

Abstract
The aim of the present study was to examine the protective effects and mechanisms of S100 calcium-binding protein A4 (S100A4) on endothelial cell apoptosis induced by oxidative stress injury. Endothelial cells were cultured and divided into control and oxidative stress injury groups, with the latter state induced by H2O2. Endothelial cells in every group were incubated with or without 50 or 100 µM S100A4. The cell viability and amounts of malondialdehyde, nitric oxide and lactate dehydrogenase in the culture medium were measured. The apoptotic index was detected by TUNEL staining. Western blot and immunoprecipitation analyses were used to detect the expression levels and the association between S100A4 and P53. H2O2 treatment led to oxidative stress injury in the cultured vascular endothelial cells, a decrease in the cell viability and an increase in the rate of apoptosis of vascular endothelial cells compared with the negative control group. Exogenous S100A4 serves a significant function against oxidative stress injury (P<0.05), increasing the viability and attenuating the apoptotic rate of endothelial cells. Western blotting results suggested that the protein levels of S100A4 and P53 increased subsequent to oxidative stress injury and that exogenous S100A4 increased the expression of P53 in the cytoplasm and decreased the expression of P53 in nucleus. The immunoprecipitation assay results revealed a protein-protein interaction between S100A4 and P53. These results suggested that rat recombinant S100A4 serves an anti-apoptotic function in oxidative stress injury. This effect of S100A4 is mediated, at least in part, via the inhibition of the translocation of P53 to the nucleus.
Keywords: rat recombinant S100 calcium-binding protein A4, oxidative stress, apoptosis, tumor protein 53
Introduction
In 2012 the World Health Organization (WHO) estimated that cardiovascular disease accounted for 30% of the total number of mortalities per year and is the leading cause of mortality worldwide (1). WHO predicts that cardiovascular mortality will rapidly increase up to 2030 (1,2). Vascular endothelial cells possess numerous biological functions, including the regulation of vascular tone, secretion of vasoactive substances and anti-platelet aggregation, serving an important role in maintaining the function of the cardiovascular system. A previous study demonstrated that the incidence of vascular disease in patients with impaired endothelial function was significantly increased compared with that in patients with normal endothelial function (3). Endothelial cell injury induced by oxidative stress is an important cause of vascular endothelial dysfunction and serves a crucial function in the occurrence of cardiovascular diseases (4). Vascular endothelial cell apoptosis is the primary form of oxidative stress injury and a prelude to the occurrence and evolution of multiple types of cardiovascular diseases (5). Wild type P53 is bound to four specific sites in the form of tetramer, which is associated with cell stress response, cell cycle and cell apoptosis (6).
The S100 protein, whose molecular weight is 11–12 kDa, belongs to the family of calcium binding proteins. It is highly homologous with calmodulin (7,8). The S100 protein family serves an important function in cell proliferation, differentiation, muscle contraction, gene expression and cell apoptosis (9,10). Dysfunction of S100 protein may cause different diseases. S100 calcium-binding protein A4 (S100A4) is a member of the S100 family and is associated with the occurrence, development and prognosis of tumors (11). Studies have indicated that the S100A4 protein not only is expressed in the cardiovascular endothelium, but also functions in promoting angiogenesis (12,13).
Previous studies have indicated that S100A4 binds the C terminal region of the P53 protein, which affects the subcellular localization and transcriptional activity of P53 (14). This combination affects apoptosis. The aim of the present study was to investigate whether the S100A4 protein exhibits a protective effect on oxidative stress injury of endothelial cells and to elucidate its mechanism. By establishing an oxidative stress damage model in vascular endothelial cells, the effect of S100A4 protein on oxidative stress injury of vascular endothelial cells was observed. The effect of S100A4 protein on cardiovascular endothelial cell apoptosis through the P53 pathway was also examined. Based on these experiments, the present study preliminarily clarified the regulatory mechanism of S100A4 protein on the apoptosis of vascular endothelial cells.
Materials and methods
Antibodies and reagents
The following antibodies were used: P53 (cat. no. 10442-1-AP; ProteinTech Group, Inc. (Chicago, IL, USA), Caspase 3 (cat. no. ab13847; Abcam, Cambridge, MA, USA), p17-specific antibody (cat. no. 25546-1-AP; Proteintech Group, Inc.), S100A4 (ab41532; Abcam) and β-actin (cat. no. 60008-1-Ig; Proteintech Group, Inc.). S100A4 (cat. no. ab41532) was purchased from Abcam. The following reagents were used: Rat Lactic Dehydrogenase (LDH; cat. no. E-0672; Meilian Shengwu, Shanghai, China), nitrous oxide (NO; cat. no. KGE001; R&D Systems, Inc. Minneapolis, MN, USA), Malondialdehyde (MDA; cat. no. ab118970; Abcam), superoxide dismutase (SOD; cat. no. DYC3419-2; R&D Systems). Rat recombinant S100A4 protein was expressed and purified in the Performance Medicine Laboratory of Tianjin Institute of Health and Environmental Medicine (Tianjin, China). Cell Death Detection kit (cat. no. G3250, Promega) was from Promega Corporation (Madison, WI, USA). Hydrogels incubated with PBS alone were used as negative controls. All the experiments were repeated at least three times. The microscopy (OLMPUS CK40, Olympus corporation, Tokyo, Japan) was preserved in cell culture room and utilized to observe endothelial cells without fixing embedding and staining at 25°C. The magnification used was ×100.
