

 m-Amidophenol derivatives were found to potently inhibit the growth of M. tuberculosis strains H37Ra and H37Rv and clinically isolated multidrug-resistant M. tuberculosis strains.
m-Amidophenol derivatives were found to potently inhibit the growth of M. tuberculosis strains H37Ra and H37Rv and clinically isolated multidrug-resistant M. tuberculosis strains.
Abstract
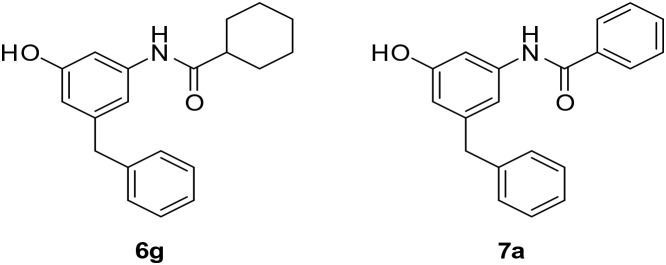
A series of m-amidophenol derivatives (6a–6l, 7a–7q, 9a, 9b, 12a–12c, 14 and 15) were designed and synthesized. Their antitubercular activities were evaluated in vitro against M. tuberculosis strains H37Ra and H37Rv and clinically isolated multidrug-resistant M. tuberculosis strains. Ten compounds displayed minimal inhibitory concentrations (MICs) against M. tuberculosis H37Ra below 2.5 μg mL–1 and 6g was the most active compound (MIC = 0.625 μg mL–1). Compounds 6g and 7a also showed potent inhibitory activity against M. tuberculosis H37Rv (MIC = 0.39 μg mL–1) and several clinically isolated multidrug-resistant M. tuberculosis strains (MIC = 0.39–3.125 μg mL–1). The compounds did not show inhibitory activity against normal Gram-positive and Gram-negative bacteria. They exhibited low cytotoxicity against HepG2 and RAW264.7 cell lines. The results demonstrated m-amidophenol as an attractive scaffold for the development of new antitubercular agents.
Introduction
Tuberculosis (TB) is a highly infectious disease caused by Mycobacterium tuberculosis (Mtb). Today tuberculosis still remains one of the leading causes of human death in developing countries. According to a report of the World Health Organization (WHO), there were an estimated 10.4 million new TB cases worldwide and over 1.6 million people died from TB in 2016. Moreover, the resistance of Mtb to clinical antitubercular agents is a continuing threat. As reported, in 2016 there were 600 000 new cases with resistance to rifampicin, one of the most effective first-line drugs. Among these cases, 490 000 were multidrug-resistant TB (MDR-TB).1 This situation brought a big challenge for clinical treatment. In the past four decades, only bedaquiline and delamanid had been approved by the Food and Drug Administration (FDA) of the United States and the European Medicines Agency (EMA) for the treatment of MDR-TB.2,3 Bedaquiline is an ATP synthase inhibitor. Delamanid inhibits the mycolic acid synthesis of Mtb. The two drugs are still beset by side effects.4,5 Therefore, antitubercular agents with innovative action mechanisms and structural scaffolds are urgently desired.6
Mycobacterial enoyl-ACP reductase (InhA) is a validated drug target against Mtb.7 Isoniazid (INH), an inhibitor of InhA, is a powerful first-line antitubercular drug.8,9 INH is a prodrug that is activated by the catalase-peroxidase KatG to form an isonicotinoyl radical which reacts with NAD to produce an INH–NAD adduct. Unfortunately, the majority of INH-resistant M. tuberculosis clinical isolates have mutations in KatG.10 Thus, direct InhA inhibitors were developed to avoid resistance.11,12 In 2015, Manjunatha, Diagana and co-workers found a new class of direct InhA inhibitors, 4-hydroxy-2-pyridones, through unbiased phenotypic screening. Several compounds showed potent bactericidal activity against INH-resistant Mtb strains.13 The lead compound NITD-916 (Scheme 1) displayed good oral bioavailability and in vivo efficacy in mouse models of Mtb infection. Based on the co-crystal structure, key interactions of 4-hydroxy-2-pyridones with the InhA–NADH complex were identified as H-bonding, π-stacking and hydrophobic interactions. Inspired by the proposed interaction model of NITD-916, we designed and synthesized a class of m-amidophenol derivatives (Scheme 1). A number of compounds were found to exert potent inhibitory activity against Mtb H37Ra, Mtb H37Rv and clinically isolated MDR-Mtb strains. In addition, the compounds showed low cytotoxicity and did not inhibit normal Gram-positive and Gram-negative bacteria.
Scheme 1. Structures of antitubercular agents.

Results and discussion
Chemistry
The synthetic pathways of m-amidophenol derivatives 6a–6l and 7a–7q are illustrated in Scheme 2. The synthesis started from 3,5-dinitrobenzoic acid 1. Compound 1 was transformed into its methyl ester with thionylchloride in methanol. Then the nucleophilic substitution of the nitro group with lithium methoxide afforded methyl 3-methoxy-5-nitrobenzoate 2. 3-Methoxy-5-nitrobenzoic acid was obtained by basic hydrolysis of 2. The reduction of the acid with sodium borohydride yielded (3-methoxy-5-nitrophenyl)-methanol 3. Friedel–Crafts alkylation of benzene with 3 gave 1-benzyl-3-methoxy-5-nitrobenzene 4. Demethylation with boron tribromide provided 3-benzyl-5-nitrophenol, which was hydrogenated over Pd/C to provide 3-amino-5-benzylphenol 5. The m-amidophenol derivatives 6a–6l and 7a–7q were obtained by the reaction of 5 with different acyl chlorides or by the coupling of 5 with the corresponding carboxylic acids.
Scheme 2. Reagents and conditions: a) SOCl2, CH3OH, reflux, 4 h, 60%; b) lithium methoxide, anhydrous methanol, reflux, 12 h, 35%; c) NaOH, CH2Cl2, CH3OH, rt, 3 h, 98%; d) NaBH4, BF3·OEt2, THF, 0 °C–rt, 1 h, 99%; e) benzene, CH2Cl2, AlCl3, reflux, 5 h, 52.6%; f) BBr3, CH2Cl2, –40–0 °C, 12 h, 60%; g) H2, 10% Pd/C, CH2Cl2/CH3OH, 4 h, 99%; h) RCOCl, Et3N, THF, 0 °C–rt, 50–80% or RCOOH, triphenyl phosphite, toluene. For the R group, see Table 1.

The synthesis of compounds 9a–9b is shown in Scheme 3. Friedel–Crafts reaction of p-xylene and mesitylene with compound 3 was achieved using hexafluoroisopropanol (HFIP) as the solvent and trifluoromethanesulfonic acid (TfOH) as the catalyst.14 Compounds 8a–8b were obtained in excellent yields. After demethylation, reduction of the nitro group and acylation with cyclohexanecarbonyl chloride, the products 9a–9b were obtained.
Scheme 3. Reagents and conditions: a) p-xylene or mesitylene, HFIP, TfOH, 100 °C, 24 h, 90–96%; b) BBr3, CH2Cl2, –40–0 °C, 12 h, 40–50%; c) H2, 10% Pd/C, CH2Cl2/CH3OH, 4 h, 99%; d) cyclohexanecarbonyl chloride, Et3N, THF, 0 °C–rt, 50–80%.

Antitubercular activity against Mtb H37Ra
The antitubercular activities of the compounds were preliminarily screened and evaluated by using a cost-efficient in vitro assay against a selectable marker-free avirulent autoluminescent Mtb H37Ra strain.15 Bacteria growth was conveniently monitored by means of the bioluminescence intensity.
The effect of the 3-amido group
The antitubercular activities of compounds 6a–6l and 7a–7q were evaluated against Mtb H37Ra and the MIC values are summarized in Table 1. INH was used as the positive control and DMSO was used as the negative control.
Table 1. In vitro antitubercular activity of compounds 6a–6l and 7a–7q against Mtb H37Ra.
