

 Novel SGLT-2 selective inhibitors, an innovative therapeutic approach for glycemia control in diabetic patients.
Novel SGLT-2 selective inhibitors, an innovative therapeutic approach for glycemia control in diabetic patients.
Abstract
Diabetes mellitus is a chronic, complex and multifactorial disease associated characteristically with hyperglycemia. One of the most recently approved antidiabetic drug classes for clinical use are sodium–glucose cotransporter type 2 (SGLT-2) inhibitors. SGLT-2 is a protein expressed in the kidneys, responsible for glucose reabsorption from the glomerular filtrate to the plasma. It is known, nowadays, that diabetic patients show an increased glucose renal reabsorption capacity, caused by the overexpression of the SGLT-2 transporter, thus contributing to hyperglycemia. From establishing this correlation, the SGLT-2 transporter started to be considered as a therapeutic target of interest, culminating in the approval of the first antidiabetic SGLT-2 inhibitor, dapagliflozin (Forxiga® or Farxiga®, Bristol-Myers Squibb & AstraZeneca), in 2012 in Europe. On the other hand, canagliflozin (Invokana®, Janssen Pharmaceutical) was the first drug in this class to be approved by the FDA, the U.S. Food and Drug Administration, in 2013. This review concerns the discovery and development of the first representatives of this class of antidiabetic drugs, and the description of new optimized analogues that are currently in the clinical and preclinical stages of development.
1. Introduction
Diabetes mellitus is a chronic, complex and multifactorial disease characteristically associated with a hyperglycemic state, which occurs as a consequence of resistance to the action of insulin or a deficiency in its secretion. Diabetes type I is characterized by the autoimmune destruction of the insulin-producing pancreatic β cells, culminating in the partial or total incapacity of secreting this hormone.1,2 On the other hand, diabetes type II is related to peripheral resistance to insulin, a result of a lower glucose uptake by the peripheral tissues, mainly muscles and fat tissues, and an increase of glucose liberation by the liver.3,4
Around 90–95% of the cases of diabetes reported nowadays are classified as type II.2 The main risk factors for the establishment of this disease include obesity, bad eating habits, sedentary lifestyle and aging.4 Considering that, it is clear that behavioral, environmental and social factors, and also the increase in life expectancy are deeply associated with the worldwide increase of diabetes type II cases.1,4,5 The projections for the future are alarming, with a prediction of 629 million affected people by 2045, according to the data released by the International Diabetes Federation.6
Although different therapeutic options are available for diabetes type II treatment, the antidiabetic drugs used do not result in a complete disease remission, but, in fact, only delay its progression. Moreover, the clinically available medicines exhibit restrictions related to the occurrence of adverse effects and/or limited efficacy in controlling the glycemia of some patients.7,8 Therefore, the search for safer and more effective alternatives that yield better treatment adherence and better control of the clinical conditions is necessary.8
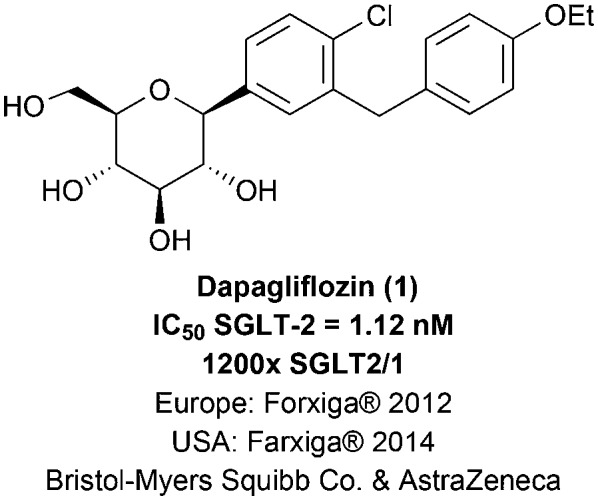
One of the most recently approved antidiabetic drug classes are sodium–glucose cotransporter type 2 (SGLT-2) inhibitors.8,9 SGLT-2 is expressed in the kidneys, modulating glucose reabsorption from the glomerular filtrate to the plasma.3,8 Nowadays, it is known that diabetic patients exhibit a higher capacity for glucose renal reabsorption, because of the overexpression of the SGLT-2 transporter, contributing to the establishment of the hyperglycemic state.8,10,11 Ever since the establishment of this correlation, the SGLT-2 transporter has become a therapeutic target of interest, culminating in the approval of the first SGLT-2 inhibitor antidiabetic drug, dapagliflozin (1; Forxiga® or Farxiga®, Bristol-Myers Squibb & AstraZeneca), through the European regulatory agency EMA (European Medicines Agency) in 2012 (Fig. 1).9,12 In this context, the scope of this review concerns the development of the first representatives of this new antidiabetic drug class and the description of new optimized analogues currently in the clinical or preclinical stages of development.
Fig. 1. Dapagliflozin (1), the first sodium–glucose cotransporter type-2 (SGLT-2) inhibitor antidiabetic drug approved for clinical use by the EMA (European Medicines Agency) in 2012.
2. The kidneys' physiological role in blood glucose control
For centuries, diabetes was considered as a kidney disease, due to some of its characteristic symptoms, such as polyuria and glycosuria. However, in the 19th century, the disease was finally characterized as a pancreatic disorder.12,13 Then, in the beginning of the 20th century, around 1930, the kidneys' function in glucose metabolism was described,12 determining their relevant role in controlling blood glucose through gluconeogenesis and glucose reabsorption, as well as glucose consumption for their operation.8,12 For that reason, the kidneys were once again seen as an organ of interest in the search for new therapeutic alternatives for diabetes treatment.
