

Human cytomegalovirus (HCMV) infections are a leading cause of brain damage, hearing loss, and other neurological disabilities in children. We report that the HCMV proteins known as IE1 and IE2 target expression of human SOX2, a central pluripotency-associated transcription factor that governs neural progenitor cell (NPC) fate and is required for normal brain development. Both during HCMV infection and when expressed alone, IE1 causes the loss of SOX2 from NPCs. IE1 mediates SOX2 depletion by targeting STAT3, a critical upstream regulator of SOX2 expression. Our findings reveal an unanticipated mechanism by which a common virus may cause damage to the developing nervous system and suggest novel targets for medical intervention.
KEYWORDS: HCMV, IE1, neural progenitor cells, SOX2, STAT3
ABSTRACT
The mechanisms underlying neurodevelopmental damage caused by virus infections remain poorly defined. Congenital human cytomegalovirus (HCMV) infection is the leading cause of fetal brain development disorders. Previous work has linked HCMV infection to perturbations of neural cell fate, including premature differentiation of neural progenitor cells (NPCs). Here, we show that HCMV infection of NPCs results in loss of the SOX2 protein, a key pluripotency-associated transcription factor. SOX2 depletion maps to the HCMV major immediate early (IE) transcription unit and is individually mediated by the IE1 and IE2 proteins. IE1 causes SOX2 downregulation by promoting the nuclear accumulation and inhibiting the phosphorylation of STAT3, a transcriptional activator of SOX2 expression. Deranged signaling resulting in depletion of a critical stem cell protein is an unanticipated mechanism by which the viral major IE proteins may contribute to brain development disorders caused by congenital HCMV infection.
IMPORTANCE Human cytomegalovirus (HCMV) infections are a leading cause of brain damage, hearing loss, and other neurological disabilities in children. We report that the HCMV proteins known as IE1 and IE2 target expression of human SOX2, a central pluripotency-associated transcription factor that governs neural progenitor cell (NPC) fate and is required for normal brain development. Both during HCMV infection and when expressed alone, IE1 causes the loss of SOX2 from NPCs. IE1 mediates SOX2 depletion by targeting STAT3, a critical upstream regulator of SOX2 expression. Our findings reveal an unanticipated mechanism by which a common virus may cause damage to the developing nervous system and suggest novel targets for medical intervention.
INTRODUCTION
Congenital human cytomegalovirus (HCMV) infection is the leading cause of birth defects worldwide. Approximately 1% of live newborns are infected in utero with the virus. At the time of birth, 5 to 10% of HCMV-infected newborn infants exhibit signs of neurological damage, such as microcephaly, cerebral calcification, and other abnormal findings (1–3). Among infected newborns who have no symptoms at birth, 10 to 15% subsequently develop central nervous system (CNS) disorders, including sensorineural hearing loss, mental retardation, and learning disability (4–6). In addition, some authors have suggested that autism, language disorders, and other, more subtle changes in brain development might be related to congenital HCMV infection (7–9).
Although the virus can infect a wide range of cell types and organs in vivo, the fetal brain is regarded as the principal target of HCMV infection that results in neurological manifestations (10–12). Due to exquisite host-specific tropism of the virus and the lack of animal models that faithfully recapitulate major characteristics of human infection, the pathogenesis of HCMV-associated disease in the developing CNS is largely unknown. However, recent advances in human neural progenitor cell (NPC) isolation and culture have provided an opportunity to study HCMV infection in a cell system relevant to fetal neuropathogenesis. Our previous studies and the work of others have shown that human NPCs are susceptible to HCMV infection and fully permissive to viral replication (13–19). HCMV infection of NPCs affects cell fate by causing premature differentiation (14, 17, 18). Whole-genome analysis demonstrated that HCMV infection modulates the expression of NPC markers (14, 19), including sex-determining region Y (SRY) box 2 (SOX2), a core transcriptional factor for stem cell self-renewal and pluripotency.
SOX2 is widely expressed in early neuroectoderm and neural progenitor cells during development (14, 19, 20), as well as in neural stem cells in the adult brain (20, 21). SOX2 missense or heterozygous loss-of-function mutations have been found to cause ocular malformations, often manifesting as anophthalmia, microphthalmia, or coloboma. These symptoms may be accompanied by hearing loss, learning disability, or brain malformation (22–24). Familial recurrence of SOX2-associated anophthalmia has been observed (24). Moreover, the level of SOX2 expression plays an important role in sensory organ, including inner ear and retina, development. Groundbreaking research has demonstrated that forced SOX2 expression in fibroblasts, with or without additional factors, can generate induced pluripotent stem cells (25–27). In fact, ectopic SOX2 expression directs reprogramming of fibroblasts into neural stem or precursor cells (25, 28–30). SOX2 is also critical for maintenance of embryonic stem cells (ESCs). The SOX2 levels in ESCs are tightly regulated (31), and even small changes in expression can lead to differentiation (32, 33).
STAT3 is a member of the signal transducer and activator of transcription (STAT) family (34) and is expressed in an activated form in the developing CNS as early as during initial NPC proliferation. The protein plays a dichotomous regulatory role in neuro- and gliogenesis (35, 36). STAT3 is activated through phosphorylation of tyrosine 705 (Y705) by receptor-associated kinases in response to various growth factors and cytokines, including interleukin 6 (IL-6). Tyrosine phosphorylation leads to the nuclear accumulation of STAT3 homodimers, which act as DNA binding transcriptional activators of numerous target genes, including the SOX2 gene. In ESCs that are differentiated into NPCs, STAT3 promotes cell fate commitment by activating the SOX2 gene promoter (37).
Here, we investigate the effects of HCMV infection on SOX2 expression in human NPCs. We demonstrate that the HCMV 72-kDa immediate early 1 (IE1) protein downregulates SOX2 transcription and mediates depletion of the SOX2 protein from HCMV-infected NPCs. IE1 exerts its effect on SOX2 expression by inactivating the upstream regulator STAT3.
RESULTS
HCMV infection of NPCs causes downregulation of SOX2 mRNA and protein.
Previous work has established that human NPCs are fully permissive to HCMV infection (15, 38). As early as 4 h postinfection (hpi), a significant decrease of SOX2 mRNA was observed in NPCs, and this decrease became more apparent as infection progressed (Fig. 1A). In contrast, SOX2 protein levels did not change in these cells at early times postinfection (up to 12 hpi), but a gradual decrease was observed at late times (from 24 hpi) compared with mock-infected cells (Fig. 1B). By 48 hpi, the SOX2 protein was clearly downregulated, and by 96 hpi, it was undetectable. The HCMV proteins IE1/IE2, UL44, and glycoprotein B (gB) were also analyzed in this experiment to monitor IE, early, and late viral gene expression, respectively.
FIG 1.
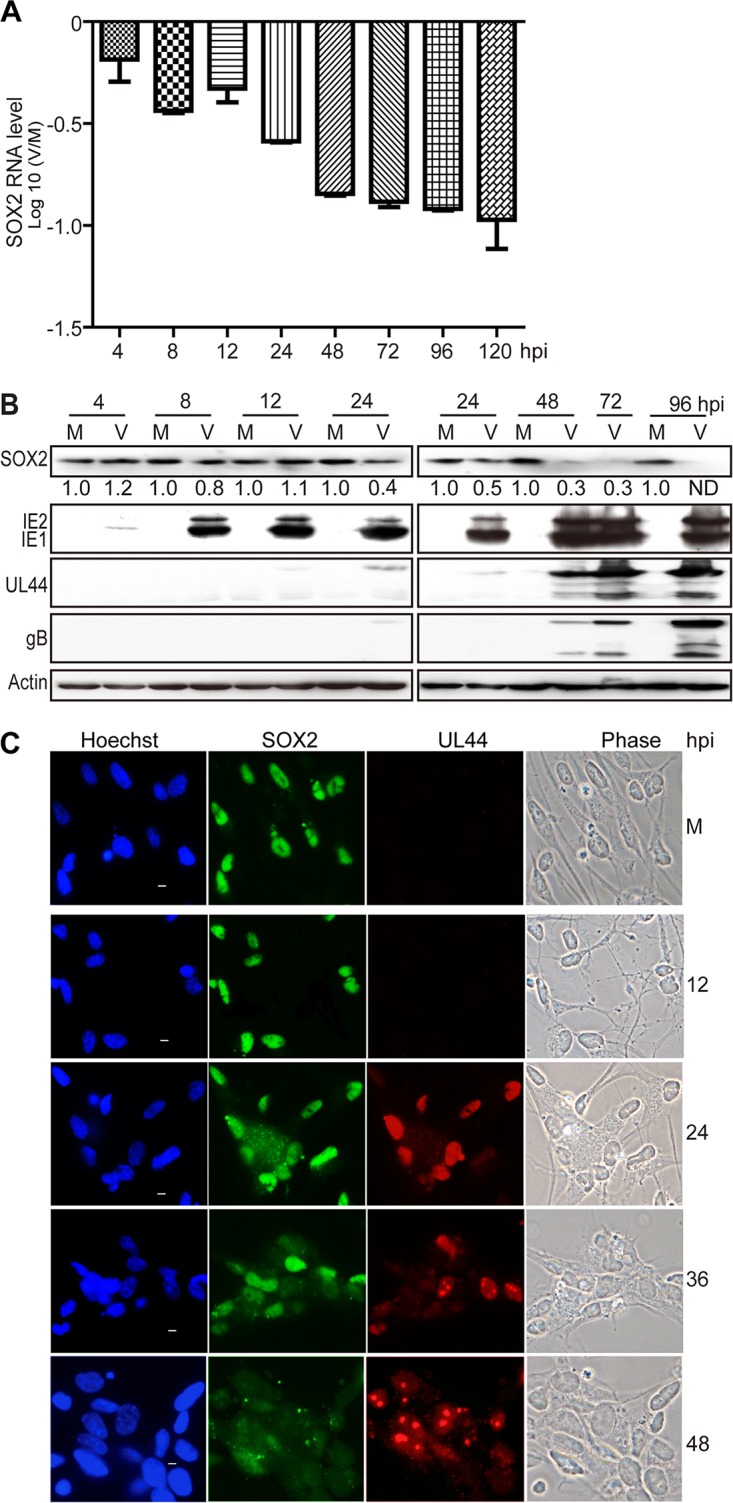
HCMV infection downregulates SOX2 at the mRNA and protein levels in NPCs. NPC monolayers were mock infected (M) or infected with HCMV (TNWT) at an MOI of 3 (V) and collected at the indicated times postinfection for mRNA or protein analyses. (A) SOX2 mRNA levels during HCMV infection of NPCs. The levels of SOX2 mRNA, normalized to GAPDH, were determined by qRT-PCR at 4 to 120 hpi. The results shown are averages and SD of data from three independent experiments, each conducted in triplicate. (B) SOX2 and viral protein levels during HCMV infection of NPCs. SOX2, IE1/IE2, UL44, and gB steady-state protein levels were determined by Western blotting at 4 to 96 hpi. Actin served as a loading control. The values listed below the SOX2 blots indicate the relative SOX2 protein levels compared to corresponding mock-infected controls following actin normalization. ND, not detectable. (C) Cellular distribution of SOX2 in relation to viral replication compartments during HCMV infection of NPCs. The distributions of SOX2 and UL44 were determined by indirect immunofluorescence assay at 12 to 48 hpi. NPCs grown on poly-d-lysine-coated coverslips were stained with antibodies against SOX2 (green) and UL44 (red), and nuclei were counterstained with Hoechst 33342 (blue). Phase-contrast images are also shown. Scale bars, 5 μm.