Cell culture and treatment
Rat aortic endothelial cells (RAECs) were isolated and cultured as described previously, with minor modifications (15). The thoracic aorta were excised from 3 male Wistar rats (150–180 g) after they were sacrificed humanely by injecting with 40 mg anesthetic (chloral hydrate) per 100 g of body weight and immediately placed in cold PBS containing 100 U/ml penicillin and 100 mg/ml streptomycin. The aorta was cut into 1 mm-wide rings subsequent to the removal of the periadventitial fat. Following transfer to a T-25 cm2 flasks (NalgeNunc International, Penfield, NY, USA), the rings were cultured in Medium 199 (cat. no. 11150059, Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 20% fetal bovine serum (FBS; cat. no. 10099141, Gibco; Thermo Fisher Scientific, Inc.), 2.5 ng/ml basic fibroblast growth factors, 100 U/ml penicillin and 100 mg/ml streptomycin. The aorta rings were placed at 37°C in a humidified atmosphere with 5% CO2 for 72–80 h without movement. All pieces of aorta rings were removed when cells migrated. In experiments, M199 medium supplemented with 1% bovine serum albumin (cat. no. B2064; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used. All experiments were performed with RAECs up to 4 passages. Male Wistar rats were gained from the Experimental Animal Room of the Institute of Health and Environmental Medicine (Tianjin, China). The rats were maintained under standard conditions (ambient temperature 21–23°C; with a 12 h dark-light cycle) with ad libitum access to food and tap water. This study was approved by the committee of the Institute of Health and Environmental Medicine, Tianjin, China.
The cells were divided into the following groups: i) A normal control group incubated in Medium 199 (NC); ii) a H2O2 injury group incubated in Medium 199 containing 100 µM H2O2 (HI); and two S100A4-treated groups incubated with iii) 50 µM or iv) 100 µM S100A4 in Medium 199 containing 100 µMH2O2 [(HI+S100A4 (50 µM)/HI+S100A4 (100 µM)]. In the MTT and ELISA analysis, two groups were added to detect whether S100A4 had damaged the cells. They were NC+S100A4 (50 µM) and NC+S100A4 (100 µM) groups.
Cell proliferation assay
Cell proliferation was determined using an MTT assay. A total of ~3×103 endothelial cells were plated into each well of a 96-well plate. Following overnight incubation at 37°C, the cells were treated with 100 µM H2O2, H2O2+S100A4 (50/100 µM) for 48 h. Then the medium was removed and MTT (20 µl of 5 mg/ml) was added to each well and incubated at 37°C for 4 h. Plates were agitated at low speed (16.77 × g) 37°C for 10 min and the purple-colored precipitates of formazan were dissolved in 150 µl dimethyl sulfoxide. Absorbance was measured at 490 nm using an ELISA plate reader. The reduction in viability of in H2O2-treated endothelial cells was expressed as a percentage compared with non-H2O2 treated control cells. Control cells were considered 100% viable.
ELISA assay
The levels of LDH (Rat LDH ELISA kit; cat. no. E-0672; Meilian Shengwu), NO (Total Nitric Oxide and Nitrate/Nitrite Parameter assay kit; cat. no. KGE001; R&D Systems), MDA (MDA assay kit; cat. no. ab118970; Abcam), and SOD (Human/Mouse/Rat Total SOD2/Mn-SOD DuoSet IC ELISA kit; cat. no. DYC3419-2; R&D Systems) from endothelial cell medium were quantified by ELISA assay according to the manufacturer's protocol.
Assessment of apoptosis by TUNEL staining
Cells apoptosis was determined by double-labeling TUNEL immunofluorescence staining. A total of 2×104 RAECs with Medium 199 (cat. no. 11150059; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS were seeded into each well of a 48-well plate. Following the aforementioned experimental treatments, cells were fixed in 4% paraformaldehyde for 30 min at room temperature. Subsequently, the cells were incubated with 3% H2O2 in methanol for 10 min at room temperature to block endogenous peroxidase activity, and were then incubated with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min at 4°C. After washing in PBS, the cells were incubated with the TUNEL reaction mixture containing 5 µl enzyme solution and 45 µl fluorochrome-labeled solution at 37°C for 60 min in the dark. For counterstaining, the cells were incubated with DAPI for 15 min at room temperature. The cells were observed using a fluorescence microscope, in which 10 fields were randomly selected. The TUNEL+ and DAPI+ nuclei in the cells were counted manually. The percentage of apoptotic cells was calculated as the ratio of the number of TUNEL-positive cells to the total number of cells, which were counted using a fluorescence microscope.
Western blot analysis
The 4 groups of cells (described above) were harvested. The cells samples were homogenized in a Radio-Immunoprecipitation Assay (RIPA) lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% TritonX-100, 1% sodium deoxycholate, 0.1% SDS]. Protein concentrations were determined with a BCA assay kit (Pierce; Thermo Fisher Scientific, Inc.). The proteins (10 µl in each line) were separated using a 10% gel with SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked with TBS-Tween-20 (0.05%) containing 5% non-fat milk for 2 h at room temperature and incubated with the following primary antibodies, anti-β-actin (1:1,000, 60008-1-Ig, Proteintech Group, Inc.), anti-P53 (1:500, 10442-1-AP, Proteintech Group, Inc.), anti-cleaved-caspase-3 (1:1,000, 25546-1-AP, Proteintech Group, Inc.), anti-S100A4 (1:250, ab41532, Abcam) overnight at 4°C. The membranes were then incubated with secondary antibody, goat anti-rabbit antibodies (1:5,000, ZB-2301, ZSGB-Bio) conjugated with horseradish peroxidase for 1 h at room temperature (25°C). A SuperSignal™-enhanced chemiluminescent substrate (Pierce; Thermo Fisher Scientific, Inc.) was applied to the probed membrane for 3 min at room temperature. The blots were quantified via densitometry using ImageJ software (version 1.37; National Institutes of Health, Bethesda, MA, USA).