| |||
| Compound | R | clog P a | MIClux (μg mL–1) |
| 6a | Me | 2.56 | >100 |
| 6b | CF3 | 3.67 | >50 |
| 6c | i-Pr | 3.39 | 5 |
| 6d |
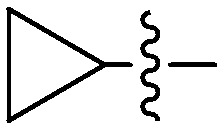
|
3.14 | 5 |
| 6e | t-Bu | 3.79 | 2.5 |
| 6f |
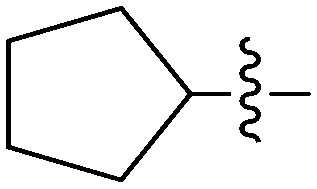
|
4.03 | 2.5 |
| 6g |
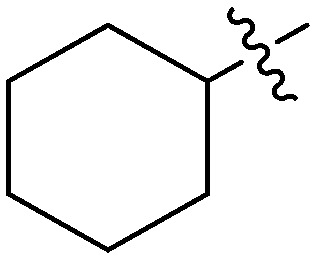
|
4.59 | 0.625 |
| 6h |
|
5.64 | 2.5 |
| 6i |
|
6.43 | >50 |
| 6j |
|
5.22 | >50 |
| 6k |
|
2.19 | 6.25 |
| 6l |
|
2.20 | 12.5 |
| 7a |
|
4.05 | 5 |
| 7b |
|
4.54 | 1.25 |
| 7c |
|
5.87 | 2.5 |
| 7d |
|
6.67 | >50 |
| 7e |
|
5.28 | 5 |
| 7f |
|
6.29 | >25 |
| 7g |
|
5.36 | 10 |
| 7h |
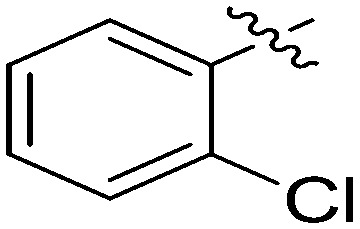
|
4.13 | 2.5 |
| 7i |
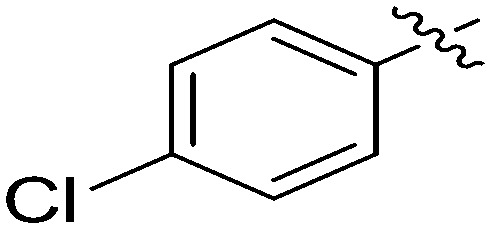
|
4.96 | 2.5 |
| 7j |
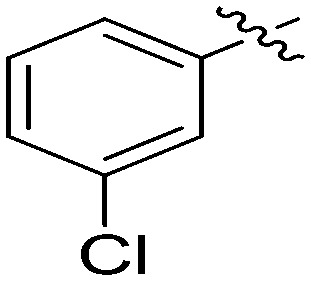
|
4.96 | 2.5 |
| 7k |
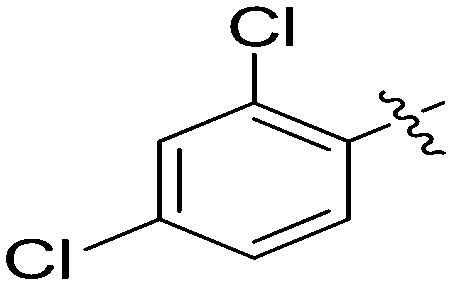
|
4.92 | 5 |
| 7l |
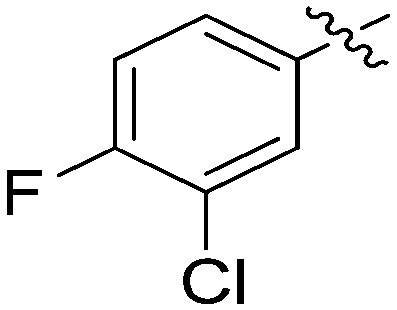
|
5.17 | 5 |
| 7m |
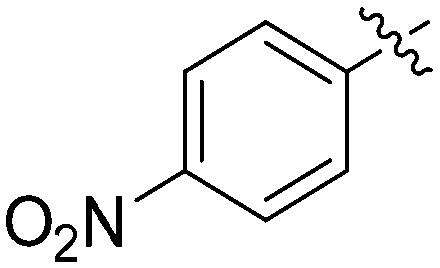
|
4.22 | >50 |
| 7n |
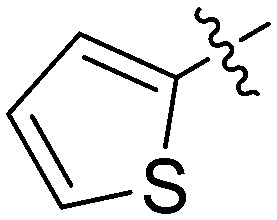
|
3.88 | 2.5 |
| 7o |
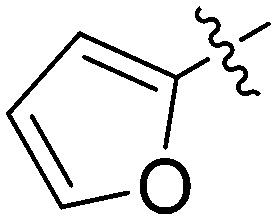
|
3.22 | 10 |
| 7p |
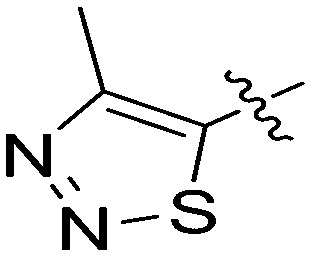
|
2.61 | 10 |
| 7q |
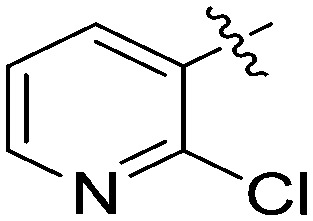
|
3.26 | 5 |
| INH | 0.1 | ||
| DMSO | — | ||
aclog P was calculated using ChemBioDraw Ultra 12.0.
The size and lipophilicity of the R group exert a strong effect on the antitubercular activity. The acetamide derivative 6a and the trifluoroacetamide derivative 6b did not show obvious activities. However, the increase in the lipophilicity of the amido group significantly improved the inhibitory activity. Compounds 6c and 6d (R = i-Pr and c-Pr, respectively) showed good inhibitory activity (MIC = 5 μg mL–1). A further increase in antitubercular activity was achieved via the introduction of more lipophilic substituents such as t-Bu, cyclopentyl, and cyclohexyl. Compound 6g with a cyclohexyl substituent showed the most potent activity (MIC = 0.625 μg mL–1). The further increase in the lipophilicity and volume of the substituent led to the gradual loss of the inhibitory activity. Compound 6h with a 4-ethyl-cyclohexyl substituent afforded a lower activity (MIC = 2.5 μg mL–1). Compounds 6i and 6j with more bulky 4-t-butyl-cyclohexyl and adamantanyl almost lost their inhibitory activity completely. The incorporation of O (6k) and N (6l) into the cyclohexyl ring resulted in lower activities. These results indicated a hydrophobic pocket binding with the amido group. The size of the pocket can best accommodate a cyclohexyl group or similar groups.
We also found that the benzoamide derivative 7a showed good inhibitory activity (MIC = 5 μg mL–1). The 4-methyl substitution (7b) afforded a 4-fold increase in activity (MIC = 1.25 μg mL–1). 4-t-Butyl substitution (7c) showed a slightly lower activity (MIC = 2.5 μg mL–1). The introduction of more bulk 4-cyclohexyl (7d) led to a loss of activity. The results are commendably consistent with the above observation. The substitution with CF3 (7e, 7f) and CF3O (7g) exerted a detrimental effect on the activity. Chloro substitution at the 2-, or 3-, or 4-position of the phenyl ring (7h, 7i, 7j) exhibited a similar efficiency. A 2-fold increase in activity was achieved in comparison with 7a. The introduction of more than one halogen atom at the phenyl ring (7k, 7l) did not afford a beneficial result. The 4-nitro substitution (7m) resulted in a loss of inhibitory activity. The replacement of the benzene ring with heteroaryl rings is applicable. Thiophene-2-carboxamide 7n, furan-2-carboxamide 7o, 1,2,3-thiadiazole-5-carboxamide 7p and 2-chloronicotinamide 7q are all active against Mtb H37Ra. Among them, thiophene-2-carboxamide 7n showed the best activity (MIC = 2.5 μg mL–1).
The effect of the 5-substitutent
The effect of the 5-substitutent was examined and the results are summarized in Table 2. The introduction of methyl substituents to the benzyl group (9a and 9b) resulted in a loss of activity. The replacement of 5-benzyl with methyl (12a), n-Pr (12b) and i-pr (12c) also led to a loss of antitubercular activity.16 The results implicated that the existence of the 5-benzyl group is important for antitubercular activity. A π–π interaction is possibly required. However, the binding pocket is relatively small. Even the methyl substitution also hampers the insertion of a benzyl group into the pocket. In addition, the benzyl oxidation product 14 showed a lower antitubercular activity compared to compound 7a. The reduction of 14 provided compound 15, which is completely inactive.17
Table 2. In vitro antitubercular activity of compounds 9a and 9b, 12a–12c, 14, and 15 against Mtb H37Ra.
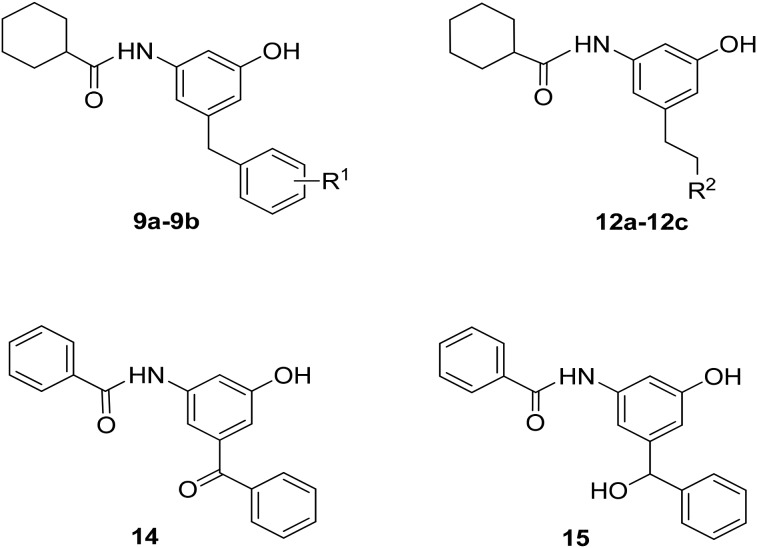
| |||||||
| Compound | R1 | clog P a | MIClux (μg mL–1) | Compound | R2 | clog P a | MIClux (μg mL–1) |
| 9a | 2,5-Dimethyl | 5.54 | >50 | 12a | Me | 4.08 | >50 |
| 9b | 2,4,6-Trimethyl | 5.98 | >50 | 12b | n-Pr | 5.14 | >50 |
| 14 | — | 6.25 | 12c | i-Pr | 5.01 | >50 | |
| 15 | — | >50 | INH | 0.1 | |||
| DMSO | — | — | |||||
aclog P was calculated using ChemBioDraw Ultra 12.0.
The effect of other structural modifications
To verify the importance of 3-amido and 5-benzyl groups, we prepared compounds 16 and 17 (Scheme 4).18,19 As expected, both compounds did not show antitubercular activity. The 1-hydroxyl group is also imperative. The methylation of 1-hydroxy (18) led to a loss of activity. Similarly, the replacement of 1-hydroxyl with a carboxylic group (19) and an amino group (20) resulted in the complete loss of or decreased activities.
Scheme 4. The effect of other structural modifications.

