

Abstract
We present a kinetic-energy density-functional theory and the corresponding kinetic-energy Kohn–Sham (keKS) scheme on a lattice and show that, by including more observables explicitly in a density-functional approach, already simple approximation strategies lead to very accurate results. Here, we promote the kinetic-energy density to a fundamental variable alongside the density and show for specific cases (analytically and numerically) that there is a one-to-one correspondence between the external pair of on-site potential and site-dependent hopping and the internal pair of density and kinetic-energy density. On the basis of this mapping, we establish two unknown effective fields, the mean-field exchange-correlation potential and the mean-field exchange-correlation hopping, which force the keKS system to generate the same kinetic-energy density and density as the fully interacting one. We show, by a decomposition based on the equations of motions for the density and the kinetic-energy density, that we can construct simple orbital-dependent functionals that outperform the corresponding exact-exchange Kohn–Sham (KS) approximation of standard density-functional theory. We do so by considering the exact KS and keKS systems and comparing the unknown correlation contributions as well as by comparing self-consistent calculations based on the mean-field exchange (for the effective potential) and a uniform (for the effective hopping) approximation for the keKS and the exact-exchange approximation for the KS system, respectively.
1. Introduction
Density-functional theory (DFT) has become over the past decades a standard approach to the quantum many-body problem. Its success comes from the fact that it combines low computational cost with a reasonable accuracy, which helps to understand and predict experimental results for systems not accessible with wave function-based methods. DFT avoids the exponential numerical costs of wave function-based methods by reformulating quantum mechanics in terms of the density. The major drawback of DFT is that the exact energy expression of the quantum system in terms of the density is not available, and in practice approximations need to be employed. Already before the rigorous formulation of DFT,1 a heuristic method based on the density instead of the wave function existed, which was called the Thomas–Fermi theory.2,3 While this theory proved to be very important for the derivation of fundamental results, for example, the stability of quantum matter,4 in practice it is not very accurate (only in the limit of atoms with arbitrarily high atomic number or for homogeneous systems) and does not provide basic properties such as the shell structure of atoms or the binding of molecules. As it was quickly realized, it is the approximation to the kinetic-energy expression that prevents Thomas–Fermi density-functional approximations from leading to accurate results. What has made DFT popular for determining properties of complex many-body systems is the Kohn–Sham (KS) construction,5 where instead of modeling the kinetic energy directly in terms of the density an auxiliary noninteracting quantum system is used that has the same density. The kinetic energy of this computationally cheap auxiliary system is then corrected by so-called Hartree-exchange-correlation (Hxc) contributions that incorporate the missing interaction and kinetic-energy contributions. Already simple approximations to this unknown expression give reasonably accurate answers. However, it is hard to systematically increase the accuracy of approximations while still keeping the favorable numerical costs.6 Moreover, it has been shown recently that numerous functionals, although accurate when it comes to total energies, fail to reproduce the true density.7 The difficulty in functional construction can be attributed to the fact that it is not easy to find the appropriate expression of Hxc contributions in terms of the auxiliary KS wave function or the density.
There are several other approaches for dealing with the quantum many-body problem that also avoid the many-body wave function, while the basic variable used makes it easier to model the desired physical quantities. Green’s function techniques can be systematically improved in accuracy by including higher-order Feynmann diagrams, but are computationally much more expensive.8,9 Reduced density-matrix (RDM) functional theories10,11 provide a compromise between accuracy and computational cost. In one-body RDM (1RDM) functional theory,11 the kinetic energy is an explicit functional of the 1RDM, thus only part of the interaction energy needs to be approximated, while in the two-body case10 even the interaction is given by an explicit functional. Although the explicit use of wave functions can be avoided in these cases, it is still necessary for the RDM to be representable by a wave function. However, the so-called N-representability conditions that guarantee an underlying wave function associated with an RDM are anything but trivial.12−15 Moreover, it is not possible to associate to every RDM an auxiliary system of noninteracting particles that would allow one to replace the N-representability conditions by a numerically simpler auxiliary wave function, like in the DFT case. The Bogoliubov–Born–Green–Kirkwood–Yvon hierarchy, where the time propagation of an RDM of certain order is related to the RDM of the next order, suffers from similar N-representability issues.16
There are now several possible ways to remedy the above-mentioned deficiencies. For 1RDM theory, it is helpful to consider the many-body problem at finite temperature and indefinite numbers of particles.17−19 In this case, the representability conditions in terms of an ensemble of wave functions are known and easy to implement, and one can even find a noninteracting auxiliary system that generates the same 1RDM. Another possibility is to construct approximate natural orbitals, which are eigenfunctions of single-particle Hamiltonians with a local effective-potential.20,21 On the DFT side, besides changing the auxiliary system for the KS construction,22−24 a possible way out is to include the kinetic-energy density as a basic functional variable along with the density, simplifying the modeling of the exchange-correlation potentials because they will not include any more kinetic-energy contributions. This however implies that an additional auxiliary potential, which couples to the kinetic-energy density, has to be introduced. A similar approach has recently appeared in a different context, that is, in thermal DFT,25,26 where the additional auxiliary potential corresponds to a proxy for local temperature variations and couples to the entire energy density, including kinetic and interaction contributions. The concept of local temperature was also introduced in the local thermodynamic ansatz of DFT.27−29 Furthermore, it is important to note that the kinetic-energy density is already used extensively in DFT, for instance, as an integral part of the so-called meta-GGAs. When treated within the generalized KS framework,30 meta-GGAs lead to a local potential coupling to the kinetic-energy density, which can be interpreted as a position-dependent mass.31
In this Article, we investigate the possibility to include the kinetic-energy density as a basic functional variable in DFT alongside the density. The idea is that by doing so one can increase the accuracy of density-functional approximations. We investigate this by constructing the exact density functionals of standard DFT and comparing them to the combined kinetic-energy density and density functionals of this extended approach we call kinetic-energy density-functional theory (keDFT). In this way, we want to assess possible advantages of such an approach when considering strongly correlated systems. The so-called kinetic contribution32 to the exchange correlation potential is important for the description of such systems. It has been shown that standard DFT functionals fail to describe the effects of this kinetic contribution such as the band narrowing due to interactions.33 By including the kinetic-energy density as a fundamental variable, this contribution is taken into account explicitly.
Further, we want to consider the quality of possible approximation schemes to keDFT based on a kinetic-energy KS (keKS) construction and test them in practice. As is clear from the extent of the proposed program, this is not possible for real systems. Similar to investigations of the exact functionals in DFT34−36 and other extensions of DFT,37 we restrict our study to a finite lattice approximation for the Hamiltonian, where the particles are only in specific states/positions. We therefore consider lattice keDFT. In this way, we not only avoid the prohibitively expensive calculation of reference data for realistic interacting many-body systems but also avoid mathematical issues connected to the continuum case, like the nonexistence of ground states and nondifferentiability of the involved functionals38,39 or having to deal with the kinetic-energy operator, which is unbounded.40 All of the operators that appear on the lattice are Hermitian matrices, which yield lowest energy eigenstates, and exact solutions can be easily calculated contrary to the continuum where one always has to resort to basis set approximations. We also highlight how simple approximations carry over from our model systems to more complex lattice systems and even to the full continuum limit. The results hint at the possibility to treat weakly and strongly correlated systems with the same simple approximation to keDFT.
This Article is structured as follows: In section 2 we introduce our lattice model, define the density and kinetic-energy density on the lattice, and highlight for a simple two-site case that the kinetic-energy density is a natural quantity to be reproduced by an extended KS construction. We then introduce the resulting keKS construction assuming the existence of the underlying maps between densities and fields. In section 3 we discuss these mappings and show how by allowing a spatially dependent mass/hopping a large gauge freedom is introduced. Still we can provide a bijective mapping between densities and fields for specific cases. In section 4 we then show how we numerically construct the mappings beyond these specific cases and hence find that keDFT on a lattice can be defined also for more general situations. In section 5 we then use the constructed mappings to determine the exact correlation expressions for the KS and the keKS construction, respectively. In section 6 we then compare the results of self-consistent calculations for similar approximations for the KS and the keKS systems, respectively. Finally, we conclude in section 7.
2. Formulation of the Lattice Problem
In the following, we consider quantum systems consisting of N Fermions (electrons) on a one-dimensional lattice of M discrete sites. We assume that these particles can move from site to site only via nearest-neighbor hopping (corresponding to a second-order finite-differencing approximation to the Laplacian) and employ zero boundary conditions for definiteness (the extension to periodic boundary conditions is straightforward). This leads to a Hamiltonian of the following type:
| 1 |
The nonlocal first term corresponds to the kinetic energy. Without loss of generality, we can assume that the hopping amplitude obeys ti > 0. Let us point out that usually the hopping amplitude is site-independent. We employ this more general form (corresponding to a site-dependent mass) to establish the necessary mappings (see eq 10). However, when we numerically consider interacting systems, we always employ a site-independent hopping, which corresponds to the standard Hubbard Hamiltonian. The second term corresponds to a local scalar electrostatic potential vi acting on the charged particles at site i. U ≥ 0 is the on-site Hubbard interaction between the Fermions, which is reminiscent of the Coulomb interaction. Further, the Fermionic creation and annihilation operators obey the anticommutation relations {ĉiσ†,ĉj} = δijδσσ′, where σ corresponds to the spin degrees of freedom of the particles, n̂iσ = ĉiĉiσ is the spin-density operator, and n̂i = n̂i + n̂i↓ is the density operator that couples to the electrostatic potential. Because we fix the number of particles, the potential vi is physically equivalent to a potential that differs by only a global constant. In the following, this arbitrary constant is fixed by requiring
| 2 |
Now,
if Ψ is the ground-state wave function
of Hamiltonian 1, we can associate to every
point in space a ground-state density ni = ⟨Ψ|n̂i|Ψ⟩. From the lattice-version
of DFT,41 we know that for every fixed
set of parameters (t,U), there is
a bijective mapping between the set of all possible potentials (in
the above gauge) to all possible densities for a fixed number of particles.
To ease notation, we introduce a vector for the density n ≡ (n1,...,nM) and accordingly for the potential v ≡ (v1,...,vM), which allows us to write the underlying
mapping as 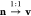 . Accordingly, for the potential of an interacting
system (U > 0) as a functional of the density,
we
write vi[n]. We further note that because the total number of particles is
fixed to N, the density is constrained by ∑i = 1Mni = N. This means that instead of
the density at every point one can equivalently use the density differences
between sites Δni = ⟨Ψ|n̂i – n̂i+1|Ψ⟩ to establish the above mapping at a fixed number
of particles. Similarly, knowing the local potential vi at every site together with the gauge
condition 2 is equivalent to knowing Δvi = vi – vi+1. In certain situations, for example, for figures,
it is more convenient to use the density and potential differences
instead of the density and potential.