Immunoprecipitation (IP)
Endothelial cells were seeded in 10-cm dishes, followed by stimulation (37°C) with or without 100 µM H2O2 for 48 h. Subsequently, the HepG2 cell (Tianjin Saierbio Biotechnology Co., Ltd., Tianjin, China) lysates were prepared with 1% Tris-Triton cell lysis buffer (Cell Signaling Technology, Inc., Danvers, MA, USA) pre-mixed with 1 mM phenylmethanesulfonyl fluoride on ice for 30 min and centrifuged (4°C) at 12,000 × g for 30 min. The supernatants were incubated (4°C) overnight with 30 µl Dynabeads protein A or protein G (Thermo Fisher Scientific, Inc.) pre-coated with anti-S100A4 (1:250, Abcam) antibodies. The immunocomplexes were subjected to western blot analysis. The normal corresponding immunoglobulin G control was assayed simultaneously.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical analysis was performed using a Student's t-test for paired samples or a single-factor analysis of variance with Student-Newman-Keuls post-hoc test as appropriate by SPSS software (version 20.0; IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
S100A4 may inhibit the cell morphological changes induced by H2O2
Visible under a phase contrast microscope, in the NC group the endothelial cells were polygonal and exhibited a clear structure, uniform size and single paving stone-like close spacing. The cell morphology of the NC+S100A4 groups was almost identical to that of the NC group. In the HI group, the endothelial cells exhibited shrinkage, roundness and clearance. There were vacuoles and nuclear chromatin condensation and aggregation in the cytoplasm of the cell. Cells demonstrated a disordered arrangement, losing the typical single layer paving stone-like arrangement. In the HI+S100A4 groups, cell morphology and structure were predominantly clear (Fig. 1A). The arrangement and morphology of the cells was not markedly changed compared with the NC group.
Figure 1.

S100A4 suppresses apoptosis. Cultured rat vascular endothelial cells 12 h after H2O2 injury with and without S100A4 treatment prior to fixing with 4% paraformaldehyde for the apoptosis assay. (A) Cell morphology was detected by light microscopy scale bar 50 µm; (B) Proliferation of cells was determined by the MTT assay. *P<0.05 vs. H2O2 group. S100A4, S100 calcium-binding protein A4; NC, normal control; HI, hypoxic injury induced by H2O2.
The MTT assay easily and effectively evaluates the activity of cells. The cell survival rate of the oxidative stress injury group was significantly decreased compared with that of the NC group (P<0.05), suggesting that H2O2 may cause endothelial cell death. There were no significant differences in cell survival rate between the NC, NC+S100A4 (50 µM) and NC+S100A4 (100 µM) groups following administration of S100A4 protein, which indicated that exogenous S100A4 protein demonstrated no significant effect on endothelial cells. In addition, the cell survival rate of the 50 and 100 µM HI+S100A4 groups was significantly increased compared with that in the HI group, but remained significantly decreased compared with that of the NC group (P<0.05) and the cell survival rate of HI+S100A4 (100 µM) group was increased compared with that of the HI+S100A4 (50 µM) group (Fig. 1B). This indicated that S100A4 protein may improve the survival rate of these cells.
Effect of S100A4 protein on LDH, NO, MDA and SOD in cultured endothelial cells induced by oxidative stress
The results indicated that the LDH activity and MDA content of HI group were significantly increased compared with those of the NC group (P<0.01), while NO content and SOD activity were significantly decreased compared with those of the NC group (P<0.01), suggesting that H2O2 oxidatively damaged these cells. However, the LDH values of the 50 and 100 µM HI+S100A4 groups were significantly decreased compared with that of the HI group, but remained significantly increased compared with that of the NC group (P<0.05). The LDH value of the HI+S100A4 (100 µM) group was decreased compared with that of the HI+S100A4 (50 µM) group (P<0.05; Fig. 2A). The NO content of 50 and 100 µM HI+S100A4 groups was significantly increased compared with that of the HI group, but remained significantly decreased compared with that of the NC group (P<0.05; Fig. 2B). The content of NO in the HI+S100A4 (100 µM) group was increased compared with that in the HI+S100A4 (50 µM) group (P<0.05; Fig. 2B). The MDA content results demonstrated that different doses of S100A4 protein may significantly reverse the increase in MDA levels induced by H2O2 injury (P<0.05; Fig. 2C). The SOD activities in the 50 and 100 µM HI+S100A4 groups were significantly increased, compared with the HI group (P<0.05; Fig. 2D). The experimental results indicated that S100A4 protein prevented oxidative damage.
Figure 2.

Effect of S100A4 protein on (A) LDH, (B) NO, (C) MDA and (D) SOD in cultured endothelial cells induced by oxidative stress. *P<0.05 vs. the H2O2 group. LDH, lactate dehydrogenase; NO, nitrous oxide; MDA, malondialdehyde; SOD, superoxide dismutase; S100A4, S100 calcium-binding protein A4; NC, normal control; HI, hypoxic injury induced by H2O2.
