

Human cytomegalovirus (HCMV) is a ubiquitous pathogen which is usually asymptomatic but that can cause serious complications and mortality in congenital infections and in immunosuppressed patients. One of the difficulties in developing novel vaccines and treatments for HCMV is its remarkable ability to evade our immune system. In particular, HCMV directs significant efforts to thwarting cells of the innate immune system known as natural killer (NK) cells. These cells are crucial for successful control of HCMV infection, and yet our understanding of the mechanisms which HCMV utilizes to elude NK cells is partial at best. In the present study, we discovered that a protein encoded by HCMV which had no known function is important for preventing NK cells from killing HCMV-infected cells. This knowledge can be used in the future for designing more-efficient HCMV vaccines and for formulating novel therapies targeting this virus.
KEYWORDS: HCMV, NK cells, immune evasion
ABSTRACT
Natural killer (NK) cells are lymphocytes of the innate immune system capable of killing hazardous cells, including virally infected cells. NK cell-mediated killing is triggered by activating receptors. Prominent among these is the activating receptor NKG2D, which binds several stress-induced ligands, among them major histocompatibility complex (MHC) class I-related chain A (MICA). Most of the human population is persistently infected with human cytomegalovirus (HCMV), a virus which employs multiple immune evasion mechanisms, many of which target NK cell responses. HCMV infection is mostly asymptomatic, but in congenitally infected neonates and in immunosuppressed patients it can lead to serious complications and mortality. Here we discovered that an HCMV protein named UL148A whose role was hitherto unknown is required for evasion of NK cells. We demonstrate that UL148A-deficient HCMV strains are impaired in their ability to downregulate MICA expression. We further show that when expressed by itself, UL148A is not sufficient for MICA targeting, but rather acts in concert with an unknown viral factor. Using inhibitors of different cellular degradation pathways, we show that UL148A targets MICA for lysosomal degradation. Finally, we show that UL148A-mediated MICA downregulation hampers NK cell-mediated killing of HCMV-infected cells. Discovering the full repertoire of HCMV immune evasion mechanisms will lead to a better understanding of the ability of HCMV to persist in the host and may also promote the development of new vaccines and drugs against HCMV.
IMPORTANCE Human cytomegalovirus (HCMV) is a ubiquitous pathogen which is usually asymptomatic but that can cause serious complications and mortality in congenital infections and in immunosuppressed patients. One of the difficulties in developing novel vaccines and treatments for HCMV is its remarkable ability to evade our immune system. In particular, HCMV directs significant efforts to thwarting cells of the innate immune system known as natural killer (NK) cells. These cells are crucial for successful control of HCMV infection, and yet our understanding of the mechanisms which HCMV utilizes to elude NK cells is partial at best. In the present study, we discovered that a protein encoded by HCMV which had no known function is important for preventing NK cells from killing HCMV-infected cells. This knowledge can be used in the future for designing more-efficient HCMV vaccines and for formulating novel therapies targeting this virus.
INTRODUCTION
Human cytomegalovirus (HCMV) is a double-stranded DNA (dsDNA) betaherpesvirus that possesses the largest genome among human herpesviruses (1). Initial infection with HCMV is generally asymptomatic and results in lifelong infection. However, HCMV is a significant cause of morbidity and mortality in congenitally infected neonates and immunosuppressed individuals (2). Moreover, chronic HCMV infection has been implicated in age-related pathologies (3). In light of the above, understanding HCMV-immune system interactions is of high importance.
Natural killer (NK) cells are innate immune lymphocytes that are able to detect and attack cells infected with viruses and other intracellular pathogens, tumor cells, and even extracellular pathogens such as fungi (4, 5). NK cells play a crucial role in combating viral infections, including HCMV infections (4), as demonstrated by the susceptibility of NK cell-deficient patients to fatal HCMV infections (6). NK cell activity is controlled by a balance of signals derived from activating and inhibitory receptors (7–9). Once activated, NK cells secrete cytokines and mediate cytotoxicity against their targets (10).