. Accordingly, for the potential of an interacting
system (U > 0) as a functional of the density,
we
write vi[n]. We further note that because the total number of particles is
fixed to N, the density is constrained by ∑i = 1Mni = N. This means that instead of
the density at every point one can equivalently use the density differences
between sites Δni = ⟨Ψ|n̂i – n̂i+1|Ψ⟩ to establish the above mapping at a fixed number
of particles. Similarly, knowing the local potential vi at every site together with the gauge
condition 2 is equivalent to knowing Δvi = vi – vi+1. In certain situations, for example, for figures,
it is more convenient to use the density and potential differences
instead of the density and potential.
Clearly a similar mapping between density and potential also holds for a noninteracting Hamiltonian, that is, U = 0. Because it is bijective, we can invert the mapping and find a potential vs (where we follow the usual convention and denote the potential of a noninteracting system with an s) for a given density n. The noninteracting mapping allows one to define vis[n], which in turn leads to
| 3 |
The noninteracting Hamiltonian reproduces the prescribed density n as its ground state by construction. This is not yet the KS construction, because we need to know the target density in advance. Only upon connecting the interacting with the noninteracting system by introducing the Hxc potential:
| 4 |
which can also be defined as a derivative of the corresponding Hxc energy functional with respect to n, we find the nonlinear KS equation for a given and fixed external potential v of the interacting system:
| 5 |
This problem has as the unique solution the noninteracting wave function that generates the density of the interacting problem without knowing it in advance.42 It is imperative at this point to understand the (often overlooked) difference between Ĥs[n] together with the density functional vis[n] and HKS together with the KS-potential functional vi[v;n] = vi + viHxc[n]. Only the latter provides an iterative scheme to predict the density of an interacting references system. Also, only at the unique fixed point of the KS iteration procedure, where vi = vi[n], do both Hamiltonians give rise to the same noninteracting wave function. When we later present results for the exact KS construction in section 5, we refer always to the results at the unique fixed point of the KS construction. In practice, however, we do not have the exact vi[v;n] available, and hence we need to devise approximations to the unknown Hxc functional. The simplest such approximation would be a mean-field ansatz of the form viHxc[n] ≈ Uni. To comply with the chosen gauge of eq 2, we could use vi[n] ≈ U(ni – N/M).
As we will see in section 5, the major problem in these approximations is that the kinetic-energy density of the KS and the interacting system become dramatically different with an increasing U. Here, the kinetic-energy density Ti at site i is defined nonlocally (because it involves the hopping) with the help of the first off-diagonal of the (spin-summed) 1RDM in site basis representation:
| 6 |
where γi,i+1 is given by
| 7 |
By analogy to the continuum case, one can also define the charge current Ji as
| 8 |
With no external magnetic field present, that is, no complex phase of the hopping amplitude, the ground-state wave functions are real valued, which implies γi,i+1 = γi+1,i, leading to zero current. We note that the current obeys the lattice version of the continuity equation:
| 9 |
in a time-dependent situation, where 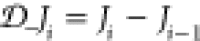 is the backward derivative of Ji. Equation 9 is an equation of motion (EOM) (see also Appendix B for further EOMs) that physical wave
functions need to adhere to. It is important to note that it is not
only the variational (minimum-energy) principle that ground states
have to fulfill, but there are many more exact relations. While for
the case of the ground state the EOM of eq 9 is trivial because both sides are individually
zero, there are many other nontrivial exact relations that can be
based on EOMs and that provide us with exact relations between the
densities, the fields, and other physical quantities. For instance,
the second-time derivative of the density provides us with the local
force balance of the equilibrium quantum system,43 which we will use in section 4. Also, while the most common way to find
approximations for the Hxc potentials is by obtaining approximate
Hxc energy expressions and then taking a functional derivative, the
EOMs provide an alternative way to construct approximate Hxc potentials
without the need to perform functional variations.44,45 In section 5 we will
show how one can obtain such approximations, for instance, the exact-exchange
approximation of standard DFT.
is the backward derivative of Ji. Equation 9 is an equation of motion (EOM) (see also Appendix B for further EOMs) that physical wave
functions need to adhere to. It is important to note that it is not
only the variational (minimum-energy) principle that ground states
have to fulfill, but there are many more exact relations. While for
the case of the ground state the EOM of eq 9 is trivial because both sides are individually
zero, there are many other nontrivial exact relations that can be
based on EOMs and that provide us with exact relations between the
densities, the fields, and other physical quantities. For instance,
the second-time derivative of the density provides us with the local
force balance of the equilibrium quantum system,43 which we will use in section 4. Also, while the most common way to find
approximations for the Hxc potentials is by obtaining approximate
Hxc energy expressions and then taking a functional derivative, the
EOMs provide an alternative way to construct approximate Hxc potentials
without the need to perform functional variations.44,45 In section 5 we will
show how one can obtain such approximations, for instance, the exact-exchange
approximation of standard DFT.
Clearly, if we could enforce
that an auxiliary noninteracting system
has the same 1RDM as the interacting one, then also the kinetic-energy
densities T of the two systems would coincide. This suggests
that one can establish a mapping between the interacting 1RDM and
a nonlocal potential, that is, a vi,j that connects any two sites of the lattice
and thus couples directly to the full 1RDM. However, in general this
is not possible as has been realized early on in 1RDM functional theory.46 A concrete example is the two-site homogeneous
Hubbard problem at half filling forming a singlet. In this case, we
have i = 1,2 and vi = 0. So we have a homogeneous density ni = 1, and we can analytically determine all
eigenfunctions of the interacting and noninteracting system. Further,
in the case of only two sites, the full spin-summed 1RDM is a 2 ×
2 matrix, where the diagonals are merely γi,i = ni = 1 and the off-diagonals are given explicitly by 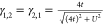 . Because the density
fixes the potential
of the interacting and KS system to be exactly zero, our only freedom
is to adopt the nonlocal potential, which is equivalent to just adopting
the hopping of the KS system (in this case, the nonlocal potential v1,2 ≡ t). Yet because
the off-diagonals for the KS system are γ1,2s = γ2,1 ≡ 1 irrespective of
the hopping amplitude, no nonlocal KS potential exists that reproduces
the interacting 1RDM. This is also true in more general lattice situations
as has been shown in, for example, ref (47). For the 1RDM, two solutions to this problem
are known. One is to include temperature and possibly an indefinite
number of particles, which introduces off-diagonals that depend on
the temperature and the hopping, that is, the nonlocal potential.19 We note that for the homogeneous two-site case,
this can still be solved analytically and verified explicitly. The
other possibility is to make the system degenerate such that we can
reproduce any density matrix.19
. Because the density
fixes the potential
of the interacting and KS system to be exactly zero, our only freedom
is to adopt the nonlocal potential, which is equivalent to just adopting
the hopping of the KS system (in this case, the nonlocal potential v1,2 ≡ t). Yet because
the off-diagonals for the KS system are γ1,2s = γ2,1 ≡ 1 irrespective of
the hopping amplitude, no nonlocal KS potential exists that reproduces
the interacting 1RDM. This is also true in more general lattice situations
as has been shown in, for example, ref (47). For the 1RDM, two solutions to this problem
are known. One is to include temperature and possibly an indefinite
number of particles, which introduces off-diagonals that depend on
the temperature and the hopping, that is, the nonlocal potential.19 We note that for the homogeneous two-site case,
this can still be solved analytically and verified explicitly. The
other possibility is to make the system degenerate such that we can
reproduce any density matrix.19
Here,
we apply a different strategy. While we cannot force the
density matrices to coincide, it is possible to require the kinetic-energy
densities to be the same. The crucial difference is that we include
the coupling in the Hamiltonian in the definition of the quantity
to be reproduced by the KS system. For example, in the two-site case,
we merely need to use an interaction-dependent hopping 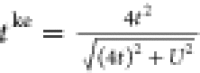 .
Thus, the auxiliary noninteracting system
reproduces now the pair (n,T) of the interacting
system. Before we move on, let us note that similarly to the continuum
case, one could use 1RDM functional theory at zero temperature also
on the lattice if one avoids the use of a noninteracting auxiliary
system and merely uses functionals based directly on the interacting
1RDM.48,49 Note that N-representability
conditions would still need to be enforced in such a scheme.
.
Thus, the auxiliary noninteracting system
reproduces now the pair (n,T) of the interacting
system. Before we move on, let us note that similarly to the continuum
case, one could use 1RDM functional theory at zero temperature also
on the lattice if one avoids the use of a noninteracting auxiliary
system and merely uses functionals based directly on the interacting
1RDM.48,49 Note that N-representability
conditions would still need to be enforced in such a scheme.
Let us now assume that similar to DFT we can establish a bijective mapping:
| 10 |
which would allow us to define hopping parameters and potentials that generate a given kinetic-energy density and density, that is, (ti[n,T], vi[n,T]). Specifically we can then consider a noninteracting auxiliary problem that generates a prescribed pair (n,T):
| 11 |
by its ground state. Whether we can construct such an auxiliary system that reproduces the density and kinetic-energy density of an interacting system is something we do not know a priori. In this Article, we provide numerical evidence as well as proofs for specific situations that suggest that such a construction is possible (see section 3). If we introduce then the corresponding mapping differences similar to eq 4 and denote them by mean-field exchange-correlation (Mxc):
| 12 |
| 13 |
we find the corresponding keKS system:
| 14 |
such that
| 15 |
and
| 16 |
where Φke is the corresponding ground state. This construction gives rise to the keKS hopping tike[t;n,T] and the keKS potential vi[v;n,T]. Similarly to standard DFT, it is important to realize the difference between Ĥs[n,T] together with (tis[n,T],vi[n,T]) and the keKS Hamiltonian Ĥke together with the keKS functionals (tike[t;n,T],vi[v;n,T]). Only the latter provides an iterative scheme to predict the physical pair (n,T) of the interacting reference system. At the unique fixed point of the keKS iteration procedure, where vi = vi[n,T] and ti = ti[n,T], both Hamiltonians coincide and give rise to the same noninteracting wave function. When we in the following present results for the exact keKS construction, we refer always to the results at the unique fixed point of the keKS construction. This also allows us in the following to only use tike and vi to highlight the difference between the usual KS and the keKS construction. To make the scheme practical, we now need two approximations: one for the Mxc potential and one for the Mxc hopping. Possible routes on how to construct approximations and how this could help to more accurately capture strongly correlated systems we consider in section 5.
At this
point, we want to make a first connection to the continuum
by considering the appropriate choice of the kinetic-energy density
for that case. There are different possible definitions for a local
kinetic-energy density, which will give rise to the same total kinetic
energy.50 For instance, we can choose the
gauge-independent definition100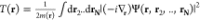 , where Ψ corresponds to the interacting
wave function, such that the kinetic-energy density is positive at
every point in space. Here, we have defined a spatially dependent
mass m(r) > 0 that takes the role
of
the site-dependent hopping in the lattice case. For the noninteracting
system, the corresponding kinetic-energy density (provided we assume
a Slater determinant) reads
, where Ψ corresponds to the interacting
wave function, such that the kinetic-energy density is positive at
every point in space. Here, we have defined a spatially dependent
mass m(r) > 0 that takes the role
of
the site-dependent hopping in the lattice case. For the noninteracting
system, the corresponding kinetic-energy density (provided we assume
a Slater determinant) reads 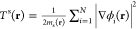 , where ms(r) > 0 is the spatially dependent
noninteracting mass and
ϕi the single particle orbitals.