2.1. Gluconeogenesis
Aiming at maintaining blood glucose levels during fasting periods, gluconeogenesis occurs in the liver and kidneys, contributing to 75–80% and 20–25% of the blood glucose levels, respectively.8 In type II diabetic patients, gluconeogenesis, especially the renal one, occurs in a greater proportion, being approximately 300% higher. The renal gluconeogenesis occurs in the proximal tubule cells and its activity is regulated by the hormones insulin and adrenaline. While insulin inhibits renal glucose release, adrenaline stimulates it.3 In the postprandial state, there is a reduction of liver gluconeogenesis, while renal gluconeogenesis increases 2-fold, contributing to 60% of the endogenous glucose release in the blood stream after meals.8
2.2. Reabsorption
The kidneys are capable of filtering approximately 180 liters of plasma daily. The plasma glucose concentration is around 5.5 mmol L–1, which corresponds to 180 grams of filtered glucose per day.10,12 In healthy patients, more than 99% of the filtered glucose is reabsorbed in the renal tubules.3,14
The glucose reabsorption process occurs through sodium–glucose cotransporters (SGLTs) and glucose transporters (GLUTs) (Fig. 2).3,15,16 The SGLT cotransporters are located in the luminal membrane and transport sodium and glucose simultaneously to the interstitial compartment.3,15 The necessary energy for active glucose transportation through the membrane is derived from the favorable gradient for sodium transport, which is continually maintained by the ATPase sodium–potassium pump present in the basolateral membrane, responsible for expelling sodium ions from the tubular cells. The GLUT transporters, on the other hand, carry glucose passively through the basolateral membrane to the plasma.3,16
Fig. 2. Glucose renal reabsorption process occurs through the sodium–glucose cotransporters (SGLTs) and the glucose transporters (GLUTs). The SGLT cotransporters are located in the luminal membrane and transport sodium and glucose simultaneously to the interstitial compartment. The necessary energy for the active glucose transport through the membrane is derived from the sodium transport favorable gradient, which is continuously maintained by the sodium potassium ATPase pump present in the basolateral membrane, responsible for expelling sodium ions from the tubular cells. The GLUT transporters, on the other hand, carry glucose passively through the basolateral membrane towards the plasma.

For each of the transporters mentioned above, there are two subtypes present in the renal proximal tubules.3,14,15 SGLT-2 is found at the beginning of the proximal region of the tubular lumen (segments 1 and 2),12,14 consisting of a high capacity and low affinity cotransporter which transports sodium and glucose in a ratio of 1 : 1,3 being responsible for reabsorbing about 90–97% of the filtered glucose in this region.12,14,16 Then, the reabsorbed glucose is transferred from the interstitial compartment to the plasma by passive transport through the GLUT-2 present in the basolateral membrane. In the distal section of the proximal tubules (segment 3),14 the remaining glucose (∼3–10%) is reabsorbed by SGLT-1, a low capacity and high affinity cotransporter, and is then transported to the plasma through GLUT-1.12,14,16 SGLT-1 transports sodium and glucose in a 2 : 1 proportion,3 and, unlike the SGLT-2 subtype, which is mostly expressed by the kidneys, SGLT-1 is not only present in this organ, but also in the heart and intestine, where it is majorly expressed, participating in the glucose and galactose absorption process throughout the gastrointestinal tract.3,14,15
The glucose renal reabsorption occurs linearly, i.e. the higher the glucose concentration in the glomerular filtrate, the greater the reabsorbed amount. However, this is a saturable process, i.e. the renal tubule has a maximum reabsorption capacity (TmG) of approximately 375 mg min–1 in healthy patients. When this threshold is exceeded under hyperglycemic conditions, due to a high amount of filtered glucose from the plasma, glucose excretion is observed in the urine, a condition known as glycosuria.3,8 Particularly in diabetic patients, the prolonged hyperglycemic state induces the increase of SGLT-2 and GLUT-2 transporter gene expression, culminating in a higher reabsorption capacity (TmG) and contributing to the maintenance of the characteristic hyperglycemia of this illness.3,10
2.3. Mutations in the SGLT cotransporter codifying genes: elucidation of its potential as a therapeutic target
Mutations in the genes involved in the SGLT cotransporters codification allowed a better comprehension of their physiological roles, contributing to the elucidation of their potential as therapeutic targets.3,8 Mutations in the SGLT-1 protein codifier genes result in poor glucose and galactose absorption during digestion, culminating in the accumulation of these sugars in the gastrointestinal tract. Fermentation of these sugars by the gut bacterial flora causes diarrhea and dehydration in individuals with this mutation. Still, these individuals exhibit low or no glycosuria at all, proving, as previously mentioned, that SGLT-1 is not the main transporter responsible for glucose reabsorption in the renal system.3,14
On the other hand, mutations in the SLC5A2 gene, which encodes the SGLT-2 cotransporter, cause a condition known as familiar renal glycosuria, in which affected patients exhibit different levels of glycosuria, depending on the type of mutation observed in the transporter protein.3,8 However, most of the affected individuals do not present relevant clinical symptoms, with hypoglycemia, dehydration, electrolyte imbalance or a significant increase in urinary tract infections not being reported.3,8 Based on this information, the SGLT-2 cotransporter started to be considered as a safe therapeutic target for the development of innovative antidiabetic drugs for treating diabetes mellitus type 2, where the selective inhibition of the SGLT-2 subtype would produce a blood glucose reducing effect totally independent from insulin secretion by the pancreas, since it would interfere with the glucose renal reabsorption from the glomerular filtrate.3,14,15
3. The discovery and development of SGLT-2 cotransporter inhibitors as a new antidiabetic drug class
The history of the development of SGLT inhibitors began in 1835, when phlorizin (2), a natural O-glycosidic product present in several plants, was isolated for the first time from the root of an apple tree.17–19 Still in the 19th century, compound 2 was discovered to be a potent inductor of glycosuria and polyuria after oral administration, mimicking diabetes characteristic clinical symptoms.9,19,20 Phlorizin (2) discovery was the determinant for the identification of sodium–glucose cotransporters (SGLTs) and for elucidating their physiological roles, especially regarding glucose renal reabsorption, with a direct influence on the glycemic index (Fig. 2).14,20,21
