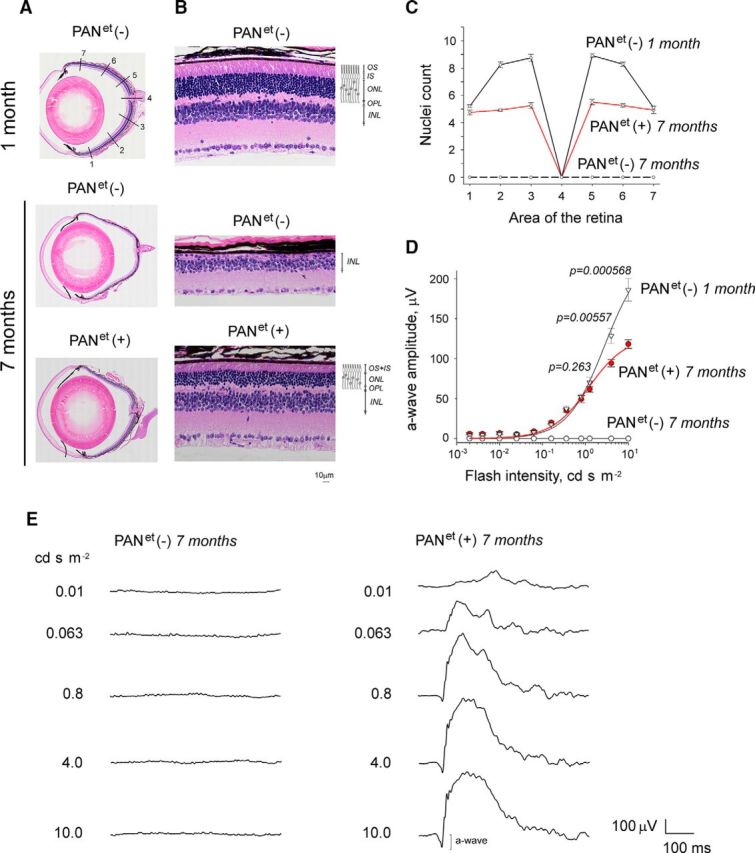
PANet improves the survival of rod photoreceptors in Gγ1 knock-out mice. Mice of PANet−/−; Gγ1−/− and PANet+/−; Gγ1−/− genotypes, designated as PANet(−) and PANet(+), respectively, were analyzed at the age indicated. A, Ocular cross-section stained with hematoxylin and eosin. B, Retina morphology at area 5 (A, top). The indicated retinal layers are as follows: OS, outer segments layer; IS, inner segments layer; ONL, outer nuclear layer containing rod nuclei; OPL, outer plexiform layer; INL, inner nuclear later. Diagram illustrates the location of rod cells within the retina. C, Photoreceptor nuclei count in areas 1–7 of the retina (A, top) of ONL. Error bars indicate SEM (n = 3). D, Visual responses of rods were analyzed by ERG and the amplitude of maximum elicited a-wave was plotted as a function of flash intensity. Error bars indicate SEM (n = 4). Each dataset was fitted with a simple rectangular hyperbola with two parameters. E, Representative ERG responses of 7-month-old mice.
Discussion
Gγ1 knock-out mice were used in this study because their neurodegenerative phenotype has been linked to protein misfolding (Lobanova et al., 2013). Gγ1 is one of the three subunits of the phototransduction heterotrimeric G-protein transducin (Gαt1β1γ1). As one might expect, knocking out the Gγ1 subunit significantly decreases the expression of its binding partner, Gβ1; however, it also decreases the amount of Gαt1 and greatly reduces the efficiency of rod phototransduction, which was observed in two independently generated knock-out lines (Lobanova et al., 2008; Kolesnikov et al., 2011). Gγ1 knock-out mice display progressive loss of rod photoreceptors (Lobanova et al., 2008; Kolesnikov et al., 2011), although knocking out the Gαt1 subunit of transducin does not cause this phenotype (Calvert et al., 2000). This apparent discrepancy was resolved by demonstrating that Gγ1-null mice have a widespread deficiency in proteasome-mediated protein degradation, due to the misfolding of the remaining Gβ1 (Lobanova et al., 2013). The folding of nascent Gβ1 is mediated by chaperonin containing t-complex subunit protein 1 (CCT/TRiC) and crucially depends on the co-chaperone phosducin like protein 1 (Lukov et al., 2005, 2006; Plimpton et al., 2015) and on Gγ1 (Schmidt and Neer, 1991; Garcia-Higuera et al., 1996; Wells et al., 2006). In the absence of Gγ1 expression, Gβ1 predominantly exists as a misfolded protein, which eventually overwhelms the UPS due to its large quantity (Lobanova et al., 2013).
