

Abstract
β-elemene (β-ELE) is a natural compound extracted from Curcuma zedoaria Roscoe that has shown promise as a novel anticancer drug to treat malignant tumors. Recent studies have demonstrated that β-ELE can reverse the drug resistance of tumor cells. To the best of our knowledge, there are no reports concerning the reversal of erlotinib resistance by β-ELE in human non-small cell lung cancer (NSCLC) cells. Therefore, the present study investigated the effects of β-ELE on erlotinib-resistant human NSCLC A549/ER cells in vitro and its possible mechanism of action. The sensitivity of A549/ER cells to erlotinib, the cytotoxicity of β-ELE on the growth of A549/ER cells and the effects of β-ELE on the reversal of drug resistance in A549/ER cells were determined by MTT assay. The cell apoptosis rate, cell cycle phase distribution and intracellular rhodamine 123 (Rh123) fluorescence intensity were detected by flow cytometry. The expression level of P-glycoprotein (P-gp) was detected by western blotting. A549/ER cells had a stable drug-resistance to erlotinib. β-ELE inhibited the proliferation of A549/ER cells in a time- and dose-dependent manner, enhanced the sensitivity of A549/ER cells to erlotinib and reversed the drug resistance in A549/ER cells. Treatment with 15 µg/ml β-ELE combined with 10 µmol/l erlotinib caused an increased rate of cell apoptosis and G0/G1 phase arrest. Furthermore, β-ELE reduced the efflux of Rh123 from A549/ER cells, increased the intracellular accumulation of Rh123 and decreased the expression of P-gp. The results of the present study indicated that β-ELE could reverse drug resistance in erlotinib-resistant human NSCLC A549/ER cells in vitro through a mechanism that may involve the decreased expression of P-gp, inhibition of P-gp dependent drug efflux and the increased intracellular concentration of anticancer drugs.
Keywords: elemene, non-small cell lung cancer, erlotinib, drug resistance, mechanism
Introduction
Lung cancer is one of the most common malignant tumors in the world; it has a low 5-year survival rate of between 16–18% (1), and ranks first globally for morbidity and mortality (2). Approximately 80% of incidences of lung cancer are of non-small cell lung cancer (NSCLC), the majority of which are middle or late stage (IIIB/IV stage) when they are diagnosed, with corresponding poor patient outcomes (3,4). The objective response rate of platinum-based chemotherapy is only 30–40%, and median progression-free survival is ~10 months (5). This treatment modality also induces severe adverse reactions, which restricts clinical use (6). Molecular targeted drugs such as erlotinib appear to demonstrate good clinical efficacy in the treatment of advanced NSCLC (7). Molecular targeted therapy has become a promising treatment strategy for Asian patients with NSCLC. Unfortunately, following 8–12 months of effective treatment, patients treated with EGFR-TKIs will inevitably develop secondary resistance to EGFR-TKIs, resulting in tumor recurrence or metastasis (8). Therefore, resistance to erlotinib results in the failure of therapy. For these aforementioned reasons, the study of EGFR-TKI resistance in lung cancer is an important direction of targeted therapy. β-elemene (β-ELE) is a novel anticancer drug extracted from Curcuma zedoaria Roscoe that has been widely used to treat malignant tumors (9). Recent studies have reported that β-ELE can reverse the drug resistance of tumor cells (9,10). To the best of our knowledge, at the time of writing there are no reports concerning the reversal of erlotinib resistance by β-ELE in human NSCLC cells. Therefore, the present study investigated the effects and possible mechanism of action of β-ELE on the erlotinib-resistant human NSCLC A549/erlotinib resistant (ER) cell line in vitro. The results of the present study describe a mechanism of β-ELE function, involving the decreased expression of P-glycoprotein (P-gp), inhibition of P-gp-dependent drug efflux and the increased intracellular concentration of anticancer drugs, leading to the reversal of drug resistance in A549/ER cells.
Materials and methods
Reagents and equipment
The human lung adenocarcinoma cell line A549 was purchased from Shanghai Fuxiang Biotechnological Co., Ltd, (Shanghai, China) and its acquired erlotinib-resistant cell line (A549/ER) was obtained from the Second Affiliated Hospital Tumor Research Institute of Third Military Medical University (Chongqing, China). The following reagents were used in the present study: β-elemene (Dalian Huali Jingang Pharmaceutical Co., Ltd. Dalian, China); erlotinib (Roche Applied Science, Penzburg, China); Roswell Park Memorial Institute (RPMI)-1640 culture medium and fetal bovine serum (FBS) (both Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA); penicillin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany); Streptomycin (Sigma-Aldrich; Merck KGaA); radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology Co., Ltd., Haimen, China); MTT cell proliferation assay kits (Ameresco, Inc., Framingham, MA, USA); dimethyl sulfoxide (DMSO; Shanghai Biological Engineering Co., Ltd., Shanghai, China); Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double dye kits (EMD Millipore, Billerica, MA, USA); enhanced chemiluminescence (ECL) reagent kits, PI and Rhodamine 123 (Rh123) (Sigma-Aldrich; Merck KGaA); mouse anti-human monoclonal antibodies against P-gp (Abcam, Cambridge, UK; cat. no., ab10333), β-actin (Abcam; cat. no., ab11003) and horseradish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin (IgG) (Abcam; cat. no., ab6789). Inverted microscope (Olympus); Western blot transfer system, Western blot electrophoresis apparatus, Microplate Reader Model 680 and Gel imaging system (Bio-Rad Laboratories, Inc., Hercules, CA, USA); and flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Cell culture
Human NSCLC A549 and A549/ER cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/l streptomycin in an atmosphere of 5% CO2 at 37°C. Cells in the exponential growth phase were used in the following experiments.