Structure–activity relationship (SAR) of m-amidophenol derivatives
The structure–activity relationship of m-amidophenol derivatives is summarized in Scheme 5. The crucial H-bonding interaction of the 1-hydroxyl group with the target is expected. The methylation or the replacement with a carboxylic group resulted in a loss of antitubercular activity. The 5-benzyl group is rather conservative. The replacement with an alkyl group or the introduction of substitutents exerted a detrimental effect on the activity. A small binding pocket and an important π–π interaction are suggested. A 3-amido group is involved in a crucial hydrophobic interaction with the target. The size of the binding pocket is best suitable for a cyclohexyl group.
Scheme 5. SAR of m-amidophenol derivatives.

In vitro inhibitive activity against Mtb H37Rv and MDR-Mtb strains
Compounds 6g and 7a with potent inhibitory activities against H37Ra were selected for further evaluation against Mtb H37Rv and six clinically isolated MDR-Mtb strains. The results are summarized in Table 3. Compounds 6g and 7a showed similar activities against H37Rv and MDR-Mtb strains P103 and P91 (0.39 μg mL–1). Compound 7a exhibited better inhibitory activity against MDR-Mtb strain R7 than 6g (MIC 0.78 μg mL–1vs. 3.125 μg mL–1). However, both compounds 6g and 7a did not inhibit MDR-Mtb strains P71, P90 and P98. The reason for the selective inhibition against different MDR-Mtb strains is unclear at present.
Table 3. Inhibitive activity against H37Rv and clinically isolated MDR-Mtb strains.
| |||||
| Strains | MIC (μg mL–1) |
||||
| 6g | 7a | INH | RIF | DMSO | |
| H37Rv | 0.39 | 0.39 | 0.41 | 0.003 | — |
| P103 a | 0.39 | 0.39 | — | — | — |
| P91 a | 0.39 | 0.39 | — | — | — |
| R7 a | 3.125 | 0.78 | — | — | — |
| P71 b | >100 | >100 | — | — | — |
| P90 a | >100 | >100 | — | — | — |
| P98 a | >100 | >100 | — | — | — |
aResistant to rifampicin and INH.
bResistant to rifampicin, INH and pyrazinamide.
Evaluation of antibacterial activity
The antibacterial activity of compounds 6g and 7a was examined against representative Gram-positive bacteria Staphylococcus aureus and Enterococcus faecalis and Gram-negative bacteria Escherichia coli. Amoxicillin and ofloxacin were used as the positive controls and DMSO was used as the negative control. The results are summarized in Table 4. Both 6g and 7a did not show antibacterial activity (MIC > 50 μg mL–1) against the tested bacteria. This observation confirmed that the inhibitory activity of 6g and 7a against Mtb is highly exclusive. Such a property is favorable considering the long treatment course for tuberculosis (typically 12–24 months). The avoidance of inhibition against normal intestinal bacteria is advantageous for antitubercular agents.
Table 4. Evaluation of antibacterial activity of compounds 6g and 7a.
| MIC (μg mL–1) |
|||||
| 6g | 7a | Amoxicillin | Ofloxacin | DMSO | |
| Staphylococcus aureus | >50 | >50 | 0.78 | 0.31 | — |
| Escherichia coli | >50 | >50 | 6.25 | 1.25 | — |
| Enterococcus faecalis | >50 | >50 | 1.25 | — | — |
Evaluation of cytotoxicity
The cytotoxicity of compounds 6g and 7a was evaluated against HepG2 and RAW264.7 cell lines. The IC50 values are listed in Table 5. Compounds 6g and 7a are almost non-cytotoxic against the two cell lines. Both compounds have a selectivity index (SI = IC50/MIC) of above 20.
Table 5. Cytotoxicity of compounds 6g and 7a.
| Entry | IC50 (μM) |
|
| HepG2 | RAW264.7 | |
| 6g | >161 | >323 |
| (SI > 80) | (SI > 160) | |
| 7a | >330 | >330 |
| (SI > 20) | (SI > 20) | |
3D-QSAR study
For further exploration of the structure–activity relationship, the CoMFA approach was employed on the test compounds to establish 3D-QSAR models. Their conformations were first generated by molecular mechanism-based structural optimization embedded in Tripos Sybyl X2.0 software. Compound 6g was selected as the template for alignment (Fig. 1).
Fig. 1. 3D-QSAR structure alignment using compound 6g as the template.

Based on these conformations with the local minimum energies and partial charges assigned using the MMFF94 force field, the CoMFA model was performed with 33 molecules as the training set and 2 molecules as the test set. The best model (CoMFA) yielded a q2 value of 0.616, an r2 value of 0.950 and an F value of 102.537 with the optimum number of components (PC) being 5 (Table 6).
Table 6. The parameters derived from the CoMFA model.
| Parameters | CoMFA model |
| q 2 | 0.616 |
| r 2 | 0.950 |
| PC | 5 |
| F | 102.537 |
| Contributions (%) | |
| Steric | 67.6 |
| Electrostatics | 32.4 |
Fig. 2 shows a scatter plot of predicted pMIC versus experimental pMIC values of the training set and test set. A contour map with rpred2 = 0.6758 provided visual and spatial information for future design and optimization of new compounds with enhanced activities.
Fig. 2. A graph of predicted versus experimental pMIC values of the molecules.

The CoMFA contour map of the steric field and electrostatic field is shown in Fig. 3. The steric contour is shown in green (more bulk favored) and yellow (less bulk favored), suggesting that a sterically bulky group is disfavored at the 5-position of the phenyl. For example, compounds 9a and 9b containing methyl groups led to the complete loss of inhibitory activity. The green contour in the o-position of cyclohexyl indicates that this space is favorable for accommodating a sterically bulky group (for example, compound 7h with 4-Cl substitution). The electrostatic contour was colored red and blue. The red contour near the 1-position of the phenyl ring showed that the electron-withdrawing groups were favored at this position. Compound 7a with 1-OH and compound 20 with 1-NH2 exhibited good anti-TB activity. Compound 7b with 4-CH3 lies near the blue contour and exhibited good anti-TB activity, while compound 7m with 4-NO2 near the blue contour lost inhibitory activity.
Fig. 3. The steric and electrostatic field distribution around 6g.

Conclusion
We have discovered m-amidophenol derivatives as a new class of antitubercular agents. Several compounds showed potent inhibitory activities against Mtb H37Ra, Mtb H37Rv and clinically isolated MDR-Mtb strains. The compounds did not inhibit normal Gram-positive and Gram-negative bacteria. Their inhibition of Mtb is highly exclusive. The compounds showed low cytotoxicity and their selectivity indexes are acceptable. Further studies toward the elucidation of the inhibition mechanism and the development of more potent candidates are currently underway.20
Experimental
Chemistry
1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE 400 or 500 spectrometer. Chemical shifts of protons are reported in parts per million downfield from tetramethylsilane. Peaks are labeled as single (s), broad singlet (br), doublet (d), triplet (t), double doublet (dd), doublet of triplets (dt), or multiplet (m). The high-resolution mass spectra were analyzed using a SHIMADZU LCMS-IT-TOF mass spectrometer. The purity of the synthesized compounds was determined by high-performance liquid chromatography (HPLC) using a TC-C18 column (250 mm × 4.6 mm, 5 μm) and a methanol/water mobile phase (0.50 mL min–1). Melting points were determined in open capillary tubes on an MPA100 Optimelt automated melting point system. All chemicals were purchased from Sigma-Aldrich and Alfa Aesar chemical companies and were used without further purification.
Synthesis of N-(3-benzyl-5-hydroxyphenyl)acetamide (6a)
To a solution of 3-amino-5-benzylphenol 5 (99.5 mg, 0.5 mmol) and triethylamine (76.3 μL, 0.55 mmol) in THF (2 mL), a solution of acetyl chloride (40 μL, 0.55 mmol) in THF (1 mL) at 0 °C was added slowly. The reaction mixture was stirred for 3 h at room temperature. The reaction was quenched with water (10 mL) and the mixture was extracted with EtOAc (15 mL × 2). The combined organic layer was dried over anhydrous Na2SO4 and filtered. After the solvent was removed in vacuo, the crude product was purified by column chromatography (petroleum/EtOAc = 20 : 1–5 : 1) to afford 6a as a colorless solid (72 mg, 60%). m.p. 158.5–160.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.72 (bs, 1H, NH), 9.26 (bs, 1H, OH), 7.28 (m, 2H, ArH), 7.19 (m, 3H, ArH), 7.06 (s, 1H, ArH), 6.76 (s, 1H, ArH), 6.29 (s, 1H, ArH), 3.78 (s, 2H, CH2), 1.98 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 168.59 (CONH), 158.01, 143.00, 141.58, 140.73, 129.20, 128.83, 126.41, 111.12, 110.65, 104.48, 41.75 (CH2), 24.50 (CH3); HRMS (ESI) calculated for C15H16NO2 [M + H]+: 242.1176, found: 242.1173. Purity: 98.1% (by HPLC).