However, due to its nonselective inhibition toward both SGLT transporter subtypes (human SGLT-2 EC50 = 36 nM and human SGLT-1 EC50 = 330 nM; with a poor selectivity of 10× SGLT2/1),14,20 the pharmacological profile of the natural product 2 was considered unsuitable for clinical use, since the inhibition of the SGLT-1 cotransporter was related to possible adverse effects in the gastrointestinal tract.9,14,21 Besides, phlorizin (2) presents an inadequate pharmacokinetic profile due to the low metabolic resistance of the labile O-glycosidic bond, which is rapidly hydrolysed by beta-glucosidase in the gastrointestinal tract, resulting in low oral bioavailability and reduced half-life.9,14,19 Lastly, the release of the correspondent aglycone after the O-glycosidic bond hydrolysis, i.e. dihydrochalcone phloretin (3) (Fig. 3),9,19 is associated with the occurrence of serious side effects, since 3 is a nephrotoxic substance and also acts as a GLUT transporter inhibitor, hindering glucose uptake by several tissues, especially the brain.20 For those reasons, the selectivity for the SGLT-2 subtype and an improvement in the pharmacokinetic profile started to be considered as basic requirements for the development of new drugs based on the structural optimization of the natural prototype 2.14,20,21
Fig. 3. First structural analogues of the natural product phlorizin (2) described by Tsujihara et al.,22–25 designed to reduce its metabolic liability and the toxicity associated with the aglycone phloretin (3) metabolite. The introduction of small substituents in the meta or para positions in phlorizin's (2) B ring (in blue) was well tolerated, such as in the bioactive compound 4 containing the methoxy substituent (in orange). The bioactive compound 5 results from the application of ring bioisosterism on the aromatic subunit B, replacing the phenyl ring for a benzofuran (in blue), while derivative 6 was designed as a prodrug of 5 by introduction of an ester group, as highlighted in green. Compound 7, on the other hand, is a methylated analogue of 5, presenting greater potency and selectivity for the SGLT-2 transporter. Finally, prototype T-1095 (8), the most promising derivative in the series, was designed as the carbonate prodrug of 7, presenting greater stability in vivo due to being more resistant against the hydrolysis by the beta-glucosidases present in the gastrointestinal tract.

In this context, several structural modifications were explored aiming at the optimization of phlorizin (2). The first structure–activity relationship studies were published by the end of the 90s (Fig. 3) by researchers from the Tanabe Seiyaku Japanese pharmaceutical company,22–25 which is currently part of the Mitsubishi Tanabe Pharma Group. In this pioneer work, modifications in the aglycone subunit of phlorizin (2) were made trying to minimize the toxic effects associated with the dihydrochalcone phloretin (3) release, maintaining, on the other hand, the glucose renal reabsorption inhibition effect. The obtained results indicate that the introduction of other substituents in the meta or para positions of phlorizin's (2) B ring was well tolerated, such as in the bioactive derivative 4.23,25 It should also be noted that the ablation of the hydroxyl group in the para position to the ketone side chain in the A ring of phlorizin (2) did not affect the SGLT transporter inhibitory activity of analogues 4–6 (Fig. 3).23,25 However, Tsujihara and co-workers found out that modifications in the ketone spacer subunit, between rings A and B, were deleterious to the desired activity, since they resulted in derivatives displaying a drastic reduction of the SGLT-2 inhibition.23,25
Then, bioisosteric substitution of the B benzene ring for other aromatic heterocyclic systems was performed, showing that benzofuran analogues, e.g.5 and 6, exhibited a more potent effect against SGLT transporter inhibition, resulting in a pronounced glucose excretion through urine.23 In compound 6, the glycoside subunit underwent hydroxyl functionalization aiming to reduce the metabolic instability of the O-glycosidic bond in the digestive tract through its protection against beta-glycosidase action. As expected, the ester prodrug 6 presented a lower affinity for the beta-glycosidase enzyme in preclinical trials, resulting in a greater in vivo resistance to hydrolysis when compared to the benzofuran analogue 5, culminating in a longer duration of the pharmacological effects in the studied animal models.24,25
Afterwards, attempts at optimizing compound 5 involved substitutions at the A aromatic ring.25 The results indicated that the mere introduction of a smaller alkyl group in the para position in relation to the carbonyl resulted in increased activity, with the methylated derivative 7 as the most potent of the series,25 besides showing a limited selectivity for the SGLT-2 subtype (thirty times more selective in regard to SGLT-1).14 Using a similar strategy to the one applied in the design of prodrug 6, compound T-1095 (8) was prepared as the correspondent carbonate prodrug of 7, aiming to reduce the hydrolysis of the O-glycosidic bond by the beta-glycosidase in the gastrointestinal tract after oral administration. During the preclinical assays in animal models, it was demonstrated that compound 8 was metabolized to its active form (7) mainly by hepatic esterases. The active metabolite (7) was able to reduce the blood glucose near control levels, however, without inducing hypoglycemia, one of the most common adverse effects associated with the antidiabetic drug of choice.25 Therefore, prodrug T-1095 (8) was selected as the most promising derivative of the family so far, being directed to human clinical trials. However, the development of this drug candidate was discontinued at phase II of the clinical trials.9
Several O-glycosidic analogues of phlorizin (2) (Fig. 4) were described by other pharmaceutical companies in the following years, reaching the clinical stages, such as sergliflozin etabonate (9)26 and remogliflozin etabonate (10),27 both from GlaxoSmithKline. Unlike the series developed by Tanabe Seiyaku, GlaxoSmithKline's compounds 9 and 10 explored modifications in the spacer between rings A and B, replacing the long 3-carbon ketone subunit by a shorter methylene spacer, which allowed the identification of more potent and selective compounds for the SGLT-2 transporter in comparison with SGLT-1, with a selectivity index of 296 and 365, respectively.20
Fig. 4. First generation of O-glycosidic structural analogues of the natural product phlorizin (2), designed as SGLT-2 transporter inhibitor antidiabetic drug candidates. Prototypes 8, 9 and 10 were discontinued because of the lack of efficacy due to pharmacokinetic limitations after oral administration during clinical trials. The O-glycosidic bond is highlighted in red and the carbonate groups of prodrugs 8, 9 and 10 are highlighted in light blue.

