

 Heterocyclic–fatty acid derivatives are a new class of compounds with a broad range of biological activities.
Heterocyclic–fatty acid derivatives are a new class of compounds with a broad range of biological activities.
Abstract
Monastrol is a small cell-permeable heterocyclic molecule that is recognized as an inhibitor of mitotic kinesin Eg5. Heterocyclic–fatty acid derivatives are a new class of compounds with a broad range of biological activities. This work describes a comparative study of the in vitro antitumoral activity of a series of new long-chain monastrol analogues against rat glioblastoma cells. The novel analogues C6-substituted monastrol and oxo-monastrol were synthesized via Biginelli multicomponent condensation of fatty β-ketoester in good yields using a simple approach catalyzed by nontoxic and free-metal sulfamic acid. Following synthesis, their in vitro antitumoral activities were investigated. Notably, all analogues tested were active against rat glioblastoma cells. Superior activity was observed by analogues derived from palmitic and stearic fatty acid chains; these compounds were the most potent molecules, showing 13-fold higher potency than monastrol with IC50 values of 5.11 and 6.85 μM, respectively. These compounds could provide promising new lead derivatives for more potent antitumor drugs.
Introduction
Multicomponent reactions (MCRs) are chemical reactions involving three or more chemical compounds in one step, generating final heterocyclic compounds that contain majority parts of precursors.1,2 Due to their atom economy and typically simple and low-energy requirements, they are powerful tools for organic synthesis and medicinal chemistry in the synthesis of diverse and complex structures organics.3,4
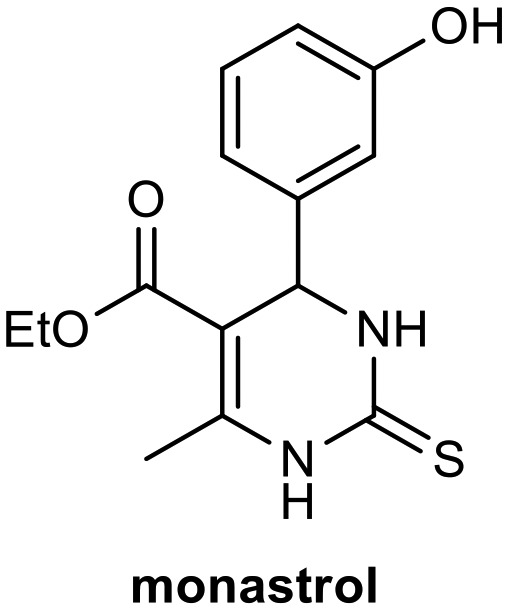
The most important MCRs are the Biginelli and Hantzsch reactions. Biginelli reactions involve simple condensation of aldehydes, urea, or thiourea and acetoacetates, generating compounds known as Biginelli compounds or dihydropyrimidinones (DHPMs).5 A wide range of biological processes are directly affected by DHPMs, which have high pharmacological potential (with anti-inflammatory, bactericidal, antiviral, antifungal, and antioxidant activities) and have important applications as biological modulators in medicinal chemistry.6–10 Due to structural similarities with dihydropyridines (DHPs) including Hantzsch adducts such as nifedipine (Fig. 1), a modulator of the calcium channel obtained from Hantzsch MCRs, DHPMs have been investigated as calcium channel blockers.11–13 However, they are also potential antitumor agents, involved in blocking mitosis by inhibiting kinesin Eg5, a motor protein.14,15 Studies using small-molecule libraries (>500 g mol–1) have demonstrated this ability of DHPMs, mainly monastrol (Fig. 1).15–19 Compounds capable of disrupting cell mitosis (ordered series of events primarily mechanical in that identical copies of the genome are moved within the cell) have shown promise for the treatment of various tumor cell lines.14
Fig. 1. Nifedipine and monastrol calcium channel blockers.

There is a great need for novel synthesized drugs with possible applications as antitumor agents, due to the large mortality rate associated with cancer and wide variety of cancer types. Cancer can be conceived as a generic term for a large group of diseases that generally involve uncontrolled cell division and subsequent tumor formation. Such tumors can progress rapidly and generate metastases in various parts of the body or affect other vital functions.20 Brain tumors are one of the most diverse and interesting categories of tumors, being challenging in their biology and pathology.21 Glioblastoma is the main and most malignant type of central nervous system (CNS) tumor, and it is difficult to treat, which limits the prognosis in such patients. Patients with this type of cancer have about 14 months of survival after diagnosis even with treatment, and this treatment presents several side effects beyond the comorbidities associated with this pathology. Hence, it is necessary to search for new treatments with greater effectiveness that can prolong the life expectancy of such individuals.22
Russowsky et al. synthesized a series of monastrol analogs and investigated antitumoral activity against several human cancer cell lines. In that work, piperastrol was considered a potent antitumor agent against breast (MCF 7), kidney (786-0), colon (HT-29), melanoma (UACC62), and ovary (OVCAR03) tumor cell lines.23 Excellent results were obtained with piperastrol and monastrol derivatives, which were more active than oxo-derivatives, suggesting the importance of the presence of sulfur for anti-proliferative activity.
According to literature, is well known that the molecular combinations derived from two biologically active molecules can result in novel hybrid molecules with enhanced biological activities.24 Biginelli and other heterocyclic compounds are interesting core structures for the development of new bioactive compounds. Fatty acids are derived from renewable raw materials and exhibit various biological activities.
In this context, several researchers are synthesizing these two bioactive components to yield bioactive hybrid molecules with specific desirable features. Heterocyclic–fatty acid hybrid derivatives are a new class of heterocyclic compounds with a broad range of biological activities and significance in the field of medicinal chemistry. Over the last few years, many studies have emphasized the significance of such derivatives.25
Building on our previous antitumoral studies using fatty acid derivatives,26–29 this work investigated the antitumoral activity of novel monastrol and oxo-monastrol fatty alkyl C6-substituted analogues against malignant rat GBM cells (C6); the compounds were synthesized via Biginelli condensation MCRs using a simple approach catalyzed by nontoxic and free-metal sulfamic acid. In this context, we selected palmitic, stearic, and oleic fatty acid derivatives to evaluate the effects of these slightly different molecular structures.