The single-particle kinetic-energy operator then becomes accordingly
, where ms(r) > 0 is the spatially dependent
noninteracting mass and
ϕi the single particle orbitals.
The single-particle kinetic-energy operator then becomes accordingly 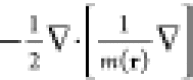 , where m(r) should be substituted with ms(r) in the noninteracting case.
, where m(r) should be substituted with ms(r) in the noninteracting case.
3. Generalized Mappings from Densities to Potentials
Similarly to fixing the constant of the local potential, one needs to fix the gauge of the hopping parameter ti(ke) (where the superscript ke in parentheses is used to denote that we refer both to interacting and to noninteracting keKS systems). One of the first things to note is that by letting ti change from site to site, we encounter a large equivalence class for the site-dependent hopping parameters. Indeed, we can arbitrarily change the signs of the hopping from ti(ke) → −ti without changing the density and the kinetic-energy density. However, the wave function and also, for example, the 1RDM change. For instance, for the noninteracting single particle Hamiltonian, we see that changing the sign locally, say at site i, will transform the single-particle wave function at this site ϕi to −ϕi (see Figure 1 for an example and Appendix D for further details). This leaves the density unchanged, as it is just a sum of the squared absolute values of the single-particle wave functions. Also, the kinetic-energy densities stay the same, because the 1RDM switches signs at the same place as the hopping amplitude. As it follows from the discussion above, the sign of ti(ke) is just a gauge choice, and we need to fix the gauge to establish the sought-after mapping. In the following, we choose ti > 0.
Figure 1.
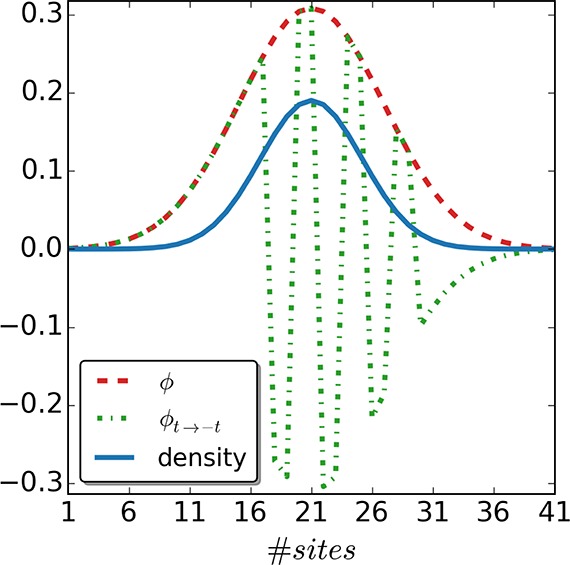
Doubly occupied orbital ϕ that corresponds to a two-electron singlet-state of a single particle Hamiltonian with all hopping parameters tike = t positive and the corresponding one ϕt→–t with alternating hopping parameters ±t from site 17 to 29. Every time we alternate t to – t at site i, the orbital ϕ changes sign from that site on. Because we replace t to −t from site to site, the orbital will recover its original sign after two sites. As one can readily see, the density stays the same in both cases, as a consequence of the sign of t being only a gauge choice.
A further complication that one encounters in establishing
the
necessary mappings is that the usual Hohenberg–Kohn approach
does not work in our case. The reason is that the control fields t now become explicitly part of the control object T. A similar problem is encountered in current-density-functional
theory, when trying to establish a mapping in terms of the gauge-independent
physical charge current.51−55 While in the time-dependent case having the field as part of the
control objective is actually an advantage and a general proof has
been established,54 these complications
unfortunately prohibit a simple general proof of the existence of
the mapping (n,T) → (v,t) for the time-independent case. However, for specific
situations, we are able to show that the discussed mapping is possible.
The most important one in our context is the case of the two-site
Hubbard model (see Appendix A for details).
In this case, we only have a single potential difference Δv and density difference Δn. So we
can simply rescale the auxiliary Hamiltonian and thus prove the existence
of the mapping in the noninteracting case by employing the Hohenberg–Kohn
results. A further simple case is two noninteracting particles, forming
a singlet, in a general M-site lattice. Here, the
density fixes the single-particle orbital (doubly occupied) up to
a sign, and thus for a given Ti only a unique site-dependent hopping ti is possible. Finally, in the homogeneous
case, where the local potential vi = 0 and periodic boundary conditions are employed, the density  and
the kinetic-energy density of the interacting
system will be constant at every site i, Ti = T. The
matrix elements γi,i+1 will also be constant from site to site, γi,i+1 = γ. In this case, the mapping
is invertible and a unique (up to a sign choice)
and
the kinetic-energy density of the interacting
system will be constant at every site i, Ti = T. The
matrix elements γi,i+1 will also be constant from site to site, γi,i+1 = γ. In this case, the mapping
is invertible and a unique (up to a sign choice)  is associated from site
to site. Note that
in this case the KS system and the keKS system yield the same wave
function and
is associated from site
to site. Note that
in this case the KS system and the keKS system yield the same wave
function and  . This last example,
although it only shows
the invertibility of the mapping (v,t) →
(n,T) at the specific points ti = t > 0 and vi = 0, has very important consequences.
It allows us in a simple yet exact way to connect the auxiliary keKS
system to the interacting system. We will use this later to construct
a first approximation to tiMxc.
. This last example,
although it only shows
the invertibility of the mapping (v,t) →
(n,T) at the specific points ti = t > 0 and vi = 0, has very important consequences.
It allows us in a simple yet exact way to connect the auxiliary keKS
system to the interacting system. We will use this later to construct
a first approximation to tiMxc.
To show that the keDFT mapping can also be defined for other, more general cases, we construct in the following the mappings numerically. Afterward, we make use of the constructed mappings to investigate the properties of the Mxc potentials and the basic functionals, which for the continuum case would be numerically prohibitively expensive.
4. Inversion of (n,T)
Because, as discussed above, it is not straightforward to show that the mapping 10 is 1:1 in general, we investigate this question numerically. Therefore, we construct sets of densities and kinetic-energy densities (n,T) by solving the interacting problem specified by the Hamiltonian given in eq 2 (with a site-independent hopping, which corresponds to the usual Hubbard Hamiltonian), and for every set we determine the potentials (v,t) of the noninteracting Hamiltonian specified in eq 14, which yields the target densities (n,T). To determine these potentials, we set up an inversion scheme by using the EOM for the density and the kinetic-energy density, respectively. These provide not only physical relations that connect the quantities (v,t) with (n,T), but they are also suitable to define correlation potentials, as we will explain in the following.
Note that in principle the inversion can be done with other techniques, which are used to find the exact local KS potential for a given interacting target density.56−59 However, it is not straightforward how to transfer these techniques to the current situation. For instance, in ref (56), an iteration scheme is introduced that adopts the potential based on the intuition that where the density is too low the potential is made more attractive and where the density is too high it is made less attractive. It is not so clear how to transfer this intuitive procedure to the kinetic-energy density Ti, which is nonlocal, and the control field is part of the observable itself. In the continuum, one could perform an inversion and define the corresponding auxiliary potentials again by EOMs.42,60 Another possibility would be to exploit techniques where the kinetic-energy density of the KS and the interacting system are used to model the exchange-correlation potential.59
Because the first-order EOM for the density, that is, the continuity eq 9, is trivially satisfied as the current is just zero in the ground state, we consider the second time derivative of the density n̈i. Because the first time derivative of the kinetic-energy density vanishes for ground-state wave functions, we use again the second-order EOM T̈i.
As examples, we give here the EOMs for n̈1 and T̈1 for two sites in the noninteracting case that we use in our numerical inversion scheme:
| 17 |
| 18 |
In Appendix B, the general expressions for any number of sites can be found. Here, we have dropped the site index because everything corresponds to site 1, Δn = (n1 – n2) is the density difference between the two sites, Δvke = (v1ke – v2) is the local potential difference, and T = −2tkeγ1,2ke. As one can readily see for the two-site case, there is no additional information in the equation for T̈, as once n̈ is 0 T̈ is also 0. Nevertheless, once we go to more sites, T̈i will also give us new equations. For a detailed discussion of this issue, see Appendix B.
The inversion scheme we employ is an iterative procedure based on the above introduced EOMs (see eqs 44 and 48 in Appendix B for the general expressions), which provide us with relations between (Δvke,tke) and the target quantities (n,T). We obtain the target quantities (n,T) by finding the ground state of the corresponding interacting Hamiltonian of eq 1, with a position-indepent hopping ti = t. We then choose as an initial guess for the auxiliary keKS system the values of the interacting system vike,0 = vi and ti = t.
(a) We solve the auxiliary noninteracting Schrödinger eq 11 with the values vike,0 and ti:
| 19 |
(b) We next calculate the density and kinetic-energy density that correspond to the state |Φke,0⟩, that is, ni0 = ⟨Φke,0|n̂i|Φke,0⟩ and Ti = −2tike,0⟨Φke,0|γ̂i,i+1|Φke,0⟩ as well as the matrix elements γi,j that enter the EOMs 44 and 48.
(c) In a last step, we then calculate the variables of the next iteration vike,1 and ti. The EOM for n̈i = 0 of eq 44 provides us with analytic expressions of vike,1 in terms of the target densities, the hopping amplitudes ti, and the reduced density matrix elements γi,j0 of the previous iteration. For calculating the ti, we use a numerical solver on all of the available EOMs for n̈i = 0 (eq 44) and T̈i = 0 (eq 48), with the target kinetic-energy densities, but updated densities ni0 and γi,j from the last iteration and the renewed local potentials vke,1. We repeat steps (a)–(c) until convergence of the calculated fields.
As an example in the two-site case, one can update in every iteration the local potential:
| 20 |
and the hopping parameter:
| 21 |
where Δn is the target density difference between the two sites and T is the target kinetic-energy density.
We want to point out that the procedure to update vke,i and tke,i is not the only one possible. For example, one could have used instead of the EOMs that we get for n̈i = 0 the ones for J̈i = 0. Further note that there are always M – 1 independent equations from n̈i = 0 because of particle number conservation, thus as many as the independent vike that we have (although it is not clear that we need all of them as we do not have a linear system of equations). The number of EOMs that we get for the kinetic-energy density T̈i is M – 2, as we explain in Appendix B. The interacting ground state was obtained using the single-site DMRG61 routine, implemented in the SyTen toolkit.62
We successfully performed inversions for systems of up to four sites with different total number of electrons for different on-site interaction strengths U and local potentials v. Some representative results for half-filling are shown in the next section, where we use the constructed mappings to consider the exact keKS system. Note that we also successfully performed inversions beyond half-filling. We also performed successful inversions for the same systems for the interacting problem; that is, we chose random values (n,T) and reproduced them with a nonzero Hubbard interaction. This makes the equations involved slightly more complex (and we refrained from showing them here explicitly), but the inversion procedure stays the same. The fact that we could indeed construct a keKS auxiliary system for these cases as well as perform inversions for the interacting problems provides us with indications for the existence of a keKS system for an arbitrary number of electrons/sites.