One key activating receptor expressed by NK cells is NKG2D (11, 12). In humans, it recognizes a family of major histocompatibility complex (MHC)-like stress-induced ligands, including UL16-binding protein 1 (ULBP1) to ULBP6 and MHC class I-related chain (MIC) molecules MICA and MICB. Under normal conditions, the NKG2D ligands are generally absent from the cell membrane (13, 14). However, their expression is rapidly induced following cellular stress such as viral infection (13).
MICA is the most polymorphic NKG2D ligand, with more than 100 alleles described to date (https://www.ebi.ac.uk/ipd/imgt/hla/). Due to its importance for NK-mediated recognition of infected cells, MICA is targeted by multiple HCMV immune evasion mechanisms. MICA was shown to be targeted in concert by the HCMV proteins US18 and US20, which leads to its lysosomal degradation (15), and by UL142, which sequesters it inside infected cells (16, 17). Finally, the truncated MICA*008 allele, which lacks the transmembrane domain and was previously considered resistant to HCMV infection, was recently shown by our group to be targeted for proteasomal degradation by US9 (18).
Clinical HCMV strains carry a genomic region, named ULb′, which was found to be the most prone to mutations (19) and in some stains to be lost during rearrangement of the viral genome in subsequent passages in fibroblast cell culture. When lost, this area is frequently replaced by duplication of the internal repeat long (IRL) sequence (20–22). The ULb′ region is a 13- to 15-kb sequence encoded at the right end of the long unique region and is considered unnecessary for viral replication. The ULb′ region has already been shown to encode immune evasion functions, including the aforementioned UL142, UL141 (which targets the NK ligand poliovirus receptor [PVR]), and UL135 (which hampers immune synapse formation) (16, 17, 23, 24).
UL148A is an HCMV protein encoded in the ULb′ genomic region. In the AD169 strain, the UL148A open reading frame (ORF) consists of 240 bp, encoding 79 amino acids. It contains a predicted transmembrane helix domain near its N terminus (http://www.uniprot.org/uniprot/B8YEF6). As part of the ULb′, UL148A is considered unnecessary for replication, but its function remains unknown. Here we demonstrate that UL148A, together with an additional unknown viral protein(s), downregulates MICA to evade NK cell-mediated killing of the HCMV-infected cell.
RESULTS
The presence of an HCMV ULb′-derived gene leads to downregulation of the NKG2D ligand MICA.
To test whether additional genes in the ULb′ genomic region are involved in the regulation of NKG2D ligands, primary human foreskin fibroblast (HFF) cells named FLS1 cells (18) were mock infected or infected with the following variants of the AD169 strain: AD169VarS (VarS), which lacks the ULb′ region, or a bacterial artificial chromosome (BAC)-cloned AD169VarL genome (25), denoted BAC2. AD169VarL (VarL) contains most of the ULb′ genomic region but harbors a UL140–144 deletion (25) (Fig. 1A). Within this region lies UL142, which targets ULBP3 and full-length MICA alleles (16, 26). FLS1 cells express the full-length MICA alleles *004 and *009:01–*049 (the latter alleles differ in a single nucleotide in exon 6 and could not be distinguished [18]). At 72 h postinfection (hpi), cells were stained for the following NKG2D ligands: MICA, MICB, ULBP1, ULBP2/5/6, and ULBP3. Interestingly, MICA surface expression was upregulated following VarS infection (Fig. 1B). No differences in the expression of the other NKG2D ligands (MICB, ULBP1, ULBP2/5/6, and ULBP3) were observed between BAC2-infected and VarS-infected cells (Fig. 1C to F). However, since both VarS and BAC2 lack the UL142 gene, ULBP3 surface expression was upregulated to similar extents following BAC2 and VarS infection (Fig. 1F). The absence of UL142 led us to hypothesize that another MICA-targeting gene, present in the ULb′ gene cluster in the VarL strain, caused the observed BAC2-specific MICA downregulation.