Synthesis of N-(3-benzyl-5-hydroxyphenyl)-2,2,2-trifluoroacetamide (6b)
To a solution of 5 (160 mg, 0.8 mmol) and trifluoroacetic acid (137 mg, 1.2 mmol) in 4 mL toluene, P(OPh)3 (314 μL, 1.2 mmol) was added. The reaction mixture was stirred at 110 °C for 12 h. After being cooled to room temperature, water (20 mL) was added. The reaction mixture was extracted with ethyl acetate (20 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by column chromatography over silica gel (petroleum/EtOAc = 4 : 1) to yield 6b as a red solid (94 mg, 40%). m.p. 95.8–98.7 °C; 1H NMR (400 MHz, CD3OD) δ 7.25 (m, 2H, ArH), 7.16 (m, 3H, ArH), 7.10 (m, 1H, ArH), 6.91 (m, 1H, ArH), 6.50 (m, 1H, ArH), 3.87 (s, 2H, CH2); 13C NMR (101 MHz, CD3OD) δ 157.68, 155.26 (q, 2J = 37.2 Hz, CONH), 143.63, 140.71, 137.19, 128.58, 128.08, 116.04 (q, 1J = 286 Hz), 125.78, 113.11, 112.44, 105.77, 41.33 (CH2); HRMS (ESI) calculated for C15H12NO2F3 [M – H]–: 294.0747, found: 294.0746. Purity: 98.9% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-isobutyramide (6c)
The compound was synthesized via a procedure similar to that of 6a. Orange solid, yield 86%, m.p. 169.8–171.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.59 (bs, 1H, NH), 9.23 (bs, 1H, OH), 7.31–7.24 (m, 2H), 7.22–7.12 (m, 3H), 7.04 (m, 1H), 6.84 (m, 1H), 6.29 (m, 1H), 3.78 (m, 2H, CH2), 2.54 (m, 1H, CH), 1.06 (s, 3H, CH3), 1.05 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ: 175.50 (CONH), 157.98, 142.93, 141.60, 140.85, 129.18, 128.82, 126.40, 111.10, 110.92, 104.69, 41.81 (CH2), 35.33 (CH), 19.97 (CH3); HRMS (ESI) calculated for C17H19NO2 [M + H]+: 270.1489, found: 270.1491. Purity: 96.2% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)cyclopropanecarboxamide (6d)
The compound was synthesized via a procedure similar to that of 6a. Orange solid, yield 63%, m.p. 163.8–165.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.95 (bs, 1H, NH), 9.24 (bs, 1H, OH), 7.28 (m, 2H), 7.23–7.14 (m, 3H), 7.01 (m, 1H), 6.82 (m, 1H), 6.29 (m, 1H), 3.78 (s, 2H, CH2), 1.73 (dq, J = 7.6, 5.0 Hz, 1H, CH), 0.82–0.64 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 171.86 (CONH), 158.02, 143.01, 141.58, 140.76, 129.19, 128.82, 126.40, 111.09, 110.74, 104.55, 41.79 (ArCH2Ar), 14.94 (cyclopropanyl-CH), 7.47 (2CH2); HRMS (ESI) calculated for C17H17NO2 [M + H]+: 268.1332, found: 268.1332. Purity: 98.456% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)pivalamide (6e)
The compound was synthesized via a procedure similar to that of 6b. Yellow solid, yield 52%, m.p. 142.6–144.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.20 (bs, 1H, NH), 8.98 (bs, 1H, OH), 7.35–7.25 (m, 2H), 7.21 (d, J = 5.1 Hz, 3H), 7.09 (m, 1H), 6.93 (m, 1H), 6.32 (m, 1H), 3.80 (s, 2H, CH2), 1.19 (s, 9H, CH3); 13C NMR (101 MHz, CD3OD) δ 178.41 (CONH), 157.33, 142.91, 141.06, 139.34, 128.55, 128.00, 125.65, 113.01, 111.68, 106.23, 41.47 (CH2), 39.11 (CMe3), 26.39 (CH3); HRMS (ESI) calculated for C18H21NO2 [M + H]+: 284.1645, found: 284.1649. Purity: 98.8% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)cyclopentanecarboxamide (6f)
The compound was synthesized via a procedure similar to that of 6a. Orange solid, yield 70%, m.p. 182.4–184.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.62 (bs, 1H, NH), 9.22 (bs, 1H, OH), 7.34–7.24 (m, 2H), 7.18 (m, 3H), 7.04 (s, 1H), 6.84 (m, 1H), 6.28 (m, 1H), 3.78 (s, 2H, ArCH2Ar), 2.78–2.65 (m, 1H, cyclopentyl-CH), 1.86–1.71 (m, 2H), 1.73–1.58 (m, 4H), 1.52 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 174.67 (CONH), 157.98, 142.93, 141.61, 140.88, 129.18, 128.82, 126.39, 111.07, 110.88, 104.65, 45.72 (cyclopentyl-CH), 41.81 (ArCH2Ar), 30.54 (cyclopentyl-CH2), 26.14 (cyclopentyl-CH2); HRMS (ESI) calculated for C19H21NO2 [M + H]+: 296.1645, found: 296.1648. Purity: 97.9% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)cyclohexanecarboxamide (6g)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 49%, m.p. 200.9–202.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.59 (bs, 1H, NH), 9.24 (bs, 1H, OH), 7.28 (m, 2H), 7.19 (m, 3H), 7.04 (m, 1H), 6.84 (m, 1H), 6.28 (m, 1H), 3.77 (s, 2H, ArCH2Ar), 2.27 (t, J = 11.4 Hz, 1H), 1.73 (d, J = 9.2 Hz, 4H), 1.63 (d, J = 9.6 Hz, 1H), 1.37 (m, 2H), 1.30–1.14 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 174.62 (CONH), 157.96, 142.93, 141.62, 140.91, 129.18, 128.82, 126.39, 111.02, 110.82, 104.55, 45.31 (cyclohexyl-CH), 41.80 (ArCH2Ar), 29.61 (cyclohexyl-CH2), 25.88 (cyclohexyl-CH2), 25.71 (cyclohexyl-CH2); HRMS (ESI) calculated for C20H24NO2 [M + H]+: 310.1802, found: 310.1800. Purity: 97.1% (by HPLC).
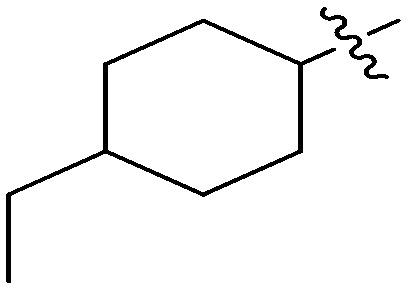
N-(3-Benzyl-5-hydroxyphenyl)-4-ethylcyclohexanecarboxamide (6h)
The compound was prepared via a procedure similar to that of 6b. White solid, yield 69%, m.p. 201.4–203.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.57 (bs, 1H, NH), 9.21 (bs, 1H, OH), 7.28 (m, 2H), 7.17 (m, 3H), 7.03 (m, 1H), 6.84 (m, 1H), 6.28 (m, 1H), 3.77 (s, 2H, ArCH2Ar), 2.21 (dd, J = 16.5, 7.7 Hz, 1H), 1.78 (d, J = 11.2 Hz, 4H), 1.45–1.31 (m, 2H), 1.20 (dt, J = 14.2, 7.3 Hz, 2H), 1.11 (m, 1H), 0.86 (m, 5H); 13C NMR (101 MHz, DMSO-d6) δ 174.64 (CONH), 157.96, 142.91, 141.61, 140.90, 129.18, 128.81, 126.39, 111.04, 110.86, 104.61, 45.51 (cyclohexyl-CH), 41.81 (ArCH2Ar), 38.72 (–CH2CH3), 32.09 (cyclohexyl-CH2), 29.91 (cyclohexyl-CH2), 29.55 (cyclohexyl-CH2), 11.74 (CH3); HRMS (ESI) calculated for C22H27NO2 [M + H]+: 338.2115, found: 338.2117. Purity: 98.8% (by HPLC).
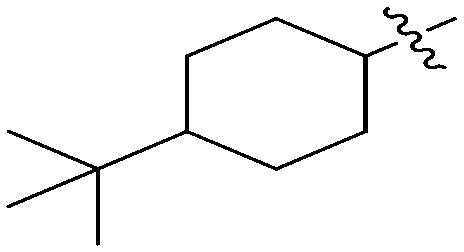
N-(3-Benzyl-5-hydroxyphenyl)-4-(tert-butyl)cyclohexanecarboxamide (6i)
This compound was prepared via a procedure similar to that of 6b. White solid, yield 76%, m.p. 227.2–229.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.56 (bs, 1H, NH), 9.21 (bs, 1H, OH), 7.32–7.23 (m, 2H), 7.17 (m, 3H), 7.02 (m, 1H), 6.84 (m, 1H), 6.27 (m, 1H), 3.77 (s, 2H, ArCH2Ar), 2.29–2.11 (m, 1H), 1.80 (m, 4H), 1.37 (m, 2H), 1.05–0.91 (m, 3H), 0.84 (s, 9H, t-Bu); 13C NMR (101 MHz, DMSO-d6) δ 174.67 (CONH), 157.96, 142.91, 141.61, 140.88, 129.17, 128.81, 126.38, 111.05, 110.87, 104.61, 47.30, 45.47, 41.82 (ArCH2Ar), 32.62, 30.00, 27.80, 26.66; HRMS (ESI) calculated for C24H31NO2 [M + H]+: 366.2428, found: 366.2445. Purity: 98.6% (by HPLC).
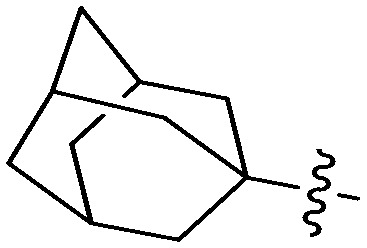
N-(3-Benzyl-5-hydroxyphenyl)adamantane-1-carboxamide (6j)
The compound was prepared via a procedure similar to that of 6a. White solid, yield 31%, m.p. 183.5–185.4 °C; 1H NMR (400 MHz, CD3OD) δ 7.23 (m, 2H), 7.15 (m, 3H), 7.00 (m, 1H), 6.83 (m, 1H), 6.37 (m, 1H), 3.79 (m, 2H, ArCH2Ar), 2.01 (s, 3H), 1.94 (m, 6H), 1.75 (s, 6H); 13C NMR (101 MHz, CD3OD) δ 177.88 (CONH), 157.31, 142.89, 141.07, 139.28, 128.55, 128.01, 125.65, 112.99, 111.64, 106.18, 41.48 (ArCH2Ar), 41.26, 38.56, 36.13, 28.31; HRMS (ESI) calculated for C24H27NO2 [M + H]+: 362.2115, found: 362.2118. Purity: 96.6% (by HPLC).