Results and discussion
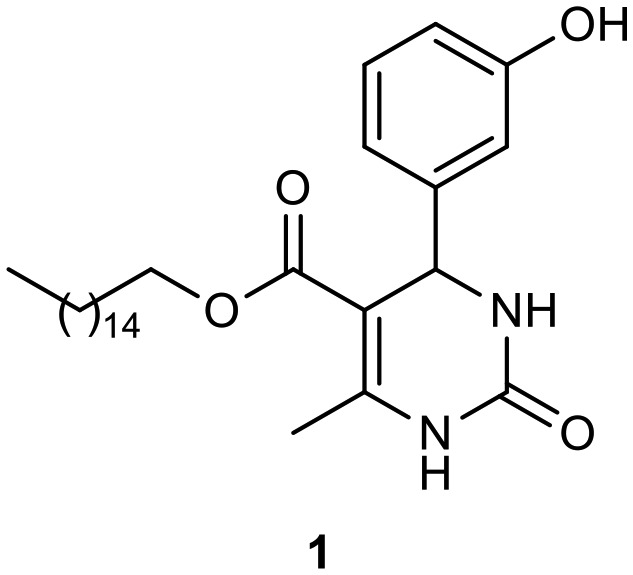
Previously, we described the synthesis of fatty analogs of dihydropyrimidinones C5-substituted using the Biginelli multicomponent protocol. After synthesis, their antitumoral activities against glioma cell lines (C6 rat and U-138-MG human) were investigated.26 The compounds derived from palmitic and oleic chains obtained from 3-hydroxybenzaldehyde and urea or thiourea reduced cell viability the most at 50 μM (ca. 70–80%) and the analogue oxo-monastrol–palmitic acid (1, Fig. 2) was the most potent, reducing cell viability by ca. 50% at 10 μM. Moreover, the DHPM–fatty acids did not increase the mortality rate of nontumor cells by at least 20-fold and 4-fold the active glioma concentration (25 mM). In addition, the compounds showed a large safety range for neural cells demonstrating non-toxicity to organotypic hippocampal culture. In another study, the antiproliferative activities of new fatty acid analogs of polyhydroquinoline (hybrid PHQ–fatty acids) against the glioma cell line were investigated.28 The fatty compounds were synthesized via the Hantzsch multicomponent protocol from several aldehydes and dimedone. The stearic fatty alkyl PHQ hybrid (2, Fig. 2) derived from 3-hydroxybenzaldehyde was found to possess the most potent biological response, reducing cell viability by 40% at 5 μM.