The observed protective effect of protein-unfolding ATPase PANet further supports the belief that retinal degeneration in Gγ1 knock-out mice is caused by protein misfolding. However, it is generally inconsistent with the notion that proteasome overload is caused by excessive amounts of proteasomal substrates because PANet has no proteolytic activity and would not degrade proteins. Therefore, we favor the hypothesis that PANet alleviates inhibition of the proteasome by preventing the formation of soluble oligomers (Fig. 4). Gβ1 is predisposed to aggregation due to a high content of β-sheets (Gaudet et al., 1996) and could potentially form oligomers. Oligomer formation could also be driven by secondary protein misfolding events caused by Gγ1 knock-out. Binding of Gγ1 has been proposed to serve as a signal that prevents mature Gβ1γ1 dimer from reentering the folding chamber of CCT (Gao et al., 2013). Consistent with this proposal, more Gβ1 co-purified with CCT in the retinal extracts of Gγ1 knock-out mice than in those of wild-type mice (Lobanova et al., 2013). Therefore, in the absence of Gγ1, Gβ1 is expected to competitively inhibit the folding of less abundant CCT substrates, resulting in misfolded proteins in addition to Gβ1. These misfolded protein substrates of CCT may eventually form oligomers that evade the recognition by the mammalian UPS. These oligomers may accumulate in rods over time and inhibit the proteasome (Thibaudeau et al., 2018). PANet, which has evolved in Archaea and does not require ubiquitin labeling to recognize misfolded proteins, may more effectively recognize and unfold the aberrant mammalian proteins. The unfolding of aggregation-prone proteins is expected to hinder their aggregation and to expose them to the innate Hsp70-Hsp40 and Hsp90 chaperone systems that cooperate with the UPS (Kettern et al., 2010). As a result, PANet helps to maintain proteostasis in Gγ1-null rods by purging the unstable nascent β-sheet-rich proteins that could oligomerize and inhibit the proteasome. Another intriguing possibility, which requires further testing, is that PANet could target amyloid oligomers directly. Given a potentially broad substrate specificity of PANet and its robust activity, it is tempting to speculate that this xenogeneic unfoldase could be equally effective against other types of neurodegenerative diseases of protein-misfolding etiology.
Figure 4.

Model: PANet averts the inhibition of the proteasome by soluble oligomers. 1, Gβ1 remains continuously misfolded due to the absence of Gγ1. 2, Other proteins also become misfolded because Gβ1 competitively inhibits the CCT-mediated protein-folding pathway. 3, Buildup of misfolded proteins leads to formation of soluble oligomers that inhibit the 26S proteasome. 4, PANet unfolds the misfolded proteins, which targets them to the innate Hsp70-Hsp40 and Hsp90 systems and the UPS.
Footnotes
This work was supported by the WVU Health Sciences Center Bridge Award (M.S.) and by the National Institutes of Health (NIH Grant R01 GM107129 to D.M.S. and Grant F31 GM115171 to A.S.). The Transgenic Animal Core Facility at WVU is supported by Centers of Biomedical Research Excellence (COBRE) Grants RR031155 and RR016440 from the NIH.
The authors declare no competing financial interests.