In addition, the change in SOX2 protein levels was examined in relation to the formation and development of HCMV intranuclear replication compartments visualized via immunofluorescence staining of UL44 (Fig. 1C). UL44 is the processivity factor of the HCMV DNA polymerase and is first expressed in the early phase of infection. Initially, UL44 was evenly distributed across the infected nucleus; then, it formed multiple small foci (36 hpi) representing early viral replication compartments; and finally, the small foci merged into bipolar foci (48 hpi) or single large late replication compartments (72 hpi) (data not shown). In mock-infected NPCs, SOX2 exhibited its typical intense and diffuse nuclear staining. As expected, the SOX2 signal decreased during the course of HCMV infection inversely related to UL44 focus development. In fact, SOX2 became dispersed and largely disappeared from late HCMV-infected NPCs.
These observations indicate the presence of a highly effective mechanism for SOX2 depletion in HCMV-infected NPCs.
SOX2 depletion from HCMV-infected NPCs requires de novo viral protein synthesis.
To determine if SOX2 downregulation is dependent on HCMV gene products expressed de novo from the infecting viral genome, NPCs were exposed to infectious HCMV or UV-inactivated virus, and SOX2 expression was analyzed. As expected, SOX2 expression was suppressed at both the RNA and protein levels in infections with the untreated virus (Fig. 2A and B). In contrast, UV-inactivated virus failed to suppress the expression of SOX2 mRNA (Fig. 2A) and protein (Fig. 2B). Similar levels of gB derived from the input virus were detected at 12 hpi with both untreated and UV-treated HCMV, confirming that cells had been exposed to equal amounts of infecting virus (Fig. 2B). However, de novo synthesis of viral proteins was undetectable in infections with UV-treated virus, confirming successful inactivation. In another experiment, NPCs were pretreated with cycloheximide (CHX) and then infected with HCMV for 4, 8, 12, and 16 h in the presence of CHX. Downregulation of SOX2 mRNA induced by HCMV infection was significantly diminished by CHX treatment at all the tested time points (Fig. 2C). No obvious change in SOX2 protein levels was observed by 16 hpi (data not shown), consistent with the results shown in Fig. 1 and previous reports (14, 19).
FIG 2.
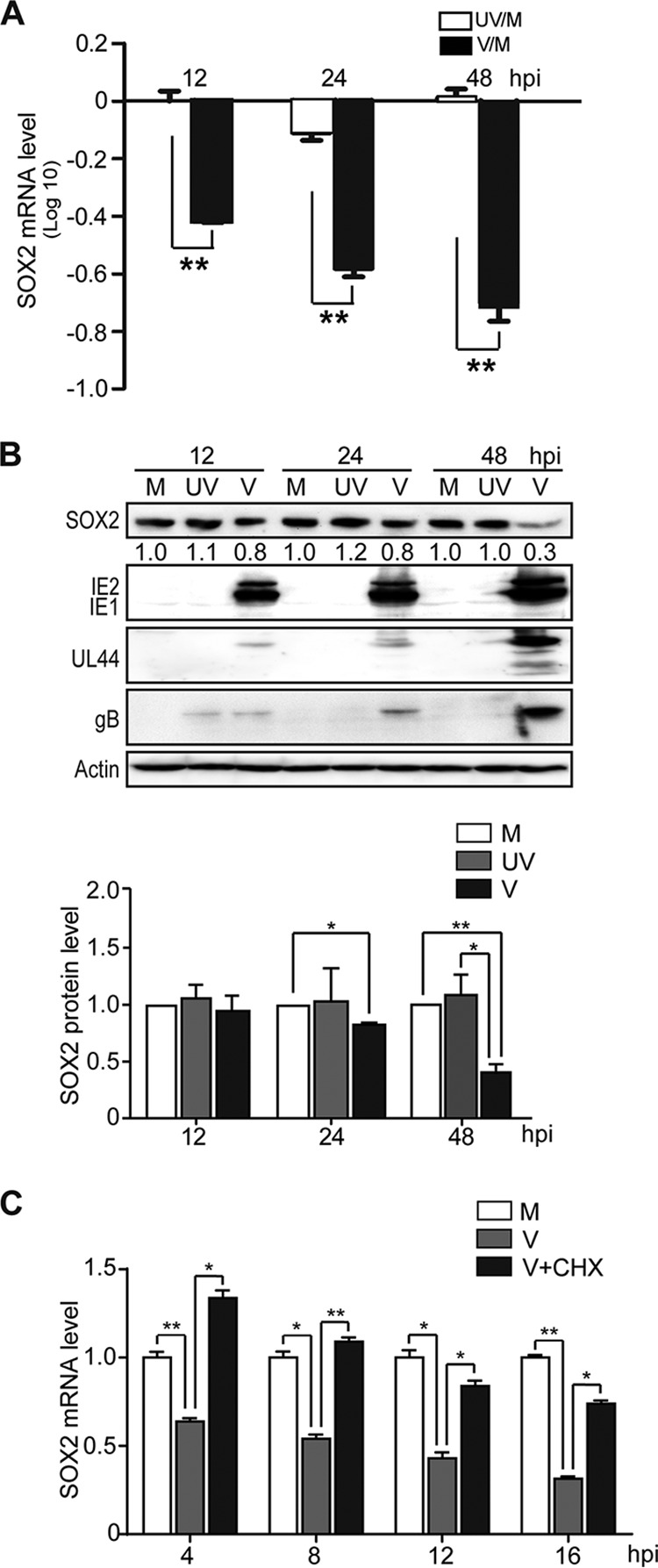
De novo synthesis of HCMV proteins is required for SOX2 downregulation in NPCs. Shown are comparisons of HCMV infections with active virus (V) and UV-inactivated virus (UV). NPCs were mock infected (M) or infected with active or UV-irradiated TNWT at an MOI of 3 and collected at the indicated times postinfection. (A) SOX2 mRNA levels normalized to GAPDH were determined by qRT-PCR. Log10 values of inactivated virus/mock-infected virus (UV/M) and active virus/mock-infected virus (V/M) ratios are shown. Data from three independent experiments were analyzed by one-way analysis of variance (ANOVA), and the results are presented as averages and SD. **, P ≤ 0.01. (B) Levels of SOX2 and representative viral proteins (IE1/IE2, UL44, and gB) were determined by Western blotting. Actin served as a loading control. The values listed below the SOX2 blots indicate the relative SOX2 protein levels compared to corresponding mock-infected controls following actin normalization. The data are from three independent experiments, and the results are presented as averages and SD. *, P ≤ 0.05; **, P ≤ 0.01. (C) Effect of protein synthesis inhibition by CHX treatment on SOX2 mRNA levels. NPCs were pretreated with CHX for 1 h prior to infection and then mock infected (M) or infected with HCMV (TNWT) at an MOI of 3 (V). Cells were collected at 4, 8, 12, and 16 hpi for analysis of SOX2 mRNA by qRT-PCR. Data from three independent experiments were analyzed by one-way ANOVA, and the results are presented as averages and SD. *, P ≤ 0.05; **, P ≤ 0.01.
These results indicate that one or more viral proteins expressed de novo upon HCMV infection are required for SOX2 downregulation in NPCs.
HCMV major IE proteins mediate SOX2 downregulation in NPCs.
Transcription of SOX2 is suppressed as early as 4 hpi, a time when virion constituents and IE gene products are the only viral components present in the infected cell nucleus. To test whether IE or tegument proteins are sufficient for SOX2 downregulation, the major IE proteins (IE1 and IE2) and the most abundant tegument protein (pp65) were individually expressed in NPCs using nucleofection. In agreement with the conclusion that newly synthesized viral proteins mediate SOX2 downregulation (Fig. 2), SOX2 levels did not change in NPCs transiently expressing pp65 compared to control cells (Fig. 3A). Thus, pp65 was used as a negative control in subsequent assays. Expression of the entire major IE transcription unit, including the IE1 and IE2 proteins, resulted in significant reduction of SOX2 protein levels (Fig. 3A). To discriminate between effects on SOX2 expression mediated by IE1 or IE2, each of the two proteins was individually expressed in NPCs. Following transfection of 2 or 5 μg IE1-expressing plasmid, the relative SOX2 mRNA levels were reduced to 61 or 23% of control levels, and the corresponding protein levels were reduced to 84 or 33%, respectively (Fig. 3B). Transfection of 2 or 5 μg IE2-expressing plasmid reduced the relative SOX2 mRNA levels to 71 or 31% of control levels and the protein levels to 88 or 54%, respectively (Fig. 3C).
FIG 3.

HCMV major IE proteins downregulate SOX2 mRNA and protein in NPCs. HCMV pp65, IE1, IE2, or IE1 and IE2 combined were transiently expressed in NPCs following nucleofection. Samples were collected at 48 h posttransfection for mRNA (qRT-PCR) or protein (Western blotting) analysis. Actin served as a loading control. Shown is the relative level of SOX2 protein compared to corresponding controls following actin normalization. Data from three independent experiments were analyzed by one-way ANOVA, and the results are presented as averages and SD. *, P < 0.05; **, P < 0.01. (A) Effects of pp65 and major IE proteins (IE1 and IE2) on SOX2 protein levels. (Left blot) NPCs were transfected with 5 μg pcDNA3.0 (Vector) or pcDNA3-pp65. (Right blot) NPCs were transfected with 5 μg pcDNA3-pp65 as a control (Vector) or pSVH (IE1 and IE2). (B) Effects of IE1 on SOX2 mRNA and protein levels. NPCs were transfected with 5 μg pcDNA3-pp65 (Vector) or 2 to 5 μg pcDNA3-IE1. (C) Effects of IE2 on SOX2 mRNA and protein levels. NPCs were transfected with 5 μg pcDNA3-pp65 (Vector) or 2 to 5 μg pcDNA3-IE2.
