

Knowledge about host factors supporting or restricting virus replication is required for a deeper understanding of virus-cell interactions and may eventually provide the basis for therapeutic intervention. This work was undertaken predominantly to explain the mechanism of A3G-mediated inhibition of MV, a negative-strand RNA virus that is not affected by the deaminase activity of A3G acting on single-stranded DNA. We found that A3G regulates the expression of several cellular proteins, which influences the capacity of the host cell to replicate MV. One of these, REDD1, which modulates the cellular metabolism in a central position by regulating the kinase complex mTORC1, was identified as the major cellular factor impairing MV replication. These findings show interesting aspects of the function of A3G and the dependence of the MV replication on the metabolic state of the cell. Interestingly, pharmacological inhibition of mTORC1 can be utilized to inhibit MV replication in Vero cells and primary human peripheral blood lymphocytes.
KEYWORDS: APOBEC3G, REDD1/DDIT4, mTORC1, measles virus
ABSTRACT
We found earlier that ectopic expression of the cytidine deaminase APOBEC3G (A3G) in Vero cells inhibits measles virus (MV), respiratory syncytial virus, and mumps virus, while the mechanism of inhibition remained unclear. A microarray analysis revealed that in A3G-transduced Vero cells, several cellular transcripts were differentially expressed, suggesting that A3G regulates the expression of host factors. One of the most upregulated host cell factors, REDD1 (regulated in development and DNA damage response-1, also called DDIT4), reduced MV replication ∼10-fold upon overexpression in Vero cells. REDD1 is an endogenous inhibitor of mTORC1 (mammalian target of rapamycin complex-1), the central regulator of cellular metabolism. Interestingly, rapamycin reduced the MV replication similarly to REDD1 overexpression, while the combination of both did not lead to further inhibition, suggesting that the same pathway is affected. REDD1 silencing in A3G-expressing Vero cells abolished the inhibitory effect of A3G. In addition, silencing of A3G led to reduced REDD1 expression, confirming that its expression is regulated by A3G. In primary human peripheral blood lymphocytes (PBL), expression of A3G and REDD1 was found to be stimulated by phytohemagglutinin (PHA) and interleukin-2. Small interfering RNA (siRNA)-mediated depletion of A3G in PHA-stimulated PBL reduced REDD1 expression and increased viral titers, which corroborates our findings in Vero cells. Silencing of REDD1 also increased viral titers, confirming the antiviral role of REDD1. Finally, pharmacological inhibition of mTORC1 by rapamycin in PHA-stimulated PBL reduced viral replication to the level found in unstimulated lymphocytes, indicating that mTORC1 activity supports MV replication as a proviral host factor.
IMPORTANCE Knowledge about host factors supporting or restricting virus replication is required for a deeper understanding of virus-cell interactions and may eventually provide the basis for therapeutic intervention. This work was undertaken predominantly to explain the mechanism of A3G-mediated inhibition of MV, a negative-strand RNA virus that is not affected by the deaminase activity of A3G acting on single-stranded DNA. We found that A3G regulates the expression of several cellular proteins, which influences the capacity of the host cell to replicate MV. One of these, REDD1, which modulates the cellular metabolism in a central position by regulating the kinase complex mTORC1, was identified as the major cellular factor impairing MV replication. These findings show interesting aspects of the function of A3G and the dependence of the MV replication on the metabolic state of the cell. Interestingly, pharmacological inhibition of mTORC1 can be utilized to inhibit MV replication in Vero cells and primary human peripheral blood lymphocytes.
INTRODUCTION
As a cytokine (predominantly type I interferon and interleukin-2 [IL-2])-induced gene product, APOBEC3G (apolipoprotein B mRNA-editing enzyme-catalytic polypeptide 3G) (A3G) is part of the antiviral innate immune and stress response (1–6). The antiviral effects of A3G, based on its deaminase activity acting on single-stranded DNA (ssDNA) as found during the replication of retroviruses, and its RNA-binding capacity have been extensively investigated (7–16). In addition, A3G interacts with a number of proteins that regulate cellular RNA metabolism and can assemble into large RNP complexes present in cytoplasmic microdomains that are associated with RNA regulation, such as P bodies, stress granules, Staufen-containing RNA granules, or Ro-RNPs (17–20). P bodies containing mRNA-protein complexes are constitutively present in eukaryotic cells, but their size and number increase during translational arrest (21). Included proteins are involved in decapping, exonuclease activities, deadenylation, microRNA-mediated silencing, and mRNA degradation and thus regulate gene expression (21–24), and a number of viruses have developed mechanisms to prevent these activities (25–27). Interestingly, the induction of stress granules and P bodies is associated with inhibition of mTORC1 signaling, reduction of protein synthesis, and induction of autophagy (28–31). One of the endogenous inhibitors of mTORC activity is REDD1/DDIT4 (regulated in development and DNA damage response-1, also called DDIT4 [DNA damage-inducible transcript 4]). REDD1 disrupts the interaction of the sensor tuberous sclerosis complex (TSC1/2) with 14-3-3 proteins, and activates the small GTPase Rheb, which in its GTP-bound form interacts with and activates mTORC1 (32–35).
We found earlier that MV replication is reduced by more than 90% in Vero cells ectopically expressing A3G (36). A3G reduced viral transcription and protein expression, but the typical A3G-specific hypermutations by RNA editing have not been detected (36). These findings suggested that A3G could alter the expression of host factors influencing the viral replication. We therefore compared the gene expression profiles of A3G-expressing and -nonexpressing Vero cells and found that the expression of several cellular genes was altered by A3G, of which REDD1 exerted a significant antiviral activity. In primary human peripheral blood lymphocytes (PBL), the expression of A3G and REDD1 is simultaneously stimulated by phytohemagglutinin (PHA) and IL-2, and A3G silencing led to a reduction of REDD1 expression and an increase of viral titers. Furthermore, REDD1 silencing also led to increased viral titers, confirming its antiviral activity also in primary lymphocytes.
RESULTS
Comparison of gene expression in A3G-expressing and -nonexpressing Vero cells by microarray analysis.
In order to define the basis of the antiviral activity of A3G in Vero cells, we used a Gene Chip rhesus macaque genome array (Affymetrix) with >47,000 transcripts to compare the gene expression in cells transduced with an A3G-expressing vector (Vero-A3G) with that in cells transduced with a control vector (Vero-023) (17). Total RNA from these cells in two independent experiments was extracted, quality controlled, and processed for hybridization to microarray gene chips. A total of 844 transcripts were significantly upregulated, and 598 were downregulated (adjusted P values of <0.05) (see Table S1 in the supplemental material). Of these transcripts, 19 were upregulated and 23 were downregulated by a factor of >2 (Fig. 1A to C), and 11 of these transcripts were upregulated annotated gene products and 9 were downregulated annotated gene products (Fig. 1D).
FIG 1.