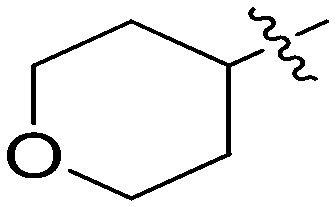
N-(3-Benzyl-5-hydroxyphenyl)tetrahydro-2H-pyran-4-carboxamide (6k)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 63%, m.p. 151.2–152.6 °C; 1H NMR (400 MHz, CD3OD) δ 7.30–7.21 (m, 1H), 7.16 (m, 1H), 7.02 (m, 1H), 6.79 (m, 1H), 6.38 (m, 1H), 3.98 (dd, J = 11.4, 2.6 Hz, 2H), 3.84 (s, 2H, ArCH2Ar), 3.45 (td, J = 11.7, 2.1 Hz, 2H), 2.57 (tt, J = 11.4, 4.0 Hz, 1H), 1.89–1.67 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 173.29 (CONH), 157.98, 142.99, 141.58, 140.73, 129.19, 128.83, 126.40, 111.20, 110.91, 104.66, 66.87 (CH2O–), 42.19, 41.79, 29.33 (CHCO); HRMS (ESI) calculated for C19H21NO3 [M + H]+: 312.1594, found: 312.1602. Purity: 97.3% (by HPLC).
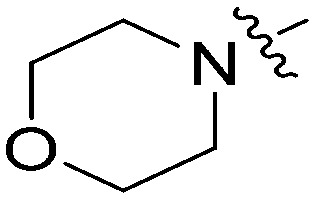
N-(3-Benzyl-5-hydroxyphenyl)morpholine-4-carboxamide (6l)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 39%, m.p. 155.6–157.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.11 (bs, 1H, NH), 8.32 (bs, 1H, OH), 7.28 (m, 2H), 7.22–7.15 (m, 3H), 6.87 (m, 1H), 6.72 (m, 1H), 6.21 (m, 1H), 3.76 (s, 2H, ArCH2Ar), 3.59–3.55 (t, J = 4 Hz, 4H, CH2), 3.40–3.35 (t, J = 1 Hz, 4H, CH2); 13C NMR (101 MHz, DMSO-d6) δ 157.82, 155.56, 142.49, 141.80, 141.74, 129.16, 128.79, 126.35, 111.40, 110.08, 105.09, 66.48 (CH2O), 44.68 (CH2N), 41.89 (ArCH2Ar); HRMS (ESI) calculated for C18H20N2O3 [M + H]+: 313.1547, found: 313.1549. Purity: 97.1% (by HPLC).
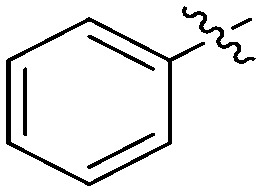
N-(3-Benzyl-5-hydroxyphenyl)benzamide (7a)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 52%, m.p. 215.8–217.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.02 (bs, 1H, NH), 9.29 (bs, 1H, OH), 7.94–7.88 (m, 1H), 7.59–7.53 (m, 1H), 7.50 (m, 1H), 7.29 (m, 1H), 7.20 (m, 2H), 7.06 (m, 1H), 6.37 (m, 1H), 3.83 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) 165.90 (CONH), 157.97, 142.91, 141.60, 140.59, 135.60, 131.86, 129.20, 128.84, 128.75, 128.09, 126.42, 112.18, 111.85, 105.90, 41.86 (ArCH2Ar); HRMS (ESI) calculated for C20H17NO2 [M + H]+: 304.1332, found: 304.1332. Purity: 98.7% (by HPLC).
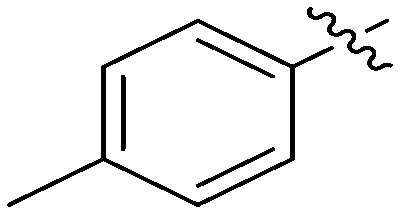
N-(3-Benzyl-5-hydroxyphenyl)-4-methylbenzamide (7b)
The compound was synthesized via a procedure similar to that of 6a. Light red solid, yield 64%, m.p. 184.5–186.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.96 (bs, 1H, NH), 9.32 (bs, 1H, OH), 7.83 (d, J = 8.1 Hz, 2H), 7.30 (t, J = 7.7 Hz, 4H), 7.20 (m, 4H), 7.05 (s, 1H), 6.36 (s, 1H), 3.82 (s, 2H, ArCH2Ar), 2.37 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 165.69 (CONH), 157.93, 142.89, 141.89, 141.63, 140.66, 132.66, 129.29, 129.20, 128.85, 128.14, 126.42, 112.10, 111.70, 105.80, 41.85 (ArCH2Ar), 21.46 (CH3); HRMS (ESI) calculated for C21H19NO2 [M + H]+: 318.1489, found: 318.1488. Purity: 99.2% (by HPLC).
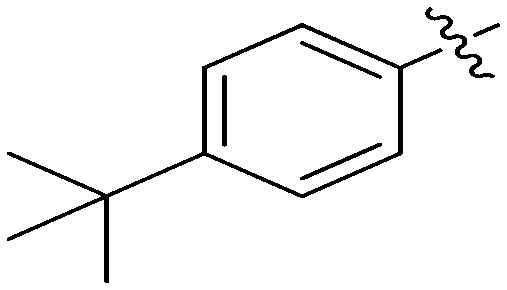
N-(3-Benzyl-5-hydroxyphenyl)-4-(tert-butyl)benzamide (7c)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 81%, m.p. 178.3–179.3 °C; 1H NMR (400 MHz, DMSO-d6) δ: 9.99 (bs, 1H, NH), 9.33 (bs, 1H, OH), 7.85 (d, J = 7.9 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.30 (m, 2H), 7.20 (m, 4H), 7.07 (m, 1H), 6.36 (m, 1H), 3.83 (s, 2H, ArCH2Ar), 1.31 (s, 9H, t-Bu); 13C NMR (101 MHz, DMSO-d6) δ 165.83 (CONH), 157.93, 154.72, 142.90, 141.63, 140.68, 132.87, 129.20, 128.85, 127.97, 126.42, 125.54, 112.03, 111.69, 105.72, 41.84 (ArCH2Ar), 35.12, 31.41 (CH3); HRMS (ESI) calculated for C24H25NO2 [M + H]+: 360.1958, found: 360.1954. Purity: 99.5% (by HPLC).
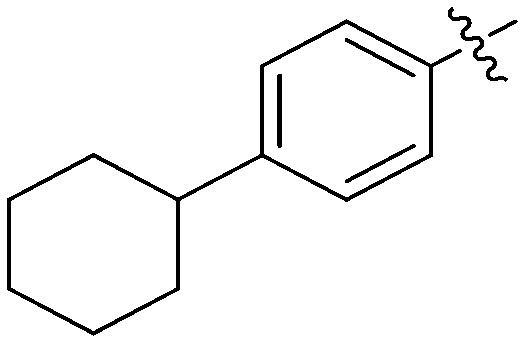
N-(3-Benzyl-5-hydroxyphenyl)-4-cyclohexylbenzamide (7d)
The compound was synthesized via a procedure similar to that of 6b. Yellow solid, yield 49%, m.p. 243.8–245.6 °C; 1H NMR (400 MHz, DMSO-d6) δ: 9.97 (bs, 1H, NH), 9.31 (bs, 1H, OH), 7.83 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 7.29 (m, 2H), 7.24–7.16 (m, 4H), 7.05 (s, 1H), 6.35 (s, 1H), 3.82 (s, 2H, ArCH2Ar), 2.58 (m, 1H, cyclohexyl-CH), 1.80 (m, 4H), 1.71 (m, 1H), 1.42 (m, 4H), 1.25 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.83 (CONH), 157.93, 151.67, 142.88, 141.62, 140.70, 133.23, 129.20, 128.84, 128.21, 127.05, 126.42, 112.01, 111.67, 105.71, 44.16, 41.84 (ArCH2Ar), 34.18, 26.73, 26.00; HRMS (ESI) calculated for C26H27NO2 [M + H]+: 386.2115, found: 386.2120. Purity: 99.8% (by HPLC).
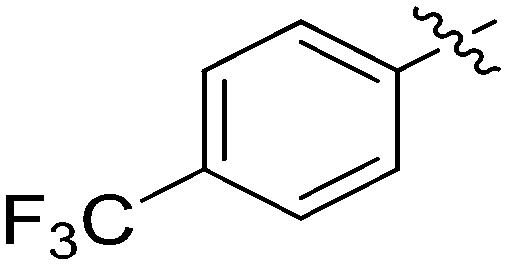
N-(3-Benzyl-5-hydroxyphenyl)-4-(trifluoromethyl)benzamide (7e)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 86%, m.p. 177.6–178.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.26 (bs, 1H, NH), 9.36 (bs, 1H, OH), 8.10 (d, J = 8.1 Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 7.30 (m, 2H), 7.20 (m, 4H), 7.05 (m, 1H), 6.40 (m, 1H), 3.84 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 164.73, 158.00, 143.08, 141.55, 140.24, 139.37, 131.71 (q, 2J = 31.8 Hz), 129.21, 129.04, 128.86, 126.44, 125.76 (q, 3J = 3.4 Hz), 124.40 (q, 1J = 270 Hz), 112.16, 112.11, 105.84, 41.79 (ArCH2Ar); HRMS (ESI) calculated for C21H16NO2F3 [M + H]+: 372.1206, found: 372.1208. Purity: 98.5% (by HPLC).