These results indicate that the HCMV IE1 and IE2 proteins both play a significant role in downregulating SOX2 expression. However, IE1 appeared to affect SOX2 more efficiently than IE2. Therefore, and because of the lethal phenotype of IE2-deficient viruses (39, 40), we focused our study on the role of IE1 in SOX2 regulation in the present study.
HCMV IE1 is required for depletion of SOX2 during infection of NPCs.
HCMV infection and IE1 expression were each sufficient to efficiently downregulate SOX2 at the mRNA and protein levels in NPCs. To confirm the requirement for IE1 for SOX2 depletion during HCMV infection, the expression of IE1 was knocked down using short hairpin RNAs (shRNAs) targeting IE1 exon 4 sequences. First, human embryonic lung (HEL) cells were transduced with lentiviruses expressing shRNAs directed against three different IE1-specific sequences, sh-1 to sh-3, or a scrambled shRNA control (scr). The transduced cells were subsequently synchronized and infected with HCMV. Analysis of IE1 protein levels determined at 24 and 48 hpi showed that sh-2 was the most efficient among the three tested shRNAs in reducing the levels of the IE1 protein (Fig. 4A). To confirm the knockdown efficiency of sh-2, another experiment was performed in which both IE1 mRNA and protein were analyzed at 24, 48, and 72 hpi. Again, sh-2 knocked down IE1 by >60% at the RNA level and 50% at the protein level through the course of infection (Fig. 4B).
FIG 4.
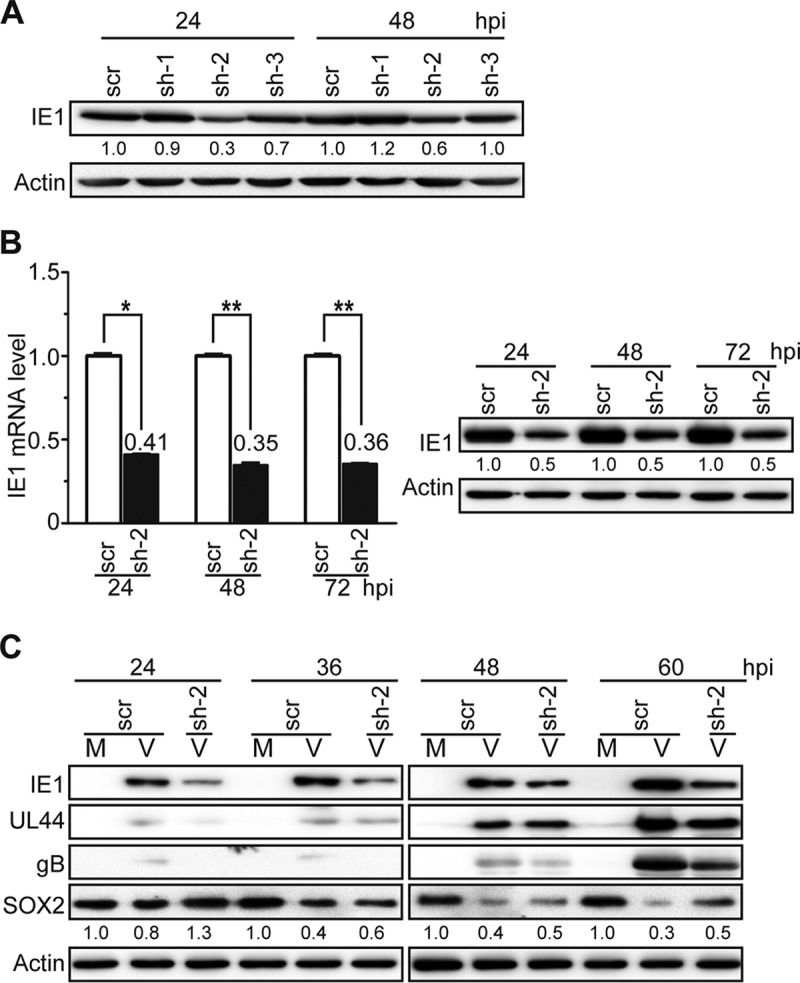
IE1 knockdown attenuates HCMV-induced SOX2 downregulation in NPCs. (A) IE1-directed knockdown efficiencies of candidate shRNAs in HEL cells. HEL cells were transduced with lentiviruses expressing shRNA-IE1-1 (sh-1), shRNA-IE1-2 (sh-2), shRNA-IE1-3 (sh-3), or shRNA-scramble (scr). At 48 h postransduction, the cells were infected with HCMV (TNWT) at an MOI of 1 and collected at 24 or 48 hpi. IE1 protein levels were determined by Western blotting. Actin served as a loading control. (B) IE1-directed knockdown efficiency of sh-2 in HEL cells. HEL cells transduced with sh-2- or scr-expressing lentiviruses were infected with HCMV (TNWT) as for panel A. (Left) IE1 mRNA levels were determined by qRT-PCR at the indicated times postinfection; data from two independent experiments were analyzed by one-way ANOVA, and the results are presented as averages and SD. *, P ≤ 0.05; **, P ≤ 0.01. (Right) Protein levels were determined at the indicated times by Western blotting. (C) Effects of sh-2 on IE1 and SOX2 expression in NPCs. NPCs were transduced with sh-2- or scr-expressing lentiviruses, cultured for 48 h, reseeded at a density of 3 × 106 cells/dish, and mock infected (M) or infected with TNWT at an MOI of 1 (V). Protein levels of IE1, UL44, gB, and SOX2 at the indicated times postinfection are shown. Actin served as a loading control.
Following the successful IE1 knockdown in HEL cells, NPCs were transduced with lentiviruses expressing sh-2 or scr. The transduced NPCs were infected with HCMV and collected at 24 to 60 hpi. Since 24 hpi was previously shown to be the “turning point” of SOX2 protein levels (Fig. 1), this time point was chosen as the earliest. As shown in Fig. 4C, SOX2 protein levels gradually decreased as infection progressed, consistent with Fig. 1. Significant knockdown of IE1 was observed at all tested time points in NPCs expressing sh-2 compared with scr-expressing cells. SOX2 downregulation was alleviated in HCMV-infected NPCs expressing sh-2 compared to cells expressing scr, most notably at 60 hpi.
Although the IE1 knockdown was incomplete and linked to reduced levels of other viral proteins (UL44 and gB), these results support the idea that IE1 contributes to SOX2 downregulation during HCMV infection of NPCs.
HCMV infection and IE1 expression inhibit STAT3 tyrosine phosphorylation and relocalize unphosphorylated STAT3 to the nuclei of NPCs.
So far, our experiments have indicated that HCMV IE1 downregulates SOX2, but a mechanism through which the viral protein alters expression of the cellular stem cell factor has not been identified. Results from coimmunoprecipitation and yeast two-hybrid assays failed to provide evidence for a physical interaction between IE1 and SOX2 (data not shown). Since SOX2 expression was affected at both the protein and RNA levels, it also seemed more likely that IE1 interferes with the transcription rather than the protein stability of SOX2. One key transcription factor regulating SOX2 expression is STAT3 (37), which has been recently identified as a physical interaction partner of IE1 (41, 42).
To investigate whether HCMV infection and IE1 expression affect SOX2 levels via modulation of STAT3 activation, the levels of total and tyrosine (Y705)-phosphorylated STAT3 (pSTAT3) were determined in infected NPCs. HCMV infection dramatically suppressed STAT3 tyrosine phosphorylation as early as 4 hpi, and pSTAT3 was maintained at low levels throughout the course of infection compared to mock-infected NPCs. In contrast, the total STAT3 levels showed little if any change during infection (Fig. 5A).
FIG 5.

HCMV infection or IE1 expression inhibits STAT3 tyrosine phosphorylation and promotes nuclear accumulation of unphosphorylated STAT3 in NPCs. (A) Inhibition of STAT3 tyrosine (Y705) phosphorylation by HCMV infection. NPCs were mock infected (M) or infected with TNWT at an MOI of 3 (V) and collected at the indicated times postinfection. The protein levels of IE1, pSTAT3, and total STAT3 were determined by Western blotting. Actin served as a loading control. (B) Nuclear trapping of STAT3 by HCMV infection. NPCs were mock infected (M) or -infected with TNWT at an MOI of 1 (V). (Left) For indirect immunofluorescence analysis, NPCs on coverslips collected at 8 hpi were stained with antibodies against STAT3 (green) or IE1/IE2 (red), and nuclei were counterstained with Hoechst 33342 (blue). Infected (IE1/IE2-positive) cells are indicated by arrows. Scale bar, 10 μm. (Right) For cellular-fractionation analysis, fractions enriched in cytoplasmic (Cyt) or nuclear (Nuc) proteins were prepared from cells collected at 4 or 8 hpi. Protein levels of pSTAT3 and total STAT3 in each fraction were determined by Western blotting. GAPDH and lamin B1 served as controls for the Cyt and Nuc fractions, respectively. (C to G) Inhibition of tyrosine phosphorylation and nuclear sequestration of unphosphorylated STAT3 by IE1. Fractions enriched in cytosolic or nuclear proteins or total cell extracts were prepared. (C) For transient-transfection analysis, NPCs were nucleofected with pcDNA3-IE1 or empty vector (Ctrl) and harvested at 48 h postnucleofection. Protein levels of IE1, pSTAT3, and total STAT3 were determined by Western blotting. GAPDH and lamin B1 served as controls for the Cyt and Nuc fractions, respectively. (D) To examine dose-dependent effects of IE1 on pSTAT3 levels, NPCs transfected with the indicated amounts of pcDNA-IE1 and empty vector (pcDNA3.0) were harvested 48 h postnucleofection. The protein levels of IE1, STAT3, pSTAT3, and SOX2 were determined by Western blotting. *, P ≤ 0.05. (E to G) For HCMV infection analysis, NPCs were mock infected (M) or infected with TNWT (WT), TNdlIE1 (dlIE1), or TNrvIE1 (rvIE1) viruses at an MOI of 10. The levels of the indicated viral and cellular proteins in whole-cell extracts at 72 hpi (E) and 24 hpi (F) or in the Cyt and Nuc fractions at 24 hpi (G) were determined by Western blotting. Actin, GAPDH, and lamin B1 served as controls for total extracts or Cyt and Nuc fractions, respectively. *, P ≤ 0.05; **, P ≤ 0.01.