FIG 1.

HCMV strain VarS fails to downregulate the NKG2D ligand MICA. (A) Scheme of HCMV genome organization. UL, unique long; US, unique short; TRL, terminal repeat long; IRS, internal repeat short; TRS, terminal repeat short; IRL, internal repeat long; RL, repeat long. (B to F) FLS1 HFFs (MICA*004/*009:01-*049) were either mock infected or infected with the indicated HCMV strains. Cells were harvested at 72 h postinfection (hpi), and the surface expression of the following NKG2D ligands was assayed by flow cytometry. (B) MICA. (C) MICB. (D) ULBP1. (E) ULBP2/5/6. (F) ULBP3. Black, blue, and red histograms represent mock-infected, BAC2-infected, and VarS-infected HFFs, respectively. Gray-filled histograms (BG [background]) represent secondary antibody staining of the mock-infected cells, which produced similar results for all cells.
Deletion of ULb′ gene UL148A abolishes MICA downregulation.
To identify the MICA-targeting ULb′ gene(s), we generated ULb′ block deletion mutants in the background of BAC2 (Fig. 2A). We then used a MICA overexpression system to test the activity of these mutants. FLS3 HFFs, which lack MICA surface expression, were transduced with MICA*004-HA (18) and infected with ULb′ block deletion mutants. MICA surface expression was assayed by flow cytometry at 72 hpi. Cells infected with parental BAC2 displayed reduced MICA surface expression compared to mock-infected cells, and a similar decrease in MICA surface expression was observed during infection with BAC2 ΔUL148–146 and with BAC2 ΔUL139–133 (Fig. 2B and C). However, little or no decrease in MICA surface expression was observed when the BAC2 ΔUL148A–150 mutant was used (Fig. 2D). The BAC2 ΔUL148A–150 mutant lacks six genes (UL148A–D, UL149, and UL150), and yet MICA downregulation was observed during infection with the BAC2 ΔUL149–150 deletion mutant (Fig. 2E), suggesting that at least one of the UL148A–D genes is involved in MICA downregulation. To identify the specific gene(s), single-deletion mutants of each of the UL148A–D genes were generated (Fig. 2F to I). MICA expression was restored only when BAC2 ΔUL148A (Fig. 2F) was used, indicating that UL148A is required for MICA downregulation.
FIG 2.

Deletion of UL148A hampers the reduction in MICA surface expression during HCMV infection. (A) Schematic representation of AD169-ULb′ ORFs. UL140 to UL144 are depicted in gray, since they are deleted in part or completely in the VarL variant. Red lines indicate gene blocks deleted in the BAC2 mutants. (B to I) FLS3 HFFs overexpressing MICA*004-HA were mock infected, BAC2 infected, or infected with the indicated BAC2 deletion mutants. Cells were harvested at 72 hpi, and MICA surface expression was assessed by flow cytometry. Black, blue, and red histograms represent mock-infected, BAC2-infected, and BAC2 deletion mutant-infected HFFs, respectively. Gray-filled histograms represent secondary antibody-only staining of mock-infected cells, which produced similar results for all cells. Histograms represent combined results of independent experiments.
UL148A targets full-length MICA alleles at an early time point.
We next investigated whether UL148A is capable of regulating both full-length MICA alleles and the truncated MICA*008 allele. FLS3 HFFs transduced either with MICA*004-HA or with MICA*008-HA were mock infected or infected with BAC2, BAC2 ΔUL148A, or BAC2 ΔUL148B (used here as a control), and cells were stained for MICA surface expression at 48 hpi (Fig. 3A to F). Downregulation of MICA was observed following BAC2 infection of FLS3 MICA*004-HA cells, confirming our observations (Fig. 3A). Infection with the viral mutant BAC2 ΔUL148B (Fig. 3B) led to similar downregulation. However, MICA expression was reduced only partly when the BAC2 ΔUL148A mutant was used (Fig. 3C). In contrast, when FLS3 MICA*008-HA cells were infected, MICA surface expression was downregulated to similar extents by all three viruses (Fig. 3D to F), indicating that the truncated MICA*008 allele is resistant to UL148A activity.