MTT drug sensitivity assay
The sensitivity of A549 and A549/ER cells to erlotinib was determined by MTT colorimetric assay. Exponentially growing cells were plated in triplicate in 96-well plates at a density of 5×103 cells per well and treated with RPMI-1640 culture medium containing different concentrations of erlotinib (0.01, 0.1, 1, 10 and 100 µmol/l) at 37°C for 24 or 48 h. Following this, 20 µl MTT dye (5 mg/ml) was added for 4 h and then 100 µl DMSO per well was added with low speed oscillation for 10 min. Spectrometric absorbance at 570 nm wavelength was conducted using a microplate reader to measure absorbance value of each well. The experiment was repeated three times and selected averaged data to generate a growth curve. The cell proliferation inhibitory rate (%)=(1-value of the experimental group/value of the control group) ×100. The half-maximal inhibitory concentration (IC50) was calculated by linear regression. The fold of drug resistance=IC50 of resistant cells/IC50 of sensitive cells (9).
MTT cytotoxicity assay
Exponentially growing cells were plated in triplicate in 96-well plates at a density of 5×103 cells per well. A final concentration of 5, 10, 20, 40 or 80 µg/ml β-ELE was added to the experimental groups (RPMI-1640 culture medium was used as vehicle control) was added to the control group at 37°C for 24 or 48 h. Subsequent MTT colorimetric assay was used to detect β-ELE cytotoxicity as aforementioned. Using the linear regression equation to calculate the IC50 and IC10 of β-ELE incubation 24 or 48 h in A549/ER cells. In general, non-cytotoxic doses are used as the reverse dose. It was identified that there was no marked cytotoxic effect on the sensitive strain and the resistant strain cells when the cell proliferation inhibitory rate was <10% (IC10) (11). Therefore, the IC10 was set as the non-toxic upper limit dose. Finally, a dose, which was lower than IC10, was determined as the optimal reversal concentration of β-ELE for subsequent experiments.
MTT drug resistance reversal assay
Exponentially growing cells were plated in triplicate in 96-well plates at a density of 5×103 cells per well. A varying final concentration of 0.01, 0.1, 1, 10 and 100 µmol/l erlotinib and 15 µg/ml β-ELE which was determined by the above method were added to the experimental group at 37°C for 24 h. The same concentration gradient of erlotinib was added to the positive control group, whereas PBS was added to the negative control group. Subsequently, an MTT assay was performed. According to IC50 to calculate reversal of drug resistance: Fold reversal, FR=IC50 of resistant cells prior to reversal/IC50 of the resistant cells following reversal.
Apoptosis assay by annexin V-FITC/PI staining
Exponentially growing A549/ER cells were plated in triplicate in 96-well plates at a density of 5×103 cells per well. The experiment groups were as follows: i) β-ELE single-drug group, in which the final using concentration of β-ELE was 15 µg/ml; ii) the erlotinib single-drug group, in which the final concentration of erlotinib was 10 µmol/l; iii) the combined β-ELE and erlotinib drug group, in which the concentrations used was the same as single drug group; iv) the negative control group containing only RPMI-1640 medium. After 24 h, the cells were collected by centrifugation at 20–25°C and 1,000 × g for 5 min, and 100 µl Binding Buffer (included in the Annexin V-FITC/PI staining apoptosis detection kit, EMD Millipore) suspension cell was added. Next, 10 µl (20 µg/ml) Annexin V-FITC and 20 µl (50 µg/ml) PI were added to cells for 5 to 15 min. Following this, 400 µl Binding Buffer was added to the cells, which were assessed by flow cytometry within 1 h. The excitation wavelength was set as 488 and 525 nm was set as the emission wavelength. The experiment was repeated three times using CellQuest Pro software (version 5.1 BD CellQuest Pro Software, BD Biosciences) to collect and analyze data.
PI staining and flow cytometric analysis of the effect of β-ELE on the cell cycle of A549/ER cells
Exponentially growing erlotinib resistant A549/ER cells were seeded in triplicate in 96-well plates at a density of 5×103 cells per well. The same aforementioned four experimental group treatments were used. After 24 h, the cells were collected by centrifugation at 20–25°C and 1,000 × g for 5 min. Subsequently, 5 ml precooled 70% ethanol was added to fix the samples overnight at 4°C. Next, the cells were incubated with 1 ml PI for 15 min at 20–25°C, and the cells were assessed by flow cytometry as aforementioned.
Rh123 retention assay
Exponentially growing A549/ER cells were plated in triplicate in 96-well plates at a density of 5×103 cells per well. The experiment groups were as follows: i) Non-toxic β-ELE dose group using the final concentration of 15 µg/ml β-ELE, which was determined as aforementioned; ii) the control group containing RPMI-1640 only. After culturing for 24 h, the cells were resuspended and 1×105/ml cell suspension was added into a BD Falcon tube at 37°C for 30 min and 5 µg/ml Rh123 was added. After incubation at 37°C for 30 min, cells were collected and centrifuged at 1,500 × g for 2 min at 20–25°C. The supernatant was aspirated and cell pellets were resuspended with the cold RPMI-1640, and assessed by flow cytometry at an excitation wavelength of 488 nm and emission wavelength of 530 nm. The mean fluorescence intensity (MFI) was calculated to detect changes in concentrations of Rh123 and thus evaluate the activity of P-glycoprotein (P-gp).