Fig. 2. Hybrids monastrol–palmitic acid (1) and PHQ–stearic acid (2) investigated against glioma cell lines.

In these studies, compared to monastrol, compounds 1 and 2 showed better potency against glioma cells (C6 rat and U-138-MG human). These results indicate that the hydroxyl group in the aromatic ring of different heterocyclic cores and the presence of a long fatty chain are essential for the antitumoral activities of the new heterocyclic–fatty acid hybrids. This also suggests that the increased lipophilicity of fatty acid DHPM and PHQs may be a promising approach for the development of new antitumor compounds.
Based on previous studies, in the present study a series of novel analogues monastrol and oxo-monastrol fatty alkyl C6-substituted were synthesized and their in vitro antitumoral activities were investigated against rat glioblastoma cells.
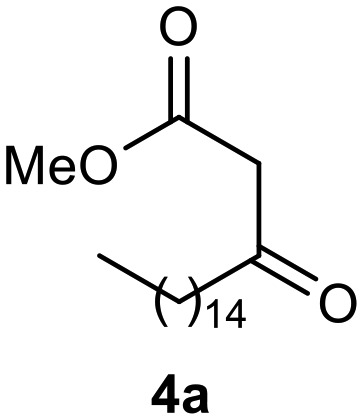
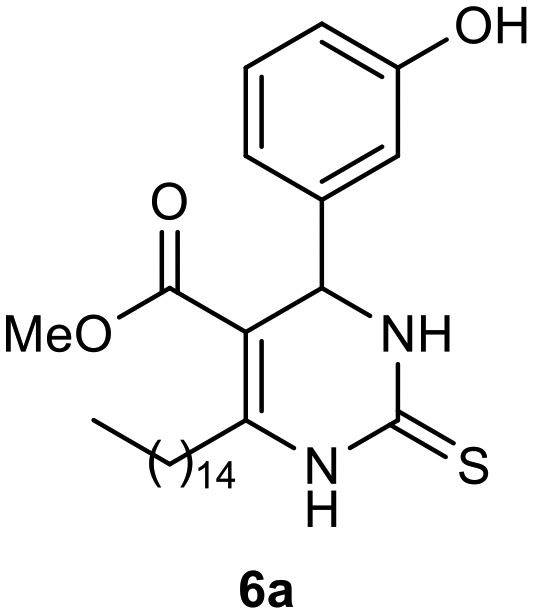
To examine their biological responses to structural variations, a positive-control monastrol (see Fig. 1) was synthesized using the Biginelli multicomponent protocol between ethyl acetoacetate, 3-hydroxybenzaldehyde (3), and thiourea in methanol using InCl3 as a catalyst. The presented spectroscopic data are in agreement with the literature.23 In addition, a fatty positive control, analogue oxo-monastrol C5-substituted (1, Fig. 2) derived from palmitic chain, was synthesized at good yields from palmitic acetoacetate in accordance with previous work.26