STAT3 is a nucleocytoplasmic shuttling protein that is efficiently exported from the nucleus in its unphosphorylated form (43). Unphosphorylated STAT3 is therefore typically located in the cytoplasm or distributed across both the nuclear and cytoplasmic compartments. However, upon tyrosine phosphorylation, pSTAT3 efficiently accumulates in the nucleus (37, 44, 45). To define the subcellular distribution of STAT3 in HCMV-infected NPCs, we performed immunofluorescence analysis. NPCs on coverslips were infected with HCMV (multiplicity of infection [MOI] = 1), collected at 8 hpi, and stained for STAT3 and IE1/IE2. STAT3 was mainly confined to the nuclei of virus-infected cells, while in mock-infected cells, the STAT3 signal was more diffuse and was cytoplasmic as well as nuclear (Fig. 5B). This observation was consistent with our results from HCMV-infected fibroblasts (41, 42). To further investigate STAT3 subcellular localization in HCMV-infected NPCs, the levels of pSTAT3 and total STAT3 in cytoplasmic and nuclear compartments separated by cellular fractionation were analyzed at 4 and 8 hpi. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and lamin B1 served as loading controls for the cytoplasmic and nuclear compartments, respectively, confirming successful fractionation. As expected, pSTAT3 was present predominantly in the nuclei of both mock- and HCMV-infected cells. Compared to mock-infected NPCs, pSTAT3 levels were reduced in both the cytoplasm and nucleus in HCMV-infected cells at 4 and 8 hpi. Total STAT3 resided mainly in the cytoplasm, but the nuclear signal was much stronger in HCMV-infected than in mock-infected cells at both tested time points (Fig. 5B). These results indicate that HCMV infection reduces the levels of activated STAT3 (pSTAT3) and promotes the nuclear accumulation of unphosphorylated STAT3 at very early times of infection in NPCs.
IE proteins are thought to be the only HCMV gene products expressed at 4 to 8 hpi, when STAT3 is relocalized to the nucleus and its phosphorylation is inhibited. To test whether IE1 was responsible for the observed effects on STAT3, the viral protein was transiently expressed in NPCs. IE1 expression was sufficient to downregulate pSTAT3 levels and to sequester an unphosphorylated form of the cellular protein in the nuclear compartment (Fig. 5C). To investigate whether IE1 has a dose-dependent inhibitory effect on STAT3 phosphorylation, pSTAT3 levels in NPCs nucleofected with different amounts of IE1-expressing plasmid were compared by Western blotting. An inverse correlation between IE1 and pSTAT3 levels was observed in these experiments, indicating that IE1 downregulates pSTAT3 in a dose-dependent manner (Fig. 5D). To further confirm the role of IE1 in altering STAT3 phosphorylation and intracellular localization, NPCs were mock infected or infected with wild-type (WT), IE1-deficient variant strain (dlIE1), or IE1-revertant variant strain (rvIE1) viruses. The levels of viral proteins (UL44 and gB) were similar, except that IE1 was absent and downregulation of SOX2 was diminished in the infection with dlIE1 (Fig. 5E). Compared with mock-infected cells, the pSTAT3 levels were reduced in WT- and rvIE1-infected cells, but not in dlIE1-infected cells. Again, the overall steady-state STAT3 levels were rather constant across all mock- and HCMV-infected samples (Fig. 5F). Finally, based on cellular-fractionation analysis, pSTAT3 was located in the nuclei of both mock- and WT- or dlIE1-infected cells. Total STAT3 localized mainly in the cytoplasm of mock- and dlIE1-infected cells, while increased nuclear localization was observed in WT-infected NPCs (Fig. 5G).
These results demonstrate that IE1 is both sufficient and required for the inhibition of tyrosine phosphorylation and nuclear sequestration of unphosphorylated STAT3 observed during HCMV infection of NPCs.
SOX2 downregulation in HCMV-infected NPCs results from IE1-mediated inhibition of STAT3 activation.
The results thus far obtained were all consistent with a mechanism in which IE1 mediates SOX2 downregulation by limiting STAT3 activation. To further test this possibility, we performed several different experiments. We first treated NPCs with cryptotanshinone (CTS), a chemical inhibitor of STAT3 tyrosine phosphorylation (46). In the presence of CTS, pSTAT3 levels (but not steady-state STAT3 levels) markedly decreased as a function of the time of treatment coinciding with gradual loss of SOX2 (Fig. 6A). Then, STAT3 expression was silenced using RNA interference. Two different shRNAs, shSTAT3-1 and shSTAT3-2, knocked down STAT3 expression with different efficiencies compared to nonspecific shRNAs (shLuci and shDsRed). The extent of STAT3 silencing correlated with differential reduction in pSTAT3 levels, in turn correlating with the levels of SOX2 (Fig. 6B).
FIG 6.
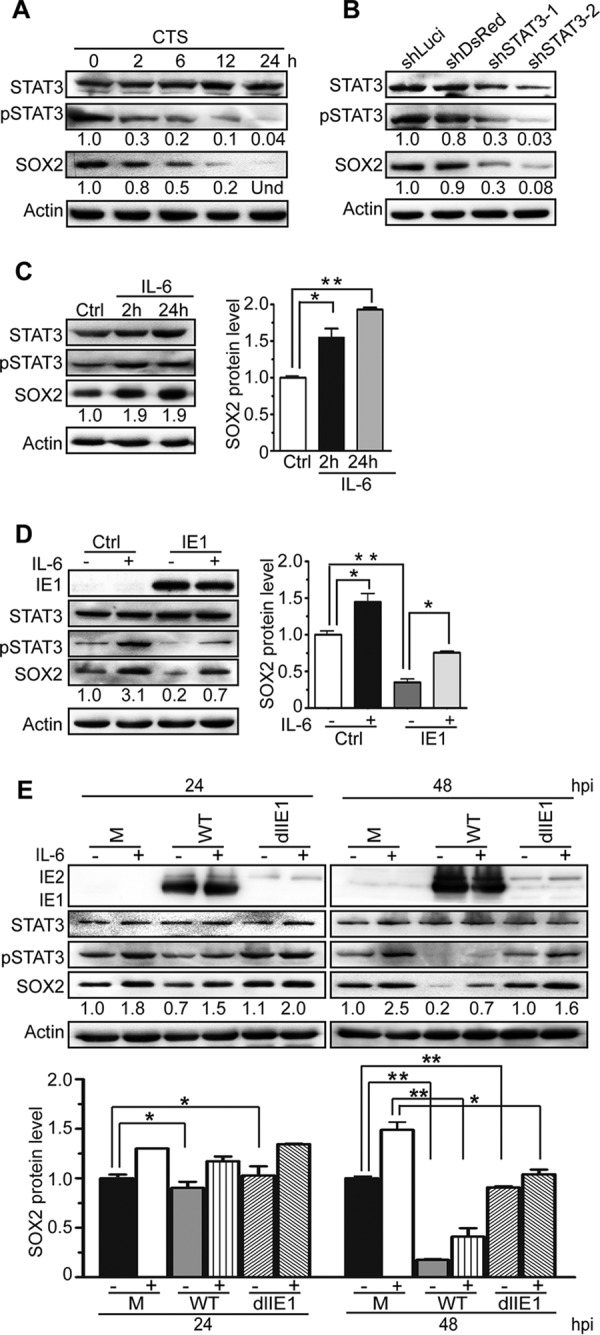
SOX2 expression strictly depends on pSTAT3, and IE1 mediates SOX2 depletion by inhibiting STAT3 activation. (A) Inhibition of STAT3 correlates with suppression of SOX2 expression. NPCs were treated with the chemical inhibitor CTS for the indicated times. The protein levels of total STAT3, pSTAT3, and SOX2 were determined by Western blotting. Actin served as a loading control. Und, undetectable. (B) Silencing of STAT3 correlates with suppression of SOX2 expression. NPCs were transduced with the lentivirus Tet-pLKO-puro-shLuci (shLuci), Tet-pLKO-puro-shDsRed (shDsRed), Tet-pLKO-puro-shSTAT3-1 (shSTAT3-1), or Tet-pLKO-puro-shSTAT3-2 (shSTAT3-2) and treated with doxycycline for 48 h to induce shRNA expression. The protein levels of total STAT3, pSTAT3, and SOX2 were determined by Western blotting. Actin served as a loading control. (C) IL-6-mediated activation of STAT3 correlates with induction of SOX2 expression. NPCs were treated with IL-6 for 2 or 24 h. The protein levels of pSTAT3, total STAT3, and SOX2 were monitored by Western blotting. Actin served as a loading control. Data from three independent experiments were analyzed by one-way ANOVA, and the results are presented as averages and SD. *, P ≤ 0.05; **, P ≤ 0.01. (D) IL-6 counteracts IE1-dependent SOX2 downregulation in transiently transfected NPCs. NPCs were transfected with pcDNA3-IE1, treated with IL-6 for 4 h or left untreated, and collected at 48 h posttransfection. The protein levels of IE1, total STAT3, pSTAT3, and SOX2 were determined by Western blotting. Actin served as a loading control. Data from three independent experiments were analyzed by one-way ANOVA, and the results are presented as averages and SD. *, P ≤ 0.05; **, P ≤ 0.01. (E) IL-6 counteracts IE1-dependent SOX2 downregulation in HCMV-infected NPCs. NPCs were infected with TNWT or TNdlIE1 virus at an MOI of 10, treated with IL-6 for 4 h or left untreated, and collected at 48 hpi. The protein levels of IE1/IE2, total STAT3, pSTAT3, and SOX2 were determined by Western blotting. Actin served as a loading control. Data from three independent experiments were analyzed by one-way ANOVA, and the results are presented as averages and SD. *, P ≤ 0.05; **, P ≤ 0.01.