Microarray analysis reveals A3G-upregulated and -downregulated genes. (A) Box plots of log2 values for quantile-quantile normalized probe signal intensities. Total RNA was isolated independently two times each from control Vero-023 cells and A3G-expressing Vero (Vero-A3G) cells and labeled. Bars 1 and 3, RNAs from Vero-023 control cells; bars 2 and 4, RNAs from Vero-A3G cells. The probes were hybridized to Gene Chip rhesus macaque genome arrays (Affymetrix) according to the manufacturer's instructions. (B) MA plot (intensity log2 fold change [M] plotted against the average log2 intensity [A]) for the comparison between treated and untreated cells. Mean intensities of both groups (x axis) were plotted against log2 fold change (y axis). Blue circles around dots show the first 15 genes with the lowest P values. (C) Numbers of all probe sets, of significantly (adjusted P values of <0.05) up- and downregulated probe sets, and of more than 2-fold-up- and downregulated probe sets. (D) List of the best up- and downregulated genes, with the alteration (ratio) of transcript expression and significance (adjusted P values) as determined by the four microarrays. (E) Total RNA was isolated from control (Vero-023) and A3G-expressing (Vero-A3G) cells. Four amounts of RNA (4, 2, 1, and 0.5 μg) were reverse transcribed into cDNA. Four microliters of each of cDNA product was amplified by PCR and analyzed on 1% agarose gels. (F) Protein lysates of Vero-023 and Vero-A3G cells were separated by 10% SDS-PAGE, blotted to nitrocellulose, incubated with antibodies to REDD1, KDELR2, MOSC2, ACY1, and PRDX2 (and GAPDH as a control), and visualized using the ECL system. Quantifications of the Western blots normalized to the value for Vero-023 cells are shown below each blot.
For further investigations, we selected genes/gene products that may be considered candidates for intracellular host factors affecting viral replication. We excluded IL-1α and -β as cytokines with well-known effects, ADRB2, which is linked to various physiological responses such as smooth muscle relaxation and the proliferation of cells, and SPINK5 and -6, which are multidomain serine protease inhibitors in stratified epithelial tissue, and we selected the A3G-downregulated gene products PRDX2, MOSC2, ACY1, and TXNIP and the upregulated gene product REDD1 for further study. The expression of corresponding mRNAs and proteins in Vero-023 and Vero-A3G cells was verified by reverse transcription-PCR (RT-PCR) and Western blotting (Fig. 1E and F, respectively). Semiquantitative RT-PCR performed with total RNA templates (from dilutions of RNA; 4, 2, 1, and 0.5 μg) supported the results obtained by microarray analysis (Fig. 1E) (PRDX1 was used as an unaffected control). Western blotting was performed to quantify the protein expression of REDD1, KDELR2, MOSC2, ACY1, and PRDX2 (Fig. 1F), while TXNIP was not detected with the available antibodies. Upregulation of REDD1 and KDELR2 expression and downregulation of MOSC2 and ACY1 expression in A3G-expressing Vero cells were confirmed on the protein level.
Analysis of potential antiviral effects of selected downregulated genes by shRNA-mediated knockdown.
In order to assess the functions of these genes individually, we silenced them by transduction of Vero-023 cells with specific short hairpin RNA (shRNA)-expressing vectors. We selected three shRNA sequences for each gene and cloned the corresponding palindromic DNA sequences in DsRed-expressing lentiviral vectors to obtain shRNA-expressing cells. Transduced Vero-023 cells were enriched by fluorescence-activated cell sorting (FACS) for DsRed expression in order to achieve cell lines highly expressing shRNA. Representative results are shown in Fig. 2A. The functionality of the expressed shRNAs was then analyzed by semiquantitative RT-PCR. The most active shRNAs (Fig. 2B) were chosen for further experiments. In order to determine the effects of the silencing of single genes on measles virus (MV) replication, transduced cells were infected with recombinant MV strain Edmonston expressing enhanced green fluorescent protein (MV-eGFP) for 2 days at a multiplicity of infection (MOI) of 0.1. The virus yields in Vero-023, Vero-A3G, and Vero-023 cells transduced with ACY1sh1, MOSC2sh3, PRDX2sh1, and TXNIPsh4 shRNAs were determined by plaque assay (Fig. 2C). Unexpectedly, silencing of ACY1, PRDX2, and TXNIP did not impair virus replication but rather showed a tendency to increase viral titers (Fig. 2C, bars 4, 6, and 7). Knockdown of MOSC2 led to a moderately reduced viral titer in comparison to the strong effect of A3G (Fig. 2C, compare bars 2 and 5).
FIG 2.

Functional characterization of A3G-downregulated genes. (A) Vero-023 cells were transduced with lentiviral vectors expressing shRNAs against ACY1, MOSC2, PRDX2, and TXNIP, and the transduction efficiency was confirmed by detection of vector-mediated DsRed expression by flow cytometry. (B) The shRNA-mediated knockdown was confirmed at the RNA level by RT-PCR. Total RNAs from transduced cells and Vero-023 control cells were prepared, and four amounts of RNA (4, 2, 1, and 0.5 μg) were reverse transcribed into cDNA. Four microliters of each of cDNA product was amplified by PCR. (C) Vero-023, Vero-A3G, and shRNA-transduced Vero-023 cells (as indicated) were infected with MV-eGFP at an MOI of 0.1, and viral titers produced by infected cultures were determined after 48 h. Data from three independent experiments were normalized to values for Vero-023 cells and are presented as percentages of control values (n = 3). Virus titers were significantly reduced in A3G-expressing cells (bar 2) and in MOSC2 shRNA-expressing cells (bar 5) compared to those in Vero-023 cells (*, P < 0.05; **, P < 0.01 [Student's t test]).
Antiviral activity of the upregulated protein REDD1 in Vero cells.
Among the upregulated gene products, we more closely investigated the most upregulated (3.5-fold [Fig. 1D]) intracellular protein, REDD1. Interestingly, it has been described earlier that REDD1 has inhibitory effects on the infection of cells with influenza virus and vesicular stomatitis virus (VSV) (37). In order to quantify the effects of this factor on MV replication, we prepared Vero cells transduced with a REDD1-expressing lentiviral vector, Vero-A3G cells transduced with a REDD1-specific shRNA-expressing vector, and control cells expressing nontargeted scrambled shRNA. Transduced Vero-A3G cells were enriched by FACS for DsRed expression (as in Fig. 2A; not shown). The expression of REDD1 in these cells was controlled by Western blotting (Fig. 3A, lane 3 and 4) and compared to the REDD1 expression in Vero-023 and Vero-A3G cells (Fig. 3A, lanes 1 and 2). When these cells were infected with the eGFP-expressing MV, the infection-induced syncytium formation was reduced by the ectopic expression of REDD1 to an extent similar to that observed in the presence of A3G (Fig. 3B). Furthermore, when Vero-A3G cells were transduced with the REDD1 shRNA-expressing vector, the inhibitory effect of A3G on the syncytium formation was abolished. Scrambled shRNA had no effect on the virus-induced syncytium formation. Transfection of Vero-A3G cells with small interfering RNA (siRNA) targeting A3G had an effect similar to that of transduction with the REDD1 shRNA-expressing vector (Fig. 3B).
FIG 3.