However, the development of this first generation of O-glycosidic analogues was discontinued due to the metabolic liability of these compounds in humans.8,9 It should be mentioned that prodrugs 8, 9 and 10 displayed, in fact, an increase in metabolic stability and half-life in animal preclinical assays when compared to direct oral administration of the corresponding active metabolites. However, despite those promising results, the observed stability gain was not reproduced in human species during clinical trials, resulting in the lack of efficacy and the consequent interruption of their development as SGLT-2 inhibitor drug candidates.9,28
The second generation of phlorizin (2) structural analogues is represented by C-glycosidic derivatives, designed in order to bypass the metabolic liability associated with the O-glycosidic precursors in the digestive tract.9,20,28 Moreover, SAR studies concerning the new C-glycosidic analogues revealed that the regioisomer containing the methylene spacer in the meta position to the C-glycosidic bond presents an increase in potency and selectivity towards the SGLT-2 cotransporter when compared to the ortho C-glycosidic regioisomer.14,20,29 The first representative of this generation of compounds was dapagliflozin (1; Forxiga® or Farxiga®), developed by the joint venture of Bristol-Myers Squibb and AstraZeneca. After a SAR study with systematic variations in the glycosidic and aglycone subunits, dapagliflozin (1) was finally identified as a potent (IC50 = 1.12 nM) and selective SGLT-2 transporter inhibitor with appropriate pharmacokinetic properties for therapeutic use. It should be highlighted that compound 1 presents a meta substitution pattern regarding the methylene spacer and the C-glycosidic bond and a hydrophobic substituent at ring A in the para position to the C-glycosidic bond (Fig. 5).14
Fig. 5. C-Glycosidic second generation of structural analogues of the natural product phlorizin (2) approved for therapeutic use as SGLT-2 inhibitors. The C-glycosidic bond is highlighted in red; the lipophilic substituent in the para position to this bond is highlighted in green in derivatives 1 and 11–15; ring B and its substituents are highlighted in pink for all the derivatives; and the modifications made in the glycosidic subunit in derivatives 13, 15 and 16 are highlighted in blue.

Drug 1 is capable of inducing glycosuria in humans in a more pronounced way than the O-glycosidic derivatives previously described, e.g. T-1095 (8), sergliflozin etabonate (9) and remogliflozin etabonate (10). This increase in in vivo potency is directly associated with the C-glycosidic stability when compared to the O-glycosidic stability in the gastrointestinal tract. Moreover, dapagliflozin (1) presents an affinity 1200 times greater for SGLT-2 in comparison with SGLT-1.14,20
Taking into account its innovative profile as a therapeutic alternative for diabetes treatment, the European regulatory agency EMA approved dapagliflozin (1) for clinical use in 2012.9,12 On the other hand, after receiving the approval application for drug 1 in 2010, the American agency FDA (Food and Drug Administration) has requested additional studies regarding possible risks of bladder and breast cancer and of hepatotoxicity for a risk–benefit analysis, resulting in the approval of dapagliflozin by this agency only in 2014.9,12,30
Canagliflozin (11, Invokana®, Janssen Pharmaceutical), another C-glycosidic antidiabetic drug (Fig. 5), was actually the first SGLT-2 selective inhibitor to be approved by the FDA for therapeutic use, which happened in 2013.9,12
Canagliflozin (11) was developed by the Japanese pharmaceutical industry Mitsubishi Tanabe Pharma, previously licensed to Janssen Pharmaceutical, a company from the Johnson & Johnson group. During the development of 11, the authors used as a basis the SAR studies previously described by Bristol-Myers Squibb researchers, who demonstrated the utility of the meta substitution pattern in the C-glycosidic derivatives for SGLT-2 transporter selective inhibition, as previously mentioned. Then, Nomura and co-workers (2010) proposed a series of C-glycosidic derivatives containing a heterocyclic system in substitution to dapagliflozin's (1) benzene ring B. On the other hand, the substitution pattern with small lipophilic groups in the para position in relation to the C-glycosidic bond, as seen in drug 1, was preserved. Among all the evaluated heterocyclic rings, e.g. furan, thiophene, pyrazole, pyridine and thiazole, the substituted thiophene derivatives stood out, culminating in the discovery of canagliflozin (11) as the most promising analogue, with elevated inhibitory potency (IC50 = 4.4 nM) and 155 times higher selectivity for SGLT-2 in comparison with SGLT-1 (Fig. 5).31
After the description and approval of drugs 1 and 11, several C-glycosidic analogues with therapeutic potential were developed and entered preclinical and clinical trials as antidiabetic drug candidates.32–36 Among them, besides dapagliflozin (1) and canagliflozin (11), empagliflozin (12, Jardiance®, Boehringer Ingelheim & Eli Lilly; IC50 SGLT-2 = 3.1 nM and a selectivity of 2680× SGLT2/1) was also approved for clinical use in Europe and the United States in 2014.10,19 More recently, the drug ertugliflozin (13, Steglatro®, Merck & Co.; IC50 SGLT-2 = 0.87 nM and a selectivity of 2250× SGLT2/1)37 was approved by the FDA (2017) and the EMA (2018) for similar clinical application. All mentioned SGLT-2 inhibitors are indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus, both as monotherapy and in combination with other antidiabetic agents.8,12,19 Additionally, derivatives ipragliflozin (14, Suglat®, Astellas & Kotobuki Pharmaceutical; IC50 SGLT-2 = 7.4 nM and a selectivity of 254× SGLT2/1),38 luseogliflozin (15, Lusefi®, Taisho Pharmaceutical Co.; IC50 SGLT-2 = 2.3 nM and a selectivity of 1770× SGLT2/1)39 and tofogliflozin (16, Deberza®, Chugai Pharma & Sanofi; IC50 SGLT-2 = 2.9 nM and a selectivity of 2912× SGLT2/1)28 have been approved for clinical use exclusively in Japan, remaining in the clinical trial stage in other countries (Fig. 5).