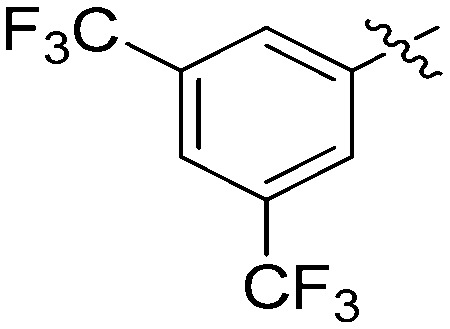
N-(3-Benzyl-5-hydroxyphenyl)-3,5-bis(trifluoromethyl)benzamide (7f)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 48%, m.p. 167.9–169.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.48 (bs, 1H, NH), 9.46 (bs, 1H, OH), 8.58 (s, 1H), 8.34 (s, 1H), 7.30 (m, 1H), 7.21 (m, 4H), 7.02 (s, 1H), 6.45 (s, 1H), 3.86 (s, 1H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 162.82 (CONH), 158.05, 143.16, 141.51, 139.89, 137.69, 130.86 (q, 2J = 33.4 Hz), 129.21, 129.01 (q, 4J = 1 Hz), 128.88, 126.47, 125.44 (q, 3J = 3 Hz), 123.60 (q, 1J = 272 Hz), 112.50, 112.26, 106.08, 41.76 (ArCH2Ar); HRMS (ESI) calculated for C22H16NO2F6 [M + H]+: 440.1080, found: 440.1092. Purity: 96.6% (by HPLC).
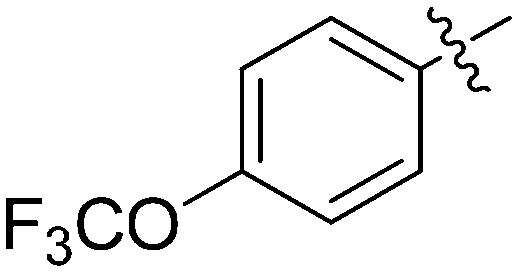
N-(3-Benzyl-5-hydroxyphenyl)-4-(trifluoromethoxy)benzamide (7g)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 80%, m.p. 129.3–131.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.15 (bs, 1H, NH), 9.34 (bs, 1H, OH), 8.08–7.96 (m, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.30 (t, J = 7.4 Hz, 2H), 7.25–7.15 (m, 4H), 7.04 (s, 1H), 6.39 (s, 1H), 3.84 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 164.82 (CONH), 157.91, 150.84, 143.11, 141.53, 140.28, 134.61, 130.46, 129.17, 128.87, 126.46, 121.09, 120.43 (q, 1J = 256 Hz), 112.14, 112.03, 105.83, 41.77 (ArCH2Ar); HRMS (ESI) calculated for C21H16NO3F3 [M – H]–: 386.1010, found: 386.1015. Purity: 99.6% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-2-chlorobenzamide (7h)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 52%, m.p. 159.2–161.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.29 (bs, 1H, NH), 9.34 (bs, 1H, OH), 7.57–7.45 (m, 3H), 7.42 (m, 1H), 7.29 (m, 2H), 7.19 (m, 3H), 7.15 (m, 1H), 6.98 (m, 1H), 6.98 (m, 1H), 6.37 (m, 1H), 3.82 (s, 1H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 163.87 (CONH), 158.12, 150.82, 146.89, 143.35, 141.46, 140.08, 138.59, 133.79, 129.23, 128.87, 126.46, 123.57, 112.20, 111.27, 105.05, 41.72 (ArCH2Ar); HRMS (ESI) calculated for C20H17NO2Cl [M + H]+: 338.0942, found: 338.0942. Purity: 99.9% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-4-chlorobenzamide (7i)
The compound was synthesized via a procedure similar to that of 6a. Red solid, yield 62%, m.p. 191.6–192.8 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.12 (bs, 1H, NH), 9.36 (bs, 1H, OH), 7.94 (d, J = 8.3 Hz, 2H), 7.58 (d, J = 8.3 Hz, 2H), 7.30 (m, 2H), 7.25–7.16 (m, 4H), 7.04 (s, 1H), 6.38 (s, 1H), 3.83 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 164.77 (CONH), 157.97, 142.98, 141.56, 140.38, 136.74, 134.25, 130.06, 129.20, 128.85, 128.83, 126.43, 112.17, 112.00, 105.91, 41.83 (ArCH2Ar); HRMS (ESI) calculated for C20H16NO2Cl [M + H]+: 338.0942, found: 338.0940. Purity: 99.3% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-3-chlorobenzamide (7j)
The compound was synthesized via a procedure similar to that of 6a. Red solid, yield 74%, m.p. 167.2–169.1 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.16 (bs, 1H, NH), 9.37 (bs, 1H, OH), 7.96 (s, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.54 (m, 1H), 7.33–7.25 (m, 2H), 7.20 (m, 4H), 7.04 (s, 1H), 6.39 (s, 1H), 3.83 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 164.38 (CONH), 157.98, 143.00, 141.56, 140.29, 137.51, 133.62, 131.71, 130.77, 129.20, 128.85, 127.86, 126.92, 126.44, 112.18, 112.10, 105.92, 41.82 (ArCH2Ar); HRMS (ESI) calculated for C20H16NO2Cl [M – H]–: 336.0797, found: 336.0797. Purity: 98.7% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-2,4-dichlorobenzamide (7k)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 55%, m.p. 130.2–132.0 °C; 1H NMR (400 MHz, CD3OD) δ 7.55 (d, J = 1.9 Hz, 1H), 7.49 (d, J = 8.2 Hz, 1H), 7.41 (dd, J = 8.2, 1.9 Hz, 1H), 7.29–7.22 (m, 2H), 7.22–7.12 (m, 5H), 6.92 (s, 1H), 3.87 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, CD3OD) δ 165.70 (CONH), 157.48, 143.41, 140.91, 139.02135.95, 135.26, 131.76, 129.69, 129.37, 128.57, 128.08, 127.14, 125.75, 112.12, 111.96, 105.11, 41.43 (ArCH2Ar); HRMS (ESI) calculated for C20H15NO2Cl2 [M + H]+: 372.0553, found: 372.0561. Purity: 991% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-3-chloro-4-fluorobenzamide (7l)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 43%, m.p. 159.3–160.9 °C; 1H NMR (400 MHz, CD3OD) δ 8.02 (d, J = 7.0 Hz, 1H), 7.92–7.80 (m, 1H), 7.33 (t, J = 8.8 Hz, 1H), 7.25 (m, 2H), 7.21–7.10 (m, 4H), 6.95 (s, 1H), 6.44 (s, 1H), 3.86 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, CD3OD) δ 164.79 (CONH), 159.93 (d, 1J = 253.0 Hz), 157.51, 143.22, 140.96, 139.17, 132.49 (d, 4J = 3.7 Hz), 130.10, 128.58, 128.11, 128.03, 125.69, 120.73 (d, 2J = 18.2 Hz), 116.35 (d, 2J = 21.9 Hz), 112.65, 112.06, 105.86, 41.45 (ArCH2Ar); HRMS (ESI) calculated for C20H15NO2FCl [M + H]+: 356.0848, found: 356.0845. Purity: 99.3% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-4-nitrobenzamide (7m)
The compound was synthesized via a procedure similar to that of 6a. Yellow solid, yield 82%, m.p. 205.9–207.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.38 (bs, 1H, NH), 9.42 (bs, 1H, OH), 8.34 (d, J = 8.8 Hz, 2H), 8.14 (d, J = 8.8 Hz, 2H), 7.30 (m, 2H), 7.21 (m, 4H), 7.05 (s, 1H), 6.41 (s, 1H), 3.84 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 164.24 (CONH), 158.01, 149.52, 143.13, 141.52, 141.22, 140.12, 129.66, 129.21, 128.87, 126.46, 123.93, 112.31, 112.12, 105.86, 41.78 (ArCH2Ar); HRMS (ESI) calculated for C20H16N2O4 [M + H]+: 349.1183, found: 349.1190. Purity: 98.3% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)thiophene-2-carboxamide (7n)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 61%, m.p. 180.6–182.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.01 (bs, 1H, NH), 9.34 (bs, 1H, OH), 7.99 (d, J = 3.3 Hz, 1H), 7.82 (d, J = 4.8 Hz, 1H), 7.36–7.25 (m, 2H), 7.20 (m, 4H), 7.16 (s, 1H), 6.99 (s, 1H), 6.38 (s, 1H), 3.83 (s, 2H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 160.21 (CONH), 157.99, 143.02, 141.56, 140.74, 140.12, 132.15, 129.42, 129.20, 128.86, 128.45, 126.44, 112.10, 111.93, 105.87, 41.81 (ArCH2Ar); HRMS (ESI) calculated for C18H15NO2S [M + H]+: 310.0896, found: 310.0892. Purity: 97.2% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)furan-2-carboxamide (7o)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 46%, m.p. 193.9–195.5 °C; 1H NMR (400 MHz, CD3OD) δ 7.70 (s, 1H), 7.30–7.23 (m, 2H), 7.21 (m, 3H), 7.19–7.12 (m, 2H), 6.95 (s, 1H), 6.61 (m, 1H), 6.43 (s, 1H), 3.88 (s, 2H, ArCH2Ar); 13C NMR (126 MHz, DMSO-d6) δ 157.95 (CONH), 156.55, 148.03, 146.08, 142.99, 141.55, 139.92, 129.21, 128.85, 126.43, 114.93, 112.49, 112.07, 111.87, 105.80, 41.80 (ArCH2Ar); HRMS (ESI) calculated for C18H15NO3 [M + H]+: 294.1125, found: 294.1118. Purity: 97.8% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (7p)
The compound was synthesized via a procedure similar to that of 6b. Yellow solid, yield 44%, m.p. 134.1–135.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.52 (bs, 1H, NH), 9.43 (bs, 1H, OH), 7.29 (m, 2H), 7.22 (m, 3H), 7.11 (s, 1H), 6.93 (s, 1H), 6.43 (s, 1H), 3.84 (s, 2H, ArCH2Ar), 2.78 (s, 3H, CH3); 13C NMR (101 MHz, CD3OD) δ 159.28 (CONH), 158.18, 157.64, 144.18, 143.49, 140.84, 138.69, 128.58, 128.07, 125.75, 112.49, 112.09, 105.40, 41.40 (ArCH2Ar), 12.01 (CH3); HRMS (ESI) calculated for C17H15N3O2S [M + H]+: 326.0958, found: 326.0956. Purity: 98.5% (by HPLC).