5. Comparing the Exact KS and keKS Construction
Next, we assess the practical implications of using the kinetic-energy density as basic functional variable along with the density. First, we use the construction of the exact keKS system and the corresponding KS system to compare the Hxc energy EHxcKS of the KS system with the corresponding quantity EMxc of the keKS system. This gives us a first indication of whether a keKS approach might help to capture also strong correlation effects more easily. For the KS system, the Hxc energy is
| 22 |
where TiKS = −2t⟨ΦKS|γ̂i,i+1|ΦKS⟩ is the kinetic-energy density of the KS system and |ΦKS⟩ its ground-state wave function. By Egs, we denote the total ground-state energy of the interacting system and vi is its external potential. The corresponding energy contribution of the keKS system reads
| 23 |
where Tike = −2ti⟨Φke|γ̂i,i+1|Φke⟩. Because the kinetic energy of the keKS system is identical to the interacting one by construction, the EMxcke ≡ Eint = U∑i = 1⟨Ψ|n̂i↑n̂i|Ψ⟩ is equal only to the interaction energy in this case. The corresponding term of the KS system includes kinetic-energy contributions as well. In Figure 2, we plot EMxcke and EHxc for a Hubbard dimer at half-filling with local potential Δv/t = 1 as a function of the interaction strength U/t. Note that the data from the numerical inversion are used. Thus, both energy quantities are exact, and there is no approximation involved.
Figure 2.
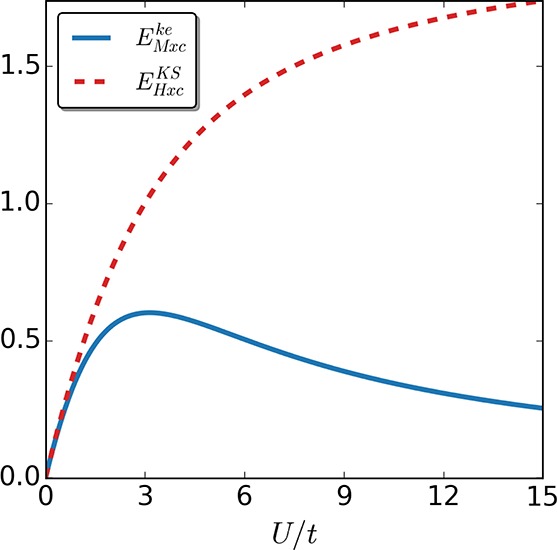
Hxc energy EHxcKS (dashed line) and the corresponding energy term of the keKS system EMxc (continuous line) for a Hubbard dimer at half-filling with local potential Δv/t = 1 as a function of U/t. We see that for U > 0 it holds that EMxcke < EHxc.
In Figure 3, we show the corresponding plot for a four-site Hubbard system at half-filling with Δv1/t = −Δv3/t = 0.625 and Δv2/t = 0.375. These two systems for two-site and four-site will serve as our test systems, and in the following we will refer to them as the two-site case and four-site case, respectively.
Figure 3.
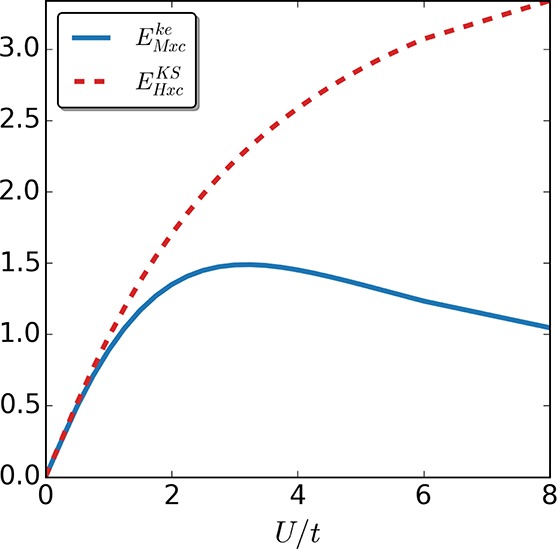
Hxc energy EHxcKS (dashed line) and EMxc (continuous line) for a four-site Hubbard model at half-filling with local potential Δv1/t = −Δv3/t = 0.625 and Δv2/t = 0.375 as a function of U/t. We again find that for U > 0 it holds that EMxcke < EHxc.
As one can readily see in Figures 2 and 3, for every interaction strength U > 0 it holds that EMxcke < EHxc. In the strong correlation limit, the kinetic-energy of the KS system is far from the interacting one. Having in mind the following relation:
| 24 |
it becomes apparent why EMxcke and EHxc are so different for strong interactions. As a consequence, the exchange-correlation potential derived from EHxcKS will need to take into account this difference in the strong interaction regime. In the keKS system, on the other hand, one needs to introduce a second field tMxc, which is responsible for reproducing the kinetic-energy density along with the potential vMxc that ensures the density is reproduced. Furthermore, due to the fact that EMxc does not contain kinetic contributions, it offers a simple scaling relation in contrast to EHxcKS.63
While the energy functionals are an interesting first indication that the keKS approach can be useful to treat also strongly correlated systems, the real quantities of interest are the effective fields that the KS and keKS constructions employ, especially those parts of the Hxc potential and of the Mxc hopping and potential that are not accessible by simple approximation strategies. Those parts, which one usually assumes to be small in practice, we will denote as correlation terms. Let us in the following, based on the EOMs we used to derive the iteration scheme, define parts of the effective fields that we can express explicitly in terms of the KS and keKS wave functions. Similar constructions based on the EOM of the density have been employed in DFT and TDDFT.44,45 For simplicity, we present the expressions only for the two-site case. The expressions for four-sites are given in Appendix C. The Hxc potential is defined as ΔvHxc[n] = Δvs[n] – Δv[n] (eq 4), where n is the target density of the interacting system, Δvs[n] is the local potential difference of the KS system, and Δv[n] is just the external potential of the interacting system. The EOMs for the noninteracting/interacting density, eq 17/53, provide expressions for the local potential Δvs[n] and Δv[n] of the noninteracting/interacting system. Thus, the Hxc potential in the two-site case reads
| 25 |
We can decompose ΔvHxc in a Hartree-exchange part ΔvHx[n,ΦKS]:
| 26 |
which corresponds to the usual Hartree plus exchange approximation in standard DFT, and a remaining correlation part:
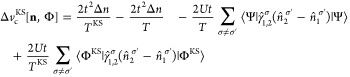 |
27 |
Here, we include the KS wave function in the functional dependencies to highlight that it is an orbital functional; that is, it depends on the KS wave function. We note, however, that in the exact case the KS wave function is uniquely determined by the density. The above decomposition is similar to that of the continuum case introduced in ref (44) and later used in, for example, refs (45) and (64). In eq 12, we have defined the Mxc potential vMxc for the keKS system, which in the two-site case (by using the same EOMs as before) reads
| 28 |
We see that the first two terms are completely determined by the keKS system, contrary to ΔvHxc of the KS system, where the second term cannot be given in terms of Δn or Φ explicitly. The first term, however, depends explicitly on the hopping in the keKS system, which has to be approximated in practice. Keeping this in mind, one can identify a mean-field exchange potential, similarly to the Hartree-exchange potential of the KS system:
| 29 |
which depends explicitly on the density, the kinetic-energy density, and the ground state of the keKS system. Let us at this point remark that if there is no approximation for the hopping parameter involved, that is, when tke = t, the expression of vMx in eq 29 is identical to the expression for vHx in eq 26. In the mean-field approximation, γi,i+1 ≈ γi,i+1ke, it follows immediately that ti = t, because we require the kinetic-energy densities to be the same, that is, tikeγi,i+1 = tγi,i+1. We note that for the exact case we consider here, that is, at the solution point of the exact keKS nonlinear equation (see also discussion about the keKS construction below eq 16), tke can be explicitly given in terms of t and the exact Φke. In practice, however, we do not know tke[t,n,T] = t + tMxc[n,T] a priori, and we need to include further an extra approximation for tMxc[n,T]. Which approximations are possible (and how accurate they are) will be discussed next, and in the following section we will see how the practical form of vMx[n,T,Φke], that is, including an approximate tMxc[n,T], performs. The remaining local potential correlation term contains now only contributions from the difference in interaction:
| 30 |
We stress that the definition of the correlation contribution to the local potential 30 only contains part of what is usually referred to as correlations in the context of KS DFT. The so-called kinetic correlation is taken care of in the hopping amplitudes tke and via definition 29 in vMx. In Figure 4 we plot for the two-site case the correlation KS and keKS potentials, which are given by eqs 27 and 30, respectively. In Figure 5 we plot the correlation potentials for the four-site case, which are given for the KS system by eqs 63–65 and for the keKS by eqs 72–74. As one can readily see for the two sites, the correlation potential is smaller in absolute value for the keKS system than in the KS one for all interaction strengths tested, apart from a small region at vanishing interaction. This follows from the fact that the kinetic contributions are included in the mean-field exchange potential ΔviMx in the keKS case. For the four sites we see the same trend. However, in the keKS construction, we have a second effective field, which we so far did not take into account in our comparison. One needs to find an analogous decomposition into a term that corresponds to the vi in the KS case, which can be approximated with a relatively simple functional, and a term that requires more advanced approximations, in correspondence to vic. Of course, if the latter part is large as compared to the former, we did not gain anything by introducing the additional field ti.
Figure 4.
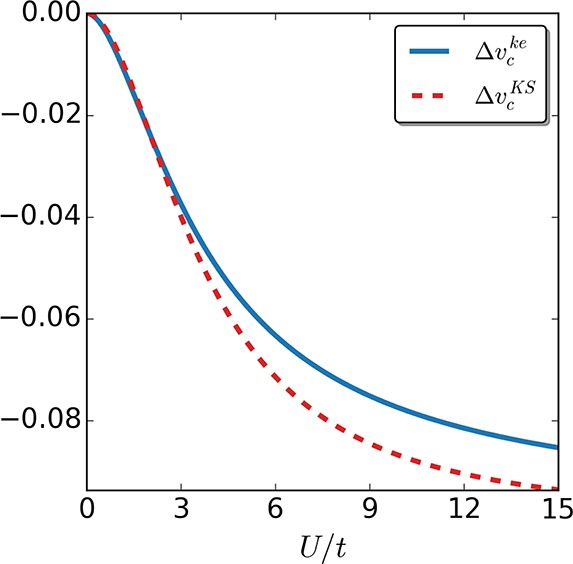
Correlation potential of the keKS Δvcke (continuous curve) and KS system Δvc (dashed curve) for the two-site case as a function of the interaction strength U/t. Apart from a small region at vanishing interaction strength U, |Δvcke| < |Δvc|.
Figure 5.