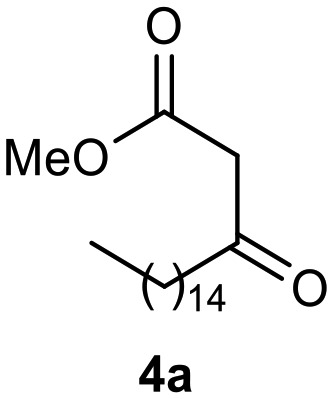
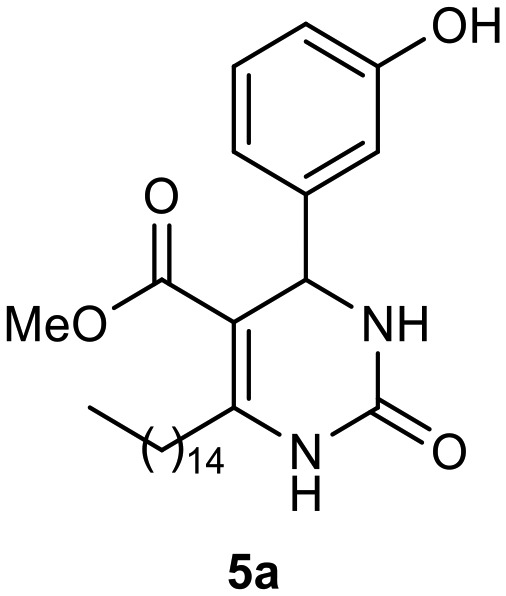
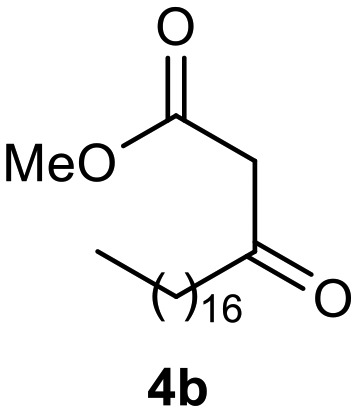
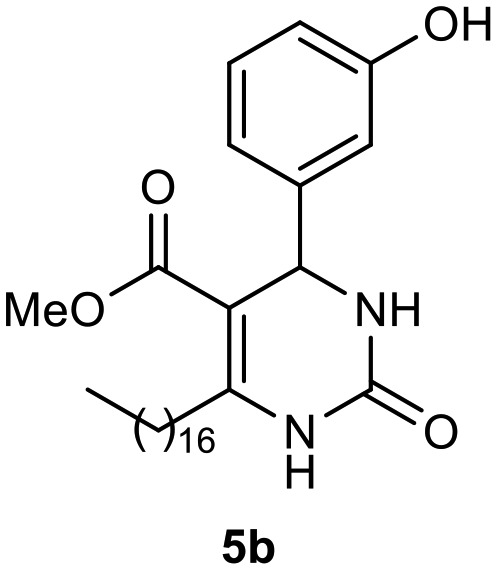
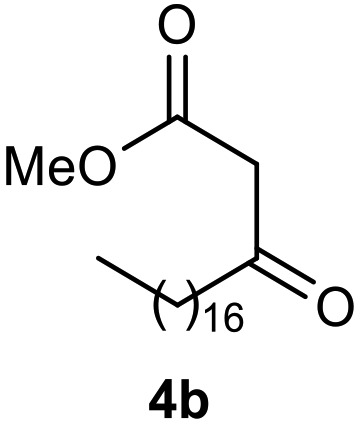
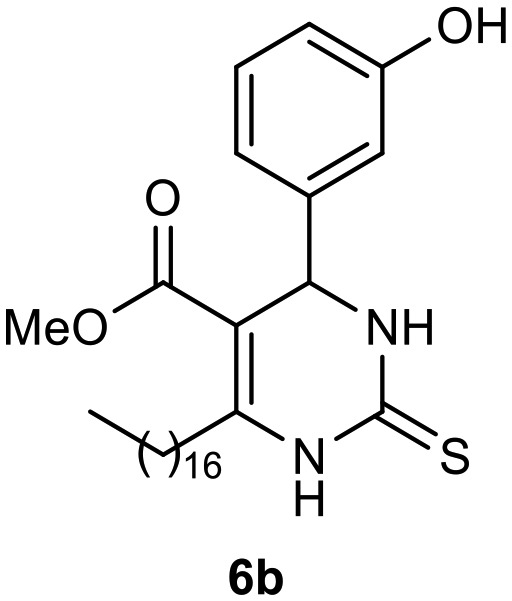
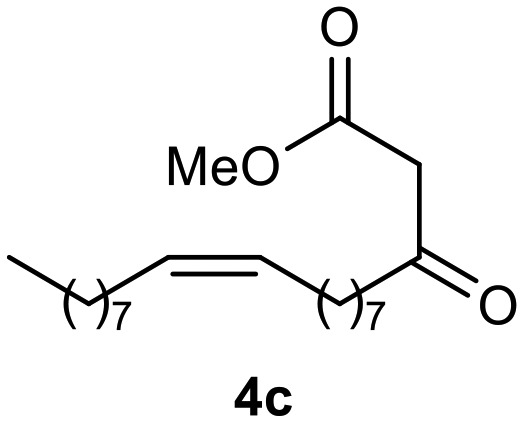
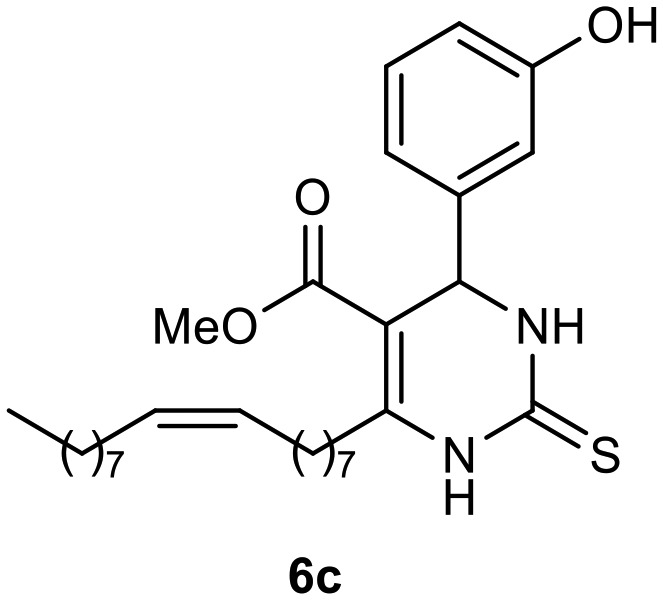
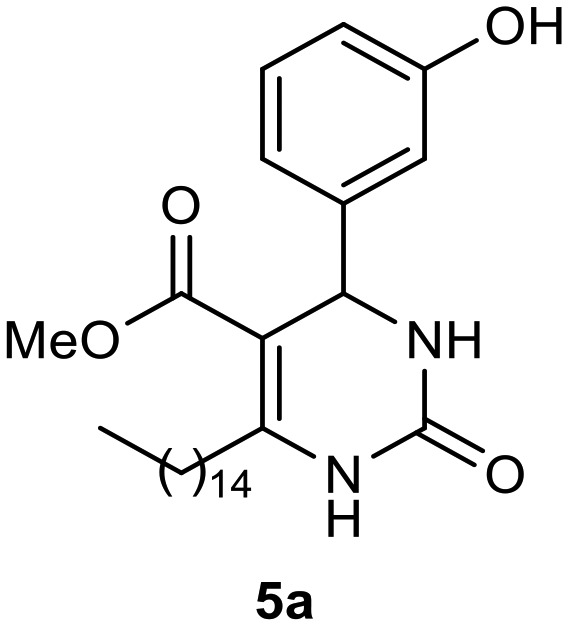
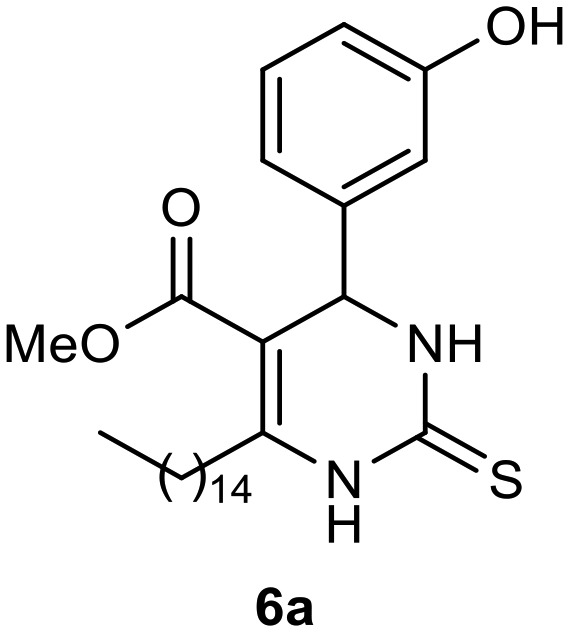
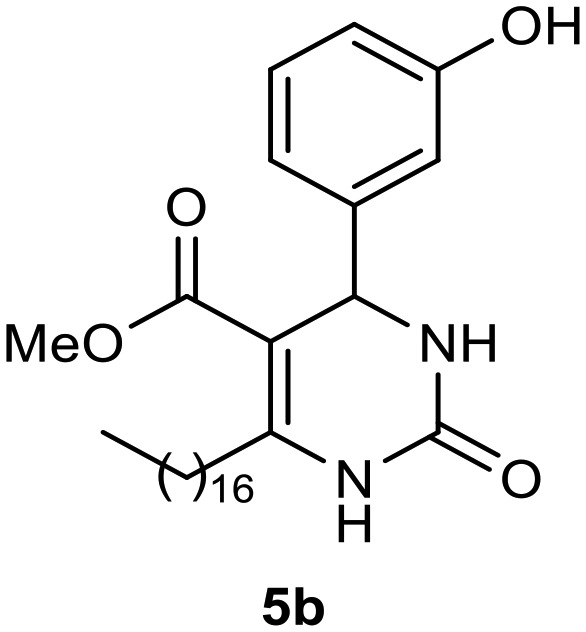
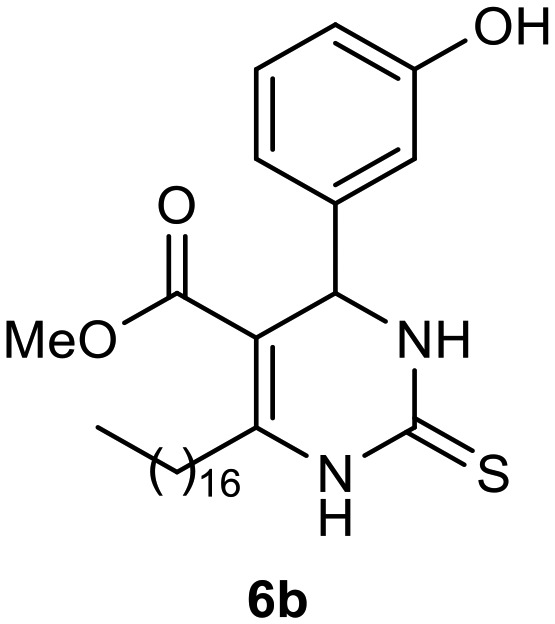
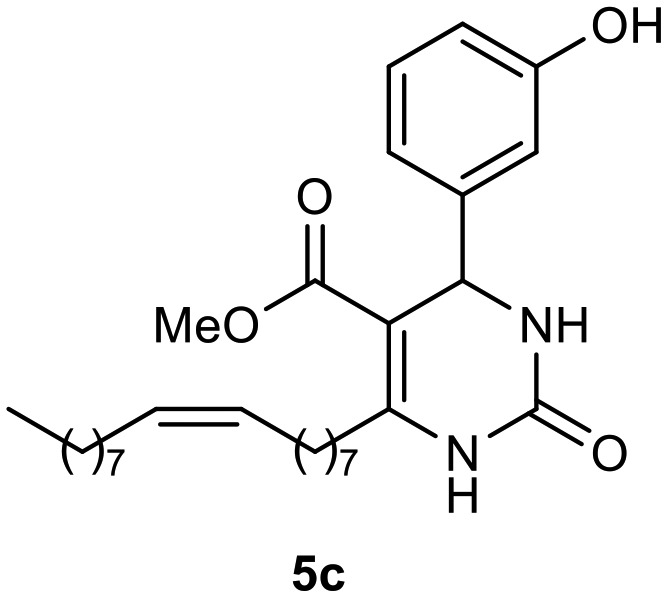
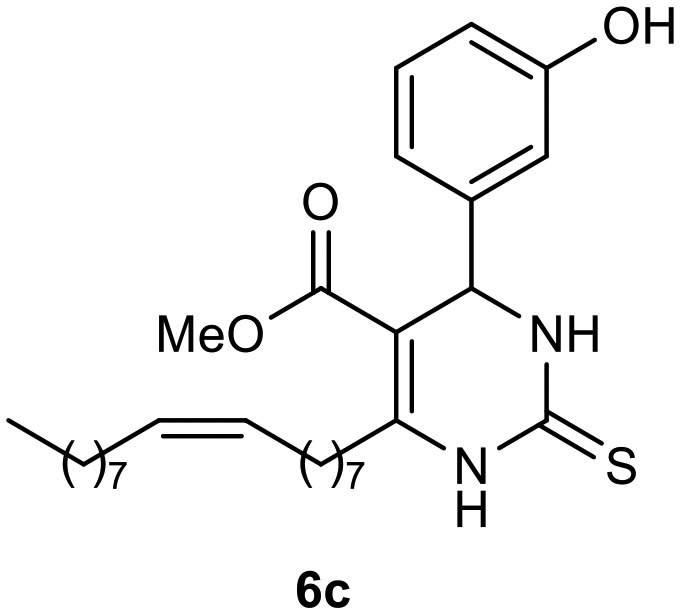
Afterward, the novel monastrol analogues fatty alkyls C6-substituted were synthesized using Biginelli multicomponent protocol. Initially, the fatty β-ketoesters 4a–c derived from palmitic (C16:0), stearic (C18:0), or oleic (cis-C18:1) acids were obtained by acylation of Meldrum acid with DCC, DMAP, and pyridine, followed by methanolysis, in accordance with literature.29 Next, the analogues 6-substituted oxo-monastrol- and monastrol–fatty acid 5a–c and 6a–c were synthesized from urea or thiourea, respectively, β-ketoesters 4a–c and 3-hydroxybenzaldehyde (3), under metal-free conditions using sulfamic acid as a catalyst (Scheme 1).