Western blot analysis
Exponentially growing A549/ER cells were plated in triplicate in 96-well plates at a density of 5×103 cells per well. The experiment was divided into two groups as aforementioned: Cells treated with non-toxic doses of β-ELE and the vehicle control group. Cells were lysed using RIPA lysis buffer following 24 h of treatment, and centrifuged at 13,000 × g and 4°C for 10 min. The protein concentration quantification was detected by the BCA method. A total of 50 µg protein per lane was separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Next, the membranes were blocked with 5% skim milk for 2 h, washed in tris-buffered saline plus 0.05% Tween-20 (TBST), and incubated with mouse anti-human monoclonal antibodies against P-gp (1:1,000 dilution) or β-actin (1:2,000 dilution) overnight at 4°C. Afterwards, HRP-labeled secondary antibodies (1:2,000 dilution) were added for 2 h at room temperature, followed visualization using an ECL kit method. The expression of P-gp protein was quantified using densitometry analysis [ChemiDoc™ XRS+ gel imaging system with Image Lab™ Software (version 2.0; Bio-Rad Laboratories, Inc.)].
Statistical analysis
Statistical analysis was performed using SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA). Data are presented as the mean ± standard deviation. Comparisons between multiple groups were performed using one-way analysis of variance, followed by a Dunnett's post-hoc test. Comparisons between the unpaired two groups were performed using Student's t-test or χ2 test. P<0.05 was considered to indicate a statistically significant difference.
Results
Drug sensitivity of A549 and A549/ER cells to erlotinib
To verify the differential drug sensitivity of A549 and A549/ER cells to erlotinib, each cell line was exposed to a gradient of erlotinib concentrations for 24 or 48 h and an MTT assay was performed. The results of this assay revealed that the various experimental concentrations of erlotinib inhibited the proliferation of A549 and A549/ER cells proliferation, regardless of the time of incubation (Fig. 1). The cell growth inhibition rate increased in a time- and dose-dependent manner (Fig. 1). Using linear regression, A549 cells incubated with erlotinib for 24 h gave an IC50 value 113.21 µmol/ml; that of A549/ER cells was 183.71 µmol/ml. The IC50 results were 50.25 and 83.81 µmol/ml for A549 and A549/ER cells incubated with erlotinib for 48 h, respectively. According to the formula: The fold of drug resistance=IC50 of resistant cells/IC50 of sensitive cells, the drug resistance of A549/ER cells were 1.62 for the 24 h erlotinib incubation and 1.67 for the 48 h erlotinib incubation, indicating that A549/ER cells were stably resistant to erlotinib.
Figure 1.

Growth inhibition of erlotinib on A549 and A549/ER cells. A549 and A549/ER cells were exposed to varying final concentrations (0.01, 0.1, 1, 10 and 100 µmol/l) of erlotinib for (A) 24 or (B) 48 h. ER, erlotinib resistant.
Effects of β-ELE on A549/ER cell toxicity
To verify the effects of β-ELE on A549/ER cells, MTT assays were performed using different β-ELE doses. According to the cell growth inhibition rate of variant β-ELE concentration, the linear regression equation for 24 h incubation was calculated as follows: x=3.064y-5.023, while the equation for 48 h incubation was: x=1.667y-1.349, where × represents the β-ELE concentration and y represents the cell proliferation inhibition rate. The results of these assays revealed that β-ELE inhibited A549/ER cell growth in a time- and dose-dependent manner (Table I and Fig. 2). When the concentration of β-ELE was <20 µg/ml, 24 or 48 h incubation of A549/ER cell inhibition rates had no significant difference (aP>0.05; Table I), whereas doses of 40 and 80 µg/ml β-ELE had a significant effect on the inhibition of A549/ER cell proliferation cP<0.01, 24 vs. 48 h; bP<0.05, 40 µg/ml group vs. 80 µg/ml group). By using the above linear regression equation, β-ELE inhibited the proliferation of A549/ER cells with an IC50=148.18 µmol/ml at 24 h, an IC50=82.00 µmol/ml at 48 h, an IC10=25.62 µmol/ml at 24 h and an IC10=15.32 µmol/ml at 48 h. Incubation of A549/ER cells with 20 µg/ml β-ELE for 24 and 48 h gave growth inhibition rates of 8.59±2.82 and 10.93±2.65% respectively. On the basis of this experimental data and according to the aforementioned principles (the dose <IC10), 15 µg/ml β-ELE was selected as the optimum concentration to reverse drug resistance for the following assays.
Table I.
Effect of β-ELE on A549/ER cell growth (n=3).
| Cell proliferation inhibition rate, % | ||
|---|---|---|
| β-ELE (µg/ml) | 24 h | 48 h |
| 5 | 2.10±0.99 | 2.80±1.49a |
| 10 | 5.75±1.49 | 7.28±1.69a |
| 20 | 8.59±2.82 | 10.93±2.65a |
| 40 | 14.96±2.27b | 32.32±2.66b,c |
| 80 | 27.38±2.98b | 43.69±2.17b,c |
The results demonstrate cell proliferation inhibition rate of A549/ER cells incubated with various concentrations of β-ELE for 24 and 48 h (
P>0.05
P<0.01, 24 h incubation vs. 48 h incubation at the same β-ELE concentration). In addition, the cell proliferation inhibition rate in 80 µg/ml concentration group was significantly higher compared with the 40 µg/ml group in the same incubation time group
(P<0.05, 40 µg/ml group vs. 80 µg/ml group for the same incubation time).
Figure 2.