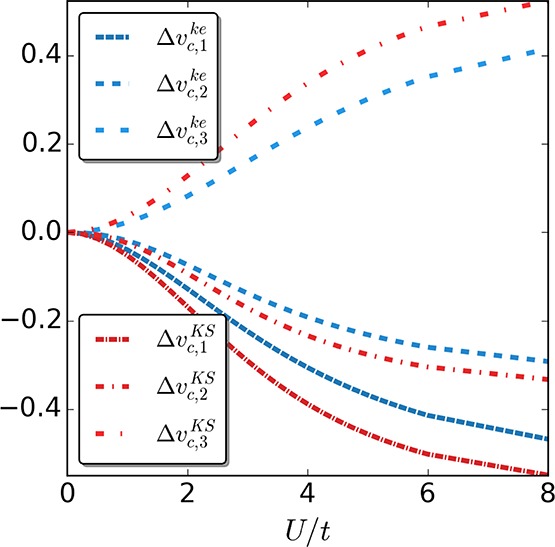
Correlation potentials of the keKS Δvc,ike (dashed) and KS system Δvc,i (dotted-dashed) for the four-site case as a function of the interaction strength U/t. Again, we find that, apart from a small region at vanishing interaction strength U, |Δvc,ike| < |Δvc,i|.
Starting from the definition of tiMxc[n,T] = ti[n,T] – ti[n,T], and using the fact that the kinetic-energy density Ti has to be the same in the interacting and keKS system, we get that
| 31 |
which by substituting Ti = ti[n,T]γi,i+1[n,T] and reordering the various terms yields the form:
| 32 |
where we have defined δγi,i+1 ≡ γi,i+1 – γi,i+1ke. Up to here, there is no approximation involved. As the term δγi,i+1 involves the solution of an interacting and noninteracting problem, an approximation based on a reference solution suggests itself. The simplest such reference solution would be to use the homogeneous case of the interacting and the keKS system, respectively, similar to the local-density approximation in standard DFT. Because in the homogeneous case with periodic boundary conditions, as discussed in section 3, the keKS and the KS density matrices are the same, we can directly use well-known results such as the Bethe-ansatz solution at half filling. In this way, it becomes also straightforward to extend the introduced approximation to the continuum case, where we can use reference calculations for interacting homogeneous continuum systems. Let us also do the same uniform approximation for the zero boundary condition case that we discuss here, although the uniform KS and keKS density matrices will not be the same apart from the two-site case. Here, we assume that we have reference data for the homogeneous problems for different local hopping parameters ti > 0, local fillings 0 < ni < 2, and for the local interactions U > 0. Further we ignore the dependence of the ti in the numerator on the internal pair (n,T) and use an explicit dependence on Φke in the denominator. In the following, we will simplify the explicit parts even further and will just take the homogeneous solution at half filling, that is, calculate δγi,i+1 for two-site and four-site cases with different U. The update formula for ti, which we will denote as tiunif to stress that the approximation comes from using uniform reference data for the 1RDM, reads:
| 33 |
Let us point out here that when 33 is used in a self-consistent loop, the tiunif is updated using the γi,i+1 of the previous iteration as the enumerator is fixed and taken from the reference calculation. In the two-site case such an ansatz seems appropriate, because despite the zero-boundary conditions the keKS and the KS systems are the same by construction. For the four-site case, however, the zero-boundary conditions make the keKS and KS density matrices different. Hence, the four-site case is a very challenging test for the accuracy of such a simple approximation. In accordance to the above introduced approximation, we will then define the remainder of the hopping field as tc,i[n,T,Φke] = tiMxc[n,T] – ti[Φke], where we emphasize that strictly speaking tc,i includes correlations beyond the correlations present in a uniform system. In Figure 6 we plot the (beyond uniform) correlation hopping field tc/t as a function of the interaction strength U/t for the two-site case. We see that the value of tc is small as compared to the chosen t for all interaction strengths and especially for weak and strong interactions. For strong interactions, the system resembles a homogeneous one as the interaction strength becomes more prominent in comparison to the local potential difference, and thus tc becomes smaller in this regime. From this we can infer that for the case of a general system with periodic boundary conditions, the homogeneous ansatz will capture not only the weak but also the strong-interaction limit accurately.
Figure 6.
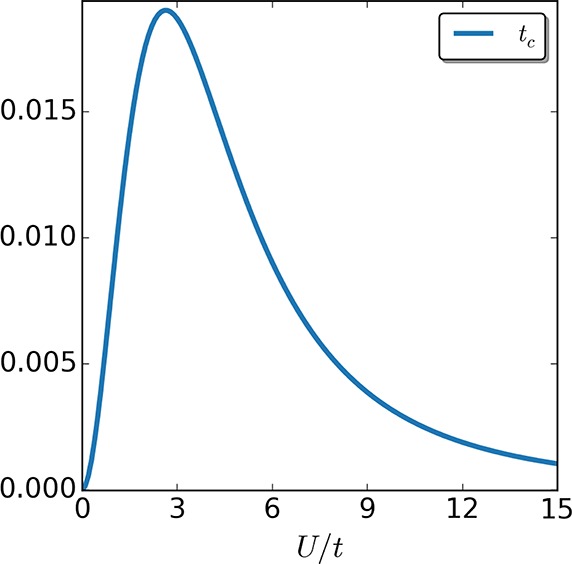
Correlation part of the hopping tc,i in units of t, for the two-site case as a function of interaction strength U/t. For strong interaction strength, the system resembles a homogeneous one so that the uniform type of approximation we employed becomes very good.
In Figure 7 we turn to the more challenging case of four sites with zero boundary conditions and plot the three different tc,i/t components as a function of the interaction strength. As one can readily see, all three tc,i are small for every interaction strength U. However, only two of them seem to converge to a value that is close to zero for strong interactions, at least for the parameter range we investigated. (We want to point out that for larger values of U we encountered some convergence issues in the four-site case. The reason is that while the density matrices are not homogeneous, the density is, which causes some problems in the iteration scheme, where we divide by the density difference between two neighboring sites in each iteration. This problem, however, can be potentially overcome by using different update equations.) We remind the reader that this difference in accuracy between the two-site and the four-site cases is not surprising. In the two-site case, the uniform KS γi,i+1KS, which is used to construct the approximation for tunif, coincides with the corresponding uniform γi,i+1 of the keKS system. For four sites this is no longer the case. Still, comparing the numerical values of the correlation hopping tc,i to those of the correlation potential vc,ike, there is an order of magnitude difference. This gives some hope that crude approximations like the ti can still lead to accurate predictions. Let us test this in the following section.
Figure 7.
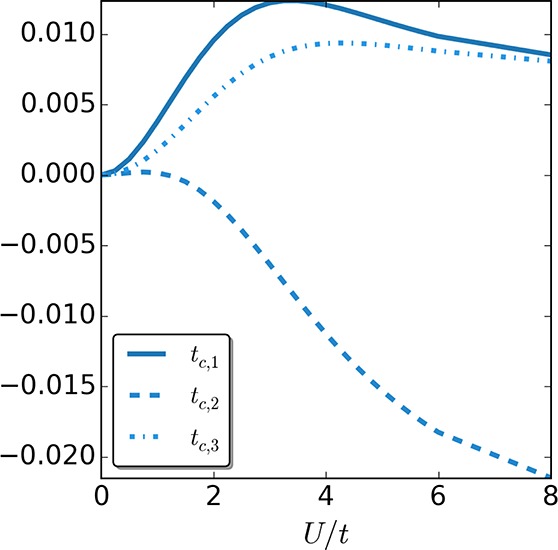
Correlation part of the hopping tc,i in units of t, for the four-site case as a function of interaction strength U/t.
6. Comparing a Self-Consistent KS and keKS Calculation
While the above considerations about the exact correlation energies, potentials, and hopping parameters are crucial to understand what the different approximations to the unknown exchange-correlation terms are able to capture, it is not their performance at the exact solutions that matters in practice. A self-consistent calculation with the approximate functionals is not at all clear that will converge to a sensible solution or even converge at all. For instance, even for the prime example of a nonlinear problem in quantum chemistry, that is, the ground-state Hartree–Fock equation, the convergence to a unique solution has not been shown except for highly unusual cases.65 To finally test whether the proposed keDFT and its keKS construction can be used in practice to predict the properties of correlated many-electron systems, we perform self-consistent calculations for our two-site and four-site Hubbard models. We use the mean-field exchange approximation of eq 29 for two sites and of eqs 69–71 for four sites together with the uniform approximation for the hopping term of eq 33. This leads to
| 34 |
where we update the involved effective fields in every iteration until convergence is achieved. We then compare the densities and kinetic-energy densities that we get with the KS ones within the exact-exchange approximation (thus ti = t and vHx given by eq 26 for two sites and eqs 60–62 for four sites). We do so as in this way we have for both the KS and the keKS construction the same level of approximation as vMx will reduce to vHx for tike = t. This allows us to judge whether including the kinetic-energy density in the modeling of many-particle systems has any advantages over the usual density-only approach.
We first quantify the density difference between the calculated quantities and the exact ones using the following measure: δnke/KS = ∑i=1M|ni – ni|, where ni is the interacting density at site i while nike/KS is the corresponding density of the keKS/KS system.
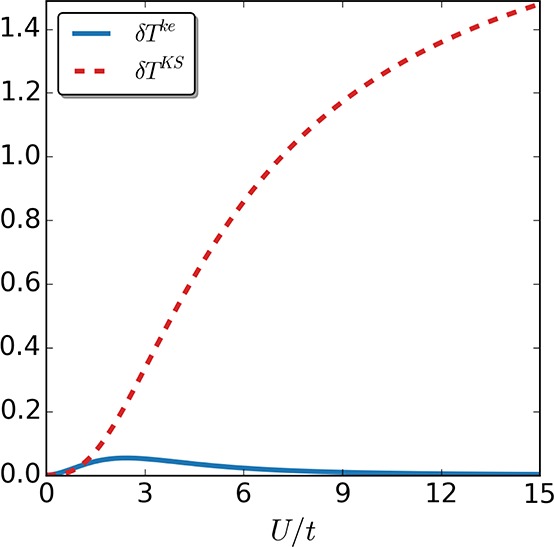
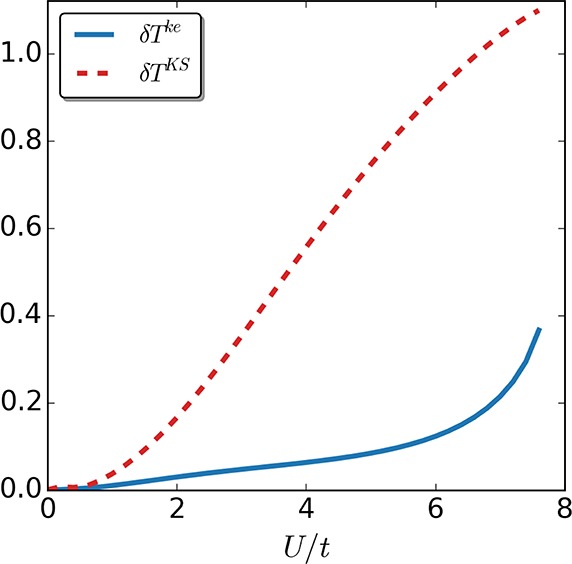
Indeed, for both the two-site case (see Figure 8) as well as the more challenging four-site case (see Figure 9), the self-consistent keKS approximation performs better than the corresponding self-consistent KS exact-exchange approximation. Because the main difference lies in the error correction to the local kinetic-energy density, we next also compare a measure for the difference in local kinetic-energy density: δTke/KS = ∑i=1M–1|Ti – Ti|, where Tike/KS is the kinetic-energy density between site i and i+1, while Ti is the corresponding interacting one. Not surprisingly, in both cases (see Figures 10 and 11), the approximate kinetic-energy density of the keKS system is much closer to the actual one than the bare KS energy density. We see that for large interaction strengths the error is basically zero for the two-site case because in this limit the interaction is much larger than the asymmetry induced by the local potential. In the four-site case, our approximation for the kinetic-energy density is not as accurate, although it is still better than the corresponding KS one. The reason for this drop in accuracy is, as discussed around eq 33, the assumption that γi,i+1 = γi,i+1unif,ke, which is violated for the four-site case. Nevertheless, for large systems (where the boundaries will not be significant) or for systems with periodic boundary conditions, this issue will essentially vanish because then the uniform reference system obeys γi,i+1 = γi,i+1unif,ke and the strong-interaction limit is captured highly accurately. Consequently, it can be expected that including the kinetic-energy density can help to treat multiparticle systems accurately from the weak to strong interaction regime.