A third generation of selective SGLT-2 transporter inhibitors was recently described by the Mitsubishi Tanabe Pharma researchers, being composed of new N-glycosidic derivatives.15,40 The N-glycosidic aniline analogue 17, designed from the C-glycosidic drug dapagliflozin (1), presented inhibitory potency (IC50 = 3.9 nM) against the therapeutic target in the same order of magnitude as prototype 1 (IC50 = 1.12 nM) (Fig. 6). However, this compound had limited efficacy in vivo in urinary glucose excretion (UGE) after oral administration. The urinary glucose excretion was measured during a 24-hour period after oral administration of compound 17 in a 30 mg kg–1 dosage. The animal's body weight was normalized to 200 g for comparison purposes, resulting in a UGE value of only 93 mg per 200 g BW per day.40 The drug dapagliflozin (1), for example, in a dosage of 10 mg kg–1 by oral administration, induces a pronounced urinary glucose excretion (UGE = 1900 mg per 200 g BW per day) in the same animal model.14
Fig. 6. Third generation of SGLT-2 inhibitors as antidiabetic drug candidates. N-Glycosidic structural analogues designed from the C-glycosidic drug dapagliflozin (1). The N-glycosidic bond is highlighted in red; the lipophilic substituent of ring A is highlighted in green in derivatives 1 and 19; ring B and its substituents are highlighted in pink for all the derivatives.

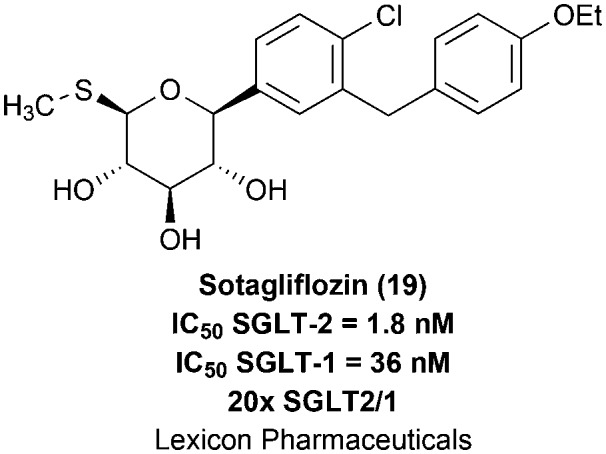
A possible explanation for the observed inefficacy of the aniline derivative 17 would be the lability of its N-glycosidic bond in vivo. Indeed, the authors described the aglycone formation from 17 in pharmacokinetic studies in rats. In this context, aiming at the design of a series of more stable N-glycosidic analogues, the authors developed new heterocyclic derivatives, among which the indolic compound 18 stood out, with an adequate in vitro inhibitory potency for SGLT-2 (IC50 = 7.1 nM) and an in vivo optimized effect on urinary glucose excretion (UGE = 1830 mg per 200 g BW per day) in rats, after an oral administration of a 30 mg kg–1 dose.40 Subsequent structural modifications in prototype 18 provided the discovery of the indolic N-glycosidic drug candidate TA-1887 (19), with optimized in vitro (IC50 = 1.4 nM) and in vivo (UGE = 2500 mg per 200 g BW per day) potencies, besides being 164 times more selective for SGLT-2 in comparison with SGLT-1 (Fig. 6).15 However, this compound has not reached the clinical trial stages yet.
Lately, nonselective SGLT-2/1 dual inhibitors have been also investigated as antidiabetic drug candidates, although there is no drug approved with this pharmacological mechanism so far.19 Even though the selective inhibition of SGLT-2 has been primarily pointed as a desirable feature for reducing gastrointestinal adverse effects and for avoiding the occurrence of hypoglycemia, some researchers are currently arguing that the partial SGLT1 inhibition might provide therapeutic benefits that could not be achieved with the selective SGLT2 inhibition alone.41 This assumption is based on the fact that SGLT-1 inhibition would decrease glucose absorption in the intestine, reducing postprandial hyperglycemia, while nonselective SGLT inhibition would block renal glucose reabsorption more potently.41,42
The prototype sotagliflozin (19), from Lexicon Pharmaceuticals, is an oral nonselective SGLT-2/1 dual inhibitor (SGLT-2 IC50 = 1.8 nM and SGLT-1 IC50 = 36 nM; with a poor selectivity of 20× SGLT2/1)37 under clinical development as an antidiabetic drug (Fig. 7).41,42 Compound 19 was able to improve glycemic control and lower body weight among patients with type 1 or 2 diabetes during a phase II clinical study. This prototype was also evaluated in a phase III trial in patients with type 1 diabetes receiving their usual insulin therapy, contributing to the reduction of glycated haemoglobin levels in comparison with insulin alone. However, the rate of diabetic ketoacidosis was higher in the sotagliflozin treated group. It is worth mentioning that no oral medication has been yet approved for its use in combination with insulin to lower the glucose level in patients with type 1 diabetes.42 Nevertheless, future studies and clinical data are demanded to demonstrate the viability and benefits of this putative therapeutic approach.
Fig. 7. Sotagliflozin (19), an oral nonselective SGLT-2/1 dual inhibitor currently under clinical development as an antidiabetic drug candidate.