References
- Balchin D, Hayer-Hartl M, Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353:aac4354. 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- Benaroudj N, Goldberg AL (2000) PAN, the proteasome-activating nucleotidase from archaebacteria, is a protein-unfolding molecular chaperone. Nat Cell Biol 2:833–839. 10.1038/35041081 [DOI] [PubMed] [Google Scholar]
- Bösl B, Grimminger V, Walter S (2006) The molecular chaperone Hsp104–a molecular machine for protein disaggregation. J Struct Biol 156:139–148. 10.1016/j.jsb.2006.02.004 [DOI] [PubMed] [Google Scholar]
- Brooks C, Murphy J, Belcastro M, Heller D, Kolandaivelu S, Kisselev O, Sokolov M (2018) Farnesylation of the transducin G protein gamma subunit is a prerequisite for its ciliary targeting in rod photoreceptors. Front Mol Neurosci 11:16. 10.3389/fnmol.2018.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicoló M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN Jr, Makino CL, Lem J (2000) Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit. Proc Natl Acad Sci U S A 97:13913–13918. 10.1073/pnas.250478897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM (1979) Rods and cones in the mouse retina. II. autoradiographic analysis of cell generation using tritiated thymidine. J Comp Neurol 188:263–272. 10.1002/cne.901880205 [DOI] [PubMed] [Google Scholar]
- Collins GA, Goldberg AL (2017) The logic of the 26S proteasome. Cell 169:792–806. 10.1016/j.cell.2017.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Bott LC (2014) The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci 7:70. 10.3389/fnmol.2014.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriziotis P, André R, Smith DM, Goold R, Kinghorn KJ, Kristiansen M, Nathan JA, Rosenzweig R, Krutauz D, Glickman MH, Collinge J, Goldberg AL, Tabrizi SJ (2011) Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J 30:3065–3077. 10.1038/emboj.2011.224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Sinha S, Belcastro M, Woodard C, Ramamurthy V, Stoilov P, Sokolov M (2013) Splice isoforms of phosducin-like protein control the expression of heterotrimeric G proteins. J Biol Chem 288:25760–25768. 10.1074/jbc.M113.486258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Fenoglio J, Li Y, Lewis C, Panchenko MP, Reiner O, Smith TF, Neer EJ (1996) Folding of proteins with WD-repeats: comparison of six members of the WD-repeat superfamily to the G protein beta subunit. Biochemistry 35:13985–13994. 10.1021/bi9612879 [DOI] [PubMed] [Google Scholar]
- Gaudet R, Bohm A, Sigler PB (1996) Crystal structure at 2.4 angstroms resolution of the complex of transducin betagamma and its regulator, phosducin. Cell 87:577–588. 10.1016/S0092-8674(00)81376-8 [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI (2006) Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311:1471–1474. 10.1126/science.1124514 [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Krupinski T, Garcia S, Morimoto RI (2009) Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet 5:e1000399. 10.1371/journal.pgen.1000399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428. 10.1152/physrev.00027.2001 [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94:73–82. 10.1016/S0092-8674(00)81223-4 [DOI] [PubMed] [Google Scholar]
- Guo Q, Lehmer C, Martínez-Sánchez A, Rudack T, Beck F, Hartmann H, Pérez-Berlanga M, Frottin F, Hipp MS, Hartl FU, Edbauer D, Baumeister W, Fernández-Busnadiego R (2018) In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell 172:696–705.e12. 10.1016/j.cell.2017.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Park SH, Hartl FU (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24:506–514. 10.1016/j.tcb.2014.05.003 [DOI] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH (1998) The major cell populations of the mouse retina. J Neurosci 18:8936–8946. 10.1523/JNEUROSCI.18-21-08936.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettern N, Dreiseidler M, Tawo R, Höhfeld J (2010) Chaperone-assisted degradation: multiple paths to destruction. Biol Chem 391:481–489. 10.1515/BC.2010.058 [DOI] [PubMed] [Google Scholar]