Effect of S100A4 protein on the rate of endothelial cell apoptosis induced by oxidative stress
The results demonstrated that the endothelial cell apoptosis rate of the oxidative stress injury group was significantly increased compared with that of the control group (P<0.01), suggesting that H2O2 may induce endothelial cell injury and cell apoptosis. The endothelial cell apoptosis rate in the HI+S100A4 (50 µM) and HI+S100A4 (100 µM) group was significantly decreased compared with that in the HI group (P<0.05; Fig. 3A and B).
Figure 3.

S100A4 causes a suppression of apoptosis induced by H2O2. (A) Images and (B) quantification of TUNEL apoptosis analysis. A total of ~4×105 rat vascular endothelial cells were cultured on the cover slips in 24-well plates and treated with 100 µM H2O2, with or without S100A4 concurrently. Following incubation, cells were washed with cold PBS (pH 7.4) and stained according to the protocol of the TUNEL assay kit. H2O2 induced endothelial cell injury and cell apoptosis. The endothelial cell apoptosis rate in the HI+S100A4 (50 µM) and HI+S100A4 (100 µM) group was significantly decreased compared with that in the HI group. (C) Western blotting and (D) quantification of protein expression of caspase 3 in rat vascular endothelial cells. The expression of cleaved caspase-3 in ECs increased significantly after H2O2 administration. S100A4 decreased caspase 3 expression in endothelial cells treated with 50 µM or 100 µM H2O2 for 12 h. *P<0.05 vs. H2O2 group. S100A4, S100 calcium-binding protein A4; NC, normal control; HI, hypoxic injury induced by H2O2.
Since mitochondrial function is associated with caspase activity (16), the function of caspase 3 was assessed. The expression of cleaved (active) caspase 3 expression was demonstrated using western blot analysis. In Fig. 3, western blot analysis of cleaved caspase 3 (Fig. 3C and D) was performed following H2O2 administration in ECs and revealed significant caspase 3 activity compared with NC (P<0.05). However, S100A4 significantly inhibited cleaved active caspase 3 activity. The results indicated that S100A4 protein exhibited prevented oxidative damage, inhibiting apoptosis.
Determination of total protein, cytoplasmic protein and nuclear protein of endothelial cell protein by western blot analysis
In Fig. 4, following S100A4 intervention, the level of intracellular S100A4 protein was significantly increased in the S100A4 groups (P<0.05). Although there was no significant change in the expression of P53 in the cell, the distribution of P53 protein in the cell was altered; the effect of S100A4 protein on P53 was dose-dependent. As the dose of S100A4 increased, the aggregation of P53 in the cytoplasm increased and the corresponding distribution in the nucleus decreased. This suggests that S100A4 protein may inhibit P53 movement from the cytoplasm into the nucleus. Therefore, we hypothesize that the function of S100A4 protein in endothelial cell apoptosis is inhibited in the oxidative stress damage model of endothelial cells and that this effect is mediated by P53.
Figure 4.
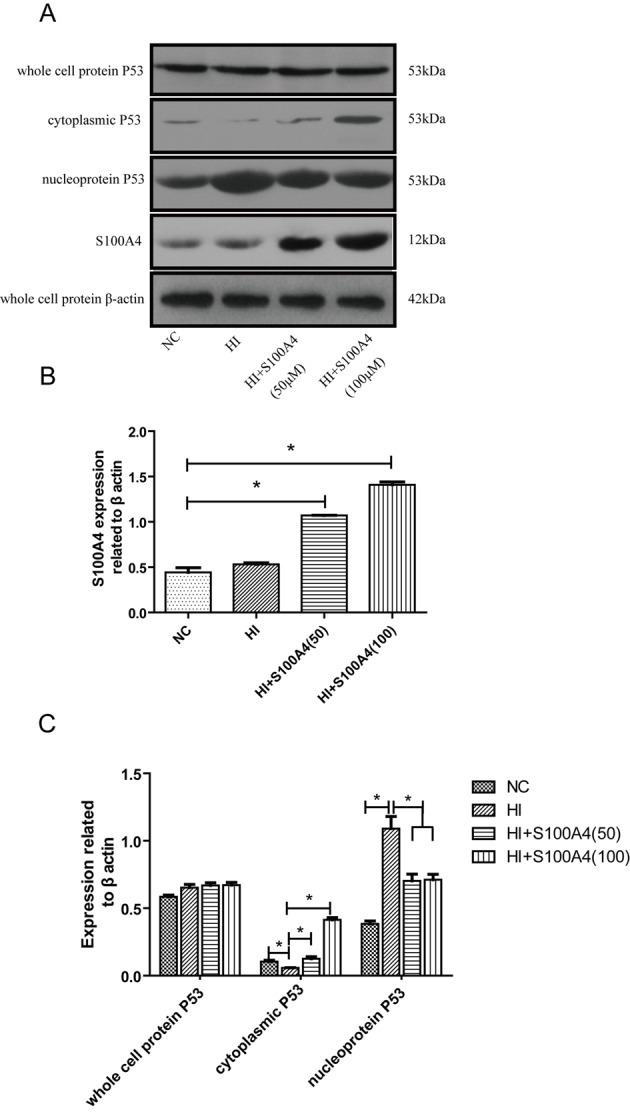
S100A4 functions in the cytoplasmic compartment. Western blot analysis of indicated proteins in nuclear, cytoplasmic and whole-cell extracts of endothelial cells treated with H2O2 with or without S100A4 for 12 h. (A) Western blotting and (B) quantification of the expression of cytoplasmic S100A4 protein could not be effected by H2O2 treatment, while it was significantly increased after exogenous S100A4 addition. (C) After H2O2 treatment, the aggregation of P53 in the cytoplasm increased and the corresponding distribution in the nucleus increased comparison with the NC group. However, exogenous S100A4 reversed the changes mentioned above. *P<0.05 vs. H2O2 group. NC, normal control; HI, hypoxic injury induced by H2O2; S100A4, S100 calcium-binding protein A4.