Scheme 1. Multicomponent synthesis of new DHPMs–fatty acid 5a–c and 6a–c under metal-free catalysis conditions.

The yields of 5a–c and 6a–c are showed in Table 1. These products were purified by column chromatography and characterized by usual spectroscopic methods.
Table 1. Yields of new analogues oxo-monastrol- and monastrol–fatty acids 5, 6a–c.
| Entry | Fatty β-ketoesters | C6-Substituted fatty alkyl DHPMs | Yield (%) |
| 1 |
|
|
78 |
| 2 |
|
|
85 |
| 3 |
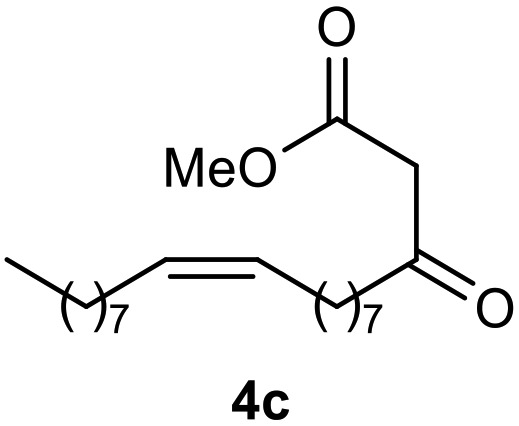
|
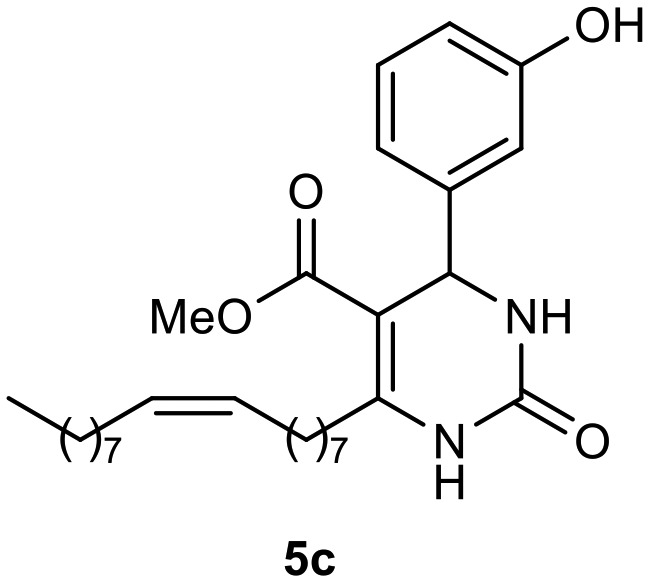
|
78 |
| 4 |
|
|
81 |
| 5 |
|
|
74 |
| 6 |
|
|
80 |
Antitumoral activity
After synthesis, the antitumoral activities of compounds 5, 6a–c were evaluated and compared to monastrol (positive control) and C5-substituted derivative 1 (fatty positive control). The MTT assay was performed 48 h after exposing rat glioma cells to each compound at selected concentrations (5, 10, 25, and 50 μM). In the control group, we used the same amount of DMSO (vehicle) in which the new molecules were solubilized.
As shown in Fig. 3, compounds 5a and 5b (palmitic and stearic oxo-monastrol derivate, respectively) reduced cell viability at concentrations as low as 5 μM while compound 5c (oleic acid oxo-monastrol derivate) was effective starting at only 10 μM. Compounds 6a and 6b (palmitic and stearic monastrol derivate, respectively) also significantly reduced cell viability in a concentration-dependent manner, showing a reduction of about 50% at a concentration of 5 μM.
Fig. 3. MTT assay results for rat glioma (C6) cells after 48 h of treatment with concentrations of 5, 10, 25, and 50 μM. C = DMSO-treated control cells. ANOVA followed by Tukey's post-test, **p < 0.01 and ***p < 0.001 with respect to the control.

These results demonstrate that these compounds were more effective than the other compounds, providing evidence that the C6 substituent influenced the biological activity of the molecule while the size of the chain did not show interference. Similar results were found in the cell-counting assay (Fig. 4).
Fig. 4. Counts of rat glioma (C6) cells after 48 h of treatment at concentrations of 5, 10, 25, and 50 μM. C = DMSO-treated control group for each compound. ANOVA followed by Tukey's post-test, **p < 0.01 and ***p < 0.001 with respect to controls.

According to Russowsky et al.,23 compounds with a thio-group are more effective at reducing tumor cell proliferation than compounds with an oxo-group. Our results support this notion in that fatty monastrol analogue showed antitumoral activity while oxo-monastrol did not (in addition, UACC.62 cells treated with monastrol showed significant changes in morphology, which were not observed with oxo-monastrol).
When we compared IC50 values (Table 2) in the MTT assay, the results indicated that adding a fatty chain to molecules increased their antitumoral activity. This may suggest that increased lipophilicity could contribute to a better Eg5 inhibition improving the transport of molecules across biological barriers by active processes mediated by fatty acid binding proteins (FABP).30,31
Table 2. IC50 and log P values from monastrol, oxo-monastrol- and monastrol–fatty acid analogues.
| Molecule | IC50 a (μM) | log P | Molecule | IC50 a (μM) | log P |
| Oxo- | Thio- | ||||
|
16.68 (14.47–19.23) | 6.06 (±0.47) |
|
87.83 (58.34–132.2) | 1.57 (±0.47) |
|
79.37 (49.76–126.6) | 5.72 (±0.47) |
|
5.11 (4.13–6.32) | 7.15 (±0.47) |
|
21.77 (18.65–25.41) | 5.47 (±0.47) |
|
6.85 (6.18–7.60) | 7.98 (±0.47) |
|
166.4 (78.40–353.00) | 6.23 (±0.47) |
|
38.36 (25.87–56.90) | 7.66 (±0.47) |
aWith their respective confidence limits (CL 95%).