- Kolesnikov AV, Rikimaru L, Hennig AK, Lukasiewicz PD, Fliesler SJ, Govardovskii VI, Kefalov VJ, Kisselev OG (2011) G-protein betagamma-complex is crucial for efficient signal amplification in vision. J Neurosci 31:8067–8077. 10.1523/JNEUROSCI.0174-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FW, Menéndez-Benito V, Dantuma NP, Portis JL, Collinge J, Tabrizi SJ (2007) Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 26:175–188. 10.1016/j.molcel.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, Tsai FT (2003) The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115:229–240. 10.1016/S0092-8674(03)00807-9 [DOI] [PubMed] [Google Scholar]
- Lem J, Applebury ML, Falk JD, Flannery JG, Simon MI (1991) Tissue-specific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron 6:201–210. 10.1016/0896-6273(91)90356-5 [DOI] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Herrmann R, Chen YM, Kessler C, Michaud NA, Trieu LH, Strissel KJ, Burns ME, Arshavsky VY (2008) Transducin gamma-subunit sets expression levels of alpha- and beta-subunits and is crucial for rod viability. J Neurosci 28:3510–3520. 10.1523/JNEUROSCI.0338-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Skiba NP, Arshavsky VY (2013) Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc Natl Acad Sci U S A 110:9986–9991. 10.1073/pnas.1305521110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukov GL, Hu T, McLaughlin JN, Hamm HE, Willardson BM (2005) Phosducin-like protein acts as a molecular chaperone for G protein betagamma dimer assembly. EMBO J 24:1965–1975. 10.1038/sj.emboj.7600673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukov GL, Baker CM, Ludtke PJ, Hu T, Carter MD, Hackett RA, Thulin CD, Willardson BM (2006) Mechanism of assembly of G protein betagamma subunits by protein kinase CK2-phosphorylated phosducin-like protein and the cytosolic chaperonin complex. J Biol Chem 281:22261–22274. 10.1074/jbc.M601590200 [DOI] [PubMed] [Google Scholar]
- Maupin-Furlow JA. (2014) Prokaryotic ubiquitin-like protein modification. Annu Rev Microbiol 68:155–175. 10.1146/annurev-micro-091313-103447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimpton RL, Cuéllar J, Lai CW, Aoba T, Makaju A, Franklin S, Mathis AD, Prince JT, Carrascosa JL, Valpuesta JM, Willardson BM (2015) Structures of the gbeta-CCT and PhLP1-gbeta-CCT complexes reveal a mechanism for G-protein beta-subunit folding and gbetagamma dimer assembly. Proc Natl Acad Sci U S A 112:2413–2418. 10.1073/pnas.1419595112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Neer EJ (1991) In vitro synthesis of G protein beta gamma dimers. J Biol Chem 266:4538–4544. [PubMed] [Google Scholar]
- Schmidt M, Finley D (2014) Regulation of proteasome activity in health and disease. Biochim Biophys Acta 1843:13–25. 10.1016/j.bbamcr.2013.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Benaroudj N, Goldberg A (2006) Proteasomes and their associated ATPases: a destructive combination. J Struct Biol 156:72–83. 10.1016/j.jsb.2006.04.012 [DOI] [PubMed] [Google Scholar]
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL (2007) Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell 27:731–744. 10.1016/j.molcel.2007.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL (2005) ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell 20:687–698. 10.1016/j.molcel.2005.10.019 [DOI] [PubMed] [Google Scholar]
- Thibaudeau TA, Anderson RT, Smith DM (2018) A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun 9:1097. 10.1038/s41467-018-03509-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher C, Garcia-Oroz L, Rubinsztein DC (2005) Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Hum Mol Genet 14:3425–3433. 10.1093/hmg/ddi372 [DOI] [PubMed] [Google Scholar]
- Vashist S, Cushman M, Shorter J (2010) Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol 88:1–13. 10.1139/O09-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Ban EU, Reid BG, Miranker AD, Horwich AL (1999) Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature 401:90–93. 10.1038/43481 [DOI] [PubMed] [Google Scholar]
- Wells CA, Dingus J, Hildebrandt JD (2006) Role of the chaperonin CCT/TRiC complex in G protein betagamma-dimer assembly. J Biol Chem 281:20221–20232. 10.1074/jbc.M602409200 [DOI] [PubMed] [Google Scholar]
- Zwickl P, Ng D, Woo KM, Klenk HP, Goldberg AL (1999) An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26 S proteasome, activates protein breakdown by 20 S proteasomes. J Biol Chem 274:26008–26014. 10.1074/jbc.274.37.26008 [DOI] [PubMed] [Google Scholar]