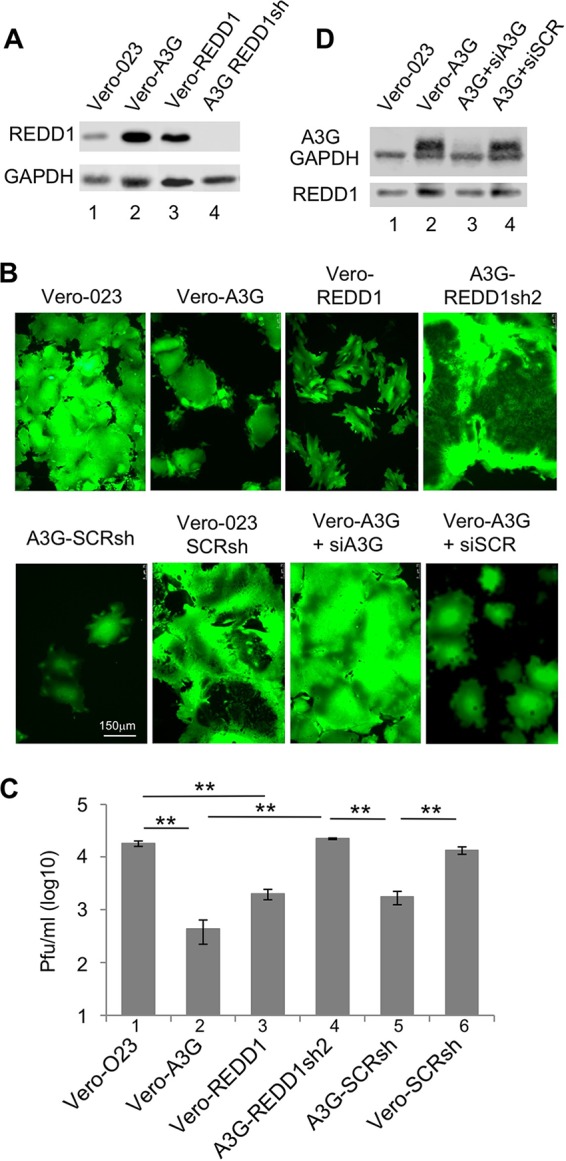
REDD1 expression and effects on MV replication. (A) The protein expression of REDD1 was assessed by Western blotting. Lysates of Vero-023 cells (lane 1), Vero-A3G cells (lane 2), REDD1 (F6gW-REDD1-Myc-DDK-tag)-transduced Vero (Vero-REDD1) cells (lane 3), and REDD1sh2-transduced Vero-A3G (REDD1sh2) cells (lane 4) were prepared, separated using 10% SDS-PAGE, and blotted on nitrocellulose, and the proteins were visualized with specific antibodies and the ECL system. (B) Vero-023 cells, Vero-A3G cells, Vero-REDD1 cells, Vero-A3G cells transduced with REDD1-specific shRNA-expressing vector (A3G-REDD1sh2), Vero-A3G cells transduced with nontargeted scrambled shRNA-expressing vector (A3G-SCRsh), Vero-023 cells transduced with nontargeted scrambled shRNA expressing vector (Vero-023-SCRsh), Vero-A3G cells transfected with A3G-specific siRNA, and Vero-A3G cells transfected with scrambled unspecific siRNA were infected with MV-eGFP at an MOI of 0.1 for 48 h. Representative photomicrographs of the eGFP fluorescence were taken to visualize the syncytium formation (magnification ×100; size bar, 150 μm). (C) Viral titers produced by these infected cells as indicated were determined 48 h after infection with MV. Mean viral titers from three independent experiments are shown (n = 3). Significances were calculated using Student's t test (**, P < 0.01). (D) Protein expression of A3G, GAPDH, and REDD1 in Vero-023 cells (lane 1), Vero-A3G cells (lane 2), Vero-A3G cells transfected with A3G-specific siRNA (lane 3), and Vero-A3G cells transfected with scrambled unspecific siRNA (lane 4) was determined by Western blotting.
In order to analyze the effect of REDD1 on MV replication, we quantified newly synthesized MV in these REDD1-expressing and -nonexpressing Vero cells. The production of infectious MV in REDD1-expressing Vero cells compared to that in Vero-023 control cells (Fig. 3C, bar 1) was efficiently reduced, by 91.2% (Fig. 3C, bar 3), similarly to what was found in the presence of A3G, which reduced MV production by 97.7% (Fig. 3C, bar 2). In REDD1 shRNA-expressing cells, MV replicated as well as in Vero-023 control cells (Fig. 3C, bar 4). Control transductions with nontargeted scrambled shRNAs did not affect virus production in comparison to that in the corresponding parental cell lines (Fig. 3C, bars 5 and 6). Thus, REDD1 inhibited the MV replication almost as efficiently as A3G.
To demonstrate that A3G expression regulates REDD1 expression as suggested by the microarray findings (Fig. 1), we transfected Vero-A3G cells with A3G-specific siRNA and analyzed the A3G and REDD1 expression by Western blotting (Fig. 3D). A3G-specific siRNA not only reduced the protein expression of A3G but also was associated with a reduction of REDD1 expression (Fig. 3D, lane 3), confirming that REDD1 expression is regulated by A3G in Vero cells.
REDD1 exerts its antiviral effect via inhibition of mTORC1.
Next, we analyzed whether the observed REDD1 effect may be mediated by inhibition of mTORC1 activity. For this purpose, MV-infected cells were treated with the mTORC1 inhibitor rapamycin throughout the incubation period. Rapamycin strongly inhibited MV replication in a dose-dependent manner in Vero-023 cells (Fig. 4A, bars 1 to 4). In contrast, when REDD1-expressing Vero cells were treated with rapamycin, no additive effect on the virus titer was detected (Fig. 4A, bars 5 to 8), suggesting that REDD1 and rapamycin act on the same pathway to exert their antiviral activity.
FIG 4.