FIG 3.

UL148A downregulates full-length MICA alleles but not the truncated allele MICA*008. The indicated HFFs were mock infected, BAC2 infected, or infected with one of the following BAC2 deletion mutants: BAC2 ΔUL148A or BAC2 ΔUL148B. Cells were harvested, and MICA surface expression was assessed by flow cytometry. (A to C) FLS3 HFFs overexpressing MICA*004-HA, harvested 48 hpi. (D to F) FLS3 HFFs overexpressing MICA*008-HA, harvested 48 hpi. (G to I) FLS1 HFFs (MICA*004/*009:01-*049), harvested 24 hpi. Black histograms represent MICA staining of mock-infected cells, red histograms represent MICA staining of cells infected with the indicated virus, and gray-filled histograms represent isotype control staining of mock-infected cells, which produced similar results for all cells.
To confirm the specificity of UL148A for full-length MICA alleles, we infected FLS1 HFFs, which endogenously express full-length MICA alleles (Fig. 3G to I). Consistent with the results seen with FLS3 MICA*004-HA cells and with the initial screen of VarS versus BAC2 (Fig. 1B to F) conducted in these cells, we observed a reduction in MICA surface expression following infection of FLS1 cells with either BAC2 or the BAC2 ΔUL148B mutant whereas BAC2 ΔUL148A-infected cells maintained MICA surface expression levels (Fig. 3G to I). These results indicate that UL148A can target only full-length MICA alleles.
To test the time course of the activity of UL148A, we assessed MICA surface expression on FLS1 HFFs at three time points: 24, 48, and 72 hpi. Cells were infected with BAC20 (a BAC-cloned AD169VarS genome), BAC2, or either of the following BAC2 deletion mutants: BAC2 ΔUL148A or BAC2 ΔUL148B. UL148A was found to have already had an effect on MICA at 24 hpi, as BAC2 or BAC2 ΔUL148B infection reduced MICA surface expression to background levels (data not shown). On the other hand, MICA levels remained similar to those seen with mock-infected cells following BAC20 or BAC2 ΔUL148A infection. This effect was also detectable at 48 hpi and 72 hpi (data not shown). These findings show that UL148A was actively downregulating MICA 24 hpi in cells endogenously expressing MICA. To detect the time point at which UL148A downregulates MICA in cells exogenously overexpressing it, MICA surface expression on FLS3 MICA*004-HA HFFs was evaluated 24 or 48 hpi. Here, at 24 hpi, cells infected with BAC2 or the BAC2 ΔUL148B mutant demonstrated reduced and yet inconsistent MICA expression whereas cells infected with BAC20 or the BAC2 ΔUL148A mutant maintained MICA levels similar to those seen with mock-infected cells (data not shown). However, at 48 hpi, MICA surface expression on BAC2 or BAC2 ΔUL148B-infected cells peaked at a close to the background level. Although MICA is also downregulated in cells infected with BAC20 or BAC2 ΔUL148A (data not shown), the UL148A effect is better distinguished at this time point than at 24 hpi. This delayed time course can be explained by the constitutive overexpression of MICA in the FLS3 MICA*004-HA cells, which requires a stronger effect for detection.
UL148A requires an additional viral partner(s) for MICA downregulation.