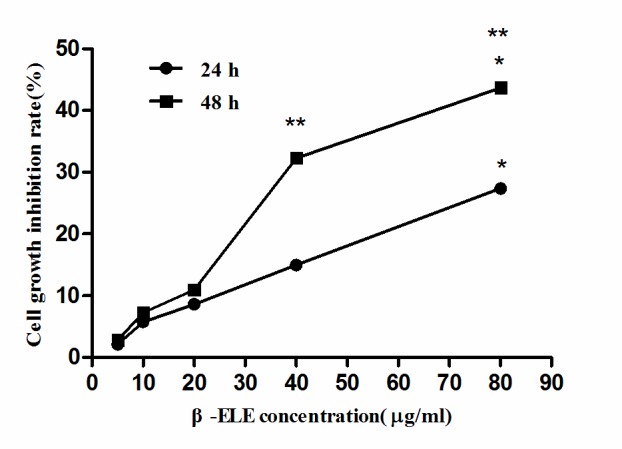
Growth inhibition effects of β-ELE on A549/ER cells. A549/ER cells were exposed to increasing concentrations (5, 10, 20, 40 and 80 µg/ml) of β-ELE for 24 or 48 h. β-ELE, β-elemene; ER, erlotinib resistant. **P<0.01 at 24 vs. 48 h at the same β-ELE concentration. *P<0.05 at 40 µg/ml vs. 80 µg/ml group at the same incubation time.
Effect of non-toxic doses β-ELE on drug-resistance of A549/ER cells
According to the cell growth inhibition rate of different concentrations erlotinib, the linear regression equation of the control group was: x=4.187y-25.64, whereas the equation for experimental group is: x=1.81y-33.778 (where × represents the erlotinib concentration and y represents the cell proliferation inhibition rate). The results of the MTT assay revealed that the rate of growth inhibition of β-ELE-treated A549/ER cells increased as erlotinib concentration increased in the control group and the experimental group (15 µg/ml β-ELE combined with erlotinib; Fig. 3). Furthermore, the IC50 value of the cells in the experimental group was significantly lower than the IC50 value of the control group, reduced from 183.71 in the control group to 56.72 µmol/l in the experimental group, a degree of drug resistance reversal of 3.24 (Table II). These results indicated that 15 µg/ml β-ELE combined with erlotinib increased the inhibition of A549/ER cell proliferation, which is likely to be associated with the β-ELE-dependent enhancement of the sensitivity of A549/ER cells to erlotinib, thereby partially reversing drug resistance in A549/ER cells.
Figure 3.
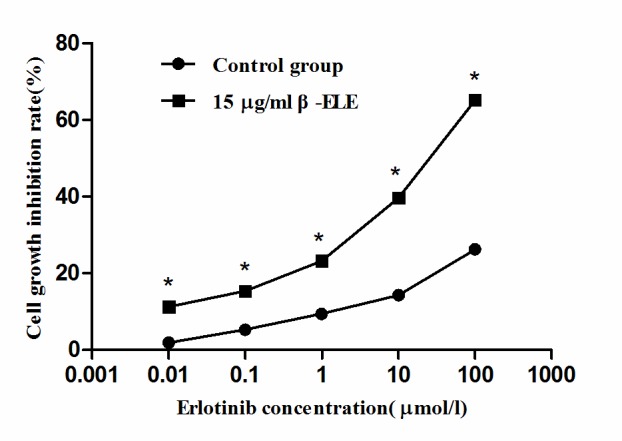
Effect of non-toxic doses β-ELE on enhancing the sensitivity of A549/ER cells to erlotinib. An MTT assay was performed to detect the A549/ER cell proliferation inhibition rate following exposure to either 15 µg/ml β-ELE combined with a range of concentrations of erlotinib or the control group consisting of a range concentrations of erlotinib alone. β-ELE, β-elemene; ER, erlotinib resistant. *P<0.01 in the experimental group vs. control group.
Table II.
Effect of non-toxic doses β-ELE on drug-resistance of A549/ER cells (n=3).
| Cell proliferation inhibition rate at 24 h, % | ||
|---|---|---|
| Erlotinib (µmol/l) | Control | 15 µg/ml β-ELE |
| 0.01 | 1.91±0.78 | 11.32±1.52a |
| 0.1 | 5.29±1.02 | 15.35±1.76a |
| 1 | 9.42±1.78 | 23.24±1.70a |
| 10 | 14.30±1.97 | 39.62±3.25a |
| 100 | 26.23±2.75 | 65.14±3.41a |
The cell proliferation inhibition rate in the experimental group was significantly higher compared with in the control group
(P<0.01 in the experimental group vs. control group).
Effect of non-toxic doses β-ELE on the apoptosis of A549/ER cells
Apoptosis was analyzed using flow cytometry following double-staining with annexin V-FITC and PI. As shown in Table III, 15 µg/ml β-ELE alone led to no significant induction of apoptosis in A549/ER cells (P>0.05 in the β-ELE single drug group vs. control group; Table III); a similar condition was also observed in the group treated with 10 µmol/l erlotinib (Fig. 4B). Although the difference of early apoptotic cells (Q3) between the erlotinib-only drug group vs. the control group was statistically significant (P<0.05), the overall rate of apoptosis (Q2+Q3) in the erlotinib-only drug group was only ~5%. β-ELE and erlotinib in combination evidently induced A549/ER apoptosis; the early apoptotic rate was 16.84±1.61% (combined drug group vs. the remaining three groups, P<0.01), and the middle-late apoptotic rate was 6.20±0.41% (combined drug group vs. the remaining three groups, P<0. 01; Fig. 4C). In conclusion, these results indicated that β-ELE combined with erlotinib may enhance apoptosis in A549/ER cells.
Table III.
Effect of non-toxic doses of β-ELE on inducing apoptosis of A549/ER cells (n=3).
| Cells in a quadrant, % | ||
|---|---|---|
| Group | Q2 | Q3 |
| β-ELE single drug group | 3.34±0.34c | 2.11±0.49c |
| Erlotinib single drug group | 4.04±0.37 | 4.22±0.28a |
| Combined drug group | 6.20±0.41b | 16.84±1.61b |
| Control group | 3.44±0.30 | 1.63±0.27 |
Q2 represents middle-late apoptotic and necrotic cells:
P<0.01, combined drug group vs. the remaining three groups
P>0.05, β-ELE single drug group vs. control group. Q3 represents early apoptotic cells:
P<0.05, Erlotinib single drug group vs. the control group; bP<0.01, Combined drug group vs. the remaining three groups; cP>0.05, β-ELE single drug group vs. control group.