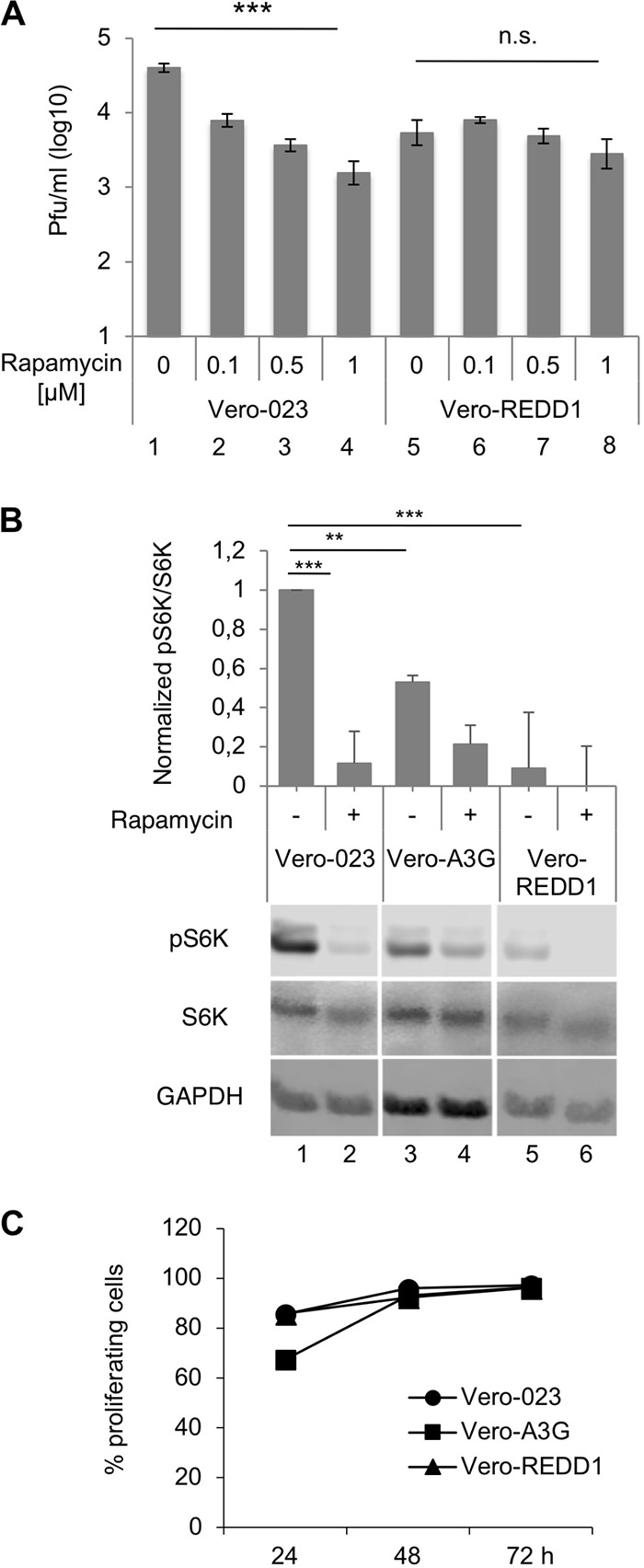
Inhibition of MV replication by rapamycin and A3G and REDD1 effects on mTORC1 activity and proliferation of cells. (A) Vero-023 and REDD1-transduced Vero-023 (Vero-REDD1) cells were infected with MV-eGFP in the absence and presence of increasing concentrations of rapamycin as indicated. Viral titers were determined 48 h after infection of cells with MV at an MOI of 0.1. Because rapamycin was dissolved in dimethyl sulfoxide (DMSO), controls received the same concentration of DMSO (0.5%) as the rapamycin-treated cultures. The mean virus production from three independent experiments is shown (n = 3). Significances (comparison over four columns each) were calculated for each cell line using the one-way ANOVA test (***, P < 0.001; n.s., not significant). (B) Phosphorylation of S6K (Thr 389) and total S6K expression were determined by Western blotting using lysates of Vero-023 (bars 1 and 2), Vero-A3G (bars 3 and 4), and Vero-REDD1 (bars 5 and 6) cells in the absence and presence of 1 μM rapamycin. Controls received 0.5% DMSO. Western blots from three experiments were quantified and evaluated for statistical significance (Student's t test). (C) The proliferation of Vero-023, Vero-A3G, and Vero-REDD1 cells was determined by flow cytometry at 48 and 72 h. Cells were stained with the cell proliferation dye eFluor 670, and the percentage of cells with reduced signal intensity due to cell division was measured.
To further demonstrate A3G and REDD1 effects on the mTORC1 activity, we quantified the phosphorylation of S6K, a downstream target of mTORC1. In comparison to that in Vero-023 cells (Fig. 4B, lane 1), the levels of phospho-S6K in Vero-A3G (lane 3) and Vero-REDD1 (lane 5) cells were reduced by approximately 50% and 80%, respectively. Addition of rapamycin further reduced the level of phosphorylated S6K in Vero-A3G cells but not significantly in Vero-REDD1 cells. Our studies revealed that ectopic A3G as well as REDD1 expression led to a strong reduction of mTORC1 signaling. Since mTORC1 signaling may influence the proliferation of the cells, we determined proliferation rates of A3G- and REDD1-expressing cells in comparison to Vero-023 cells. Cells were stained with the cell proliferation dye eFluor 670, and the percentage of cells with reduced signal intensity due to cell division was measured by flow cytometry after 48 and 72 h (Fig. 4C). The proliferation of the Vero-A3G and Vero-REDD1 cells was moderately reduced at 48 h but was similar to that of Vero-023 cells at 72 h in culture, indicating that there is no major effect on the proliferative capacity of the Vero cells.
Antiviral activity of A3G and effect on REDD1 expression in primary human PBL.
A3G and REDD1 proteins are expressed at a low level in unstimulated primary human PBL, and the level is increased 2- to 3-fold by PHA and IL-2 treatment (Fig. 5A). A comparison of the A3G expression in Vero-A3G cells with that in IL-2-stimulated PBL revealed that A3G levels in PBL reach approximately 40% of the levels in Vero-A3G cells (Fig. 5B).
FIG 5.

Expression of A3G and REDD1 in primary human PBL and comparison to that in Vero cells. (A) The expression of A3G and REDD1 in unstimulated PBL and in PBL stimulated with PHA or IL-2 48 h was determined by Western blotting. Western blots from three independent experiments (with PBL from three independent donors) were quantified, and mean values relative to GAPDH normalized to those for unstimulated PBL are presented. (B) Comparison of A3G expression in primary human PBL (lane 1, unstimulated; lane 2, IL-2 stimulated), Vero-023 cells (lane 3), and Vero-A3G cells (lane 4).
To further investigate the effect of A3G on MV replication in primary human PBL, PBL were transfected (electroporated) with A3G-specific and nontargeted (NT) siRNAs, and newly synthesized MV was titrated in unstimulated, PHA-stimulated, and IL-2-stimulated PBL (Fig. 6). MV replication in unstimulated PBL is generally low and varies considerably among blood donors. PHA and IL-2 stimulation increased the obtained viral titers by approximately 1 log in comparison to that in unstimulated cells. Furthermore, MV replication in PHA-stimulated PBL was found to be significantly better after A3G siRNA transfection than after nontargeted control siRNA transfection, indicating that A3G reduces MV titers (Fig. 6A). A tendency toward similar results, which were not significant due to large variations between individual blood donors, was found in IL-2-stimulated transfected PBL.
FIG 6.