In addition to the experiments involving inhibition of STAT3, we set out to study the IE1-STAT3-SOX2 axis by activating STAT3 signaling. To this end, we first treated NPCs with IL-6, a major agonist of STAT3 signaling (47). IL-6 treatment led to a marked increase in pSTAT3 without significantly affecting the overall STAT3 protein levels. Concurrent with increased STAT3 tyrosine phosphorylation, there was also a rise in SOX2 levels. These data were consistent with the finding that pSTAT3 promotes SOX2 expression in NPCs (Fig. 6C). Next, we transiently expressed IE1 in NPCs; treated the cells with IL-6 for 4 h or left them untreated; and analyzed the levels of IE1, pSTAT3, total STAT3, and SOX2. Both pSTAT3 and SOX2 were strongly upregulated by IL-6 but downregulated by IE1, and IL-6 efficiently counteracted IE1-mediated downregulation of pSTAT3 and SOX2. Again, the total steady-state STAT3 levels did not significantly change upon IL-6 treatment or IE1 expression (Fig. 6D). Finally, to further investigate the link between IE1 expression, STAT3 activation, and SOX2 regulation in the context of HCMV infection, NPCs were mock infected or infected with HCMV WT or dlIE1. The infected NPCs were treated with IL-6 for 4 h or left untreated prior to collection at 24 and 48 hpi. As expected, IL-6 treatment led to elevated levels of pSTAT3 and SOX2 in mock-, WT-, and dlIE1-infected NPCs. HCMV WT infection reduced the pSTAT3 levels slightly at 24 hpi and dramatically at 48 hpi. In contrast, no obvious decrease of pSTAT3 was observed following dlIE1 infection. Concordantly, SOX2 protein levels fell substantially in WT infection at 48 hpi but remained constant in dlIE1 infection at the same time point. Again, IL-6 treatment counteracted IE1-dependent downregulation of both pSTAT3 and SOX2 (Fig. 6E).
In summary, these data demonstrate that SOX2 expression in NPCs strictly depends on pSTAT3 and that there is a causal link between SOX2 downregulation by HCMV and inhibition of STAT3 activation by IE1, including inhibition of tyrosine phosphorylation and trapping of unphosphorylated STAT3 in the nucleus.
DISCUSSION
Proliferation, differentiation, and migration of NPCs, as well as synapse formation among mature neurons, are all critical factors for fetal brain development and function (48–50). HCMV infection has been shown to induce neural cell loss and abnormal differentiation of NPCs (14, 15, 19). This was not only demonstrated in vitro, but also confirmed in a mouse model of congenital infection, where the virus appeared to affect NPCs in the subventricular zone (12). Although HCMV is the leading cause of neurological damage in children, the mechanisms by which the virus perturbs NPC proliferation and differentiation have remained unclear.
SOX2 is a master controller of stem cells, which is strikingly illustrated by the fact that its overexpression can reprogram terminally differentiated fibroblasts into induced pluripotent stem cells (25–30). More specifically, SOX2 is a transcription factor essential for maintaining self-renewal and pluripotency of ESCs and NPCs. SOX2 mutations cause symptoms resembling damage resulting from congenital HCMV infection, including neural development disorders accompanied by ocular malformation (22–24). In this study, we demonstrate that HCMV significantly downregulates SOX2 mRNA levels in human primary NPCs from as early as 4 hpi, extending our previous observations (14, 38). The reduced mRNA levels subsequently lead to almost full depletion of the SOX2 protein from HCMV-infected cells at later times (24 to 96 hpi). The temporal delay between mRNA and protein downregulation suggests that the SOX2 protein may be very stable in NPCs, in contrast to what has been reported for ESCs (51). The timing of changes in SOX2 mRNA levels and the observation that the downregulation depends on de novo viral protein synthesis pointed us to the HCMV major IE proteins as potential regulators of this stem cell factor. The 72-kDa IE1 and the 86-kDa IE2 proteins subsequently proved to be sufficient to decrease SOX2 mRNA and protein levels in the absence of other viral proteins, together and individually. IE1 and IE2 are nuclear-localized key regulators of viral and cellular transcription during infection (52–54). They have been extensively studied in fibroblasts, but there is little information on how the proteins behave in cell types more relevant to HCMV pathogenesis.
Our results demonstrate that the HCMV IE1 protein is not only sufficient, but also necessary, for the reduction of SOX2 levels through the early and late stages of infection. As noted above, the downregulation of SOX2 RNA and protein was also observed with ectopically expressed HCMV IE2, but that protein appeared to be less efficient than IE1 in this respect. We propose that IE2 also contributes to SOX2 downregulation in the context of HCMV infection, although this remains to be formally shown. Due to the difficulties in working with IE2-deficient viruses (IE2 is essential for HCMV replication and is difficult to complement), we focused on IE1 in this study. Our findings may come as a surprise in the light of two previous reports that concluded that HCMV infection or IE1 expression leads to increased instead of decreased SOX2 levels in human glioma stem-like cells, human glioblastoma cells, and mouse glioma tissue (55, 56). The seemingly disparate findings might be due to differences in cell types or virus strains. However, in our hands, IE1 induced robust inhibition of STAT3 activation in all cell types tested so far, including glioblastoma-derived cell lines (data not shown). Notably, the previous studies did not discriminate between the levels of SOX2 in IE1-expressing cells and IE1-negative cells in the same culture or tumor. Even if the STAT3 pathway is inhibited in HCMV-infected cells or cells ectopically expressing IE1, the secretome associated with these cells may trigger STAT3 signaling in IE1-negative bystander cells. In population analyses, this bystander effect may be detected as an overall increase in STAT3-dependent gene expression (42).
Our work also determines the molecular events underlying IE1-dependent SOX2 downregulation, which appear to be intimately linked to JAK-STAT signaling. JAK-STAT (STAT1 and STAT3) signaling pathways play an important role in neural development by regulating NPC neurogenesis and gliogenesis (35, 36). IE1 markedly affects the activation state and subcellular localization of STAT3, a key regulatory protein known to affect the pluripotency of ESCs and to initiate the commitment to NPC fate via transcriptional activation of SOX2 (37). IE1 inhibits tyrosine (Y705) phosphorylation and promotes nuclear accumulation of STAT3 without altering the protein's overall steady-state levels. These observations closely recapitulate previous results in fibroblasts and are likely the consequence of a direct physical interaction between IE1 and STAT3 (41, 42). Although paradoxical on its face, decreased tyrosine phosphorylation and increased nuclear localization of STAT3 may be coupled. STAT proteins, including STAT3, continually shuttle between the cytoplasm and the nucleus. In fact, STAT3 is imported to the nucleus independently of tyrosine phosphorylation, and this is normally followed by export to the cytoplasm. Tyrosine phosphorylation transiently increases STAT3 nuclear accumulation due to sequestration by DNA binding or heterodimerization with other phosphorylated STAT proteins (43). Likewise, IE1 may bind to STAT3 passing through the nucleus. Nuclear sequestration may reduce the amounts of STAT3 undergoing export to the cytoplasm and, consequently, the pools of cytoplasmic STAT3 available for tyrosine phosphorylation by the corresponding (cytoplasmic) JAK family kinases. The expected consequences of depleting activatable STAT3 by nuclear sequestration are in line with the reduced pSTAT3 levels, restricted responsiveness to IL-6, and diminished expression of SOX2 we observed in HCMV-infected NPCs.
In summary, this study for the first time links the interaction between IE1 and STAT3 to SOX2 depletion and thereby identifies a novel pathway predicted to contribute to developmental neuropathogenesis caused by congenital HCMV infection.
MATERIALS AND METHODS
Ethics statement.
Postmortem fetal brain tissues from different gestational age cases were obtained according to an approval notice from the Institutional Review Board (IRB) (WIVH10201202) and the Guidelines for Biomedical Research Involving Human Subjects at Wuhan Institute of Virology, Chinese Academy of Sciences. The original source of the anonymized tissues was Zhongnan Hospital of Wuhan University (China). The cell isolation procedures and research plans were approved by the IRB (WIVH10201202) according to the Guidelines for Biomedical Research Involving Human Subjects at Wuhan Institute of Virology, Chinese Academy of Sciences. The need for written or oral consent was waived by the IRB (57).
Cells and cell culture.
All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. NPCs were isolated as described previously (38, 58) and cultured in a 1:1 mixture of growth medium and conditioned medium (14, 15, 19). The NPC growth medium was Dulbecco's modified Eagle medium (DMEM)-F12 (Thermo Fisher Scientific) supplemented with 2 mM GlutaMax (Thermo Fisher Scientific), 100 U/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific), 50 μg/ml gentamicin (Sigma), 1.5 μg/ml amphotericin B (Thermo Fisher Scientific), 10% BIT 9500 (Stem Cell Technologies), 20 ng/ml epidermal growth factor (EGF) (Prospec), and 20 ng/ml basic fibroblast growth factor (FGF) (Prospec). Conditioned medium was collected from cultured NPCs and stored at −20°C after cell debris had been removed by centrifugation. NPCs were maintained as monolayers in fibronectin-coated dishes and seeded at a defined density into dishes coated with poly-d-lysine (50 μg/ml; Millipore) prior to infection. In order to induce STAT3 activation, NPCs were treated with carrier-free recombinant human IL-6 (183 ng/ml; BioLegend) for the indicated times before being collected for protein analysis. To inhibit STAT3 activation, NPCs were treated for the indicated times with 10 μM CTS (Sigma), a small molecular inhibitor of STAT3 tyrosine (Y705) phosphorylation.
HEL cells maintained in the laboratory and HEL cells immortalized by transfection with pCI-neo-hTERT (HELf cells, kindly provided by J. Chen, Columbia University), were cultured in minimal essential medium (MEM) (Thermo Fisher Scientific). Human embryonic kidney (HEK) 293T cells (CRL-11268) were grown in DMEM. Both MEM and DMEM were supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific), penicillin and streptomycin, and 2 mM l-glutamine (Thermo Fisher Scientific). For the establishment of an IE1-expressing HELf cell line, a lentivirus stock was prepared from plasmid pCDH-puro-IE1. A volume of 2 ml of this stock was used to transduce 1 × 106 HELf cells. The transduced cells were cultured in normal medium for 2 days to allow transgene expression and then switched to selection medium containing 8 μg/ml puromycin (Sigma) for 2 weeks with medium changes every other day. Expression of IE1 was confirmed by Western blotting. The resulting HELf cell line stably expressing IE1 was designated HELf-IE1 and maintained in medium containing puromycin (4 μg/ml).
Plasmids and nucleofection.