4. SGLT-2 inhibitors' pharmacological profiles
The SGLT-2 cotransporter inhibitors, introduced in the pharmaceutical market in 2012 as a new antidiabetic drug class, have been well tolerated by most patients, presenting efficacy in blood glucose control and reducing the occurrence of microvascular damage related to the disease.3,10,12 Moreover, they also induce daily caloric loss (approximately 200–300 kcal per day)8 through the elevation of glucose elimination rates, contributing to body weight reduction during treatment, an additional therapeutic benefit for obese diabetic individuals. These drugs still promote a subtle decrease in blood pressure, since they cause osmotic diuresis, and are associated with a reduction in cardiovascular accidents, being beneficial to diabetic patients with hypertension.8,43,44
One of the main advantages attributed to this antidiabetic drug class is that the blood glucose control is totally independent from the insulin hormone.3 Therefore, these drugs are useful in monotherapy or in association with other antidiabetic drugs,8,12,19 being able to be used in different evolutionary stages of the disease, including in patients with impaired insulin secretion. Besides, once the SGLT-2 inhibitors are not able to suppress the glucose renal reabsorption completely due to a secondary increase in glucose reabsorption by SGLT-1 in segment S3,19 the treatment with SGLT-2 inhibitors promotes urinary elimination of less than 50% of the daily filtered glucose (approximately 180 g) and the risk of hypoglycemia is consequently reduced.3,8,19
Due to their favorable pharmacological profiles, the popularity of SGLT-2 inhibitors is growing exponentially in the world's pharmaceutical market. According to the FDA, in the period between October 2014 and September 2015, there were approximately 1.7 million prescriptions of dapagliflozin (1) and canagliflozin (11) just in the U.S.45 However, the use of SGLT-2 inhibitors is also associated with important adverse effects that need to be considered regarding the risk–benefit for the patient.3,8,10,45
The main adverse effects described are related to glycosuria, which results in a significant increase in genital and urinary infections, mainly female genital mycotic infections.3,8,10 Moreover, in 2015, the FDA released a warning stating that the treatment with SGLT-2 inhibitors was associated with an increased risk of diabetic ketoacidosis, a severe clinical condition caused by elevated blood levels of ketone bodies, being considered as a medical emergency.45 However, it is still not clear if this is a class side effect or not.46,47 Other studies also indicate possible adverse effects such as dehydration and a rise in plasmatic levels of cholesterol.3,8,10
Conclusions
The new SGLT-2 selective inhibitor drug class represents an innovative therapeutic approach for blood glucose control in type II diabetic patients, as its action is independent from the insulin hormone. In addition, this new class displays other advantages such as body weight and blood pressure reduction, which are highly desirable in several related diseases. For this reason, this therapeutic class has been approved in the past years for diabetes treatment both as a monotherapy and associated with other antidiabetic agents all over the world.
From the drug design and medicinal chemistry points of view, there are also a lot of possibilities to be explored, once a restricted chemical space has been investigated towards this specific molecular target. It seems that there is still a lot of room for discovery and design in terms of potential pharmacophore subunits that might present high affinity for SGLT-2, thus opening possibilities for novel generation inhibitors.
Finally, the clinical results to be obtained in the next years on ongoing large trials will play a critical role in defining the precise risk–benefit relation associated with this new antidiabetic class, determining the bases for its consolidation as a valuable therapeutic alternative for diabetes treatment, especially for obese patients or for the ones also affected by cardiovascular disorders.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgments
The authors thank the Faculty of Pharmacy of the Federal University of Rio de Janeiro (FF-UFRJ, BR), the Laboratory of Evaluation and Synthesis of Bioactive Substances of the Federal University of Rio de Janeiro (LASSBio-UFRJ, BR) and the funding agencies CAPES (BR), CNPq (BR) and FAPERJ (BR).
Biographies

Paula Nogueira da Silva
Paula Nogueira da Silva studied pharmacy at Fluminense Federal University, Brazil. As a pharmacy student, she joined in 2014 the research team from the Laboratory of Organic Synthesis and Medicinal Chemistry (LaSOQuiM), at the Faculty of Pharmacy of the Federal University of Rio de Janeiro, as a scholarship holder of the Carlos Chagas Filho Foundation for Support of Research in the State of Rio de Janeiro (FAPERJ-BR). Paula was engaged in projects aiming at the identification of drug candidates for cardiovascular and metabolic disorders. Currently, she is doing her residency in the Multiprofessional Oncology Residency Program at the Brazilian National Cancer Institute.

Raissa Alves da Conceição
Raissa Alves da Conceição studied pharmacy at the Federal University of Rio de Janeiro (UFRJ), Brazil. From 2014 until 2017, she was a scholarship holder of the Institutional Scientific Initiation Scholarship Program (PIBIC) at the Laboratory of Organic Synthesis and Medicinal Chemistry (LaSOQuiM), in the Faculty of Pharmacy of UFRJ. During this period, she was engaged in projects aiming at the development of novel drug candidates for neurodegenerative and metabolic disorders. Currently, she is doing her master's in pharmaceutical sciences at the same university, developing a project on the design, synthesis, and in silico and in vitro evaluation of anti-prion drug candidates.

Rodolfo do Couto Maia
Rodolfo Maia is a pharmacist and a young researcher who tries to build his background as broad as possible. He obtained his M.Sc. and D.Sc. degrees in medicinal chemistry in Brazil while working at LASSBio, a major Brazilian medicinal chemistry group at the Federal University of Rio de Janeiro (UFRJ). After this, he obtained some experience in the intellectual property area and then returned to the medicinal chemistry field as a post-doc at University College London for a year. He also acted as a lecturer in medicinal and organic chemistry. Nowadays, he is back at UFRJ working mainly on chemical libraries and neuropathic pain projects.

Maria Leticia de Castro Barbosa
Maria Leticia Barbosa studied pharmacy at the Federal University of Juiz de Fora, Brazil. She obtained her M.Sc. (2009) and D.Sc. degrees in chemistry (2013) at the Federal University of Rio de Janeiro (UFRJ), Brazil, under the supervision of Prof. Eliezer Barreiro and Prof. Lidia Lima, working in the medicinal chemistry field. One year of her PhD was spent at Eberhard Karls Universität Tübingen, Germany, under the supervision of Prof. Stefan Laufer, with a DAAD scholarship. Currently, she is a permanent professor of medicinal chemistry at the Faculty of Pharmacy of UFRJ and head of the Laboratory of Organic Synthesis and Medicinal Chemistry (LaSOQuiM).