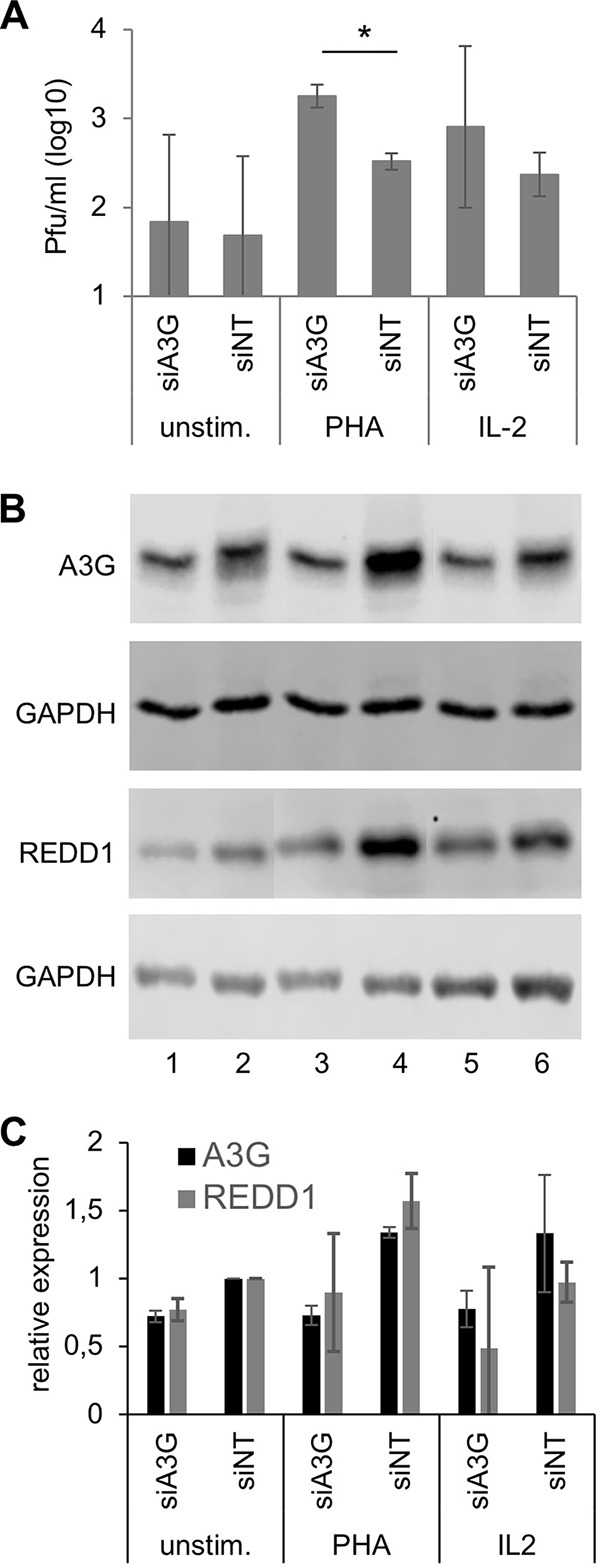
Antiviral activity of A3G and effect on REDD1 expression in primary human PBL. Primary human PBL were cultivated in medium and transfected (nucleofected) with A3G-specific and nontargeted (NT) siRNAs. Cells were kept in medium (unstim.) or stimulated with 2.5 μg/ml PHA or 25 ng/ml IL-2. (A) At 24 h after stimulation, cells were infected with rMVIC323eGFP at an MOI of 0.1 for 48 h, and the newly synthesized virus was titrated using Vero-hSLAM cells. Mean values from three experiments with PBL from three independent donors are shown (significances were determined using Student's t test; *, P < 0.05). (B) Lysates were prepared from parallel uninfected cultures, and proteins were fractionated by SDS-PAGE and blotted on nitrocellulose membranes. A3G and REDD1 expression was visualized with specific antibodies and secondary antibodies on Western blots and the amount of protein on the blots controlled by staining with an antibody to GAPDH. A representative example is shown. (C) Quantification of A3G and REDD1 protein expression on Western blots from PBL from three independent blood donors. The relative protein levels of A3G and REDD1 were normalized and the expression in siNT-transfected PBL set to 1.
A3G and REDD1 protein expression in the transfected PBL was controlled by Western blotting (Fig. 6B). A quantification of three blots obtained from three experiments with cells from independent blood donors is shown in Fig. 6C. In addition to the expected reduction of A3G expression by A3G-specific siRNA, these data revealed that silencing of A3G also affected the expression of REDD1. The data suggest that also in primary PBL, A3G upregulates the REDD1 expression as initially observed in Vero cells (Fig. 1).
Antiviral activity of REDD1 and rapamycin in primary PBL.
In order to investigate the potential antiviral role of REDD1 in primary PBL, we transfected (electroporated) unstimulated and PHA-stimulated lymphocytes with REDD1-specific siRNA and with nontargeted control siRNA and infected them with MV for 2 days. The titers of newly synthesized virus indicated that downregulation of REDD1 led to improved viral replication (Fig. 7A). The expression of REDD1 was controlled by Western blotting (Fig. 7A) and the stimulation of lymphocytes by flow cytometry (Fig. 7B).
FIG 7.

REDD1 and rapamycin reduce MV titers in primary human PBL. (A) PBL were transfected (electroporated) with REDD1-specific (siREDD1) and unspecific nontargeted (NT) siRNAs. At 24 h after stimulation with 2.5 μg/ml of PHA, cells were infected with rMVIC323eGFP at an MOI of 0.1 for 48 h and the newly synthesized virus titrated using Vero-hSLAM cells. Mean values from three experiments with cells from three independent blood donors are shown. Significances were determined using Student's t test (***, P = 0.005; *, P = 0.019). The expression of REDD1 was controlled by Western blotting and compared to the expression of GAPDH. (B) The stimulation of the PBL was controlled in all experiments by flow cytometry. A representative evaluation is shown. (C) Representative photomicrographs of rMVIC323eGFP-infected PHA-stimulated primary human PBL at 48 h in the absence (CTRL) and presence of 0.5 and 1 μM rapamycin (Rapa) (size bar, 150 μm). (D) MV titers in unstimulated and PHA-stimulated PBL in the absence and presence of rapamycin were determined using Vero-hSLAM cells. Mean values from three experiments with PBL from independent donors are shown. Because rapamycin was solved in DMSO, the controls for panels C and D received the same concentration of DMSO (0.5%) as the rapamycin-treated cultures (*, P < 0.05 [Student's t test]; **, P < 0.01 [Student's t test] or P = 0.0012 [one-way ANOVA]).
In a final experiment, we wanted to investigate whether the replication of MV also can be reduced by pharmacological inhibitors of the mTORC1 activity, such as rapamycin, in primary human PBL. The mTORC1 inhibitor efficiently reduced giant cell formation (virus-induced cell fusion) (Fig. 7C) and the levels of newly synthesized MV (Fig. 7D). Approximately 50% inhibition was obtained with 0.5 μM rapamycin, and 1 μM rapamycin reduced viral titers to the level measured in unstimulated PBL. This rapamycin-mediated reduction of MV titers by more than one log (approximately 20-fold) demonstrates that MV replication in PBL is strongly enhanced by mTORC1 activity.
DISCUSSION
It is known that A3G restricts viruses by cytidine deaminase-dependent editing of retroviral genomes. However, several reports also suggested editing-independent mechanisms, mainly by binding to reverse transcription products (ssDNA) or viral RNA. In addition to retroviruses, A3G has antiviral activity against other viruses, such as, for example, blocking of hepatitis B virus (HBV) replication involving effects on RNA packaging rather than any mutations in viral cDNA (38), direct binding to hepatitis C virus (HCV) NS3 protein resulting in reduced HCV replication (39, 40), and reducing the replication of MV, mumps virus, and respiratory syncytial virus (36), and may affect viral replication by still-unknown mechanisms (10). In addition, A3G has been under investigation for its potential ability to edit more than hundreds of cellular mRNA transcripts (41). Here we report a novel mechanism for how A3G impairs the replication of viruses by regulating the expression of host cell factors. By analyzing A3G-induced antiviral activities in Vero cells, we found a strong antiviral effect of REDD1 on MV replication. It is known that REDD1 disrupts the interaction of 14-3-3 proteins with TSC1/2 and inhibits the function of mTORC1 (for reviews, see references 28, 32, and 42), while its stability is affected in a feedback loop by mTORC1 activity (43). The expression of REDD1 is induced by hypoxia and cellular stress and regulated by cellular microRNAs (miRs) (44). In this paper, we describe that silencing of A3G not only reconstituted the full capacity of Vero cells to replicate MV but also led to a reduction of the REDD1 expression, confirming that A3G promotes REDD1 expression. Furthermore, we observed that stimulation of primary human PBL with PHA and IL-2 is associated with increased A3G and REDD1 expression and that siRNA-mediated knockdown of A3G also led to reduced REDD1 expression in these cells. In addition, we demonstrated that REDD1 impairs MV replication by inhibiting mTORC1 in Vero cells and primary human PBL. Finally, we found that inhibition of the mTORC1 activity by rapamycin reduced the amount of newly synthesized MV to the level in unstimulated cells, indicating the strong influence of mTORC1 on viral replication.