Plasmids pcDNA3-IE1 and pcDNA3-IE2 were constructed by subcloning a BamHI-BamHI fragment containing the full-length coding sequence of the HCMV 72-kDa IE1 or 86-kDa IE2 protein, respectively, from pSG-IE1 or pSG-IE2 (kindly provided by E. A. Fortunato, University of Idaho), respectively, into the backbone of pcDNA3.0. Plasmid pcDNA3-pp65 was generated by cloning a fragment PCR amplified from HCMV (Towne) cDNA between the BamHI and EcoRI sites of pcDNA3.0 (57). Plasmid pSVH (kindly provided by Q. Tang, Howard University) containing the entire HCMV major IE transcription unit (including IE1, IE2, and the major IE promoter-enhancer) was also used.
Plasmids expressing shRNAs directed against HCMV (Towne) IE1 sequences were constructed based on lentiviral vector pLKO.1 puro (Addgene; plasmid 8453). Three shRNAs targeting IE1 exon 4 and a scrambled shRNA not targeting any viral or human gene were designed (http://jura.wi.mit.edu/bioc/siRNAext), synthesized, and inserted between the AgeI and EcoRI sites of pLKO.1 puro to generate pLKO.1-shRNA-IE1-1 (sh-1), pLKO.1-shRNA-IE1-2 (sh-2), pLKO.1-shRNA-IE1-3 (sh-3), and pLKO.1-scramble (scr), respectively. The sequences are listed in Table 1. For the establishment of HELf-IE1 cells (described above), a fragment containing the IE1 coding sequence was recovered from pSG-IE1 and cloned into the BamHI site of pCDH-CMV-MCS-EF1-puro (System Biosciences). The resulting construct was designated pCDH-puro-IE1.
TABLE 1.
Oligonucleotides used in this study
| Purpose | Name | Sequence (5′–3′) |
|---|---|---|
| IE1 knockdown | scr F | CCGGCCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTTG |
| scr R | AATTCAAAAACCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG | |
| sh-1 F | CCGGGCATGTATGAGAACTACATTGCTCGAGCAATGTAGTTCTCATACATGCTTTTTG | |
| sh-1 R | AATTCAAAAAGCATGTATGAGAACTACATTGCTCGAGCAATGTAGTTCTCATACATGC | |
| sh-2 F | CCGGGCTGTGCTGCTATGTCTTAGACTCGAGTCTAAGACATAGCAGCACAGCTTTTTG | |
| sh-2 R | AATTCAAAAAGCTGTGCTGCTATGTCTTAGACTCGAGTCTAAGACATAGCAGCACAGC | |
| sh-3 F | CCGGGCCTGAGGTTATCAGTGTAATCTCGAGATTACACTGATAACCTCAGGCTTTTTG | |
| sh-3 R | AATTCAAAAAGCCTGAGGTTATCAGTGTAATCTCGAGATTACACTGATAACCTCAGGC | |
| STAT3 knockdown | #1102 | CCGGGCCTCAAGATTGACCTAGACTCGAGTCTAGGTCAATCTTGAGGCTTTTT |
| #1103 | AATTAAAAAGCCTCAAGATTGACCTAGACTCGAGTCTAGGTCAATCTTGAGGC | |
| #1134 | CCGGAGTCAGGTTGCTGGTCAAACTCGAGTTTGACCAGCAACCTGACTTTTTT | |
| #1135 | AATTAAAAAAGTCAGGTTGCTGGTCAAACTCGAGTTTGACCAGCAACCTGACT | |
| #1098 | CCGGGTGCGTTGCTAGTACCAACCTCGAGGTTGGTACTAGCAACGCACTTTTT | |
| #1099 | AATTAAAAAGTGCGTTGCTAGTACCAACCTCGAGGTTGGTACTAGCAACGCAC | |
| #1138 | CCGGGTGGGAGCGCGTGATGAACCTCGAGGTTCATCACGCGCTCCCACTTTTT | |
| #1139 | AATTAAAAAGTGGGAGCGCGTGATGAACCTCGAGGTTCATCACGCGCTCCCAC | |
| PCR | pp65 F | CGCGGATCCATGGAGTCGCGCGGTCGCCGT |
| pp65 R | CCGGAATTCTCAACCTCGGTGCTTTTTGGG | |
| qRT-PCR | SOX2 F | GCCGAGTGGAAACTTTTGTCG |
| SOX2 R | GCAGCGTGTACTTATCCTTCTT | |
| GAPDH F | GAGTCAACGGATTTGGTCGT | |
| GAPDH R | GACAAGCTTCCCGTTCTCAG |
Plasmids expressing shRNAs directed against human STAT3 sequences were constructed based on the lentiviral vector Tet-pLKO-puro (Addgene; plasmid 21915). Two shRNA plasmids targeting STAT3, Tet-pLKO-puro-shSTAT3-1 and Tet-pLKO-puro-shSTAT3-2, were generated by inserting annealed oligonucleotides 1102 and 1103 or 1134 and 1135, respectively, between the EcoRI and AgeI sites of Tet-pLKO-puro. Likewise, plasmids Tet-pLKO-puro-shLuci and Tet-pLKO-puro-shDsRed expressing shRNAs directed against Photinus luciferase or Discosoma red fluorescent protein sequences, respectively, not present in human cells were constructed by inserting annealed oligonucleotides 1098 and 1099 or 1038 and 1039, respectively, between the EcoRI and AgeI sites of Tet-pLKO-puro. The oligonucleotide sequences are listed in Table 1.
To transiently overexpress individual viral proteins, NPCs were transfected with plasmids pSVH, pcDNA3-IE1, pcDNA3-IE2, pmaxGFP (provided by Lonza to assess transfection efficiency), pcDNA3-pp65, or empty vector (pcDNA3.0) using Nucleofector technology (Lonza) according to the manufacturer's instructions. In brief, 5 × 106 NPCs were mixed with 100 μl mesenchymal stem cell (MSC) nucleofector solution (82 μl nucleofector solution with 18 μl supplement 1 [Lonza]) and a total of 5 μg pSVH, pcDNA3-IE1, pcDNA3-IE2, pmaxGFP, pcDNA3-pp65, pcDNA3.0, or pcDNA3-IE1, along with pcDNA3.0. The cell-DNA suspension was transferred to certified cuvettes, which were inserted into a Nucleofector II (Lonza), and program A-033 was applied. Immediately following nucleofection, 500 μl NPC growth medium was added to the cuvette, and the sample was gently transferred to poly-d-lysine-coated dishes. After 24 h, the culture medium was replaced, and cells were analyzed at 48 h postnucleofection.
HCMV preparation and infection.
Enhanced green fluorescent protein (EGFP)-expressing bacterial artificial chromosome-derived wild-type (T-BAC; referred to here as TNWT), IE1-deficient (TNdlIE1), and revertant (TNrvIE1) variants of the HCMV Towne strain (ATCC VR977) and the parental virus HCMV Towne strain (ATCC VR977) were used in this study. The construction of TNdlIE1 and TNrvIE1 was described previously (59). The TNWT and TNrvIE1 viruses were propagated in HEL cells and titrated by plaque assay as described previously (60, 61). The TNdlIE1 mutant was propagated and titrated in HELf-IE1 cells. HCMV particles from infected cell supernatants were concentrated by ultracentrifugation, after removing the cell debris by high-speed centrifugation, and resuspended in NPC growth medium to avoid any potential undesired effects induced by components (including FBS) in the medium used for virus preparation (61). UV-inactivated HCMV was prepared by exposure to 6,000 J/cm2 in a CL-1000 UV cross-linker (UVP), and sodium pyruvate was added to a final concentration of 5 mM to prevent damage from free radicals induced by UV radiation (62).
For NPC infections, confluent cell monolayers on fibronectin-coated dishes were dissociated using Accutase (Millipore), 3 × 106 cells were reseeded in poly-d-lysine-coated 100-mm dishes or uncoated dishes with poly-d-lysine-coated coverslips, and the cells were allowed to attach overnight. The cells were exposed to HCMV at the indicated MOI. For the evaluation of IE1-directed shRNAs, NPCs or HEL cells were infected with HCMV at an MOI of 1. To overcome the delayed infection process of TNdlIE1, an MOI of 10 was used. After incubation for 3 h to allow virus adsorption, the inoculum was removed and the cells were refed with fresh medium. Cells were collected and analyzed at the indicated times postinfection. To study infection in the absence of de novo protein synthesis, NPCs were pretreated with 10 μg/ml CHX (Sigma) for 1 h prior to infection, infected with HCMV in the presence of CHX (10 μg/ml), and collected at 16 hpi for RNA and protein analysis.
Lentivirus preparation and transduction.
Stocks of replication-defective lentiviruses were prepared as described previously (57, 63). Briefly, 1.5 × 106 HEK 293T cells were seeded in a 100-mm dish. The next day, calcium phosphate precipitation was applied to cotransfect the cells with the packaging plasmids pML-Δ8.9 (12 μg) and pVSV-G (8 μg) (System Biosciences), along with 15 μg of one of the following expression plasmids: pCDH-puro-IE1, pLKO.1-scramble, pLKO.1-shRNA-IE1-1, pLKO.1-shRNA-IE1-2, pLKO.1-shRNA-IE1-3, Tet-pLKO-puro-shDsRed, Tet-pLKO-puro-shLuci, Tet-pLKO-puro-shSTAT3-1, or Tet-pLKO-puro-shSTAT3-2. Following a medium change, the lentivirus-containing supernatants were collected 72 h after transfection and stored at −80°C.
The lentivirus stocks were used to transduce HEL cells, HELf cells, or NPCs. To this end, 5 × 106 NPCs were seeded in fibronectin-coated dishes and infected with equal volumes (2 ml) of lentivirus stock on the following day. Lentivirus stock was added to the NPCs again on the next day. The inoculum was replaced with fresh culture medium 3 h after each transduction. Likewise, 5 × 105 HEL cells were infected with 2 ml lentivirus stock on the day following seeding, and the inoculum was replaced with fresh culture medium 5 h after infection. The transduced cells were cultured for 3 days to allow transgene expression before they were subjected to HCMV infection and/or RNA or protein analysis. The transduction procedure used to establish HELf-IE1 cells is described above.
Gene silencing with shRNAs.