S100A4 interacts with P53 in the nucleus
S100 family proteins have no known enzymatic activity; therefore, it may be hypothesized that S100 proteins function through interaction with other proteins to regulate the function of interacting proteins. Previous studies have focused on non-muscle myosin IIA and P53 as potential S100A4-interacting proteins. Therefore, the present study investigated the potential interaction between S100A4 and P53. IP of endogenous S100A4 in endothelial cells resulted in the co-precipitation of endogenous P53 in untreated cells (Fig. 5). Following H2O2 treatment, the total cell P53 did not change significantly. However, P53 in the nucleus was significantly increased. P53 binding to S100A4 was also significantly increased in the nucleus (P<0.05). Therefore, we hypothesized that S100A4 may combine with P53 in the nucleus to increase the degradation of P53, thereby inhibiting the apoptosis of cells.
Figure 5.
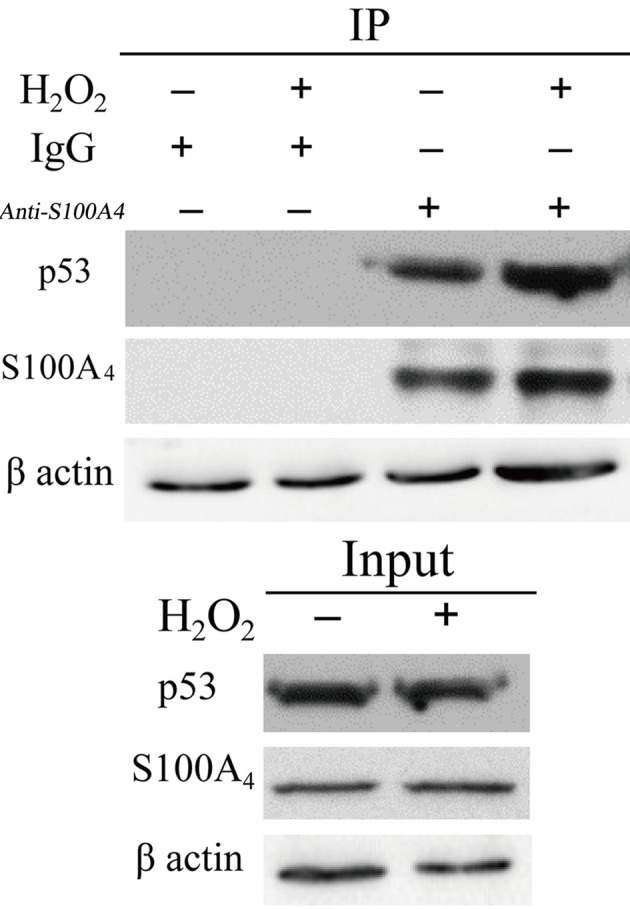
S100A4 interacts with P53, as indicated by IP. The whole-cell extracts of ECs were treated with H2O2. Immunoblot analysis of P53 and S100A4 protein in S100A4 IP experiments in untreated and H2O2-treated ECs. IP, immunoprecipitation; ECs, endothelial cells; S100A4, S100 calcium-binding protein A4; Ig, immunoglobulin.
Discussion
The vascular endothelium is composed of a monolayer of vascular endothelial cells, which is a barrier between circulating blood and vascular smooth muscle. The vascular endothelium participates in the contraction and relaxation of blood vessels, the formation of blood vessels, thrombosis and the inflammatory response (17,18). Oxidative stress is the key factor that causes changes in endothelial function and cell damage (19,20). Oxygen free radicals, collectively known as reactive oxygen species (ROS), are one of the primary factors that cause oxidative damage of endothelial cells (18,19). Free radical reactions cause cell membrane lipid peroxidation, and then alterations to the cell transport and enzyme functions (19,20). Oxidative stress is associated with the pathological process of the endothelial cells, including increasing vascular endothelial cell permeability, affecting cell proliferation, increasing leukocyte infiltration and interfering with the signal transduction in the cell, which causes cell death and apoptosis (21). It serves a key function in the occurrence and development of vascular diseases (22). Therefore, it is important to prevent the occurrence of cardiovascular disease through the protection of vascular endothelial cells from oxidative damage.
The S100 protein family is one of the largest families of calcium-binding proteins. Including the S100A4 protein, at present, there are 21 members of the S100 family (23). Previous studies have demonstrated abnormal expression of S100 in numerous diseases, including psoriasis, Alzheimer's disease, cystic fibrosis, cardiomyopathy, muscle atrophy (spinal cord) lateral sclerosis and cancer (24,25). The S100A4 protein belongs to the S100 protein family and is composed of 101 amino acids with a molecular weight of 11.5 kD (23). Studies have confirmed that the S100A4 protein may promote the invasion and metastasis of tumors and regulate the function of the cell, including cell growth and cell signal transduction (26,27). Multiple studies demonstrate that the S100A4 protein is associated with vascular regeneration and extracellular matrix remodeling and may even be used as an independent vascular-stimulating factor (13,28). Schmidt-Hansen et al (29) identified that intracellular S100A4 stimulates endothelial cells to produce matrix metalloproteinases, which promotes the remodeling of the extracellular matrix and the degradation of matrix remodeling is a necessary step in angiogenesis. An additional study indicated that the release of S100A4 from the cornea of the implanted rat cornea may induce novel blood vessel formation (30). Furthermore, the interaction of S100A4 protein and membrane protein may promote the activation of plasminogen activator, which is induced by the activation of plasminogen activator (31).