Figure 4.

Annexin V-FITC/PI staining and flow cytometry analysis of the effect of β-ELE on apoptosis of A549/ER cells. (A) β-ELE single-drug group (15 µg/ml). (B) erlotinib single drug group (10 µmol/l). (C) β-ELE (15 µg/ml) combined with erlotinib (10 µmol/l) group; (D) control group. FITC, fluorescein isothiocyanate; PI, propidium iodide; β-ELE, β-elemene; ER, erlotinib resistant.
Effect of non-toxic doses β-ELE on A549/ER cell cycle
To determine whether β-ELE could influence the cell cycle distribution of A549/ER cells, flow cytometry was performed (Table IV). The number of A549/ER cells in S phase increased following treatment with 15 µg/ml β-ELE alone for 24 h. However, there was no statistically significant difference compared with the control group, which contained only RPMI-1640 medium (P>0.05). The combination of β-ELE and erlotinib induced significantly higher rates G0/G1 arrest than the other groups (P<0.01).
Table IV.
Effect of non-toxic doses β-ELE on A549/ER cell cycle (n=3).
| Cell cycle distributions, % | |||
|---|---|---|---|
| Group | G0/G1 | S | G2/M |
| 15 µg/ml β-ELE | 53.16±2.89 | 30.47±2.85a | 16.37±2.86 |
| 10 µmol/l erlotinib | 62.71±3.74b | 18.46±2.15 | 18.83±3.39 |
| 15 µg/ml β-ELE and 10 µmol/l erlotinib | 77.50±3.36c | 12.57±1.95 | 9.93±2.65 |
| Control group | 55.60±2.26 | 25.39±1.83 | 19.01±2.10 |
P>0.05 compared with S phase of the control group
P<0.05 compared with G0/G1 phase of control group
P<0.01, compared with G0/G1 phase of the rest groups. β-ELE, β-elemene; ER, erlotinib resistant.
Effect of non-toxic doses β-ELE on A549/ER cells P-gp function
Rh123 retention assays were used to assess P-gp function in A549/ER cells (Fig. 5). Following treatment with 15 µg/ml β-ELE for 24 h, the A549/ER MFI increased 6.8 times compared with the control group, and MFI increased 6.8 times relative to the control group (P<0.01), indicating that non-toxic doses of β-ELE can effectively reduce Rh123 efflux and increase the concentration of Rh123 in A549/ER cells. The results of this assay indicated that non-toxic doses β-ELE can reduce drug efflux in A549/ER cells, inhibiting the transmembrane pumping function of P-gp.
Figure 5.

β-ELE increases intracellular accumulation of Rh123 in A549/ER cells. The MFI of intracellular Rh123 was detected by flow cytometry, indirectly reflecting the function of P-gp following exposure of A549/ER cells to 15 µg/ml β-ELE for 24 h. (A) Control group; (B) experimental group (15 µg/ml β-ELE). β-ELE, β-elemene; Rh123, rhodamine 123; ER, erlotinib resistant; MFI, mean fluorescence intensity; P-gp, P-glycoprotein.
Effect of non-toxic doses β-ELE on A549/ER cells P-gp expression
The results of western blot analysis revealed that levels of P-gp protein expression decreased significantly in A549/ER cells incubated with 15 µg/ml β-ELE for 24 h, to approximately one-third of the control group (0.25 vs. 0.74; Fig. 6), indicating that 15 µg/ml β-ELE serves a notable role in reversing drug resistance by downregulating the expression of P-gp.
Figure 6.

β-ELE decreases P-gp expression in A549/ER cells. (A) Western blot analysis detected P-gp protein expression level after exposure of A549/ER cells to 15 µg/ml β-ELE for 24 h, β-actin was used as a loading control. (B) The relative expression intensity of P-gp protein in each group was calculated by analyzing the gray value of bands. Values represented averages ± standard deviation of triplicate wells and the experiment was repeated three times. *P<0.01 vs. control group. β-ELE, β-elemene; ER, erlotinib resistant; P-gp, P-glycoprotein.
Discussion
Erlotinib is a commonly used molecular targeted drug for the treatment of NSCLC. However, it is efficacious for a limited time period as resistance will occur, resulting in the failure of therapy (8). Therefore, methods of overcoming erlotinib resistance and identification of novel drugs that can delay or reverse the drug resistance have become the subject of research in recent years (12,13). Acquired drug resistance limits the long-term clinical success of targeted therapies including EGFR inhibitors in patients with EGFR mutant NSCLC. The most common mechanism of acquired resistance, detected in 60% of patients, is a secondary mutation in EGFR at position T790, which is located in exon 20 (14).
ELEs are novel anticancer drugs isolated from Curcuma zedoaria Roscoe, commonly known as zedoary, and are made up of the α, β, γ and δ forms (9). β-ELE has a chemical structure of 1-methyl-1-vinyl-2,4-diisopropenyl-cyclohexane, a molecular formula C15H24, and a molecular weight of 204, β-ELE confers the primary anti-tumor effect (9,15–17).