N-(3-Benzyl-5-hydroxyphenyl)-2-chloronicotinamide (7q)
The compound was synthesized via a procedure similar to that of 6a. White solid, yield 48%, m.p. 157.6–159.3 °C; 1H NMR (400 MHz, DMSO-d6) δ: 10.42 (bs, 1H, NH), 9.39 (bs, 1H, OH), 8.51 (d, J = 4.7 Hz, 1H), 8.02 (d, J = 7.4 Hz, 1H), 7.54 (dd, J = 7.3, 4.9 Hz, 1H), 7.30 (m, 1H), 7.21 (m, 1H), 7.14 (s, 1H), 6.94 (s, 1H), 6.40 (s, 1H), 3.84 (s, 1H, ArCH2Ar); 13C NMR (101 MHz, DMSO-d6) δ 163.87 (CONH), 158.12, 150.82, 146.89, 143.35, 141.46, 140.08, 138.59, 133.79, 129.23, 128.87, 126.46, 123.57, 112.20, 111.27, 105.05, 41.72 (ArCH2Ar); HRMS (ESI) calculated for C19H16N2O2Cl [M + H]+: 339.0895, found: 339.0896. Purity: 98.9% (by HPLC).
Synthesis of N-(3-(2,5-dimethylbenzyl)-5-hydroxyphenyl)cyclohexanecarboxamide (9a)
A solution of (3-methoxy-5-nitrophenyl) methanol 3 (460 mg, 2.5 mmol), p-xylene (1.5 mL, 12.5 mmol), TfOH (38 mg, 0.25 mmol) and hexafluoroisopropanol (5 mL) in a 10 mL glass pressure tube was stirred at 100 °C for 24 h. After being cooled to room temperature, water (50 mL) was added. The reaction mixture was extracted with EtOAc (50 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by flash column chromatography over silica (petroleum ether/EtOAc = 50 : 1) to give 2-(3-methoxy-5-nitrobenzyl)-1,4-dimethylbenzene 8a. 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H), 7.54 (s, 1H), 7.04 (m, 1H), 6.98 (m, 2H), 6.91 (s, 1H), 3.97 (s, 2H, ArCH2Ar), 3.82 (s, 3H, CH3O), 2.29 (s, 3H), 2.17 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.13, 149.34, 143.76, 136.89, 135.81, 133.30, 130.76, 130.56, 127.84, 121.80, 116.19, 105.52, 55.79 (CH3O), 39.21 (ArCH2Ar), 20.99, 19.21.
To a solution of compound 8a (271 mg, 1 mmol) in methylene chloride (2 mL), a solution of BBr3 in dichloromethane (1.0 M, 5 mL, 5 mmol) was added slowly at –80 °C. The resulting red solution was warmed to 0 °C and stirred for 12 h. Saturated aqueous sodium bicarbonate (5 mL) was added at 0 °C. The solution was extracted with dichloromethane (5 mL × 3). The combined organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by flash chromatography over silica gel (petroleum/EtOAc = 20 : 1 ∼ 5 : 1) to give 3-(2,5-dimethylbenzyl)-5-nitrophenol (141 mg, 55%). 1H NMR (400 MHz, CD3OD) δ 7.40 (m, 2H), 7.02 (m, 1H), 6.95 (m, 2H), 6.88–6.84 (m, 1H), 3.93 (s, 2H, ArCH2Ar), 2.26 (s, 3H), 2.12 (s, 3H).
A solution of 3-(2,5-dimethylbenzyl)-5-nitrophenol (141 mg, 0.55 mmol) and 10% Pd/C (28 mg) in a mixed solvent (CH3OH/CH2Cl2 = 1 : 1 (V/V), 5 mL) was purged with H2 three times. The reaction mixture was stirred with a balloon of H2 at room temperature for 4 h. The reaction mixture was filtered and concentrated in vacuo. 3-Amino-5-(2,5-dimethylbenzyl)phenol was obtained and directly used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.04 (m, 1H), 7.00–6.91 (m, 2H), 6.04 (m, 1H), 6.01 (t, J = 2.1 Hz, 1H), 5.98 (m, 1H), 3.79 (s, 2H, ArCH2Ar), 2.29 (s, 3H, CH3), 2.19 (s, 3H, CH3).
A solution of cyclohexanecarbonyl chloride (73.3 μL, 0.55 mmol) in THF (1 mL) was slowly added at 0 °C to a solution of 3-amino-5-(2,5-dimethylbenzyl)phenol (113.5 mg, 0.5 mmol) and triethylamine (76.3 μL, 0.55 mmol) in THF (2 mL). After the reaction mixture was stirred for 3 h at room temperature, the reaction was quenched with water (10 mL) and the mixture was extracted with EtOAc (15 mL × 2). The combined organic phase was dried over anhydrous Na2SO4 and filtered. After the solvent was removed under reduced pressure, the crude product was purified by column chromatography (petroleum/EtOAc = 20 : 1–2 : 1) to afford 9a as a white solid. m.p. 179.5–181.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.56 (bs, 1H, NH), 9.19 (bs, 1H, OH), 7.09–7.00 (m, 2H), 6.93 (m, 2H), 6.72 (s, 1H), 6.18 (s, 1H), 3.75 (s, 2H, ArCH2Ar), 2.25 (m, 4H), 2.14 (s, 3H), 1.73 (m, 4H), 1.63 (m, 1H), 1.36 (m, 2H), 1.26–1.12 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 176.27 (CONH), 157.34, 142.53, 139.56, 138.33, 134.94, 133.17, 130.51, 129.78, 126.70, 111.38, 110.97, 104.75, 45.78 (cyclohexyl-CH), 38.99 (ArCH2Ar), 29.27, 25.49, 25.38, 19.65, 17.91; HRMS (ESI) calculated for C22H27NO2 [M – H]–: 336.1969, found: 336.1955. Purity: 99.4% (by HPLC).
N-(3-Hydroxy-5-(2,4,6-trimethylbenzyl)phenyl)cyclohexanecarboxamide (9b)
This compound was synthesized via a procedure similar to that of 9a. Yellow solid, yield 44%, m.p. 248.5–249.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.49 (bs, 1H, NH), 6.96 (bs, 1H, OH), 6.84 (s, 2H), 6.60 (s, 1H), 6.06 (s, 1H), 3.80 (s, 2H, ArCH2Ar), 2.32–2.19 (m, 4H), 2.15 (s, 6H), 1.72 (m, 3H), 1.63 (m, 1H), 1.36 (m, 2H), 1.28–1.10 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 174.56 (CONH), 158.28, 141.65, 140.83, 136.84, 135.17, 134.24, 129.09, 110.13, 109.61, 104.58, 45.28 (cyclohexyl-CH), 34.74 (ArCH2Ar), 29.61, 25.88, 25.72, 20.99, 20.25; HRMS (EI) calculated for C23H29NO2 [M + H]+: 352.2271, found: 352.2261. Purity: 96.4% (by HPLC).
Biological assays
Determination of minimum inhibitory concentration (MIC) against Mtb H37Ra21
The assay was conducted over a range of 4-fold increasing concentrations prepared in 1 mL of 7H9 broth containing 0.2 mL of a 1/100 dilution of an autoluminescent Mtb H37Ra broth culture (OD600, 0.8) grown in 7H9 broth. The test compound was prepared as 10-point two-fold serial dilutions in DMSO and diluted in 7H9-Tw-OADC medium in 96-well plates with a final DMSO concentration of 2%. The highest concentration of the compound was 100 μg mL–1. To each well, 196 μL of a 1/100 dilution of an autoluminescent Mtb H37Ra broth culture (OD600 = 0.8, grown in 7H9 broth) and 4 μL of the solution of the test compound were added. For potent compounds, assays were repeated at a lower starting concentration.
Each plate included a control for zero growth (INH, 1 μg mL–1) and maximum growth (DMSO only), for generating the INH dose–response curve. 200 μL medium was added into holes on the inside edge of the plate to prevent evaporation. RLU counts were determined daily in triplicate for 0–5 days. Growth was measured by fluorescence using a GloMax reader. For MIC, the 10-point dose response curve was plotted as the percentage (%) of growth and fitted to the Gompertz model using GraphPad Prism 5. The MIC was defined as the minimum concentration at which growth was completely inhibited and was calculated from the inflection point of the fitted curve to the lower asymptote (zero growth).