References
- Skyler J. S. J. Med. Chem. 2004;47:4113–4117. doi: 10.1021/jm0306273. [DOI] [PubMed] [Google Scholar]
- Kim M. J., Lee J., Kang S. Y., Lee S. H., Son E. J., Jung M. E., Lee S. H., Song K. S., Lee M., Han H. K., Kim J., Lee J. Bioorg. Med. Chem. Lett. 2010;20:3420–3425. doi: 10.1016/j.bmcl.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Wilding J. P. Metabolism. 2014;63:1228–1237. doi: 10.1016/j.metabol.2014.06.018. [DOI] [PubMed] [Google Scholar]
- Global Report on Diabetes, http://www.who.int, (accessed May 2016).
- Kahn S. E., Cooper M. E., Del Prato S. Lancet. 2014;383:1068–1083. doi: 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDF Diabetes Atlas 8th edition, 2017, http://www.diabetesatlas.org, (accessed May 2018).
- Donath M. Y. Nat. Rev. Drug Discovery. 2014;13:465–476. doi: 10.1038/nrd4275. [DOI] [PubMed] [Google Scholar]
- Hasan F. M., Alsahli M., Gerich J. E. Diabetes Res. Clin. Pract. 2014;104:297–322. doi: 10.1016/j.diabres.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Bauer A., Brönstrup M. Nat. Prod. Rep. 2014;31:35–60. doi: 10.1039/c3np70058e. [DOI] [PubMed] [Google Scholar]
- Miller E. M. J. Fam. Pract. 2017;66:S3–S16. [PubMed] [Google Scholar]
- Mende C. W. Curr. Med. Res. Opin. 2017;33:541–551. doi: 10.1080/03007995.2016.1271779. [DOI] [PubMed] [Google Scholar]
- Diamant M., Morsink L. M. Lancet. 2013;382:917–918. doi: 10.1016/S0140-6736(13)60902-2. [DOI] [PubMed] [Google Scholar]
- Sanders L. J. Diabetes Spectr. 2002;15:56–60. [Google Scholar]
- Meng W., Ellsworth B. A., Nirschl A. A., McCann P. J., Patel M., Girota R. N., Wu G., Sher P. M., Morrison E. P., Biller S. A., Zahler R., Deshpande P. P., Pullockaran A., Hagan D. L., Morgan N., Taylor J. R., Obermeier M. T., Humphreyes W. G., Khanna A., Discenza L., Robertson J. G., Wang A., Han S., Wetterau J. R., Janovitz E. B., Flint O. P., Whaley J. M., Washburn W. N. J. Med. Chem. 2008;51:1145–1149. doi: 10.1021/jm701272q. [DOI] [PubMed] [Google Scholar]
- Nomura S., Yamamoto Y., Matsumura Y., Ohba K., Sakamaki S., Kimata H., Nakayama K., Kuriyama C., Matsushita Y., Ueta K., Tsuda-Tsukimoto M. ACS Med. Chem. Lett. 2014;5:51–55. doi: 10.1021/ml400339b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V. Am. J. Physiol. 2011;300:C6–C8. doi: 10.1152/ajpcell.00444.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen C. Ann. Acad. Sci. Fr. 1835;15:178. [Google Scholar]
- Ehrenkranz J. R. L., Lewis N. G., Kahn C. R., Roth J. Diabetes/Metab. Res. Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- Blaschek W. Planta Med. 2017;83:985–993. doi: 10.1055/s-0043-106050. [DOI] [PubMed] [Google Scholar]
- Washburn W. N. J. Med. Chem. 2009;52:1785–1794. doi: 10.1021/jm8013019. [DOI] [PubMed] [Google Scholar]
- Yao C.-H., Song J.-S., Chen C.-T., Yeh T.-K., Hung M.-S., Chang C.-C., Liu Y.-W., Yuan M.-C., Hsieh C.-J., Huang C.-Y., Wang M.-H., Chiiu C.-H., Hsieh T.-C., Wu S.-H., Hsiao W.-C., Chu K.-F., Tsai C.-H., Chao Y.-S., Lee J.-C. J. Med. Chem. 2011;54:166–178. doi: 10.1021/jm101072y. [DOI] [PubMed] [Google Scholar]
- Tsujihara K., Hongu M., Saito K., Inamasu M., Arakawa K., Oku A., Matsumotu M. Chem. Pharm. Bull. 1996;44:1174–1180. doi: 10.1248/cpb.44.1174. [DOI] [PubMed] [Google Scholar]
- Hongu M., Tanaka T., Funami N., Saito K., Arakawa K., Matsumotu M., Tsujihara K. Chem. Pharm. Bull. 1998;46:22–33. doi: 10.1248/cpb.46.22. [DOI] [PubMed] [Google Scholar]
- Hongu M., Funami N., Takahashi Y., Saito K., Arakawa K., Matsumotu M., Yamakita H., Tsujihara K. Chem. Pharm. Bull. 1998;46:1545–1555. doi: 10.1248/cpb.46.1545. [DOI] [PubMed] [Google Scholar]
- Tsujihara K., Hongu M., Saito K., Kawanishi H., Kuriyama K., Matsumoto M., Oku A., Ueta K., Tsuda M., Saito A. J. Med. Chem. 1999;42:5311–5324. doi: 10.1021/jm990175n. [DOI] [PubMed] [Google Scholar]
- Katsuno K., Fujimori Y., Takemura Y., Hiratochi M., Itoh F., Komatsu Y., Fujikura H., Isaji M. J. Pharmacol. Exp. Ther. 2007;320:323–330. doi: 10.1124/jpet.106.110296. [DOI] [PubMed] [Google Scholar]
- Fujimori Y., Katsuno K., Nakashima I., Ishikawa-Takemura Y., Fujikura H., Isaji M. J. Pharmacol. Exp. Ther. 2008;327:268–276. doi: 10.1124/jpet.108.140210. [DOI] [PubMed] [Google Scholar]
- Ohtake Y., Sato T., Kobayashi T., Nishimoto M., Taka N., Takano K., Yamamoto K., Ohmori M., Yamaguchi M., Takami K., Yeu S.-Y., Ahn K.-H., Matsuoka H., Morikawa K., Suzuki M., Hagita H., Ozawa K., Yamaguchi K., Kato M., Ikeda S. J. Med. Chem. 2012;55:7828–7840. doi: 10.1021/jm300884k. [DOI] [PubMed] [Google Scholar]
- Ellsworth B. A., Meng W., Patel M., Girotra R. N., Wu G., Sher P. M., Hagan D. L., Obermeier M. T., Humphreys W. G., Robertson J. G., Wang A., Han S., Waldron T. L., Morgan N. N., Whaley J. M., Washburn W. N. Bioorg. Med. Chem. Lett. 2008;18:4770–4773. doi: 10.1016/j.bmcl.2008.07.109. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration (FDA) Center for drug evaluation and research: Final Risk Evaluation and Mitigation Strategy (REMS) Review. Application number: 202293Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/202293orig1s000riskr.pdf, (accessed May 2018).