As shown in Table 2, compounds 6a and 6b were the most potent molecules, with IC50 values of 5.11 (4.13–6.32) and 6.85 (6.18–7.60) μM, respectively, making them 13 times more potent than monastrol. These molecules were also 2.5 times more potent than analogue C5-substituted fatty DHPM 1 (fatty positive control).
In conclusion, this work described the synthesis of novel analogues C6-substituted monastrol and oxo-monastrol fatty acids from fatty acid families at good yields (60–85%). We tested the antitumoral activities of each compound against C6 rat glioma cells using the MTT assay. All of the molecules tested exhibited antitumoral effects, and the molecules of saturated fat chains derived from thiourea (monastrol–fatty acids derivatives), induced a significant drop in cell viability at low concentrations. The effect followed a concentration–response curve, qualifying the molecules 6a and 6b as the most potent. Cell-counting tests demonstrated the action of monastrol–fatty acids in that all such compounds decreased the number of tumor cells after treatment. In addition, these results suggest that all C6-substituted monastrol- and oxo-monastrol–fatty acid analogues may have higher antitumoral activities than monastrol itself, except for molecule 5c derived from oleic acid and urea.
Finally, our results indicate that compounds 6a and 6b, derived from palmitic (C16:0) and stearic acid (C18:0), were the most potent molecules with IC50 values of 5.11 and 6.85 μM, respectively, making them promising compounds for future experiments in vivo. The actions of these compounds are better than monastrol itself, a kinesin-inhibiting agent.
Experimental section
Apparatus and chemistry
Sulfamic acid (98 wt%), urea and thiourea were purchased from Sigma-Aldrich Chemical and 3-hydroxybenzaldehyde was bought from Alfa Aesar. All organic solvents used for synthesis were of analytical grade. Solvents were dried and freshly distilled. Column chromatography was performed on a silica gel 60 Å (ACROS Organics, 0.035–0.070 mesh). Reactions were monitored using thin-layer chromatography (TLC) on silica gel plates (Merck 60GF245) with hexane:ethyl acetate as eluent. Spots were visualized using iodine. Yields refer to chromatographically and spectroscopically homogeneous materials. Melting points were obtained on a Fisatom 430D. NMR spectra were recorded using a Brucker AVANCE III 400 spectrometer (1H at 400 MHz and 13C at 100 MHz) in deuterochloroform (CDCl3) as the solvent. The chemical shift data are reported in units of δ (ppm) downfield from tetramethylsilane (TMS), which was used as an internal standard. The coupling constants (3J) are reported in Hz and refer to apparent peak multiplicities.
Synthesis
General procedure for the synthesis of fatty acid oxo-monastrol and monastrol analogues 5, 6a–c
β-Ketoesters (1 mmol) 4a–c, 3-hydroxybenzaldehyde (3.1 mmol), urea or thiourea (1.3 mmol), and sulfamic acid (0.6 mmol) were added to a round-bottom flask of 25 mL, and the reaction contents were dissolved in methanol (3 mL). The reaction was refluxed for 24 h and monitored by TLC. After this interval, the methanol was removed and the crude reaction was purified by column chromatography (7 : 3 hexane : ethyl acetate).
Methyl 4-(3-hydroxyphenyl)-2-oxo-6-pentadecyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5a)
C27H42N2O4; molecular weight 458.63: g mol–1; yield: 78%; 1H NMR (400 MHz, CDCl3) δ 7.93 (br. s., 1H), 6.99–7.19 (m, 1H), 6.80 (br. s., 1H), 6.71 (d, J = 7.82 Hz, 1H), 6.75 (d, J = 7.83 Hz, 1H), 5.27 (br. s., 1H), 3.62 (br. s., 3H), 2.52–2.75 (m, 2H), 1.45–1.64 (m, 2H), 1.16–1.38 (m, 24H), 0.89 (t, J = 6.85 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.0, 156.8, 154.1, 150.8, 144.9, 129.9, 118.0, 115.3, 113.3, 100.9, 55.1, 51.3, 50.6, 31.9, 29.7, 29.7, 29.6, 29.6, 29.4, 28.3, 22.7, 14.1.
Methyl 6-heptadecyl-4-(3-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5b)
C29H46N2O4; molecular weight 486.69 g mol–1; yield: 85%; 1H NMR (400 MHz, CDCl3) δ 8.03 (br. s., 1H), 7.09 (t, J = 7.95 Hz, 1H), 6.83 (s, 1H), 6.75 (d, J = 7.82 Hz, 1H), 6.71 (d, J = 8.07 Hz, 1H), 6.49 (br. s., 1H), 5.27 (d, J = 2.69 Hz, 1H), 3.63 (s, 3H), 2.49–2.71 (m, 2H), 1.44–1.61 (m, 2H), 1.19–1.35 (m, 24H), 0.90 (t, J = 7.10 Hz, 3H). NMR 13C (100 MHz, CDCl3) δ 166.0, 156.7, 154.4, 150.7, 144.9, 129.9, 118.1, 115.4, 113.2, 100.9, 55.1, 51.3, 31.9, 29.8, 29.7, 29.7, 29.6, 29.4, 29.4, 28.4, 22.7, 14.1.
(Z)-Methyl 6-(heptadec-8-en-1-yl)-4-(3-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5c)
C29H44N2O4; molecular weight: 484.67 g mol–1; yield: 68%; 1H NMR (400 MHz, CDCl3) δ 8.21 (br. s., 1H), 7.25–7.35 (m, 6H), 5.99 (br. s., 1H), 5.40 (d, J = 2.93 Hz, 1H), 5.31–5.39 (m, 2H), 3.63 (s, 3H), 2.66–2.78 (m, 2H), 1.98–2.09 (m, 4H), 1.57–1.67 (m, 2H), 1.24–1.42 (m, 21H), 0.90 (t, J = 6.80 Hz, 3H). NMR 13C (100 MHz, CDCl3) δ 165.8, 153.5, 151.1, 143.7, 130.0, 129.8, 128.8, 127.9, 126.5, 100.7, 55.6, 51.1, 31.9, 31.9, 31.9, 29.8, 29.7, 29.5, 29.4, 29.4, 29.3, 29.3, 29.3, 29.2, 28.3, 27.3, 27.2, 22.7, 14.1.