To assess the function of S100A4 protein on endothelial cell damage induced by oxidative stress and the potential mechanism, the present study selected 100 µM H2O2-induced injury for 12 h to establish a model of endothelial cell oxidative stress injury (32). The results indicated that the oxidative stress caused the loss of the typical morphology of the endothelial cells. The cells exhibited shrinkage and roundness, the gap increased and vacuoles in the cytoplasm of cells were not arranged in order. The activity of LDH was increased, the content of NO was decreased and the content of MDA was increased, while the activity of SOD was decreased. The survival rate of endothelial cells was decreased and the apoptosis rate was increased. The experiment suggested that oxidative stress resulted in the injury of endothelial cells. S100A4 prevented oxidative stress to protect endothelial cells.
The important mechanism of oxygen free radical-induced cell and tissue damage is to induce peroxidation of the polyunsaturated fatty acids in the biological membrane, consequently producing lipid peroxide. Lipid peroxidation may be associated with the occurrence of a chain reaction of polyunsaturated fatty acids (28). As one of the unsaturated fatty acids, linoleic acid can induce both NF-κB and AP-1 transcriptional activation (33). Lipid free radicals and their degradation products (MDA) are formed, causing membrane fluidity, permeability and integrity damage and then the cell membrane is destroyed (34,35). ROS mainly includes oxygen ions and peroxides. As a superoxide scavenging enzyme, active SOD is able to neutralize O2− and presents antioxidative effect (36). With the aid of SOD, superoxide radical anion or hyperoxide in biological tissues can be changed into hydrogen peroxide (HO) and singlet oxygen (1O2). HO may be changed into water by catalase or glutathione peroxidase (36). Therefore, the content of MDA may reflect the degree of lipid peroxidation and indirectly reflect the degree of cell damage. SOD activity indirectly reflects the ability of the body to clear oxygen free radicals. Oxidative stress may increase the secretion of endothelin-1 and diminish the bioavailability of nitric oxide (NO) (3,37). These vasoactive molecules promote the contraction of blood vessels and then initiate a series of post-injury reactions, resulting in the occurrence of cardiovascular disease (38,39).
Oxidative stress may lead to endothelial dysfunction (40,41), resulting in decreased NO levels and increased LDH release (42). The results indicated that H2O2 may increase the content of MDA, the activity of LDH was increased and that the activity of SOD was decreased, suggesting that the endothelial cells were damaged by lipid peroxidation. S100A4 protein intervention may be effective against oxygen free radical damage.
Therefore, we hypothesized that the protective mechanism of S100A4 on the endothelial cells may be associated with the protection of cell mitochondria, increased cell activity.
Endothelial cell activity is key to the maintenance of endothelial function. If the apoptosis rate of endothelial cells exceeds the normal level, it will affect the integrity of the blood vessels and induce the injury of endothelial cells. The P53 gene is an important anti-cancer gene. A previous study demonstrated that wild type P53 may induce the apoptosis of leukocytes (43). Three members of the S100 protein family, namely S100B, S100A2 and S100A4, physiologically interact with P53 in a calcium dependent manner. Phosphorylation and acetylation of P53 may be affected by the interaction of S100 protein, which results in the regulation of the subcellular localization and transcriptional activity of P53 (44). Previously, studies have focused on the role of the P53 gene in cardiovascular diseases and have indicated that P53 serves an important function in the oxidative stress of blood vessels as an important type of transcription protein (45,46). The results of the present study suggested that oxidative stress causes an increased rate of endothelial cell apoptosis and P53 may be the target protein of S100A4. Subsequent to the cells being administered S100A4 protein, the total level of P53 protein was not increased but the distribution of P53 protein in the cells was changed, primarily with an increase in the aggregation of P53 in the cytoplasm and a decrease in the distribution of P53 in the nucleus. As the dose of S100A4 increased, the aggregation of P53 in the cytoplasm increased and the corresponding distribution in the nucleus decreased. This suggested that S100A4 protein may inhibit P53 movement from the cytoplasm into the nucleus.