To determine the IE1-specific silencing efficiency, equal amounts of shRNA-expressing lentiviruses (sh-1, sh-2, sh-3, and scr) were used to transduce HEL cells in parallel. The resulting HEL cells were cultured for 2 days to allow shRNA expression, and this was followed by 2 days of serum starvation with serum-free medium to synchronize the cells. The synchronized cells were reseeded in 60-mm dishes (1 × 106 cells/dish), allowed to attach, and infected with HCMV (MOI = 1). The infected cells were collected at 24 and 48 hpi, and the levels of IE1 were determined by protein analysis. After selection of the most efficient shRNA (sh-2), NPCs were transduced with sh-2 and scr lentiviruses in parallel. The transduced NPCs were cultured for 48 h to allow shRNA expression, reseeded in poly-d-lysine-coated dishes (3 × 106 cells/dish), and infected with HCMV (MOI = 1) on the following day. Cells were collected at the indicated times postinfection and subjected to protein and RNA analysis.
NPCs transduced with the shRNA-expressing lentivirus Tet-pLKO-puro-shDsRed, Tet-pLKO-puro-shLuci, Tet-pLKO-puro-shSTAT3-1, or Tet-pLKO-puro-shSTAT3-2 (described above) were treated with 1 μg/ml doxycycline hyclate (Dox) (Aladdin) for 48 h, with a medium change after 24 h, to induce shRNA expression. Cells were collected and analyzed at the indicated times.
Quantitative reverse transcriptase (qRT) PCR.
Transfected or infected NPCs were collected at the indicated times. Total RNA was isolated using the RNAiso Plus reagent (TaKaRa), followed by RNase-free DNase I (Thermo Fisher Scientific) treatment to remove residual genomic DNA. Equal amounts (500 ng) of DNA-free RNA were reverse transcribed using the PrimeScript RT reagent kit (Perfect Real Time; TaKaRa) according to the manufacturer's instructions. Then, quantitative PCR (qPCR) was performed in a CFX96 Connect real-time PCR detection system (Bio-Rad). Each 20-μl qPCR mixture contained 2 μl reverse transcription product, 10 μl 2× iTaq Universal SYBR green Supermix (Bio-Rad), and 200 nM forward (F) and reverse (R) primers. The primer sequences are shown in Table 1. Amplification was performed by denaturation at 95°C for 5 min, followed by 35 two-step cycles of 95°C for 10 s and 60°C for 30 s. Melting curve analysis was carried out at 95°C for 1 min, 55°C for 1 min, and 55 to 95°C for 10 s. Each reaction was performed in triplicate, and the results for the target gene mRNA were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the 2ΔΔCT method. Three independent experiments were performed, and the results are presented as means and 1 standard deviation (SD). Data were statistically evaluated using the Student t test. A P value of ≤0.05 was considered statistically significant.
Western blotting.
At the indicated times, cells were washed in phosphate-buffered saline (PBS), detached with Accutase, collected, counted, and centrifuged. Cell pellets were snap-frozen in liquid nitrogen and stored at −80°C until completion of the time course. Then, the cell pellets were lysed in radioimmunoprecipitation assay (RIPA) buffer. Equal amounts of cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). After incubation with the indicated primary and corresponding secondary antibodies, signals were detected using a chemiluminescence instrument and analyzed by densitometry (ImageJ; National Institutes of Health). At least three sets of independent experiments were performed, and representative results are shown. HCMV proteins were detected using mouse monoclonal antibodies to IE1 (clone p63-27; IgG2a), IE1/IE2 (CH16), UL44 (IgG1; Virusys), gB (clone 27-156; IgG2b), or pp65 (IgG1; Virusys). Cellular proteins were detected using a goat polyclonal antibody to SOX2 (clone L1D6A2; IgG1), a mouse monoclonal antibody to STAT3 (clone 124H6; IgG2a; Cell Signaling Technology), a rabbit polyclonal antibody to pSTAT3 (Y705) (IgG; Cell Signaling Technology), a mouse monoclonal antibody to β-actin (IgG; Santa Cruz Biotechnology), a rabbit polyclonal antibody to GAPDH (IgG; Proteintech), or a rabbit polyclonal antibody to lamin B1 (IgG; Proteintech). Secondary antibodies used were horseradish peroxidase-conjugated sheep anti-mouse IgG (Amersham Bioscience), donkey anti-rabbit IgG (Amersham Bioscience), and donkey anti-goat IgG (Proteintech).
Immunofluorescence assay.
Viral and cellular proteins in cells grown on coverslips were detected by indirect immunofluorescence analysis as described previously (64). Briefly, NPCs were seeded on poly-d-lysine-coated coverslips in uncoated dishes and mock or HCMV infected (MOI = 3). Coverslips were collected at the indicated times postinfection. UL44, SOX2, and STAT3 were stained with the respective primary antibodies: mouse monoclonal anti-UL44 (IgG1; Virusys), goat polyclonal anti-SOX2 (Santa Cruz Biotechnology), or mouse monoclonal anti-STAT3 (clone 124H6; IgG2a; Cell Signaling Technology). The secondary antibodies included fluorescein-isothiocyanate (FITC)-conjugated donkey anti-goat IgG (Jackson ImmunoResearch), Alexa Fluor 488-conjugated goat anti-mouse IgG1 (Molecular Probes), and Alexa Fluor 488-conjugated goat anti-mouse IgG2a (Molecular Probes). Nuclei were counterstained with Hoechst 33342 dye, and coverslips were mounted with antifade mounting solution containing paraphenylenediamine (65). Images were obtained using a Nikon Eclipse 80i or Nikon Eclipse Ti-S epifluorescence microscope equipped with a Nikon DS-Ri1 camera and processed using NIS Elements F3.0 software.
Cellular fractionation.
Nucleofected or infected NPCs were washed in precooled PBS and collected by scraping. Cytosolic and nuclear fractions were prepared using a nuclear-cytosol extraction kit (Applygen Technologies) following the manufacturer's instructions. Briefly, cell pellets were lysed in precooled cytosol extraction buffer A (CEB-A), vortexed, reacted with cytosol extraction buffer B (CEB-B), vortexed, and centrifuged. The resulting supernatant contained the cytosolic fraction. The precipitate was washed with CEB-A, lysed in nuclear extraction buffer (NEB), vortexed, and centrifuged. The resulting supernatant contained the nuclear fraction.
ACKNOWLEDGMENTS
We appreciate the critical reading by Tom Shenk. We thank the colleagues mentioned in Materials and Methods for providing important reagents.
M.H.L. was supported by the Ministry of Science and Technology of China (National Program on Key Basic Research Project 2015CB755600), the National Natural Science Foundation of China (81620108021, 31170155, and 81427801), the Sino-Africa Joint Research Centre (SAJC201605), and a seed grant from the University of Idaho (YDP-764). M.N. and C.P. were supported by the Wellcome Trust Institutional Strategic Support Fund, M.N. was supported by the Medical Research Council (MR/P022146/1) and Tenovus Scotland (T15/38), and C.P. was supported by the Deutsche Forschungsgemeinschaft (PA 815/2-1).
REFERENCES
- 1.Bale JF., Jr 1984. Human cytomegalovirus infection and disorders of the nervous system. Arch Neurol 41:310–320. doi: 10.1001/archneur.1984.04050150092023. [DOI] [PubMed] [Google Scholar]
- 2.Stagno S, Pass RF, Cloud G, Britt WJ, Henderson RE, Walton PD, Veren DA, Page F, Alford CA. 1986. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA 256:1904–1908. [PubMed] [Google Scholar]
- 3.Bale JF., Jr 2014. Congenital cytomegalovirus infection. Handb Clin Neurol 123:319–326. doi: 10.1016/B978-0-444-53488-0.00015-8. [DOI] [PubMed] [Google Scholar]
- 4.Conboy TJ, Pass RF, Stagno S, Britt WJ, Alford CA, McFarland CE, Boll TJ. 1986. Intellectual development in school-aged children with asymptomatic congenital cytomegalovirus infection. Pediatrics 77:801–806. [PubMed] [Google Scholar]
- 5.Pass RF, Stagno S, Myers GJ, Alford CA. 1980. Outcome of symptomatic congenital cytomegalovirus infection: results of long-term longitudinal follow-up. Pediatrics 66:758–762. [PubMed] [Google Scholar]
- 6.Rosenthal LS, Fowler KB, Boppana SB, Britt WJ, Pass RF, Schmid SD, Stagno S, Cannon MJ. 2009. Cytomegalovirus shedding and delayed sensorineural hearing loss: results from longitudinal follow-up of children with congenital infection. Pediatr Infect Dis J 28:515–520. doi: 10.1097/INF.0b013e318198c724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sweeten TL, Posey DJ, McDougle CJ. 2004. Brief report: autistic disorder in three children with cytomegalovirus infection. J Autism Dev Disord 34:583–586. doi: 10.1007/s10803-004-2552-y. [DOI] [PubMed] [Google Scholar]
- 8.Yamashita Y, Fujimoto C, Nakajima E, Isagai T, Matsuishi T. 2003. Possible association between congenital cytomegalovirus infection and autistic disorder. J Autism Dev Disord 33:455–459. doi: 10.1023/A:1025023131029. [DOI] [PubMed] [Google Scholar]
- 9.Zhang XW, Li F, Yu XW, Shi XW, Shi J, Zhang JP. 2007. Physical and intellectual development in children with asymptomatic congenital cytomegalovirus infection: a longitudinal cohort study in Qinba mountain area, China. J Clin Virol 40:180–185. doi: 10.1016/j.jcv.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. 1992. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J 11:93–99. doi: 10.1097/00006454-199202000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Dahle AJ, Fowler KB, Wright JD, Boppana SB, Britt WJ, Pass RF. 2000. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J Am Acad Audiol 11:283–290. [PubMed] [Google Scholar]
- 12.Tsutsui Y, Kosugi I, Kawasaki H. 2005. Neuropathogenesis in cytomegalovirus infection: indication of the mechanisms using mouse models. Rev Med Virol 15:327–345. doi: 10.1002/rmv.475. [DOI] [PubMed] [Google Scholar]
- 13.Cheeran MC, Hu S, Ni HT, Sheng W, Palmquist JM, Peterson PK, Lokensgard JR. 2005. Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J Neurosci Res 82:839–850. doi: 10.1002/jnr.20682. [DOI] [PubMed] [Google Scholar]
- 14.Luo MH, Hannemann H, Kulkarni AS, Schwartz PH, O'Dowd JM, Fortunato EA. 2010. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol 84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarthy M, Auger D, Whittemore SR. 2000. Human cytomegalovirus causes productive infection and neuronal injury in differentiating fetal human central nervous system neuroepithelial precursor cells. J Hum Virol 3:215–228. [PubMed] [Google Scholar]
- 17.Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Soderberg-Naucler C. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol 80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odeberg J, Wolmer N, Falci S, Westgren M, Sundtrom E, Seiger A, Soderberg-Naucler C. 2007. Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J Neurosci Res 85:583–593. doi: 10.1002/jnr.21144. [DOI] [PubMed] [Google Scholar]