Methyl 4-(3-hydroxyphenyl)-6-pentadecyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (6a)
C27H42N2O3S; molecular weight: 474.70 g mol–1; yield: 81%; 1H NMR (400 MHz, CD3OD) δ 7.14 (t, J = 7.83 Hz, 1H), 6.76–6.79 (m, 1H), 6.75–6.76 (m, 1H), 6.69–6.72 (m, 1H), 5.28 (s, 1H), 3.66 (s, 3H), 2.69–2.84 (m, 2H), 1.56–1.69 (m, 2H), 1.31 (s, 24H), 0.92 (t, J = 7.10 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 175.2, 166.1, 157.4, 148.9, 144.6, 129.3, 117.4, 114.5, 113.1, 101.3, 54.8, 50.3, 31.7, 30.3, 29.4, 29.4, 29.3, 29.3, 29.3, 29.1, 29.1, 28.2, 22.3, 13.0.
Methyl 6-heptadecyl-4-(3-hydroxyphenyl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (6b)
C29H46N2O3S; molecular weight: 502.75 g mol–1; yield: 74%; 1H NMR (400 MHz, CDCl3) δ 7.81 (br. s., 1H), 7.34 (br. s., 1H), 7.19 (t, J = 7.95 Hz, 1H), 6.83 (d, J = 7.58 Hz, 1H), 6.78 (s, 1H), 5.36 (d, J = 3.42 Hz, 1H), 3.67 (s, 3H), 2.66–2.77 (m, 2H), 1.54–1.67 (m, 2H), 1.28 (s, 24H), 0.90 (t, J = 6.85 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.8, 165.6, 156.3, 147.4, 143.8, 130.2, 118.7, 115.6, 113.6, 102.2, 55.8, 51.5, 31.9, 31.6, 29.7, 29.6, 29.6, 29.5, 29.5, 29.3, 29.3, 28.2, 22.7, 14.1.
(Z)-Methyl 6-(heptadec-8-en-1-yl)-4-(3-hydroxyphenyl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (6c)
C29H44N2O3S; molecular weight: 500.74 g mol–1; yield: 60%; 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 7.90 (br. s., 1H), 7.25–7.36 (m, 5H), 5.39 (d, J = 3.42 Hz, 1H), 5.34–5.39 (m, 2H), 3.64 (s, 3H), 2.76–2.85 (m, 1H), 2.62–2.74 (m, 1H), 1.96–2.11 (m, 4H), 1.56–1.67 (m, 2H), 1.22–1.44 (m, 20H), 0.90 (t, J = 6.80 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.8, 165.6, 156.3, 147.4, 143.8, 130.2, 118.7, 115.6, 113.6, 102.2, 55.8, 51.5, 31.9, 31.6, 29.7, 29.6, 29.6, 29.5, 29.5, 29.3, 29.3, 28.2, 22.7, 14.1.
Biological assays
General cell culture procedures
The experiments were performed using C6 rat cells obtained from American Type Culture Collection (ATCC®CCL-107™). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and containing penicillin/streptomycin (1%) and Fungizone® (1%). They were kept in a humidified atmosphere with 5% CO2 at a temperature of 37 °C.
Treatment and experimental groups
The compounds were prepared as described above and dissolved in cell culture-grade DMSO. Glioma cells were seeded at a density of 1 × 104 cell per well in DMEM/10% FBS in 96-well plates. Cell cultures were exposed to concentrations of 5, 10, 25, and 50 μM of the molecules for 48 h, while the control group was treated with vehicle (1% of DMSO).
Cell viability assay
After 48 h of treatment, cell viability was evaluated using an MTT assay. MTT solution (sterile stock solution of 5 mg mL–1) was added to the incubation medium at a final concentration of 0.5 mg mL–1. The cells were left for 60 min at 37 °C in a humidified 5% CO2 atmosphere. Then the medium was removed and the plates were shaken for 30 min using DMSO as a solvent. The optical density of each well was measured at 492 nm. The results of the MTT assay are expressed as percentages of control group values.
Cell proliferation assay
C6 cells were cultured at 5 × 103 cells per well in DMEM containing 10% FBS, in 96-well plates for 48 h. Then the cells were trypsinized, and 100 μL aliquots were taken from each well for counting in a Neubauer chamber, using a microscope stereoscope.
Lipophilicity calculations
The physicochemical parameter clog P (the logarithm of n-octanol/water partition coefficient P), based on established chemical interactions, was calculated using ChemDraw, Level: Ultra, Version: 12.0.2.1076 (CambridgeSoft, Cambridge, MA, USA).
Calculating the half maximal inhibitory concentration (IC50)
The half maximal inhibitory concentration (IC50) was calculated using GraphPad PRISM® Prism 5 software for Windows Version 5.3. IC50 was determined from a linear regression, which was related to the inhibition percentage as a function of the logarithm of the concentrations tested, assuming a confidence interval of 95% (p < 0.05).
Statistical analysis
Data were analyzed for statistical significance by one-way analysis of variance (ANOVA) followed by a post hoc test for multiple comparisons (Tukey test) using GraphPad Prism® software. Data are expressed as the mean ± standard deviation. Differences were considered significant at p < 0.05.
Conflicts of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
The authors are thankful for financial support from Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES), Fundação de Amparo a Pesquisa do Estado do Rio Grande do Sul (FAPERGS/PRONEM), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). Fellowships from CAPES are also acknowledged.