To test whether UL148A is sufficient for reduction of MICA expression, we transduced RKO cells with the full-length MICA*004-HA allele and then cotransduced these cells with an empty vector (EV) or with UL148A, either untagged or Flag tagged at its N or C terminus. Western blot validation revealed that UL148A was expressed with a C-terminal tag but not with an N-terminal tag (data not shown). Cells were stained for stress-induced ligands MICA, MICB, ULBP1, ULBP2/5/6, and ULBP3. Surprisingly, UL148A expression had no effect on the surface expression of any of the ligands, including MICA (data not shown). To test whether this was due to the ectopic expression of MICA, we transduced A549 (MICA*001/*004) and HCT116 (MICA*001/*009:02b) cells, which endogenously express MICA, with EV or with UL148A, either untagged or tagged, and stained for the stress-induced ligands. Again, no MICA reduction was observed (data not shown). These observations suggest that UL148A by itself is not sufficient for MICA downregulation and that it might require another viral factor to downregulate MICA.
UL148A's viral partner is encoded externally to the ULb′ region.
To further explore the hypothesis that UL148A has an additional viral partner, we conducted a complementation assay in which FLS1 (MICA*004/*009:01-*049) HFFs transduced with EV or with UL148A (either untagged or tagged at its C terminus) were mock infected or VarS infected. MICA surface expression was assayed at 24 hpi. In EV-transduced cells, MICA levels following VarS infection remained similar to the levels seen with mock-infected cells (Fig. 4A), since the virus lacked UL148A. However, FLS1 cells overexpressing UL148A (either untagged or tagged at its C terminus) demonstrated a significant reduction in MICA expression when infected with VarS (Fig. 4B and C). Cells were assessed for total MICA protein expression by Western blotting (Fig. 4D; quantified in Fig. 4E). UL148A expression was confirmed by blotting with a tag-specific antibody (Ab) (bottom panel). Vinculin served as the loading control (upper panel). No differences in total MICA quantity were observed in mock-infected cells overexpressing UL148A (Fig. 4D and E). The quantity of MICA in EV-expressing cells increased following VarS infection (Fig. 4D and E). However, the quantity of MICA in VarS-infected cells overexpressing UL148A was lower than the basal quantity in mock-infected cells (Fig. 4D and E). In BAC2-infected cells, a nonsignificant UL148A-dependent reduction in MICA quantity was observed, probably due to some augmentation of endogenous UL148A levels (Fig. 4D and E). Results similar to those seen with the VarS infection were obtained in BAC2 ΔUL148A infection (Fig. 4F). Taken together, these results indicate that UL148A requires another viral partner to downregulate MICA and that this viral partner is encoded externally to the ULb′ region.