To conclude, exogenous S100A4 protein may reverse oxidative stress damage, improve the survival rate of cells and inhibit the apoptosis of endothelial cells. The protective mechanism of S100A4 protein on the apoptosis of endothelial cells may be mediated by inhibiting P53. This provides a reference for the additional exploration of the application of S100A4 protein in endothelial cell damage induced by oxidative stress.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant nos. 81373108 and 30971421).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
TW and BS developed the project. XM, XG and ZZ performed experiments and wrote the manuscript. XZ, LW and MY analyzed the data. KW and HR helped with computational analysis. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the committee of the Institute of Health and Environmental Medicine, Tianjin, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Shanthi M, Tim A, Douglas B, Francesco B, Jeremy L, Cecile M, Vladimir P, Leanne R, Vera Da Costa E Silva, Gretchen S. Global status report on concommunicable diseases 2014[R] Geneva: World Health Organization 9; 2014. [Google Scholar]
- 2.World Health Organization, corp-author. Fact Sheet no. 317. Geneva: WHO; 2011. [Google Scholar]
- 3.Crimi E, Ignarro LJ, Napoli C. Microcirculation and oxidative stress. Free Radi Res. 2007;41:1364–1375. doi: 10.1080/10715760701732830. [DOI] [PubMed] [Google Scholar]
- 4.Loscalzo J. Redox dysregulation in vascular pathobiology. Free Radic Biol Med. 2014;75(Suppl 1):S2. doi: 10.1016/j.freeradbiomed.2014.10.597. [DOI] [PubMed] [Google Scholar]
- 5.Marchenko ND, Moll UM. Mitochondrial death functions of p53. Mol Cell Oncol. 2014;1:e955995. doi: 10.1080/23723548.2014.955995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarabykina S, Griffiths TR, Tulchinsky E, Mellon JK, Bronstein IB, Kriajevska M. Metastasis-associated protein S100A4: Spotlight on its role in cell migration. Curr Cancer Drug Targets. 2007;7:217–228. doi: 10.2174/156800907780618329. [DOI] [PubMed] [Google Scholar]
- 7.Moroz OV, Antson AA, Murshudov GN, Maitland NJ, Dodson GG, Wilson KS, Skibshøj I, Lukanidin EM, Bronstein IB. The three-dimensional structure of human S100A12. Acta Crystallogr D Biol Crystallogr. 2001;57:20–29. doi: 10.1107/S090744490001458X. [DOI] [PubMed] [Google Scholar]
- 8.Ridinger K, Schäfer BW, Durussel I, Cox JA, Heizmann CW. S100A13 biochemical characterization and subcellular localization in different cell lines. J Biol Chem. 2000;275:8686–8694. doi: 10.1074/jbc.275.12.8686. [DOI] [PubMed] [Google Scholar]
- 9.Mazzuccheli L. Protein S100A4: Too long overlooked by pathologists? Am J Pathol. 2002;160:7–13. doi: 10.1016/S0002-9440(10)64342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donato R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracelluar functions roles. Int J Biochem Cell Biol. 2001;33:637–668. doi: 10.1016/S1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 11.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a mediator of metastasis. J Biol Chem. 2006;281:677–680. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 12.Ambartsumian N, Grigorian M, Lukanidin E. Genetically modified mouse models to study the role of metasasis-promoting S100A4(mts1) protein in metastatic mammary cancer. J Dairy Res. 2005;72:27–33. doi: 10.1017/S0022029905001093. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt-Hansen B, Klingelhöfer J, Grum-Schwensen B, Christensen A, Andresen S, Kruse C, Hansen T, Ambartsumian N, Lukanidin E, Grigorian M. Functional significance of metastasis-including S100A4(Mts1) in tumor-stroma interplay. J Biol Chem. 2004;279:24498–24504. doi: 10.1074/jbc.M400441200. [DOI] [PubMed] [Google Scholar]
- 14.Orre LM, Panizza E, Kaminskyy VO, Vernet E, Gräslund T, Zhivotovsky B, Lehtiö J. S100A4 interacts with p53 in the nucleus and promotes p53 degradation. Oncogene. 2013;32:5531–5540. doi: 10.1038/onc.2013.213. [DOI] [PubMed] [Google Scholar]
- 15.Wang XH, Chen SF, Jin HM, Hu RM. Differential analyses of angiogenesis and expression of growth factors in micro- and macrovascular endothelial cells of type 2 diabetic rats. Life Sci. 2009;84:240–249. doi: 10.1016/j.lfs.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 16.Hao J, Shen W, Tian C, Liu Z, Ren J, Luo C, Long J, Sharman E, Liu J. Mitochondrial nutrients improve immune dysfunction in the type 2 diabetic Goto-Kakizaki rats. J Cell Mol Med. 2009;13:701–11. doi: 10.1111/j.1582-4934.2008.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chesterman CN. Vascular endothelium, haemostasis and thrombosis. Blood Rev. 1988;2:88–94. doi: 10.1016/0268-960X(88)90029-X. [DOI] [PubMed] [Google Scholar]
- 18.Tang X, Yang X, Peng Y, Lin J. Protective effects of lycopene against H2O2-induced oxidative injury and apoptosis in human endothelial cells. Cardiovasc Drugs Ther. 2009;23:439–448. doi: 10.1007/s10557-009-6206-3. [DOI] [PubMed] [Google Scholar]
- 19.Kadl A, Leitinger N. The role of endothelial cells in the resolution of acute inflammation. Antioxid Redox Signal. 2005;7:1744–1754. doi: 10.1089/ars.2005.7.1744. [DOI] [PubMed] [Google Scholar]