Footnotes
†Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of the compounds. See DOI: 10.1039/c8md00169c
References
- Ugi I., Domling A., Horl W. Endeavour. 1994;18:115–122. [Google Scholar]
- Zarganes-Tzitzikas T., Chandgude A. L., Domling A. Chem. Rec. 2015;15:981–996. doi: 10.1002/tcr.201500201. [DOI] [PubMed] [Google Scholar]
- Domling A., Wang W., Wang K. Chem. Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioc R. C., Ruijter E., Orru R. V. A. Green Chem. 2014;16:2958–2975. [Google Scholar]
- Alvim H. G. O., Lima T. B., de Oliveira A. L., de Oliveira H. C. B., Silva F. M., Gozzo F. C., Souza R. Y., da Silva W. A., Neto B. A. D. J. Org. Chem. 2014;79:3383–3397. doi: 10.1021/jo5001498. [DOI] [PubMed] [Google Scholar]
- Kim J., Park C., Ok T., So W., Jo M., Seo M., Kim Y., Sohn J. H., Park Y., Ju M. K., Kim J., Han S. J., Kim T. H., Cechetto J., Nam J., Sommer P., No Z. Bioorg. Med. Chem. Lett. 2012;22:2119–2124. doi: 10.1016/j.bmcl.2011.12.090. [DOI] [PubMed] [Google Scholar]
- Mokale S. N., Shinde S. S., Elgire R. D., Sangshetti J. N., Shinde D. B. Bioorg. Med. Chem. Lett. 2010;20:4424–4426. doi: 10.1016/j.bmcl.2010.06.058. [DOI] [PubMed] [Google Scholar]
- Trivedi A. R., Bhuva V. R., Dholariya B. H., Dodiya D. K., Kataria V. B., Shah V. H. Bioorg. Med. Chem. Lett. 2010;20:6100–6102. doi: 10.1016/j.bmcl.2010.08.046. [DOI] [PubMed] [Google Scholar]
- Sharma P., Rane N., Gurram V. K. Bioorg. Med. Chem. Lett. 2004;14:4185–4190. doi: 10.1016/j.bmcl.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Stefani H. A., Oliveira C. B., Almeida R. B., Pereira C. M. P., Braga R. C., Cella R., Borges V. C., Savegnago L., Nogueira C. W. Eur. J. Med. Chem. 2006;41:513–518. doi: 10.1016/j.ejmech.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Atwal K. S., Rovnyak G. C., Schwartz J., Moreland S., Hedberg A., Gougoutas J. Z., Malley M. F., Floyd D. M. J. Med. Chem. 1990;33:1510–1515. doi: 10.1021/jm00167a035. [DOI] [PubMed] [Google Scholar]
- Atwal K. S., Swanson B. N., Unger S. E., Floyd D. M., Moreland S., Hedberg A., Oreilly B. C. J. Med. Chem. 1991;34:806–811. doi: 10.1021/jm00106a048. [DOI] [PubMed] [Google Scholar]
- Rovnyak G. C., Atwal K. S., Hedberg A., Kimball S. D., Moreland S., Gougoutas J. Z., Oreilly B. C., Schwartz J., Malley M. F. J. Med. Chem. 1992;35:3254–3263. doi: 10.1021/jm00095a023. [DOI] [PubMed] [Google Scholar]
- Kapoor T. M., Mayer T. U., Coughlin M. L., Mitchison T. J. J. Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowicz R., Finer J. T., Beraud C., Crompton A., Lewis E., Fritsch A., Lee Y., Mak J., Moody R., Turincio R., Chabala J. C., Gonzales P., Roth S., Weitman S., Wood K. W. Cancer Res. 2004;64:3276–3280. doi: 10.1158/0008-5472.can-03-3839. [DOI] [PubMed] [Google Scholar]
- Heald R. Cell. 2000;102:399–402. doi: 10.1016/s0092-8674(00)00044-1. [DOI] [PubMed] [Google Scholar]
- Brier S., Lemaire D., DeBonis S., Forest E., Kozielski F. Biochemistry. 2004;43:13072–13082. doi: 10.1021/bi049264e. [DOI] [PubMed] [Google Scholar]
- Cochran J. C., Gatial J. E., Kapoor T. M., Gilbert S. P. J. Biol. Chem. 2005;280:12658–12667. doi: 10.1074/jbc.M413140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran J. C., Gatial J. E., Maliga Z., Kapoor T. M., Mitchison T. J., Gilbert S. P. Biophys. J. 2005;88:648A. [Google Scholar]
- Levitzki A., Klein S. Mol. Aspects Med. 2010;31:287–329. doi: 10.1016/j.mam.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Ligon K. L., Wilkinson K. and Stiles C. D., in Pathology and Epidemiology of Cancer, Springer, 2017, pp. 291–311. [Google Scholar]
- Van Tellingen O., Yetkin-Arik B., De Gooijer M., Wesseling P., Wurdinger T., De Vries H. Drug Resist. Updates. 2015;19:1–12. doi: 10.1016/j.drup.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Russowsky D., Canto R. F. S., Sanches S. A. A., D'Oca M. G. M., de Fatima A., Pilli R. A., Kohn L. K., Antonio M. A., de Carvalho J. E. Bioorg. Chem. 2006;34:173–182. doi: 10.1016/j.bioorg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Meunier B. Acc. Chem. Res. 2008;41:69–77. doi: 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- Venepally V., Jala R. C. R. Eur. J. Med. Chem. 2017;141:113–137. doi: 10.1016/j.ejmech.2017.09.069. [DOI] [PubMed] [Google Scholar]
- Treptow T. G. M., Figueiro F., Jandrey E. H. F., Battastini A. M. O., Salbego C. G., Hoppe J. B., Taborda P. S., Rosa S. B., Piovesan L. A., Montes D'Oca C. D., Russowsky D., Montes D'Oca M. G. Eur. J. Med. Chem. 2015;95:552–562. doi: 10.1016/j.ejmech.2015.03.062. [DOI] [PubMed] [Google Scholar]
- dos Santos D. S., Piovesan L. A., D'Oca C. R. M., Hack C. R. L., Treptow T. G., Rodrigues M. O., Vendramini-Costa D. B., Ruiz A. L. T., de Carvalho J. E., D'Oca M. G. M. Bioorg. Med. Chem. 2015;23:340–347. doi: 10.1016/j.bmc.2014.11.019. [DOI] [PubMed] [Google Scholar]
- da Costa Cabrera D., Rosa S. B., de Oliveira F. S., Marinho M. A., D'Oca C. R. M., Russowsky D., Horn A. P., D'Oca M. G. M. MedChemComm. 2016;7:2167–2176. [Google Scholar]
- Brinkerhoff R. C., Tarazona H. F., de Oliveira P. M., Flores D. C., D'Oca C. D. R. M., Russowsky D., D'Oca M. G. M. RSC Adv. 2014;4:49556–49559. [Google Scholar]
- Liu X. L., Testa B., Fahr A. Pharm. Res. 2011;28:962–977. doi: 10.1007/s11095-010-0303-7. [DOI] [PubMed] [Google Scholar]
- Mota A. A. R., Carvalho P. H. P. R., Guido B. C., de Oliveira H. C. B., Soares T. A., Corrêa J. R., Neto B. A. D. Chem. Sci. 2014;5:3995–4003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.