FIG 4.
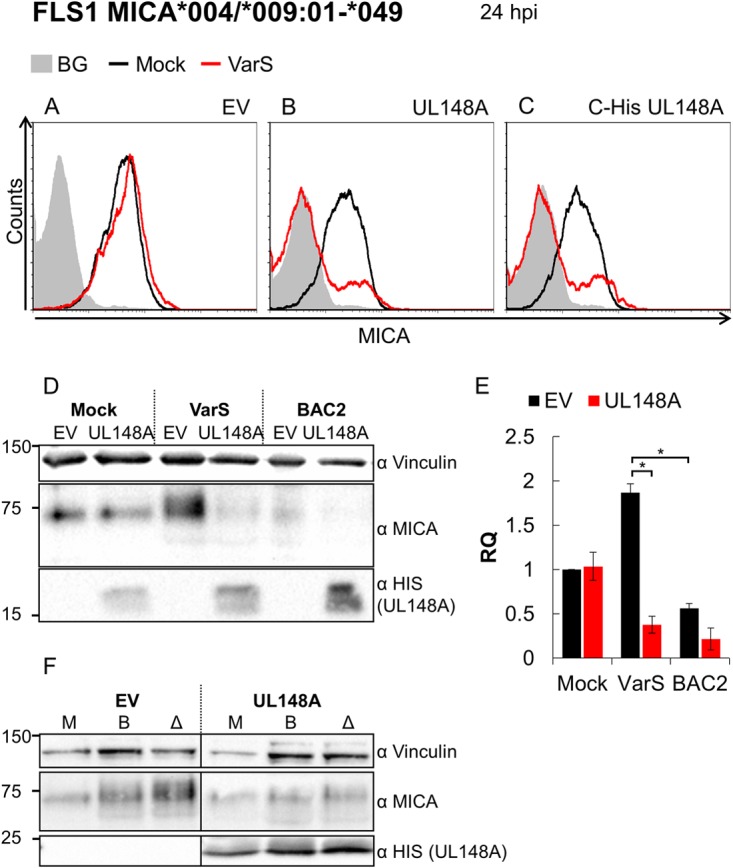
UL148A requires a viral interaction partner external to the ULb′ region. (A to C) FLS1 HFFs (MICA*004/*009:01-*049) transduced with an EV (A), with UL148A (B), or with C-His UL148A (C) were either mock infected (black histograms) or VarS infected (red histograms). Cells were harvested 24 hpi, and MICA surface expression was assessed by flow cytometry. Gray-filled histograms represent isotype control staining of mock-infected cells, which produced similar results for all cells. (D and E) FLS1 HFFs (MICA*004/*009:01-*049) transduced with an EV or with C-His UL148A were either mock infected or infected with VarS or BAC2. (D) Cells were lysed 24 hpi, and Western blotting was performed using anti-MICA antibody for detection of MICA, anti-HIS antibody for detection of UL148A, and anti-vinculin antibody as a loading control. (E) Quantification of two independent experiments. MICA protein levels were quantified relative to the loading control (vinculin). RQ, relative quantification. Error bars represent SEM. Student's t test was performed to evaluate significance. *, P < 0.05. (F) FLS1 HFFs (MICA*004/*009:01-*049) transduced with an EV or with C-His UL148A were either mock infected or infected with mutant BAC2 ΔUL148A or BAC2. (F) Cells were lysed 24 hpi, and Western blotting was performed using anti-MICA antibody for detection of MICA, anti-HIS antibody for detection of UL148A, and anti-vinculin antibody as a loading control.
UL148A-mediated MICA reduction diminishes NK cell killing of HCMV-infected cells.
To test whether the UL148A-mediated MICA reduction was functional and resulted in reduced NK cell-mediated killing, we performed NK cell killing assays. FLS3 cells, transduced with MICA*004-HA, were infected with BAC2 or the BAC2 ΔUL148A mutant. At 48 hpi, cells were labeled with [35S]methionine and incubated for 5 h with bulk primary NK cells (Fig. 5A). Elevated levels of killing were observed when NK cells were incubated with infected cells compared to the mock-infected cells (Fig. 5A). Interestingly, the killing of the infected cells was further elevated when they were infected with the BAC2 ΔUL148A deletion mutant. To demonstrate that athe increased killing of cells infected with a virus lacking UL148A was due to NKG2D recognition, we blocked NKG2D prior to the incubation with the target cells, using a specific monoclonal antibody (MAb). As can be seen in Fig. 5A, when NKG2D was blocked, all cells, infected or uninfected, were killed to similar extents, and killing levels were significantly lower than those seen with BAC2- and BAC2 ΔUL148A-infected cells without the blocking. This indicates that the UL148A-mediated decrease in MICA surface expression was functional and NKG2D dependent.
FIG 5.