- Nomura S., Sakamaki S., Hongu M., Kawanishi E., Koga Y., Sakamoto T., Yamamoto Y., Ueta K., Kimata H., Nakayama K., Tsuda-Tsukimoto M. J. Med. Chem. 2010;53:6355–6360. doi: 10.1021/jm100332n. [DOI] [PubMed] [Google Scholar]
- Kang S. Y., Kim M. J., Lee J. S., Lee J. Bioorg. Med. Chem. Lett. 2011;21:3759–3763. doi: 10.1016/j.bmcl.2011.04.063. [DOI] [PubMed] [Google Scholar]
- Lee S. H., Song K. S., Kim J. Y., Kang M., Lee J. S., Cho S. H., Park H. J., Kim J., Lee J. Bioorg. Med. Chem. 2011;19:5813–5832. doi: 10.1016/j.bmc.2011.08.014. [DOI] [PubMed] [Google Scholar]
- Xu B., Feng Y., Cheng H., Song Y., Lv B., Wu Y., Wang C., Li S., Xu M., Du J., Peng K., Dong J., Zhang W., Zhang T., Zhu L., Ding H., Sheng Z., Welihinda A., Roberge J. Y., Seed B., Chen Y. Bioorg. Med. Chem. Lett. 2011;21:4465–4470. doi: 10.1016/j.bmcl.2011.06.032. [DOI] [PubMed] [Google Scholar]
- Ikegai K., Imamura M., Suzuki T., Nakanishi K., Murakami T., Kurosaki E., Noda A., Kobayashi Y., Yokota M., Koide T., Kosakai K., Ohkura Y., Takeuchi M., Tomiyama H., Ohta M. Bioorg. Med. Chem. 2013;21:3934–3948. doi: 10.1016/j.bmc.2013.03.067. [DOI] [PubMed] [Google Scholar]
- Zhang S., Wang Y. L., Wei Q.-C., Xu W.-R., Tang L.-D., Zhao G.-L., Wang J.-W. Chin. Chem. Lett. 2013;24:429–432. [Google Scholar]
- Cinti F., Moffa S., Impronta F., Cefalo C. M. A., Sun V. A., Sorice G. P., Mezza T., Giaccari A. Drug Des., Dev. Ther. 2017;11:2905–2919. doi: 10.2147/DDDT.S114932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki E., Ogasawara H. Pharmacol. Ther. 2013;139:51–59. doi: 10.1016/j.pharmthera.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Kakinuma H., Oi T., Hashimoto-Tsuchiya Y., Arai M., Kawakita Y., Fukasawa Y., Iida I., Hagima N., Takeuchi H., Chino Y., Asami J., Okumura-Kitajima L., Io F., Yamamoto D., Miyata N., Takahashi T., Uchida S., Yamamoto K. J. Med. Chem. 2010;53:3247–3261. doi: 10.1021/jm901893x. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Kawanishi E., Koga Y., Sakamaki S., Sakamoto T., Ueta K., Matsushita Y., Kuriyama C., Tsuda-Tsukimoto M., Nomura S. Bioorg. Med. Chem. Lett. 2013;23:5641–5645. doi: 10.1016/j.bmcl.2013.08.042. [DOI] [PubMed] [Google Scholar]
- Lapuerta P., Zambrowicz B., Strumph P., Sands A. Diabetes Vasc. Dis. Res. 2015;12:101–110. doi: 10.1177/1479164114563304. [DOI] [PubMed] [Google Scholar]
- Garg S. K., Henry R. R., Banks P., Buse J. B., Davies M. J., Fulcher G. R., Pozzilli P., Gesty-Palmer D., Lapuerta P., Simó R., Danne T., McGuire D. K., Kushner J. A., Peters A., Strumph P. N. Engl. J. Med. 2017;377:2337–2348. doi: 10.1056/NEJMoa1708337. [DOI] [PubMed] [Google Scholar]
- Kramer C. K., Zinman B. Eur. Heart J. 2016;37:3201–3202. doi: 10.1093/eurheartj/ehw158. [DOI] [PubMed] [Google Scholar]
- Imprialos K., Faselis C., Boutari C., Stavropoulos K., Athyros V., Karagiannis A., Doumas M. Curr. Pharm. Des. 2017;23:1510–1521. doi: 10.2174/1381612823666170124123927. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration (FDA) Drug Safety Communications: FDA revises labels of SGLT2 inhibitors for diabetes to include warnings about too much acid in the blood and serious urinary tract infections. https://www.fda.gov/downloads/Drugs/DrugSafety/UCM475487.pdf, (accessed May 2018).
- Kum-Nji J. S., Gosmanov A. R., Steinberg H., Dagogo-Jack S. J. Diabetes Complications. 2017;31:611–614. doi: 10.1016/j.jdiacomp.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Monami M., Nreu B., Zannoni S., Lualdi C., Mannucci E. Diabetes Res. Clin. Pract. 2017;130:53–60. doi: 10.1016/j.diabres.2017.04.017. [DOI] [PubMed] [Google Scholar]