REDD1 has been identified previously as an antiviral host defense factor in association with chemical inhibition of RNA viruses, including influenza virus and vesicular stomatitis virus (37), and the involvement of mTORC1 in viral replication and protein expression has been demonstrated for viruses such as Andes virus (45), West Nile virus (46), and Epstein-Barr virus (47). Interestingly, mTORC has been identified as a factor controlling HIV latency, and mTORC inhibitors suppress HIV transcription (48). Recently, influenza virus was found to activate mTORC1 and mTORC2 signaling and AKT phosphorylation in order to maximize its replication in late stages of the infection (49). These findings underline the relevance of such regulators of the cellular metabolism and cell survival in viral replication and may help to identify novel targets for pharmacological intervention.
Mammalian TORC1 is a serine/threonine kinase, which in its active state is located at the cytoplasmic side of lysosomal membranes and regulates cellular homeostasis by integrating signals from nutrients, energy, and oxygen controlling cell growth and protein synthesis. The control of mTORC1 activity is exerted through the tuberous sclerosis complex (TSC1/TSC2), a GTPase-activating protein that negatively regulates the Rheb G protein, which in turn positively regulates mTOR (for a review, see reference 28). Our findings that REDD1 expression and rapamycin treatment have similar effects on MV replication and that REDD1 expression and rapamycin treatment in combination had no further additive antiviral effect indicated that both act on the same pathway by inhibiting mTORC activity. This additionally suggests that by this mechanism A3G contributes to the establishment of a general antiviral state of the cell. Interestingly, artificial downregulation of TXNIP by shRNA led to approximately 6-fold-higher MV titers than in control cells (Fig. 2). This may result from the fact that TXNIP stabilizes REDD1 and thus reduces mTORC1 activity (50, 51). In A3G-expressing Vero cells, REDD1 mRNA was 3.5-fold upregulated and TXNIP mRNA 0.44-fold downregulated, which may partially dampen REDD1 upregulation and its antiviral effect.
mTORC activity differs across cell subsets of primary lymphocytes: whereas it is high in plasma B cells, TH1, TH2, TH17, and CD8+ effector cells, and NK cells, it is low in regulatory T cells and memory B and T cells (52, 53). This is independent from the expression of the cellular receptor CD150, which determines the tropism of the MV infection (54). According to our findings, high mTORC1 activity enables a 10- to 100-fold-higher replication of MV than in infected lymphocytes with low mTORC activity. Infection of cells with low mTORC activity, such as memory T cells, and the resulting limited MV replication also contribute to the observed pathogenesis as demonstrated in a macaque model (54). The effect of rapamycin has already been investigated in infections with a number of pathogens, and it turned out to promote responses by more metabolically inactive regulatory T, follicular helper T, and memory cells but would inhibit T helper, CD8 effector, and antibody responses (52). Thus, the potential outcome of therapeutic intervention is difficult to predict and depends on the exact type of infection and the target cells and organs (52).
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney control Vero (Vero-023) cells were transduced with the empty vector (pCMS28), and Vero cells expressing human A3G (Vero-A3G cells, the same as Vero-024-2 cells in reference 36) were transduced with pCMS28 expressing A3G (a kind gift from M. Malim, King's College, London, United Kingdom). Transduced Vero cells were grown under selection pressure with 5 μg/ml puromycin as a bulk culture (17). Human embryonic kidney 293T cells and Vero and Vero-hSLAM cells were cultivated in Dulbecco modified Eagle medium (DMEM) containing 5% fetal bovine serum (FBS), 100 U/ml penicillin-streptomycin, and nonessential amino acids.
All experiments involving human cells were conducted according to the principles expressed in the Declaration of Helsinki and approved by the Ethical Committee of the Medical Faculty of the University of Würzburg. Primary human peripheral blood mononuclear cells (PBMC) obtained from leuko-reduction chambers of thrombocyte donations from anonymous healthy adult volunteers were diluted 1:6 in Versene (GIBCO) and layered on Histopaque 1077. PBMC were purified by density gradient centrifugation. Isolated PBMC were washed three times with phosphate-buffered saline (PBS) and suspended in RPMI 1640 (GIBCO) medium containing 10% FBS. Mononuclear cells were reduced by adherence to tissue culture plasticware surfaces to obtain peripheral blood lymphocytes (PBL). Cells were stimulated with phytohemagglutinin L (PHA) (2.5 μg/ml; Roche Diagnostics) or with recombinant human IL-2 (Proleukin) (25 ng/ml; Novartis). Stimulation was controlled by measuring CD69 expression by flow cytometry.
The recombinant wild-type MV rMVIC323eGFP (55) was propagated using Vero-hSLAM cells. The attenuated vaccine strain MV-Edmonston and the recombinant Edmonston-based MV strain rMV-eGFP expressing eGFP (56) were propagated using Vero cells. For assessing the effects of individual genes on viral replication, transduced Vero cells were seeded in 6-well plates and infected with rMV-eGFP at an MOI of 0.1. For infection in PBL, unstimulated or stimulated PBL were infected with rMVIC323eGFP. The virus was harvested after indicated times by freezing and thawing the complete culture, and thus cell-associated and supernatant viruses were harvested together.
Microarray analysis.
Total RNA was isolated independently two times each from control Vero-023 and A3G-expressing Vero-024-2 cells using the RNeasy kit (Qiagen) and analyzed using a Gene Chip rhesus macaque genome array (>47,000 transcripts; Affymetrix). Probe sets were combined and the resulting signal intensities normalized using rma and quantile-quantile normalization. Differential gene expression was calculated using the Limma package (R/Bioconductor). A list of results is available in Table S1 in the supplemental material.
Semiquantitative RT-PCR.
Total RNA from 1.5 × 105 cells was extracted using the GenElute mammalian total RNA miniprep kit (Sigma). Four amounts (0.5, 1, 2, and 4 μg) of RNA were used for first-strand cDNA synthesis with the RevertAid first-strand cDNA synthesis kit (Fermentas). The cDNA was then amplified by PCR in 25 cycles (the sequences of all primers used can be provided on request). The amplified products were analyzed on 1% agarose gels.
Cloning of lentiviral expression plasmids and production of pseudotyped particles.
For shRNA expression, we used the vector F6gW-DsRed, also expressing DsRed2 as a control for transduction efficiency, as described previously (57). For cloning of shRNA-expressing vectors, we selected three siRNA sequences from published mRNA sequences using the program provided by Block-iT RNAi designer (Invitrogen) and used DNA oligonucleotides for cloning into pF6gW-DsRed (the sequences of all primers used can be provided on request). The oligonucleotides were aligned and cloned into the HpaI and XhoI sites of F6gW-DsRed. The sequences of the clones were confirmed by sequencing. The REDD1-specific shRNA expression construct 2 (REDD1sh2) silenced the expression of REDD1 most efficiently and therefore was used for further experiments. A lentiviral expression vector for REDD1 (F6gW-REDD1-Myc-DDK-tag) was generated using the BamHI-SacII fragment of REDD1 expression plasmid RC202847 (OriGene) and cloned into the BamHI-EcoRI sites of F6gW. Vero cells were transduced with F6gW-REDD1-Myc-DDK-tag to obtain REDD1-overexpressing Vero (Vero-REDD1) cells. The sequences of the cloned plasmids were confirmed by sequencing. VSV G-pseudotyped viral particles were produced by transfection of 293T cells with plasmids pVSV-G, pRSVrev, and pMDLg/pRRE and pF6gW-based retroviral vectors, using polyethylenimine (PEI) (25 K; Polysciences Inc.) as described previously (36).