Figure 8.
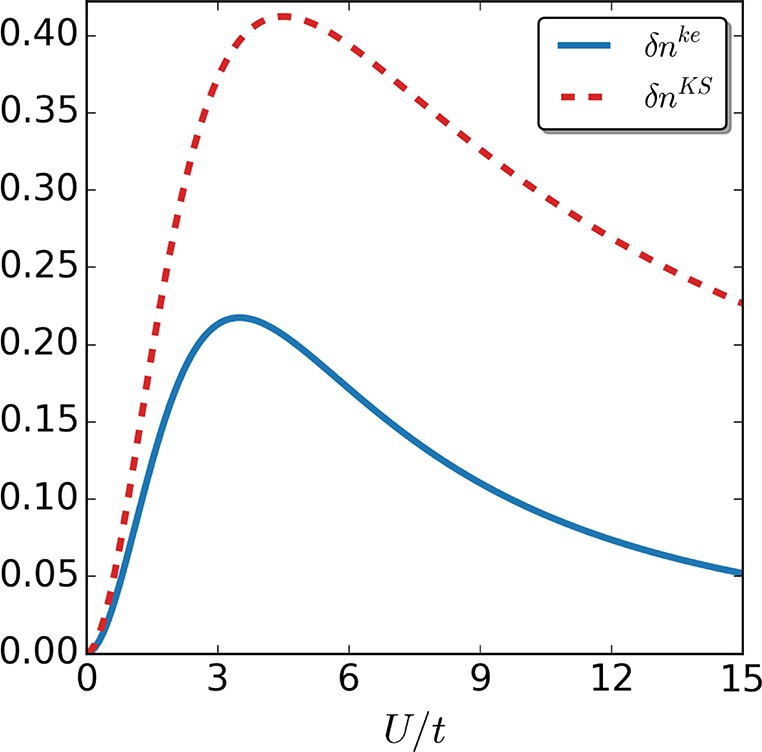
Density difference δnke/KS between the self-consistent calculations in the keKS system and the exact one (continuous, blue line), as well as for the self-consistent solution in the KS system and the exact one (dotted, red line), for the two-site case as a function of interaction strength U/t.
Figure 9.
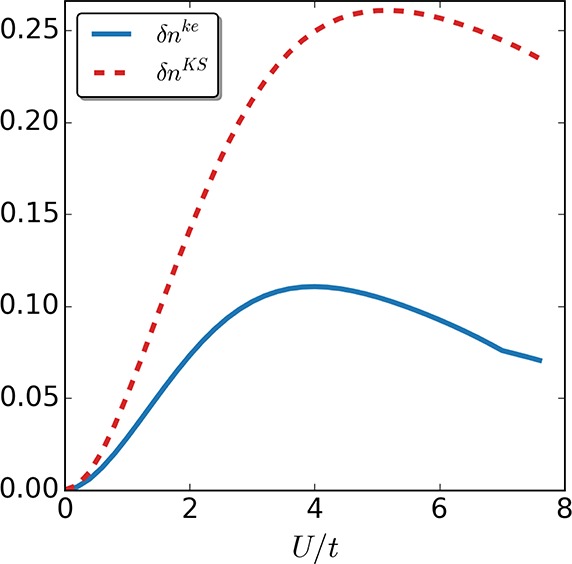
Density difference δnke/KS between the self-consistent calculations in the keKS system and the exact one (continuous, blue line), as well as for the self-consistent solution in the KS system and the exact one (dotted, red line), for the four-site case as a function of interaction strength U/t.
Figure 10.
Kinetic-energy density difference δTke/KS between the self-consistent calculations in the keKS system and the exact one (continuous line), as well as for the self-consistent solution in the KS system and the exact one (dotted line), for the two-site case as a function of interaction strength U/t.
Figure 11.
Kinetic-energy density difference δTke/KS between the self-consistent calculations in the keKS system and the exact one (continuous line), as well as for the self-consistent solution in the KS system and the exact one (dotted line), for the four-site case as a function of interaction strength U/t.
7. Conclusion and Outlook
In this work, we have introduced a kinetic-energy density-functional theory (keDFT) and the resulting kinetic-energy Kohn–Sham (keKS) scheme on a lattice. The idea was that by lifting the kinetic-energy density T to a fundamental variable along with the density n, the resulting effective theory becomes easier to approximate because more parts are known explicitly. Because the new external field, a site-dependent hopping t, is part of the kinetic-energy density, the usual Hohenberg–Kohn-type proof strategy to establish the necessary one-to-one correspondence between (v,t) and (n,T), where v is the usual on-site potential, does not work. However, besides giving proofs for specific cases and discussing the gauge freedom of the approach, we provided an indication that the necessary bijectivity holds by numerically constructing the inverse maps from a given pair (n,T) to (v,t) for two- to four-site Hubbard models. We did so by introducing an iterative scheme based on the equations of motion (EOMs) of the density and the kinetic-energy density. We then introduced a decomposition of the two unknown effective fields of the keKS scheme, the mean-field-exchange-correlation potential viMxc[n,T] and the mean-field exchange-correlation hopping tMxc[n,T], into explicitly known mean-field exchange (for the effective potential) and uniform (for the effective hopping) as well as unknown correlation parts. By comparing the unknown parts of the standard Kohn–Sham (KS) approach to the keKS approach, we saw that including the kinetic-energy density in the fundamental variables reduced the unknown parts considerably. Finally, we tested the keKS approach in practice by solving the resulting nonlinear equations with the introduced approximations. We found that the mean-field exchange and uniform keKS outperform the corresponding exact-exchange KS from weak to strong interactions and hence hold promise to become an alternative approach to treat many-particle systems efficiently and accurately.
While the presented approach was thoroughly investigated only for simple few-sites problems, its extension to many sides, arbitrary dimensions, and even the continuum is straightforward. Following ref (50) in the continuum, we can choose a gauge-independent and strictly positive definition of the kinetic-energy density with a spatially dependent mass term. The main reason why the keKS scheme can be more accurate than the usual KS scheme also in the continuum is that we can model explicitly the kinetic-energy density in this case. Because the simple kinetic-energy density approximations we introduced proved to be already quite reasonable, the extension to the continuum seems especially promising. For homogeneous systems, many reference calculations exist that can be used to derive a universal local kinetic-energy density approximation that resembles the uniform approximation introduced in this work.
Appendix A: Two-Sites Proof of (Δvke,tke) ↔ (Δn,T)
In this Appendix, we provide a proof of the bijectiveness of the mapping between density and kinetic-energy density and the corresponding fields in the case of a noninteracting system of up to N ≤ 3 electrons on a two-site lattice. For this case, the noninteracting Hamiltonian reads:
| 35 |
where 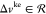 and tke > 0.
The mapping that we wish to show that is bijective is the following:
and tke > 0.
The mapping that we wish to show that is bijective is the following:
| 36 |
with T > 0 and Δn∈[–N,N]. Here, Δvke ≡ v1ke – v2 and Δn ≡ n1 – n2, where the lower indexes refer to different site points. Note that tke ≥ 0 is a certain gauge choice as {(tke,Δvke),(−tke,Δvke)} → (T,Δn). Moreover, due to particle hole symmetry, it holds also that {(−tke,−Δvke),(tke,−Δvke)} → (T,Δn). Thus, from now on, when we refer to different potentials and hopping parameters, we will mean that they differ by more than a sign change. For the local potential, the gauge choice is v1ke + v2 = 0 as throughout the document.
Proof: We are going to prove eq 36 through different cases.
Case 1: Two Hamiltonians Ĥ, Ĥ′ have the same hopping parameters tke = tke′ but different local potentials Δvke ≠ Δvke′.
From the Hohenberg–Kohn theorem, we then have that the corresponding wave functions are different, that is, Φ ≠ Φ′, and the same holds also for the densities Δn ≠ Δn′ and consequently (T,Δn) ≠ (T′,Δn′).
Case 2: Two Hamiltonians Ĥ, Ĥ′ have different hopping parameters tke ≠ tke′ but the same local potentials Δvke ≠ Δvke′.
Assume that (T,Δn) = (T,Δn′). We then have two wave functions Φ and Φ′ that are ground states of the corresponding Hamiltonians:
| 37 |
We can multiply Ĥ′ with a scaling factor 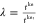 so that we get a Hamiltonian Ĥ″, which has the same hopping as the Ĥ.
so that we get a Hamiltonian Ĥ″, which has the same hopping as the Ĥ.
| 38 |
We assume
that Δn = Δn′,
which means that we have two different ground-state wave
functions with different local potentials 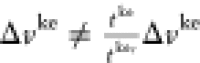 that still give the same densities. This
is clearly in contradiction with Case 1.
that still give the same densities. This
is clearly in contradiction with Case 1.
Case 3: Two Hamiltonians Ĥ, Ĥ′ have different hopping parameters tke ≠ tke′ and different local potentials Δvke ≠ Δvke′.
Again, we assume that (T,Δn) = (T′,Δn′), and we scale such that we find:
| 39 |
This requires that Δn = Δn′ can only hold if also the local potentials are the same as we showed in Case 1. Thus, it has to hold that
| 40 |
which means that the two Hamiltonians Ĥ and Ĥ′ are connected through the scaling relation Ĥ = λĤ′. Thus, we have
| 41 |
which contradicts our initial assumption.
Appendix B: Equations of Motion
In this Appendix, we derive the EOMs that we use in our numerical inversion scheme and to define the vHxc and vMxc potentials. Furthermore, we provide the explicit expressions of vHxc and vMxc for four sites. Finally, we discuss how the number of useful EOMs for our numerical inversion scheme depends on the number of sites.