Determination of minimum inhibitory concentration (MIC) against Mtb H37Rv and MDR-Mtb
The MICs of the test compounds were determined by the well-established microplate alamar blue assay (MABA) against H37Rv and clinically isolated MDR-Mtb strains (provided by the Guangzhou Chest Hospital).22 Mtb H37Rv and MDR-Mtb strains in 7H9-Tw-OADC were cultivated in a 50 mL tube containing glass beads at 37 °C. The strains were transferred into a 250 mL flask containing 50 mL 7H9-Tw-OADC while the OD600 value reached 0.3–0.8. The test compounds were prepared as in 4.2.1. Each well contained 196 μL broth culture of a 1/100 dilution of Mtb H37Rv or MDR-Mtb (OD600 = 0.8, grown in 7H9 broth) and 4 μL of the solution of the test compound. For potent compounds, assays were repeated at lower starting concentrations. INH and RIF were used as the positive controls. The plates were incubated at 37 °C. On the 7th day, 12.5 μL of 20% Tween 80 and 20 μL of alamarBlue (Bio-Rad) were added to the test plate. A change in color from blue (oxidized state) to pink (reduced) indicated the growth of bacteria after incubation at 37 °C for 16–24 h. The MIC was defined as the lowest concentration of drug that prevented this change in color.
Determination of the antibacterial activity
The minimum inhibitory concentration (MIC) of compounds 6g and 7a was defined as the lowest concentration (the highest dilution) of each compound that completely inhibited the growth of bacteria after incubation at 37 °C for 18–24 h, by means of the standard twofold serial dilution method in 96-well microtest plates. Amoxicillin and ofloxacin were used as the positive controls. The bacterial suspension was adjusted with sterile saline to a concentration of 1 × 105 CFU. Initially the compounds were dissolved in DMSO to prepare the stock solutions (10 mg mL–1), and then the test compounds and reference drugs were prepared in BHI broth to obtain the required concentrations 50–1.5125 μg mL–1. These dilutions were inoculated and incubated at 37 °C for 24 h.
Cytotoxicity studies
The cytotoxicities of compounds 6g and 7a were assayed against RAW 264.7 and HepG2 cell lines at concentrations from 100 to 6.25 μg mL–1. The cells were seeded in 96-well plates and then allowed to recover for 24 h. Different concentrations of the test compound were added to the plate and each experiment was repeated three times. After being incubated for 72 h, cells were harvested and cell viability was assessed by MTT assay. The cytotoxicities were reported as IC50 values, which were calculated by GraphPad Prism Software version 5.23
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank the National Science and Technology Major Project of the Ministry of Science and Technology of China (2018ZX09735010 and 2017ZX10302301-003-002), the Chinese Academy of Sciences Grants (154144KYSB20150045, KFZD-SW-207 and YJKYYQ20170036), the National Mega-project of China for Innovative Drugs (2018ZX09721001-003-003), the Science and Technology Innovation Leader of Guangdong Province (2016TX03R095), and the Guangzhou Science Technology and Innovation Commission (201707010210, 201604020019) for the financial support for this study.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00212f
References
- World Health Organization, Global tuberculosis report, 2017.
- Palomino J. C., Martin A. Future Microbiol. 2013;8:1071–1080. doi: 10.2217/fmb.13.85. [DOI] [PubMed] [Google Scholar]
- Ryan N. J., Lo J. H. Drugs. 2014;74:1041–1045. doi: 10.1007/s40265-014-0241-5. [DOI] [PubMed] [Google Scholar]
- Hoagland D. T., Liu J., Lee R. B., Lee R. E. Adv. Drug Delivery Rev. 2016;102:55–72. doi: 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field S. K. Clin. Med. Insights: Ther. 2013;5:137–149. [Google Scholar]
- For the reviews of antitubercular agents, see: ; (a) Beena, Rawat D. S. Med. Res. Rev. 2013;33:693–764. doi: 10.1002/med.21262. [DOI] [PubMed] [Google Scholar]; (b) Fernandes G. F. D. S., Jornada D. H., De Souza P. C., Chin C. M., Pavan F. R., Dos Santos J. L. Curr. Med. Chem. 2015;22:3133–3161. doi: 10.2174/0929867322666150818103836. [DOI] [PubMed] [Google Scholar]; (c) Mehra R., Khan I. A., Nargotra A. Eur. J. Pharm. Sci. 2017;104:1–15. doi: 10.1016/j.ejps.2017.03.028. [DOI] [PubMed] [Google Scholar]; (d) Chiarelli L. R., Mori G., Esposito M., Orena B. S., Pasca M. R. Curr. Med. Chem. 2016;23:3813–3846. doi: 10.2174/1389557516666160831164925. [DOI] [PubMed] [Google Scholar]
- RoŽman K., Sosič I., Fernandez R., Young R. J., Mendoza A., Gobec S., Encinas L. Drug Discovery Today. 2017;22:492–502. doi: 10.1016/j.drudis.2016.09.009. [DOI] [PubMed] [Google Scholar]
- Bernstei J., Lott W. A., Steinberg B. A., Yale H. L. Am. Rev. Tuberc. 1952;65:357–364. doi: 10.1164/art.1952.65.4.357. [DOI] [PubMed] [Google Scholar]
- Dessen A., Quémard A., Blanchard J. S., Jacobs W. R., Sacchettini J. C. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S. V., Reich R., Dou S. J., Jasperse L., Pan X., Wanger A., Quitugua T., Graviss E. A. Antimicrob. Agents Chemother. 2003;47:1241–1250. doi: 10.1128/AAC.47.4.1241-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the reviews of InhA inhibitors, see: ; (a) Lu X. Y., You Q. D., Chen Y. D. Mini-Rev. Med. Chem. 2010;10:182–193. [Google Scholar]; (b) AlMatar M., Makky E. A., Var I., Kayar B., Köksal F. Pharmacol. Rep. 2018;70:217–226. doi: 10.1016/j.pharep.2017.09.001. [DOI] [PubMed] [Google Scholar]
- For the recent studies of InhA inhibitors as the antitubercular agents, see: ; (a) Spagnuolo L. A., Eltschkner S., Yu W. X., Daryaee F., Davoodi S., Knudson S. E., Allen E. K. H., Merino J., Pschibul A., Moree B., Thivalapill N., Truglio J. J., Salafsky J., Slayden R. A., Kisker C., Tonge P. J. J. Am. Chem. Soc. 2017;139:3417–3429. doi: 10.1021/jacs.6b11148. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Štular T., Lešnik S., RoŽman K., Schink J., Zdouc M., Ghysels A., Liu F., Aldrich C. C., Haupt V. J., Salentin S., Daminelli S., Schroeder M., Langer T., Gobec S., JaneŽič D., Konc J. J. Med. Chem. 2016;59:11069–11078. doi: 10.1021/acs.jmedchem.6b01277. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chollet A., Stigliani J. L., Pasca M. R., Mori G., Lherbet C., Constant P., Quémard A., Bernadou J., Pratviel G., Bernardes-Génisson V. Chem. Biol. Drug Des. 2016;88:740–755. doi: 10.1111/cbdd.12804. [DOI] [PubMed] [Google Scholar]; (d) Chollet A., Mori G., Menendez C., Rodriguez F., Fabing I., Pasca M. R., Madacki J., Korduláková J., Constant P., Quémard A., Bernardes-Génisson V., Lherbet C., Baltas M. Eur. J. Med. Chem. 2015;101:218–235. doi: 10.1016/j.ejmech.2015.06.035. [DOI] [PubMed] [Google Scholar]
- (a) Manjunatha U. H., Rao S. P. S., Kondreddi R. R., Noble C. G., Camacho L. R., Tan B. H., Ng S. H., Ng P. S., Ma N. L., Lakshminarayana S. B., Herve M., Barnes S. W., Yu W., Kuhen K., Blasco F., Beer D., Walker J. R., Tonge P. J., Glynne R., Smith P. W., Diagana T. T. Sci. Transl. Med. 2015;7:269ra3. doi: 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ng P. S., Manjunatha U. H., Rao S. P. S., Camacho L. R., Ma N. L., Herve M., Noble C. G., Goh A., Peukert S., Diagana T. T., Smith P. W., Kondreddi R. R. Eur. J. Med. Chem. 2015;106:144–156. doi: 10.1016/j.ejmech.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Vuković V. D., Richmond E., Wolf E., Moran J. Angew. Chem., Int. Ed. 2017;56:3085–3089. doi: 10.1002/anie.201612573. [DOI] [PubMed] [Google Scholar]
- Zhang T. Y., Li S. Y., Nuermberger E. L. PLoS One. 2012;7:e29774. doi: 10.1371/journal.pone.0029774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the synthesis of compounds 12a-12c, See SI for the details.
- For the synthesis of compounds 14, 15, See SI for the details.
- Brendan P. O., Justin T. E., Andrew D. H. J. Am. Chem. Soc. 2001;123:5382–5383. [Google Scholar]
- Ueda S., Nagasawa N. H. J. Org. Chem. 2009;74:4272–4277. doi: 10.1021/jo900513z. [DOI] [PubMed] [Google Scholar]
- The inhibitive activity of compound 6g against Mtb-InhA was examined. Unexpectedly, 6g did not exert inhibitive activity against InhA. The result indicated that Mtb-InhA is not the target of m-amidophenol derivatives. The action mechanism remains to be investigated. See SI for the experiment details.
- Yang F., Njire M. M., Liu J., Wu T., Wang B. X., Liu T. Z., Cao Y. Y., Liu Z. Y., Wan J. T., Tu Z. C., Tan Y. J., Tan S. Y., Zhang T. Y. PLoS One. 2015;10:e0119341. doi: 10.1371/journal.pone.0119341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., Wang B. X., Wu T., Wan J. T., Tu Z. C., Njire M., Wan B. J., Franzblauc S. G., Zhang T. Y., Lu X. Y., Ding K. ACS Med. Chem. Lett. 2015;6:814–818. doi: 10.1021/acsmedchemlett.5b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa J., Khan S. I., Ma G., Walker L. A., Khan I. A. Lipids. 2004;39:167–172. doi: 10.1007/s11745-004-1215-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.