The current clinical use of elemene injection and oral liquid is based on β-ELE as main ingredients, containing small amounts of δ-ELE and γ-ELE. β-ELE injection has been widely used in treatment of a variety of malignancies, including lung cancer (18,19), liver cancer (20) and stomach cancer (21). β-ELE may elicit a good therapeutic effect and has several advantages, including a broad antitumor spectrum, mild side effects, low in liver and kidney toxicity and no bone marrow suppression, meaning it is well tolerated by patients (15). β-ELE can reverse multidrug resistance (MDR) via the following mechanisms: Killing the tumor cells directly, reducing tumor cell proliferation, inhibiting tumor growth, inducing tumor cell apoptosis and cell cycle arrest, inhibiting tumor angiogenesis and inhibiting tumor cell migration (15). Using a β-ELE and chemotherapy drugs in combination may prevent bone marrow suppression caused by chemotherapy and improve the sensitivity of tumor cells to chemotherapy drugs, potentially even reversing resistance to chemotherapy drugs (15,18).
A prior study demonstrated that NP chemotherapy combined with elemene can improve the efficacy of advanced NSCLC (18). Gao et al (22) reported that β-ELE could reverse drug resistance in the gefitinib-resistant human lung adenocarcinoma PC9/ZD cell line. Yao et al (9,10) investigated the potential mechanism by which β-ELE inhibited the proliferation of the cisplatin-resistant human NSCLC A549/DDP cell line; they revealed that the combination of β-ELE and cisplatin can improve the sensitivity of A549/DDP cells to cisplatin and reverse drug resistance. The mechanism may be associated with decreased mitochondrial membrane potential and P-gp expression activated intracellular redox system, regulation of apoptosis-signaling proteins by increasing the protein expression of cytochrome c, caspase-3, Bcl-2-associated death promoter and reducing protein levels of B-cell lymphoma-2 and procaspase-3 in the A549/DDP cells (9,10). Zhao et al (23) revealed that β-ELE can inhibit the growth of NSCLC cells via extracellular signal-regulated kinase 1/2 and AMP-activated protein kinase-α-mediated inhibition of transcription factor Sp1, followed by reduction in DNA methyltransferase 1 expression.
β-ELE has been observed to reverse drug resistance not only in lung cancer cells but also in other types of cancer cells. Guo et al (24) demonstrated that β-ELE inhibited the overexpression of P-gp and the proliferation KB-C2 cells and enhanced the sensitivity of tumor cells to colchicine, mitoxantrone, vincristine and taxane, reversing drug resistance. Dong et al (25) identified that β-ELE may inhibit breast cancer stem cell growth and reduce Breast cancer resistance protein expression. In a previous study, Li et al (26) observed that β-ELE improve the sensitivity of cisplatin-resistant human ovarian cancer MCAS and A2780/CP70 cells to cisplatin. A recent study revealed that β-ELE can reverse chemoresistance in breast cancer cells by altering the expression of MDR-associated microRNAs, including Phosphatase and tensin homolog and P-gp in adriacin- and docetaxel-resistant MCF-7 cells, consequently regulating the corresponding target genes through the gene regulatory network to induce the development of drug resistance (27).
The results of the present study revealed that β-ELE induced inhibition of A549/ER cell proliferation in a time- and dose-dependent manner in A549/ER cells. According to the IC10, 15 µg/ml β-ELE was determined to be the non-toxic dose and ultimately the test concentration for reversal of resistance. A549/ER cell growth inhibition rates were significantly increased (P<0.05) in each group following a 24-h incubation with β-ELE and erlotinib, with IC50 values decreasing significantly and the degree of drug resistance reversal was 3.24. The aforementioned experimental results indicated that non-toxic doses of β-ELE can enhance the sensitivity of A549/ER cells to erlotinib, reversing drug resistance.
Following incubation with 15 µg/ml β-ELE for 24 h, the rate of proliferation inhibition in A549/ER cells was negligible, at ~6.5%. The results indicated that 15 µg/ml β-ELE combined with erlotinib increased the inhibition of A549/ER cell proliferation, which could be due to the enhancement that β-ELE brings about in the sensitivity of A549/ER cells to erlotinib, thereby partially reversing the drug resistance of A549/ER cells. The cell apoptosis rate and cell cycle distribution were analyzed by flow cytometry; the results indicated that treatment with 15 µg/ml β-ELE alone failed to induce A549/ER cell apoptosis, but in combination with erlotinib an effect was observed (the overall apoptosis rate increased from 5 to 23%). Similarly, treatment of A549/ER cells with 15 µg/ml β-ELE alone for 24 h did not significantly increase the degree of S phase arrest compared with the control group (P>0.05). Treatment with 10 µmol/l erlotinib alone resulted in a significantly higher degree of G0/G1 phase arrest compared with the control (P<0.05), with β-ELE in combination with erlotinib could cause a more significant G0/G1 phase arrest (P<0.01). The non-toxic dose of β-ELE may increase the drug concentration of erlotinib within A549/ER cells, improving the cytotoxic effect; thereby inducing tumor cells apoptosis and cell cycle arrest in G0/G1 phase.