Antibodies and flow cytometry.
The following unlabeled or labeled primary antibodies were used in immunoblotting and flow cytometry: rabbit monoclonal anti-ACY1 (Epitomics, 5879-1), rabbit polyclonal anti-MOSC2 (Epitomics, T3362), rabbit anti-DDIT4/REDD1 (ProteinTech, 10638-1-AP), rabbit anti-PRDX2 (Sigma, SAB2101878), rabbit anti-KDELR2 (Sigma, SAB1401554), rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Santa Cruz, sc-25778), rabbit anti-phospho-p70 S6 kinaseThr 389 (Cell Signaling, 9205), rabbit anti-p70S6 kinase (Cell Signaling, 9202), phycoerythrin (PE)–anti-human CD3 (BioLegend, 300408), allophycocyanin (APC)–anti-human CD69 (BioLegend, 310910), and rabbit polyclonal anti-A3G (a kind gift from M. Malim, King's College, London, United Kingdom). The following labeled secondary antibodies were used: Alexa 488–anti-mouse (Life Technology, A11001), Alexa 594–anti-rabbit (Life Technology, A11012), APC–anti-mouse (BioLegend, 405308), horseradish peroxidase (HRP)–anti-mouse (Cell Signaling, 7076S), and HRP–anti-rabbit (Cell Signaling, 7074S). For flow cytometric determination of the proliferation of cells, 1 × 105 cells were stained with the respective antibodies and resuspended in FACS buffer (PBS containing 0.4% bovine serum albumin [BSA] and 0.02% sodium azide). Cells were acquired immediately using an LSR II flow cytometer (BD), and data were evaluated using FlowJo (Cytek Development) software.
SDS-PAGE and immunoblotting.
Cells (5 × 106) were lysed at 4°C for 1 h in 1 ml of lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM sodium chloride [NaCl], 1.0% Igepal CA-630 [NP-40], 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) containing complete protease inhibitor cocktail (Sigma, P8340) and 1 mM dl-dithiothreitol (DTT). The protein quantification was done using the bicinchoninic acid (BCA) assay. Equal amounts of proteins were heated at 95°C for 5 min in reducing Laemmli sample buffer (50 mM Tris HCl [pH 6.8], 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.02% bromophenol blue) and subjected to 10% SDS-PAGE. Proteins were blotted semidry on nitrocellulose membranes (Amersham), followed by blocking with 5% dry milk (AppliChem) or 5% BSA in PBS with 0.05% Tween 20. The membranes were then incubated with specific primary antibodies and HRP-conjugated secondary antibodies. Signals were visualized using chemiluminescent FemtoMax supersensitive HRP substrate (Rockland). The densitometric quantification of protein bands of target genes and respective housekeeping genes was done using ImageJ (National Institutes of Health, Bethesda, MD). The fold changes in target proteins were normalized to band densities of GAPDH, and fold changes in phosphorylated proteins were normalized to the band densities of total protein and GAPDH levels. All Western blotting experiments were repeated 2 or 3 times, and representative images are shown.
Transfection of primary human PBL and Vero cells.
Human SMARTpool siRNA duplexes targeting A3G (M-013072-00-0005), REDD1 (L-010855-01-0005), and a nontargeting control siRNA (D-001206-13-05) were purchased from Dharmacon. PBL were transfected using Nucleofection (nucleofected) according to the manufacturer's protocol (Amaxa) with 400 pmol of siRNA twice with a 48-h interval. Nucleofected cells were then stimulated with PHA (2.5 μg/ml) or IL-2 (25 ng/ml) for 24 h. Stimulated cells were routinely stained for surface expression of CD3 for detection of T cells and CD69 as an activation marker. The siRNA silencing efficiency for specific proteins was controlled by Western blotting. For determination of viral replication 2 × 106 cells were infected with rMVIC323eGFP at an MOI of 0.2, and the virus was harvested after the indicated times by freezing and thawing the complete culture; thus, cell-associated and supernatant viruses were harvested together and titrated on Vero hSLAM cells. Vero cells were transfected with human SMARTpool siRNA duplexes targeting A3G (M-013072-00-0005) and a nontargeting control siRNA (D-001206-13-05) from Dharmacon using DharmaFECT2 (Dharmacon, T-2002-01) according to the manufacturer's instructions. At 48 h after transfection, cells were infected with rMV-eGFP at an MOI of 0.1 and incubated for 2 days. The virus was harvested and titrated on Vero hSLAM cells, and cell lysates were used for Western blot analysis.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 6. Comparisons of two groups were done using the unpaired two-tailed Student t test, and those of more than two groups were done with one-way analysis of variance (ANOVA). P values lower than or equal to 0.05 were considered statistically significant (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). The data represent means ± standard deviations (SD) from at least three independent experiments.
Accession number(s).
The microarray data have been deposited at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE85102.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the German Research Foundation (DFG) and a grant from the German Excellence Initiative to the Graduate School of Life Sciences (GSLS), University of Würzburg.
We thank Sabine Kendl, Anika Grafen, and Johannes Siewert for technical assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00835-18.
REFERENCES
- 1.Bonvin M, Achermann F, Greeve I, Stroka D, Keogh A, Inderbitzin D, Candinas D, Sommer P, Wain-Hobson S, Vartanian JP, Greeve J. 2006. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43:1364–1374. doi: 10.1002/hep.21187. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Wang LW, Huang YQ, Gong ZJ. 2010. Interferon-alpha induces high expression of APOBEC3G and STAT-1 in vitro and in vivo. Int J Mol Sci 11:3501–3512. doi: 10.3390/ijms11093501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM. 2006. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med 203:41–46. doi: 10.1084/jem.20051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarkis PT, Ying S, Xu R, Yu XF. 2006. STAT1-independent cell type-specific regulation of antiviral APOBEC3G by IFN-alpha. J Immunol 177:4530–4540. doi: 10.4049/jimmunol.177.7.4530. [DOI] [PubMed] [Google Scholar]
- 5.Stopak KS, Chiu YL, Kropp J, Grant RM, Greene WC. 2007. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem 282:3539–3546. doi: 10.1074/jbc.M610138200. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka Y, Marusawa H, Seno H, Matsumoto Y, Ueda Y, Kodama Y, Endo Y, Yamauchi J, Matsumoto T, Takaori-Kono A, Ikai I, Chiba T. 2006. Anti-viral protein APOBEC3G is induced by interferon-alpha stimulation in human hepatocytes. Biochem Biophys Res Commun 341:314–319. doi: 10.1016/j.bbrc.2005.12.192. [DOI] [PubMed] [Google Scholar]
- 7.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 8.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog 4:e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chelico L, Sacho EJ, Erie DA, Goodman MF. 2008. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J Biol Chem 283:13780–13791. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes RK, Malim MH, Bishop KN. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci 32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res 35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol 15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 13.Wedekind JE, Gillilan R, Janda A, Krucinska J, Salter JD, Bennett RP, Raina J, Smith HC. 2006. Nanostructures of APOBEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J Biol Chem 281:38122–38126. doi: 10.1074/jbc.C600253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol 79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol 78:12058–12061. doi: 10.1128/JVI.78.21.12058-12061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. 2009. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog 5:e1000330. doi: 10.1371/journal.ppat.1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH. 2007. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol 81:2165–2178. doi: 10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D. 2006. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem 281:29105–29119. doi: 10.1074/jbc.M601901200. [DOI] [PubMed] [Google Scholar]