The EOM for a generic operator A, which has no explicit time dependence, is given by
| 42 |
We are interested in obtaining EOM for a non interacting Hamiltonian of type 11. For a state Φ it follows that
| 43 |
which is an EOM for the observable associated with the operator Â. When Ψ is the ground state, eq 43 equals zero. Let us take now this operator to be the density. Because the first-order EOM for the density, that is, the continuity eq 9, is trivially satisfied as the current is just zero in the ground state, we consider the second time derivative of the density:
| 44 |
where we have introduced the forward (backward)
difference operators 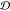 (
( ), which act on one-index objects:
), which act on one-index objects: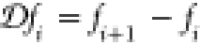 ,
, 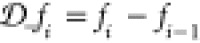 . Here we note that in
this Appendix we
stray from the convention that we only use tike and vi because we use the same EOMs for the
KS and the keKS scheme as well. In eq 44, we furthermore introduced
. Here we note that in
this Appendix we
stray from the convention that we only use tike and vi because we use the same EOMs for the
KS and the keKS scheme as well. In eq 44, we furthermore introduced
| 45 |
which, by analogy to the continuum case, can be identified as the kinetic contribution to the momentum-stress tensor. The time derivative of the kinetic energy density also leads to
| 46 |
where we introduced the kinetic-energy current
| 47 |
Both Ξi and Ji vanish trivially for real-valued wave functions such as the ground state, so eq 46 is fulfilled trivially. Taking yet another time derivative leads to
| 48 |
In eq 48, there are two more EOMs involved. The one for Ξi is
| 49 |
with
| 50 |
The EOM for the current is
| 51 |
Moreover, we use the second-order EOM for the interacting density, obtained from a Hamiltonian of Case 2, to define the vHxc and vMxc potentials:
| 52 |
As an example, let us show here the EOM for n̈1 for the two-site interacting Hamiltonian:
| 53 |
Formally, eq 44 defines M equations, that is, one for every site. However, because the sum of densities at every site has to give the total number of electrons, it holds that
| 54 |
We get M – 1 nontrivial equations. Now for M sites we do have M – 1 different Ti of the form eq 48. Yet as the total energy of the keKS system is fixed to some value E, that is:
| 55 |
taking the second time derivative of the above equation shows us that we have M – 2 nontrivial equations for Ti. Taking into account the gauge choice of vis (eq 2) and the particle number conservation eq 54, we see that the equations for n̈i and T̈i connect through the following relation:
| 56 |
At this point, we have to mention that eq 54 still holds for the interacting system; however, eqs 55 and 56 will not hold anymore because the on-site repulsion term enters the energy expression in this case.
Appendix C: Exchange-Correlation Potentials for Four Sites
From the EOMs 44 and 52, one can derive the vHxc and vMxc for any number of sites. We give here their expressions for four sites because it is one of our test cases in this Article.
| 57 |
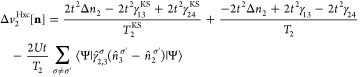 |
58 |
| 59 |
Similarly to the two-site case, we can decompose vHxc to a vHx and a vc contribution:
| 60 |
| 61 |
| 62 |
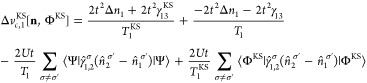 |
63 |
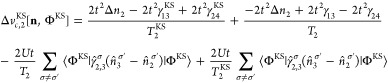 |
64 |
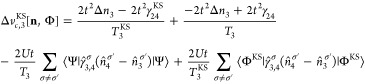 |
65 |
Next, we give the corresponding expressions for the keKS system:
| 66 |
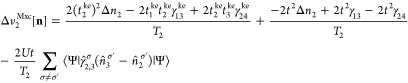 |
67 |
| 68 |
Similarly to vHxc, we decompose vMxc in a vMx and a correlation part vc. This is analogous to the decomposition for the two-site case presented in section 5:
| 69 |
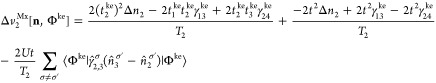 |
70 |
| 71 |
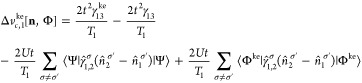 |
72 |
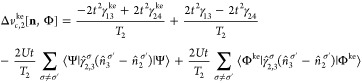 |
73 |
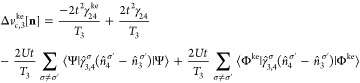 |
74 |
Appendix D: Gauge Choice for Hopping
In this Appendix, we demonstrate that the sign of the hopping is just a gauge choice. Let us consider a single-particle Hamiltonian H. In the site basis, it corresponds to the matrix:
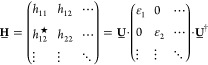 |
75 |
where we also introduced its spectral decomposition with the unitary matrix U̲ having its eigenvectors as columns. Suppose we transform the Hamiltonian by
| 76 |
where 1̲n is the n × n unit matrix and N is the total number of sites. Note that G̲† = G̲–1; that is, G̲ is unitary. It is straightforward to verify that the transformed Hamiltonian is given by
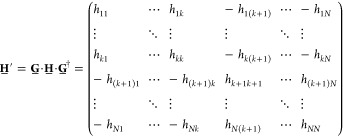 |
77 |
We can see that the effect of the transformation is to change the sign of all matrix elements in the “off-diagonal” parts determined by the site k after which the sign in G̲ changes. For a one-dimensional nearest-neighbor tight-binding Hamiltonian with “zero boundary conditions”, like the one we consider in this Article, this corresponds to flipping the sign of the hopping amplitude between site k and k + 1. Obviously the eigenvalues of H̲′ are the same as the eigenvalues of H̲, and the eigenstates are simply given by U̲′ = G̲U̲, which means that the signs of the wave function in position representation are flipped from site k + 1 on. All Hamiltonians, which can be connected by such a transformation, are to be considered equivalent.
Financial support from the European Research Council (ERC-2015-AdG-694097), by the European Unions H2020 program under GA no. 676580 (NOMAD), is acknowledged. F.G.E. has received funding from the European Unions Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie Skłodowska-Curie Grant agreement no. 701796.
The authors declare no competing financial interest.
References
- Hohenberg P.; Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. 10.1103/PhysRev.136.B864. [DOI] [Google Scholar]
- Thomas L. H. The calculation of atomic fields. Math. Proc. Cambridge Philos. Soc. 1927, 23, 542548. 10.1017/S0305004100011683. [DOI] [Google Scholar]
- Fermi E. Eine statistische Methode zur Bestimmung einiger Eigenschaften des Atoms und ihre Anwendung auf die Theorie des periodischen Systems der Elemente. Eur. Phys. J. A 1928, 48, 73–79. 10.1007/BF01351576. [DOI] [Google Scholar]
- Lieb E. H. The stability of matter. Rev. Mod. Phys. 1976, 48, 553–569. 10.1103/RevModPhys.48.553. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Burke K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. 10.1063/1.4704546. [DOI] [PubMed] [Google Scholar]
- Medvedev M. G.; Bushmarinov I. S.; Sun J.; Perdew J. P.; Lyssenko K. A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. 10.1126/science.aah5975. [DOI] [PubMed] [Google Scholar]
- Fetter J. D. W.Quantum Theory of Many-Particle Systems; McGraw-Hill Book Co.: New York, 1971. [Google Scholar]
- Gianluca Stefanucci R. v. L.Nonequilibrium Many-Body Theory of Quantum Systems; Cambridge University Press: Cambridge, 2013. [Google Scholar]
- Mazziotti D. A. Quantum Chemistry without Wave Functions: Two-Electron Reduced Density Matrices. Acc. Chem. Res. 2006, 39, 207–215. 10.1021/ar050029d. [DOI] [PubMed] [Google Scholar]
- Pernal K.; Giesbertz K. J. H. In Density-Functional Methods for Excited States; Ferré N., Filatov M., Huix-Rotllant M., Eds.; Springer International Publishing: Cham, 2016; pp 125–183. [Google Scholar]
- Mazziotti D. A. Structure of Fermionic Density Matrices: Complete N-Representability Conditions. Phys. Rev. Lett. 2012, 108, 263002. 10.1103/PhysRevLett.108.263002. [DOI] [PubMed] [Google Scholar]
- Klyachko A. A. Quantum marginal problem and N-representability. J. Phys.: Conf. Ser. 2006, 36, 72. 10.1088/1742-6596/36/1/014. [DOI] [Google Scholar]
- Theophilou I.; Lathiotakis N. N.; Marques M. A. L.; Helbig N. Generalized Pauli constraints in reduced density matrix functional theory. J. Chem. Phys. 2015, 142, 154108. 10.1063/1.4918346. [DOI] [PubMed] [Google Scholar]
- Theophilou I.; Lathiotakis N. N.; Helbig N. Structure of the first order reduced density matrix in three electron systems: A generalized Pauli constraints assisted study. J. Chem. Phys. 2018, 148, 114108. 10.1063/1.5020978. [DOI] [PubMed] [Google Scholar]
- Bonitz M.Quantum Kinetic Theory, 2nd ed.; Springer: New York, 2016. [Google Scholar]
- Baldsiefen T.; Cangi A.; Gross E. Reduced-density-matrix-functional theory at finite temperature: Theoretical foundations. Phys. Rev. A: At., Mol., Opt. Phys. 2015, 92, 052514. 10.1103/PhysRevA.92.052514. [DOI] [Google Scholar]
- Baldsiefen T.; Cangi A.; Eich F. G.; Gross E. K. U. Exchange-correlation approximations for reduced-density-matrix-functional theory at finite temperature: Capturing magnetic phase transitions in the homogeneous electron gas. Phys. Rev. A: At., Mol., Opt. Phys. 2017, 96, 062508. 10.1103/PhysRevA.96.062508. [DOI] [Google Scholar]
- Giesbertz K. J. H.; Ruggenthaler M.. One-body reduced density-matrix functional theory in finite basis sets at elevated temperatures. ArXiv e-prints 2017. [Google Scholar]
- Lathiotakis N. N.; Helbig N.; Rubio A.; Gidopoulos N. I. Local reduced-density-matrix-functional theory: Incorporating static correlation effects in Kohn-Sham equations. Phys. Rev. A: At., Mol., Opt. Phys. 2014, 90, 032511. 10.1103/PhysRevA.90.032511. [DOI] [Google Scholar]
- Theophilou I.; Lathiotakis N. N.; Gidopoulos N. I.; Rubio A.; Helbig N. Orbitals from local RDMFT: Are they Kohn-Sham or natural orbitals?. J. Chem. Phys. 2015, 143, 054106. 10.1063/1.4927784. [DOI] [PubMed] [Google Scholar]