Drug resistance refers to the resistance of cancer cells to a certain antitumor drug. MDR is a phenomenon wherein cancer cells develop resistance to a number of antitumor drugs (28). MDR is characterized by cancer cells becoming resistant to a variety of drugs that have distinct structures and mechanisms of action (29). The expression levels of P-gp serve an important role in a number of resistance mechanisms. P-gp was the first transmembrane protein to be identified to mediate drug transport in the cell. There are two primary types of human P-gp, P-gp I and P-gp II, which are encoded by the MDR1 and MDR2 genes, respectively. The overexpression of P-gp in tumor cells is one of the important contributing factors of MDR (30). P-gp pump the drugs to outside of the cell via ATP hydrolysis (31), exporting drugs away from their target, causing the tumor to be insensitive to the anticancer drug. High expression of P-gp is always associated with MDR. P-gp is an ATP-depended drug efflux pump that is located on the cell membrane; it can reduce the intracellular concentration of drugs by pumping drugs over the cell membrane and lead to drug resistance (32). P-gp may efflux anticancer agents out of cells, therefore decreasing their intracellular accumulation (33). Yao et al (9) confirmed that β-ELE may significantly downregulate the expression of P-gp in the A549/DDP cells membrane, inhibit P-gp-mediated Rh-123 efflux and enhance the intracellular anti-cancer drug accumulation in drug-resistant cancer cells, ultimately leading to a reversal of drug resistance (9). The drug resistance of tumor cells can be reversed by inhibiting the function of P-gp or downregulating the expression of P-gp (34,35). Rh123 is a fluorescent substrate of P-gp, and is widely used as an indicator of P-gp activity. Rh123 is a cationic fluorescent dye that may penetrate the cell membrane, and can be used to assess the expression and the activity levels of P-gp on the surface of the cell via the detection of changes in Rh123 fluorescence intensity in the tumor cells. The higher fluorescence intensity of Rh123, the lower the activity of P-gp on cell surface and the lower the rate of drug efflux. Using the intracellular Rh123 aggregation test, it was found that intracellular Rh123 fluorescence intensity was enhanced following treatment of A549/ER cells with 15 µg/ml β-ELE for 24 h. The results of western blot analysis also indicated that 15 µg/ml β-ELE could significantly reduce P-gp protein levels at the A549/ER cell membrane. These results indicated that 15 µg/ml β-ELE could increase the intracellular accumulation of erlotinib in A549/ER cells by downregulating the levels of P-gp at cell membrane, reducing the efflux of erlotinib and improving the cytotoxic effect of erlotinib on A549/ER cells, thereby inducing apoptosis in the tumor cell and cell cycle arrest.
In conclusion, the results of the present study indicated that treatment with β-ELE could reverse drug resistance in erlotinib-resistant human NSCLC A549/ER cells in vitro. Further analysis revealed that the mechanism of action of β-ELE may involve the decreased expression of P-gp, the inhibition of P-gp dependent drug efflux and the increase in concentration of anticancer drugs in the cells. The present preliminary study provides a potential mechanism of action for β-ELE that may explain its ability to overcome erlotinib resistance. If the reversal of drug resistance induced by β-ELE in erlotinib-resistant human NSCLC cells can be supported by data from clinical trials, it could improve the therapeutic effects in presently erlotinib-resistant NSCLC patients.
Acknowledgements
The authors would like to thank the laboratory of Fujian Medical University for their assistance in completing the study.
Funding
The present study was supported by the Joint Funds for the Innovation of Science and Technology, Fujian province, China (grant no. 2017Y9031); Fujian Provincial Natural Science Foundation, China (grant no. 2017J01295); Startup Fund for scientific research, Fujian Medical University (grant no. 2016QH033) and the Key Specialty Discipline Construction Program of Fujian and Nation.
Availability of data and materials
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
LL designed and performed the experiments and was the major contributor in writing the manuscript. LBL performed the experiments and performed analysis and interpretation of data. XC made substantial contributions to conception and design the study and aided in drafting the manuscript. BZ made contributions to the analysis and interpretation of data. TL made contributions to conception and design the study, assisted with drafting and revising important content of the manuscript, and gave final approval of the version to be published. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Tanoue LT, Tanner NT, Gould MK, Silvestri GA. Lung cancer screening. Am J Respir Crit Care Med. 2015;191:19–33. doi: 10.1164/rccm.201410-1777CI. [DOI] [PubMed] [Google Scholar]
- 2.Stewart B, Wild CP, editors. World Cancer Report 2014. IARC Nonserial Publication; [Google Scholar]
- 3.Szasz A. Current status of oncothermia therapy for lung cancer. Korean J Thorac Cardiovasc Surg. 2014;47:77–93. doi: 10.5090/kjtcs.2014.47.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 5.Ichite N, Chougule M, Patel AR, Jackson T, Safe S, Singh M. Inhalation delivery of a novel diindolylmethane derivative for the treatment of lung cancer. Mol Cancer Ther. 2010;9:3003–3014. doi: 10.1158/1535-7163.MCT-09-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Ruysscher D, Van Meerbeeck J, Vande casteele K, Oberije C, Pijls M, Dingemans AM, Reymen B, van Baardwijk A, Wanders R, Lammering G, et al. Radiation-induced oesophagitis in lung cancer patients. Is susceptibility for neutropenia a risk factor? Strahlenther Onkol. 2012;188:564–567. doi: 10.1007/s00066-012-0098-z. [DOI] [PubMed] [Google Scholar]