- 19.Pan X, Li XJ, Liu XJ, Yuan H, Li JF, Duan YL, Ye HQ, Fu YR, Qiao GH, Wu CC, Yang B, Tian XH, Hu KH, Miao LF, Chen XL, Zheng J, Rayner S, Schwartz PH, Britt WJ, Xu J, Luo MH. 2013. Later passages of neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J Virol 87:10968–10979. doi: 10.1128/JVI.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wegner M. 2011. SOX after SOX: SOXession regulates neurogenesis. Genes Dev 25:2423–2428. doi: 10.1101/gad.181487.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brazel CY, Limke TL, Osborne JK, Miura T, Cai J, Pevny L, Rao MS. 2005. Sox2 expression defines a heterogeneous population of neurosphere-forming cells in the adult murine brain. Aging Cell 4:197–207. doi: 10.1111/j.1474-9726.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- 22.Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. 2003. Mutations in SOX2 cause anophthalmia. Nat Genet 33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 23.Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JR, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, Fitzpatrick DR. 2005. SOX2 anophthalmia syndrome. Am J Med Genet A 135:1–8. doi: 10.1002/ajmg.a.30642. [DOI] [PubMed] [Google Scholar]
- 24.Schneider A, Bardakjian TM, Zhou J, Hughes N, Keep R, Dorsainville D, Kherani F, Katowitz J, Schimmenti LA, Hummel M, Fitzpatrick DR, Young TL. 2008. Familial recurrence of SOX2 anophthalmia syndrome: phenotypically normal mother with two affected daughters. Am J Med Genet A 146A:2794–2798. doi: 10.1002/ajmg.a.32384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ring KL, Tong LM, Balestra ME, Javier R, Andrews-Zwilling Y, Li G, Walker D, Zhang WR, Kreitzer AC, Huang Y. 2012. Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 11:100–109. doi: 10.1016/j.stem.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, Lipton SA, Zhang K, Ding S. 2011. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci U S A 108:7838–7843. doi: 10.1073/pnas.1103113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lujan E, Chanda S, Ahlenius H, Sudhof TC, Wernig M. 2012. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc Natl Acad Sci U S A 109:2527–2532. doi: 10.1073/pnas.1121003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maucksch C, Firmin E, Butler-Munro C, Montgomery J, Dottori M, Connor B. 2012. Non-viral generation of neural precursor-like cells from adult human fibroblasts. J Stem Cells Regen Med 8:162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizzino A. 2013. Concise review: the Sox2-Oct4 connection: critical players in a much larger interdependent network integrated at multiple levels. Stem Cells 31:1033–1039. doi: 10.1002/stem.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopp JL, Ormsbee BD, Desler M, Rizzino A. 2008. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 26:903–911. doi: 10.1634/stemcells.2007-0951. [DOI] [PubMed] [Google Scholar]
- 33.Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, Ko MS, Niwa H. 2007. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 9:625–635. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- 34.Imada K, Leonard WJ. 2000. The Jak-STAT pathway. Mol Immunol 37:1–11. doi: 10.1016/S0161-5890(00)00018-3. [DOI] [PubMed] [Google Scholar]
- 35.Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. 1997. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME. 2001. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 104:365–376. doi: 10.1016/S0092-8674(01)00224-0. [DOI] [PubMed] [Google Scholar]
- 37.Foshay KM, Gallicano GI. 2008. Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev 17:269–278. doi: 10.1089/scd.2007.0098. [DOI] [PubMed] [Google Scholar]
- 38.Pan W, Jin Y, Chen J, Rottier RJ, Steel KP, Kiernan AE. 2013. Ectopic expression of activated notch or SOX2 reveals similar and unique roles in the development of the sensory cell progenitors in the mammalian inner ear. J Neurosci 33:16146–16157. doi: 10.1523/JNEUROSCI.3150-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heider JA, Bresnahan WA, Shenk TE. 2002. Construction of a rationally designed human cytomegalovirus variant encoding a temperature-sensitive immediate-early 2 protein. Proc Natl Acad Sci U S A 99:3141–3146. doi: 10.1073/pnas.052710599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marchini A, Liu H, Zhu H. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 75:1870–1878. doi: 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reitsma JM, Sato H, Nevels M, Terhune SS, Paulus C. 2013. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J Virol 87:10763–10776. doi: 10.1128/JVI.01197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harwardt T, Lukas S, Zenger M, Reitberger T, Danzer D, Ubner T, Munday DC, Nevels M, Paulus C. 2016. Human cytomegalovirus immediate-early 1 protein rewires upstream STAT3 to downstream STAT1 signaling switching an IL6-type to an IFNgamma-like response. PLoS Pathog 12:e1005748. doi: 10.1371/journal.ppat.1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reich NC. 2013. STATs get their move on. JAKSTAT 2:e27080. doi: 10.4161/jkst.27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, ten Hoeve J, Shuai K, Sun YE. 2005. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 132:3345–3356. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- 45.Miller FD, Gauthier AS. 2007. Timing is everything: making neurons versus glia in the developing cortex. Neuron 54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 46.Shin DS, Kim HN, Shin KD, Yoon YJ, Kim SJ, Han DC, Kwon BM. 2009. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res 69:193–202. doi: 10.1158/0008-5472.CAN-08-2575. [DOI] [PubMed] [Google Scholar]
- 47.Onishi K, Zandstra PW. 2015. LIF signaling in stem cells and development. Development 142:2230–2236. doi: 10.1242/dev.117598. [DOI] [PubMed] [Google Scholar]
- 48.Biran J, Tahor M, Wircer E, Levkowitz G. 2015. Role of developmental factors in hypothalamic function. Front Neuroanat 9:47. doi: 10.3389/fnana.2015.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guerrini R, Parrini E. 2010. Neuronal migration disorders. Neurobiol Dis 38:154–166. doi: 10.1016/j.nbd.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Hofman MA. 2014. Evolution of the human brain: when bigger is better. Front Neuroanat 8:15. doi: 10.3389/fnana.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y, Li J, Du JX, Wong J. 2014. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol Cell 55:537–551. doi: 10.1016/j.molcel.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 52.Noris E, Zannetti C, Demurtas A, Sinclair J, De Andrea M, Gariglio M, Landolfo S. 2002. Cell cycle arrest by human cytomegalovirus 86-kDa IE2 protein resembles premature senescence. J Virol 76:12135–12148. doi: 10.1128/JVI.76.23.12135-12148.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spector DH. 1996. Activation and regulation of human cytomegalovirus early genes. Intervirology 39:361–377. doi: 10.1159/000150507. [DOI] [PubMed] [Google Scholar]
- 54.Stinski MF, Meier JL. 2007. Immediate-early viral gene regulation and function, Chapter 17. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, UK. [PubMed] [Google Scholar]
- 55.Soroceanu L, Matlaf L, Khan S, Akhavan A, Singer E, Bezrookove V, Decker S, Ghanny S, Hadaczek P, Bengtsson H, Ohlfest J, Luciani-Torres MG, Harkins L, Perry A, Guo H, Soteropoulos P, Cobbs CS. 2015. Cytomegalovirus immediate-early proteins promote stemness properties in glioblastoma. Cancer Res 75:3065–3076. doi: 10.1158/1538-7445.AM2015-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fornara O, Bartek J Jr, Rahbar A, Odeberg J, Khan Z, Peredo I, Hamerlik P, Bartek J, Stragliotto G, Landazuri N, Soderberg-Naucler C. 2016. Cytomegalovirus infection induces a stem cell phenotype in human primary glioblastoma cells: prognostic significance and biological impact. Cell Death Differ 23:261–269. doi: 10.1038/cdd.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu YR, Liu XJ, Li XJ, Shen ZZ, Yang B, Wu CC, Li JF, Miao LF, Ye HQ, Qiao GH, Rayner S, Chavanas S, Davrinche C, Britt WJ, Tang Q, McVoy M, Mocarski E, Luo MH. 2015. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J Virol 89:1070–1082. doi: 10.1128/JVI.01740-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwartz PH, Bryant PJ, Fuja TJ, Su H, O'Dowd DK, Klassen H. 2003. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J Neurosci Res 74:838–851. doi: 10.1002/jnr.10854. [DOI] [PubMed] [Google Scholar]
- 59.Knoblach T, Grandel B, Seiler J, Nevels M, Paulus C. 2011. Human cytomegalovirus IE1 protein elicits a type II interferon-like host cell response that depends on activated STAT1 but not interferon-gamma. PLoS Pathog 7:e1002016. doi: 10.1371/journal.ppat.1002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen ZZ, Pan X, Miao LF, Ye HQ, Chavanas S, Davrinche C, McVoy M, Luo MH. 2014. Comprehensive analysis of human cytomegalovirus microRNA expression during lytic and quiescent infection. PLoS One 9:e88531. doi: 10.1371/journal.pone.0088531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu XJ, Yang B, Huang SN, Wu CC, Li XJ, Cheng S, Jiang X, Hu F, Ming YZ, Nevels M, Britt WJ, Rayner S, Tang Q, Zeng WB, Zhao F, Luo MH. 2017. Human cytomegalovirus IE1 downregulates Hes1 in neural progenitor cells as a potential E3 ubiquitin ligase. PLoS Pathog 13:e1006542. doi: 10.1371/journal.ppat.1006542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fortunato EA, Dell'Aquila ML, Spector DH. 2000. Specific chromosome 1 breaks induced by human cytomegalovirus. Proc Natl Acad Sci U S A 97:853–858. doi: 10.1073/pnas.97.2.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tiscornia G, Singer O, Verma IM. 2006. Production and purification of lentiviral vectors. Nat Protoc 1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 64.Duan Y, Miao L, Ye H, Yang C, Fu B, Schwartz PH, Rayner S, Fortunato EA, Luo MH. 2012. A faster immunofluorescence assay for tracking infection progress of human cytomegalovirus. Acta Biochim Biophys Sin 44:597–605. doi: 10.1093/abbs/gms041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo MH, Rosenke K, Czornak K, Fortunato EA. 2007. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol 81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]