- Gori-Giorgi P.; Seidl M.; Vignale G. Density-Functional Theory for Strongly Interacting Electrons. Phys. Rev. Lett. 2009, 103, 166402. 10.1103/PhysRevLett.103.166402. [DOI] [PubMed] [Google Scholar]
- Malet F.; Mirtschink A.; Giesbertz K. J.; Wagner L. O.; Gori-Giorgi P. Exchange–correlation functionals from the strong interaction limit of DFT: applications to model chemical systems. Phys. Chem. Chem. Phys. 2014, 16, 14551–14558. 10.1039/C4CP00407H. [DOI] [PubMed] [Google Scholar]
- Grossi J.; Kooi D. P.; Giesbertz K. J.; Seidl M.; Cohen A. J.; Mori-Sánchez P.; Gori-Giorgi P. Fermionic statistics in the strongly correlated limit of Density Functional Theory. J. Chem. Theory Comput. 2017, 13, 6089–6100. 10.1021/acs.jctc.7b00998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eich F. G.; Di Ventra M.; Vignale G. Density-Functional Theory of Thermoelectric Phenomena. Phys. Rev. Lett. 2014, 112, 196401. 10.1103/PhysRevLett.112.196401. [DOI] [PubMed] [Google Scholar]
- Eich F. G.; Ventra M. D.; Vignale G. Functional theories of thermoelectric phenomena. J. Phys.: Condens. Matter 2017, 29, 063001. 10.1088/1361-648X/29/6/063001. [DOI] [PubMed] [Google Scholar]
- Ghosh S. K.; Berkowitz M.; Parr R. G. Transcription of ground-state density-functional theory into a local thermodynamics. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 8028–8031. 10.1073/pnas.81.24.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers P. W.; Parr R. G.; Nagy A. Local kinetic energy and local temperature in the density-functional theory of electronic structure. Int. J. Quantum Chem. 2002, 90, 309–326. 10.1002/qua.989. [DOI] [Google Scholar]
- Nagy g. Thermodynamical transcription of the density functional theory with constant temperature. Int. J. Quantum Chem. 2017, 117, e25396–e25396. 10.1002/qua.25396. [DOI] [Google Scholar]
- Seidl A.; Görling A.; Vogl P.; Majewski J. A.; Levy M. Generalized Kohn-Sham schemes and the band-gap problem. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 53, 3764–3774. 10.1103/PhysRevB.53.3764. [DOI] [PubMed] [Google Scholar]
- Eich F. G.; Hellgren M. Derivative discontinuity and exchange-correlation potential of meta-GGAs in density-functional theory. J. Chem. Phys. 2014, 141, 224107. 10.1063/1.4903273. [DOI] [PubMed] [Google Scholar]
- Buijse M. A.; Baerends E. J.; Snijders J. G. Analysis of correlation in terms of exact local potentials: Applications to two-electron systems. Phys. Rev. A: At., Mol., Opt. Phys. 1989, 40, 4190–4202. 10.1103/PhysRevA.40.4190. [DOI] [PubMed] [Google Scholar]
- Ying Z.-J.; Brosco V.; Lopez G. M.; Varsano D.; Gori-Giorgi P.; Lorenzana J. Anomalous scaling and breakdown of conventional density functional theory methods for the description of Mott phenomena and stretched bonds. Phys. Rev. B: Condens. Matter Mater. Phys. 2016, 94, 075154. 10.1103/PhysRevB.94.075154. [DOI] [Google Scholar]
- Dimitrov T.; Appel H.; Fuks J. I.; Rubio A. Exact maps in density functional theory for lattice models. New J. Phys. 2016, 18, 083004. 10.1088/1367-2630/18/8/083004. [DOI] [Google Scholar]
- Carrascal D. J.; Ferrer J.; Smith J. C.; Burke K. The Hubbard dimer: a density functional case study of a many-body problem. J. Phys.: Condens. Matter 2015, 27, 393001. 10.1088/0953-8984/27/39/393001. [DOI] [PubMed] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P. Landscape of an exact energy functional. Phys. Rev. A: At., Mol., Opt. Phys. 2016, 93, 042511. 10.1103/PhysRevA.93.042511. [DOI] [Google Scholar]
- Dimitrov T.; Flick J.; Ruggenthaler M.; Rubio A. Exact functionals for correlated electronphoton systems. New J. Phys. 2017, 19, 113036. 10.1088/1367-2630/aa8f09. [DOI] [Google Scholar]
- Lammert P. E. Well-behaved coarse-grained model of density-functional theory. Phys. Rev. A: At., Mol., Opt. Phys. 2010, 82, 012109. 10.1103/PhysRevA.82.012109. [DOI] [Google Scholar]
- Kvaal S.; Ekstrm U.; Teale A. M.; Helgaker T. Differentiable but exact formulation of density-functional theory. J. Chem. Phys. 2014, 140, 18A518. 10.1063/1.4867005. [DOI] [PubMed] [Google Scholar]
- Lieb E. H. Density functionals for coulomb systems. Int. J. Quantum Chem. 1983, 24, 243–277. 10.1002/qua.560240302. [DOI] [Google Scholar]
- Chayes J. T.; Chayes L.; Ruskai M. B. Density functional approach to quantum lattice systems. J. Stat. Phys. 1985, 38, 497–518. 10.1007/BF01010474. [DOI] [Google Scholar]
- Ruggenthaler M.; Penz M.; Van Leeuwen R. Existence, uniqueness, and construction of the density-potential mapping in time-dependent density-functional theory. J. Phys.: Condens. Matter 2015, 27, 203202. 10.1088/0953-8984/27/20/203202. [DOI] [PubMed] [Google Scholar]
- Tokatly I. V. Quantum many-body dynamics in a Lagrangian frame: II. Geometric formulation of time-dependent density functional theory. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71, 165105. 10.1103/PhysRevB.71.165105. [DOI] [Google Scholar]
- Ruggenthaler M.; Bauer D. Local Hartree-exchange and correlation potential defined by local force equations. Phys. Rev. A: At., Mol., Opt. Phys. 2009, 80, 052502. 10.1103/PhysRevA.80.052502. [DOI] [Google Scholar]
- Fuks J. I.; Nielsen S. E.; Ruggenthaler M.; Maitra N. T. Time-dependent density functional theory beyond Kohn–Sham Slater determinants. Phys. Chem. Chem. Phys. 2016, 18, 20976–20985. 10.1039/C6CP00722H. [DOI] [PubMed] [Google Scholar]
- Gilbert T. L. Hohenberg-Kohn theorem for nonlocal external potentials. Phys. Rev. B 1975, 12, 2111. 10.1103/PhysRevB.12.2111. [DOI] [Google Scholar]
- Schindlmayr A.; Godby R. W. Density-functional theory and the v-representability problem for model strongly correlated electron systems. Phys. Rev. B: Condens. Matter Mater. Phys. 1995, 51, 10427–10435. 10.1103/PhysRevB.51.10427. [DOI] [PubMed] [Google Scholar]
- López-Sandoval R.; Pastor G. M. Density-matrix functional theory of strongly correlated lattice fermions. Phys. Rev. B: Condens. Matter Mater. Phys. 2002, 66, 155118. 10.1103/PhysRevB.66.155118. [DOI] [Google Scholar]
- Kamil E.; Schade R.; Pruschke T.; Blöchl P. E. Reduced density-matrix functionals applied to the Hubbard dimer. Phys. Rev. B: Condens. Matter Mater. Phys. 2016, 93, 085141. 10.1103/PhysRevB.93.085141. [DOI] [Google Scholar]
- Eich F. G.; Ventra M. D.; Vignale G. Functional theories of thermoelectric phenomena. J. Phys.: Condens. Matter 2017, 29, 063001. 10.1088/1361-648X/29/6/063001. [DOI] [PubMed] [Google Scholar]
- This implies that that the derivative has to be replaced −i∇r → −i∇r + A(r).
- Vignale G.; Rasolt M. Density-functional theory in strong magnetic fields. Phys. Rev. Lett. 1987, 59, 2360–2363. 10.1103/PhysRevLett.59.2360. [DOI] [PubMed] [Google Scholar]
- Diener G. Current-density-functional theory for a nonrelativistic electron gas in a strong magnetic field. J. Phys.: Condens. Matter 1991, 3, 9417. 10.1088/0953-8984/3/47/014. [DOI] [Google Scholar]
- Vignale G. Mapping from current densities to vector potentials in time-dependent current density functional theory. Phys. Rev. B: Condens. Matter Mater. Phys. 2004, 70, 201102. 10.1103/PhysRevB.70.201102. [DOI] [Google Scholar]
- Tokatly I. V. Time-dependent current density functional theory on a lattice. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 83, 035127. 10.1103/PhysRevB.83.035127. [DOI] [Google Scholar]
- Ullrich C. A.Time-Dependent Density-Functional Theory: Concepts and Applications. Oxford Graduate Texts; Oxford University Press: Oxford, 2012. [Google Scholar]
- van Leeuwen R.; Baerends E. J. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1994, 49, 2421–2431. 10.1103/PhysRevA.49.2421. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Yang W. A direct optimization method for calculating density functionals and exchangecorrelation potentials from electron densities. J. Chem. Phys. 2003, 118, 2498–2509. 10.1063/1.1535422. [DOI] [Google Scholar]
- Jensen D. S.; Wasserman A. Numerical methods for the inverse problem of density functional theory. Int. J. Quantum Chem. 2018, 118, e25425–e25425. 10.1002/qua.25425. [DOI] [Google Scholar]
- Ryabinkin I. G.; Kohut S. V.; Staroverov V. N. Reduction of Electronic Wave Functions to Kohn-Sham Effective Potentials. Phys. Rev. Lett. 2015, 115, 083001. 10.1103/PhysRevLett.115.083001. [DOI] [PubMed] [Google Scholar]
- Nielsen S. E. B.; Ruggenthaler M.; van Leeuwen R. Many-body quantum dynamics from the density. EPL (Europhysics Letters) 2013, 101, 33001. 10.1209/0295-5075/101/33001. [DOI] [Google Scholar]
- Hubig C.; McCulloch I. P.; Schollwöck U.; Wolf F. A. Strictly single-site DMRG algorithm with subspace expansion. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 91, 155115. 10.1103/PhysRevB.91.155115. [DOI] [Google Scholar]
- Hubig C.Symmetry-Protected Tensor Networks. Ph.D. Thesis, LMU Mnchen, 2017. [Google Scholar]
- Levy M.; Perdew J. P. Hellmann-Feynman, virial, and scaling requisites for the exact universal density functionals. Shape of the correlation potential and diamagnetic susceptibility for atoms. Phys. Rev. A: At., Mol., Opt. Phys. 1985, 32, 2010–2021. 10.1103/PhysRevA.32.2010. [DOI] [PubMed] [Google Scholar]
- Liao S.-L.; Ho T.-S.; Rabitz H.; Chu S.-I. Time-Local Equation for the Exact Optimized Effective Potential in Time-Dependent Density Functional Theory. Phys. Rev. Lett. 2017, 118, 243001. 10.1103/PhysRevLett.118.243001. [DOI] [PubMed] [Google Scholar]
- Griesemer M.; Hantsch F. Unique solutions to Hartree–Fock equations for closed shell atoms. Arch. Ration. Mech. Anal. 2012, 203, 883–900. 10.1007/s00205-011-0464-5. [DOI] [Google Scholar]