- 7.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et al. Gefitinib versus cisplatin plus docetaxel in patients with non- small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJ-TOG3405): An open-label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Du Y, Liu H, Ma T, Shen Y, Pan Y. Study of efficacy and safety of pulsatile administration of high-dose gefitinib or erlotinib for advanced non-small cell lung cancer patients with secondary drug resistance: A single center, single arm, phase II clinical trial. Thorac Cancer. 2016;7:663–669. doi: 10.1111/1759-7714.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao C, Jiang J, Tu Y, Ye S, Du H, Zhang Y. β-elemene reverses the drug resistance of A549/DDP lung cancer cells by activating intracellular redox system, decreasing mitochondrial membrane potential and P-glycoprotein expression, and inducing apoptosis. Thorac Cancer. 2014;5:304–312. doi: 10.1111/1759-7714.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao CC, Tu YR, Jiang J, Ye SF, Du HX, Zhang Y. β-elemene reverses the drug resistance of lung cancer A549/DDP cells via the mitochondrial apoptosis pathway. Oncol Rep. 2014;31:2131–2138. doi: 10.3892/or.2014.3083. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Ying W, Jia-li Y, Yan-Ting S, Chun-ying L. Effects of Buzhong Yiqi decoction-medicated serum on drug resistance of human lung adenocarcinoma cell line A549/DDP to cisplatin. Chinese J Pathophysiol. 2014;30:223–238. [Google Scholar]
- 12.Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, Zhang T, Marrano P, Whitehead M, Squire JA, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26:4268–4275. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 13.Brevet M, Johnson ML, Azzoli CG, Ladanyi M. Detection of EGFR mutations in plasma DNA from lung cancer patients by mass spectrometry genotyping is predictive of tumor EGFR status and response to EGFR inhibitors. Lung cancer. 2011;73:96–102. doi: 10.1016/j.lungcan.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Jänne PA. EGFR mutations and resistance to Irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21:3913–3923. doi: 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, HU X. Research progress on the anti-tumor mechanism and reversal mechanism of drug resistance of β-elemene. J Mod Oncol. 2014;22:1711–1714. [Google Scholar]
- 16.Jianfeng JU, Weiping YU, Chunsheng FU, Liming MA. Modern research and clinical application of β-elemene. Qilu Pharmaceutical Affairs. 2008;27:546–548. [Google Scholar]
- 17.Guangchao LI. Basic research and clinical applications of β-elemene. J Changchun Univ Tradit Chin Med. 2009;25:185–186. [Google Scholar]
- 18.Lihua W, Fang S. Elemene emulsion combined with NP chemotherapy in the treatment of advanced NSCLC patients. China Oncol. 2010;20:547–550. [Google Scholar]
- 19.Li T, Yuean C, Chaosheng P, Wei W, Nanzhan L. Elemene emulsion combined with chemotherapy in the treatment of patients with advanced non-small cell lung cancer. Chinese J Clin Med. 2009;16:725–727. [Google Scholar]
- 20.Qirong P, Bi X, Long M, Tao C, Yongcai T, Caixia H, Pengfei L. Short-term clinical observation of Elemene injection used to treat metaphase and advanced primary liver cancer. Chin J Integ Tradit West Med Liv Dis. 2010;20:274–276. [Google Scholar]
- 21.Dan Y, Guangyu A, Hong D. Efficacy observation of advanced gastric cancer treated with elemene and Tegafur. World J Integ Tradit West Med. 2013;8:264–266. [Google Scholar]
- 22.Gao FY, Zhang AQ, Sun Y. Reversing Effect of β-elemene on human lung adenocarcinoma cell line PC9. Chin Arch Tradit Chin Med. 2014;32:131–133. [Google Scholar]
- 23.Zhao S, Wu J, Zheng F, Tang Q, Yang L, Li L, Wu W, Hann SS. β-elemene inhibited expression of DNA methyltransferase 1 through activation of ERK1/2 and AMPKa signalling pathways in human lung cancer cells: The role of Sp1. J Cell Mol Med. 2015;19:630–641. doi: 10.1111/jcmm.12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo HQ, Zhang GN, Wang YJ, Zhang YK, Sodani K, Talele TT, Ashby CR, Jr, Chen ZS. β-elemene, a compound derived from Rhizoma zedoariae, reverses multidrug resistance mediated by the ABCB1 transporter. Oncol Rep. 2014;31:858–866. doi: 10.3892/or.2013.2870. [DOI] [PubMed] [Google Scholar]
- 25.Dong Y, Li L, Wang L, Zhou T, Liu JW, Gao YJ. Preliminary study of the effects of β-elemene on MCF-7/ADM breast cancer stem cells. Genet Mol Res. 2015;14:2347–2355. doi: 10.4238/2015.March.27.20. [DOI] [PubMed] [Google Scholar]
- 26.Li QQ, Lee RX, Liang H, Wang G, Li JM, Zhong Y, Reed E. β-elemene enhances susceptibility to cisplatin in resistant ovarian carcinoma cells via downregulation of ERCC-1 and XIAP and inactivation of JNK. Int J Oncol. 2013;43:721–728. doi: 10.3892/ijo.2013.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Zhang HD, Yao YF, Zhong SL, Zhao JH, Tang JH. β-elemene reverses chemoresistance of breast cancer cells by reducing resistance transmission via exosomes. Cell Physiol Biochem. 2015;36:2274–2286. doi: 10.1159/000430191. [DOI] [PubMed] [Google Scholar]
- 28.Gottesman MM. How cancer cells evade chemotherapy: Sixteenth Richard and Hinda Rosenthal Foundation Award Lecture. Cancer Res. 1993;53:747–754. [PubMed] [Google Scholar]
- 29.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 30.Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P- glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA, Montrose MH, Beck WT, Erickson LC. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J Pharmacol Exp Ther. 2008;324:95–102. doi: 10.1124/jpet.107.127704. [DOI] [PubMed] [Google Scholar]
- 32.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura T, Oka M, Aizawa K, Soda H, Fukuda M, Terashi K, Ikeda K, Mizuta Y, Noguchi Y, Kimura Y, et al. Direct interaction between a quinoline derivative, MS-209, and multidrug resistance protein (MRP) in human gastric cancer cells. Biochem Biophys Res Commun. 1999;255:618–624. doi: 10.1006/bbrc.1999.0245. [DOI] [PubMed] [Google Scholar]
- 34.Jovelet C, Bénard J, Forestier F, Farinotti R, Bidart JM, Gil S. Inhibition of P-glycoprotein functionality by vandetanib may reverse cancer cell resistance to doxo-rubicin. Eur J Pharm Sci. 2012;46:484–491. doi: 10.1016/j.ejps.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 35.Angelini A, Conti P, Ciofani G, Cuccurullo F, Di Ilio C. Modulation of multidrug resistance p-glycoprotein activity by antiemetic compounds in human doxorubicin-resistant sarcoma cells (MES-SA/Dx-5): Implications on cancer therapy. J Biol Regul Homeost Agents. 2013;27:1029–1037. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.