- 19.Beckham C, Hilliker A, Cziko AM, Noueiry A, Ramaswami M, Parker R. 2008. The DEAD-box RNA helicase Ded1p affects and accumulates in Saccharomyces cerevisiae P-bodies. Mol Biol Cell 19:984–993. doi: 10.1091/mbc.e07-09-0954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wichroski MJ, Robb GB, Rana TM. 2006. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog 2:e41. doi: 10.1371/journal.ppat.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eulalio A, Behm-Ansmant I, Izaurralde E. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8:9–22. [DOI] [PubMed] [Google Scholar]
- 23.Sheth U, Parker R. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lloyd RE. 2012. How do viruses interact with stress-associated RNA granules? PLoS Pathog 8:e1002741. doi: 10.1371/journal.ppat.1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol 85:6882–6892. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci U S A 104:9041–9046. doi: 10.1073/pnas.0703348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhowmick R, Mukherjee A, Patra U, Chawla-Sarkar M. 2015. Rotavirus disrupts cytoplasmic P bodies during infection. Virus Res 210:344–354. doi: 10.1016/j.virusres.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Dibble CC, Manning BD. 2013. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desantis A, Bruno T, Catena V, De Nicola F, Goeman F, Iezzi S, Sorino C, Ponzoni M, Bossi G, Federico V, La Rosa F, Ricciardi MR, Lesma E, De Meo PD, Castrignano T, Petrucci MT, Pisani F, Chesi M, Bergsagel PL, Floridi A, Tonon G, Passananti C, Blandino G, Fanciulli M. 2015. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J 34:1214–1230. doi: 10.15252/embj.201489920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krisenko MO, Higgins RL, Ghosh S, Zhou Q, Trybula JS, Wang WH, Geahlen RL. 2015. Syk is recruited to stress granules and promotes their clearance through autophagy. J Biol Chem 290:27803–27815. doi: 10.1074/jbc.M115.642900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L. 2013. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 152:791–805. doi: 10.1016/j.cell.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 32.Bar-Peled L, Sabatini DM. 2012. SnapShot: mTORC1 signaling at the lysosomal surface. Cell 151:1390–1390.e1. doi: 10.1016/j.cell.2012.11.038. [DOI] [PubMed] [Google Scholar]
- 33.Reiling JH, Sabatini DM. 2006. Stress and mTORture signaling. Oncogene 25:6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 34.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr. 2004. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellisen LW. 2005. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle 4:1500–1502. doi: 10.4161/cc.4.11.2139. [DOI] [PubMed] [Google Scholar]
- 36.Fehrholz M, Kendl S, Prifert C, Weissbrich B, Lemon K, Rennick L, Duprex PW, Rima BK, Koning FA, Holmes RK, Malim MH, Schneider-Schaulies J. 2012. The innate antiviral factor APOBEC3G targets replication of measles, mumps and respiratory syncytial viruses. J Gen Virol 93:565–576. doi: 10.1099/vir.0.038919-0. [DOI] [PubMed] [Google Scholar]
- 37.Mata MA, Satterly N, Versteeg GA, Frantz D, Wei S, Williams N, Schmolke M, Pena-Llopis S, Brugarolas J, Forst CV, White MA, Garcia-Sastre A, Roth MG, Fontoura BM. 2011. Chemical inhibition of RNA viruses reveals REDD1 as a host defense factor. Nat Chem Biol 7:712–719. doi: 10.1038/nchembio.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turelli P, Mangeat B, Jost S, Vianin S, Trono D. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 39.Zhu YP, Peng ZG, Wu ZY, Li JR, Huang MH, Si SY, Jiang JD. 2015. Host APOBEC3G protein inhibits HCV replication through direct binding at NS3. PLoS One 10:e0121608. doi: 10.1371/journal.pone.0121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng ZG, Zhao ZY, Li YP, Wang YP, Hao LH, Fan B, Li YH, Wang YM, Shan YQ, Han YX, Zhu YP, Li JR, You XF, Li ZR, Jiang JD. 2011. Host apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology 53:1080–1089. doi: 10.1002/hep.24160. [DOI] [PubMed] [Google Scholar]
- 41.Sharma S, Patnaik SK, Taggart RT, Baysal BE. 2016. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci Rep 6:39100. doi: 10.1038/srep39100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 43.Tan CY, Hagen T. 2013. mTORC1 dependent regulation of REDD1 protein stability. PLoS One 8:e63970. doi: 10.1371/journal.pone.0063970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canal M, Romani-Aumedes J, Martin-Flores N, Perez-Fernandez V, Malagelada C. 2014. RTP801/REDD1: a stress coping regulator that turns into a troublemaker in neurodegenerative disorders. Front Cell Neurosci 8:313. doi: 10.3389/fncel.2014.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McNulty S, Flint M, Nichol ST, Spiropoulou CF. 2013. Host mTORC1 signaling regulates andes virus replication. J Virol 87:912–922. doi: 10.1128/JVI.02415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shives KD, Beatman EL, Chamanian M, O'Brien C, Hobson-Peters J, Beckham JD. 2014. West Nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J Virol 88:9458–9471. doi: 10.1128/JVI.01323-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamson AL, Le BT, Siedenburg BD. 2014. Inhibition of mTORC1 inhibits lytic replication of Epstein-Barr virus in a cell-type specific manner. Virol J 11:110. doi: 10.1186/1743-422X-11-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J, Svensson JP, Gramatica A, Conrad RJ, Ott M, Greene WC, Krogan NJ, Siliciano RF, Weissman JS, Verdin E. 2016. The mTOR complex controls HIV latency. Cell Host Microbe 20:785–797. doi: 10.1016/j.chom.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuss-Duerkop SK, Wang J, Mena I, White K, Metreveli G, Sakthivel R, Mata MA, Munoz-Moreno R, Chen X, Krammer F, Diamond MS, Chen ZJ, Garcia-Sastre A, Fontoura BMA. 2017. Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathog 13:e1006635. doi: 10.1371/journal.ppat.1006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. 2008. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin HO, Seo SK, Kim YS, Woo SH, Lee KH, Yi JY, Lee SJ, Choe TB, Lee JH, An S, Hong SI, Park IC. 2011. TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene 30:3792–3801. doi: 10.1038/onc.2011.102. [DOI] [PubMed] [Google Scholar]
- 52.Keating R, McGargill MA. 2016. mTOR regulation of lymphoid cells in immunity to pathogens. Front Immunol 7:180. doi: 10.3389/fimmu.2016.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. 2009. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Vries RD, McQuaid S, van Amerongen G, Yuksel S, Verburgh RJ, Osterhaus AD, Duprex WP, de Swart RL. 2012. Measles immune suppression: lessons from the macaque model. PLoS Pathog 8:e1002885. doi: 10.1371/journal.ppat.1002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J Virol 76:6743–6749. doi: 10.1128/JVI.76.13.6743-6749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. 1999. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol 73:9568–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zinke M, Kendl S, Singethan K, Fehrholz M, Reuter D, Rennick L, Herold MJ, Schneider-Schaulies J. 2009. Clearance of measles virus from persistently infected cells by short hairpin RNA. J Virol 83:9423–9431. doi: 10.1128/JVI.00846-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.