- 20.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 21.Ramagopal UA, Dulyaninova NG, Varney KM, Wilder PT, Nallamsetty S, Brenowitz M, Weber DJ, Almo SC, Bresnick AR. Structure of the S100A4/myosin-IIA complex. BMC Struct Biol. 2013;20:13–31. doi: 10.1186/1472-6807-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toya SP, Malik AB. Role of endothelial injury in disease mechanisms and contribution of progenitor cells in mediating endothelial repair. Immunobiolog. 2012;217:569–580. doi: 10.1016/j.imbio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Boye K, Maelandsmo GM. S100A4 and metastasis: A small actor playing many roles. Am J Pathol. 2010;176:528–535. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 25.Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. 2006;396:201–214. doi: 10.1042/BJ20060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider M, Kostin S, Strøm CC, Aplin M, Lyngbaek S, Theilade J, Grigorian M, Andersen CB, Lukanidin E, Hansen Lerche J, Sheikh SP. S100A4 is regulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc Res. 2007;75:40–50. doi: 10.1016/j.cardiores.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 27.Chaabane C, Otsuka F, Virmani R, Bochaton-Piallat ML. Biological responses in stented arteries. Cardiovasc Res. 2013;99:353–563. doi: 10.1093/cvr/cvt115. [DOI] [PubMed] [Google Scholar]
- 28.Nowak JZ. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: Focus on age-related macular degeneration. Pharmacol Rep. 2013;65:288–304. doi: 10.1016/S1734-1140(13)71005-3. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt-Hansen B, Ornås D, Grigorian M, Klingelhöfer J, Tulchinsky E, Lukanidin E, Ambartsumian N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene. 2004;23:5487–5495. doi: 10.1038/sj.onc.1207720. [DOI] [PubMed] [Google Scholar]
- 30.Ambartsumian N, Klingelhöfer J, Grigorian M, Christensen C, Kriajevska M, Tulchinsky E, Georgiev G, Berezin V, Bock E, Rygaard J, et al. The metastasis associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene. 2001;20:4685–4695. doi: 10.1038/sj.onc.1204636. [DOI] [PubMed] [Google Scholar]
- 31.Semov A, Moreno MJ, Onichtchenko A, Abulrob A, Ball M, Ekiel I, Pietrzynski G, Stanimirovic D, Alakhov V. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J Biol Chem. 2005;280:20833–20841. doi: 10.1074/jbc.M412653200. [DOI] [PubMed] [Google Scholar]
- 32.Sakamoto T, Repasky WT, Uchida K, Hirata A, Hirata F. Modulation of cell death pathways to apoptosis and necrosis of H2O2-treated rat thymocytes by lipocortin I. Biochem Biophys Res Comunun. 1996;220:643–647. doi: 10.1006/bbrc.1996.0457. [DOI] [PubMed] [Google Scholar]
- 33.Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. 2002;75:119–125. doi: 10.1093/ajcn/75.1.119. [DOI] [PubMed] [Google Scholar]
- 34.Mazière C, Conte MA, Degonville J, Ali D, Mazière JC. Cellular enrichment with polyunsaturated fatty acids induces an oxidative stress and activates the transcription factors AP1 and NFkappaB. Biochem Biophys Res Commun. 1999;265:116–122. doi: 10.1006/bbrc.1999.1644. [DOI] [PubMed] [Google Scholar]
- 35.Alexander-North LS, North JA, Kiminyo KP, Buettner GR, Spector AA. Polyunsaturated fatty acids increase lipid radical formation induced by oxidant stress in endothelial cells. J Lipid Res. 1994;35:1773–1785. [PubMed] [Google Scholar]
- 36.Nowak JZ. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: Focus on age-related macular degeneration. Pharmacol Rep. 2013;65:288–304. doi: 10.1016/S1734-1140(13)71005-3. [DOI] [PubMed] [Google Scholar]
- 37.Takeda N, Manabe I. Cellular interplay between cardiomyocytes and nonmyocytes in cardiac remodeling. Int J Inflam. 2011;2011:535241. doi: 10.4061/2011/535241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raij L. Hypertension, endothelium, and cardiovascular risk factors. Am J Med. 1991;90(Supple 2A):13S–18S. doi: 10.1016/0002-9343(91)90029-W. [DOI] [PubMed] [Google Scholar]
- 39.Smith EJ, Rothman MT. Antiproliferative coatings for the treatment of coronary heart disease: What are the targets and which are the tools? J Interv Cardiol. 2003;16:475–483. doi: 10.1046/j.1540-8183.2003.01058.x. [DOI] [PubMed] [Google Scholar]
- 40.Onat D, Brillon D, Colombo PC, Schmidt AM. Human vascular endothelial cells: A model system for studying vascular inflammation in diabetes and atherosclerosis. Curr Diab Rep. 2011;11:193–202. doi: 10.1007/s11892-011-0182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parnell E, Smith BO, Palmer TM, Terrin A, Zaccolo M, Yarwood SJ. Regulation of the inflammatory response of vascular endothelial cells by EPAC1. Br J Pharmacol. 2012;166:434–446. doi: 10.1111/j.1476-5381.2011.01808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma S, Yao S, Tian H, Jiao P, Yang N, Zhu P, Qin S. Pigment epithelium-derived factor alleviates endothelial injury by inhibiting Wnt/β-catenin pathway. Lipids Health Dis. 2017;16:31. doi: 10.1186/s12944-017-0407-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yonish-Rouach E, Grunwald D, Wilder S, Kimchi A, May E, Lawrence JJ, May P, Oren M. p53-mediated cell death: Relationship to cell cycle control. Mol Cell Biol. 1993;13:1415–1423. doi: 10.1128/MCB.13.3.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mueller A, Schäfer BW, Ferrai S, Weibel M, Makek M, Höchli M, Heizmann CW. The calcium-binding protein S100A2 interact with p53 and modulates its transcriptional activity. J Biol Chem. 2005;280:29186–29193. doi: 10.1074/jbc.M505000200. [DOI] [PubMed] [Google Scholar]
- 45.Ma JQ, Ding J, Zhang L, Liu CM. Ursolic acid protects mouse liver against CCl4-induced oxidative stress and inflammation by the MAPK/NF-κB pathway. Environ Toxicol Pharmacol. 2014;37:975–983. doi: 10.1016/j.etap.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 46.Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122:2601–2612. doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.