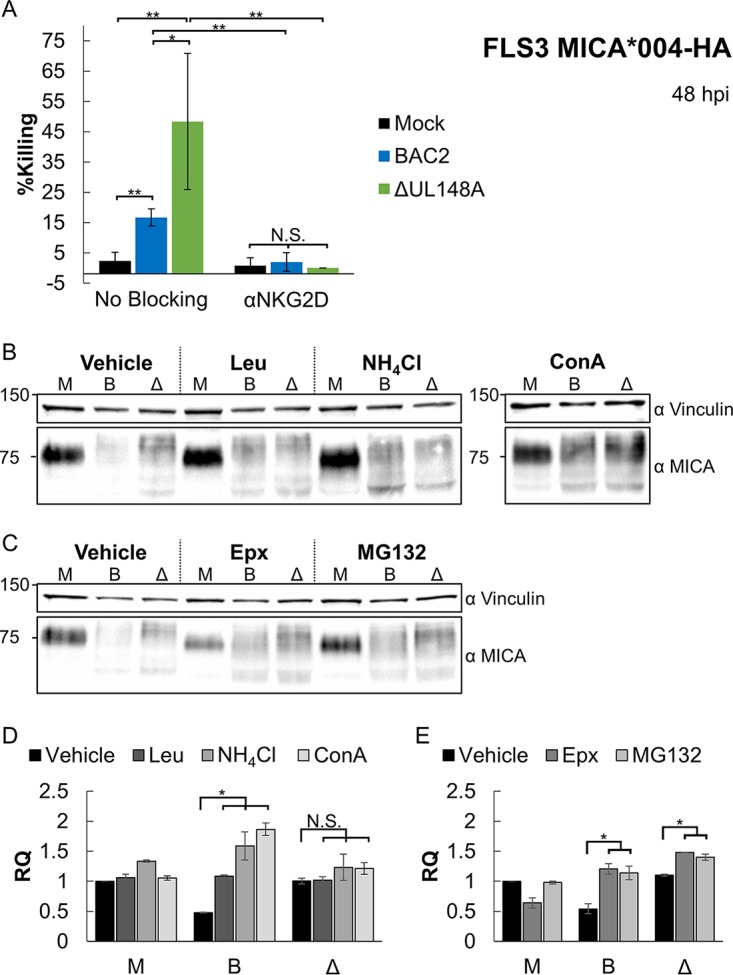
UL148A's downregulation of MICA is functional, and UL148A targets MICA to lysosomal degradation. (A) FLS3 MICA*004-HA cells were mock infected or infected with BAC2 or the BAC2 ΔUL148A mutant. NK cells were incubated 48 hpi with a blocking anti-NKG2D MAb or with no antibody and then incubated with the aforementioned HFFs. Data represent results from two independent experiments. Error bars represent SEM. Student's t test was performed to evaluate significance. *, P < 0.05; **, P < 0.005; N.S., nonsignificant. (B to E) FLS3 MICA*004-HA cells were mock infected (M) or infected with BAC2 (B) or with BAC2 ΔUL148A (Δ) and were incubated for 12 h with a vehicle-only treatment (Vehicle), lysosome inhibitors (in panel B, leupeptin [Leu], NH4Cl, or concanamycin A [ConA]), or proteasome inhibitors (in panel C, epoxomicin [Epx] or MG132). Cells were lysed 48 hpi, and Western blotting was performed using anti-MICA antibody for detection and anti-vinculin antibody as a loading control. (D and E) Quantification of two independent lysosome (D) or proteasome (E) inhibition experiments. MICA protein levels were quantified relative to the loading control (Vinculin). RQ, relative quantification. Error bars represent SEM. Student's t test was performed to evaluate significance. *, P < 0.05. The ConA panel is derived from a separate gel with a vehicle-only control.
UL148A utilizes the lysosomal pathway for MICA degradation.
Finally, we wanted to identify the mechanism by which UL148A downregulates MICA. For that, FLS3 MICA*004 cells were mock infected or infected with BAC2 or the BAC2 ΔUL148A mutant and incubated for 12 h with either vehicle only or the following lysosomal inhibitors: leupeptin (Leu), NH4Cl, and concanamycin A (ConA) (Fig. 5B). A parallel experiment was conducted either with vehicle only or with the following proteasomal inhibitors: epoxomicin (Epx) or MG132 (Fig. 5C). The cells were then harvested at 24 hpi. Lysates were prepared, Western blotting was performed (Fig. 5B and C), and total MICA protein levels were quantified relative to the vinculin loading control (see Fig. 5D and E for lysosomal and proteasomal inhibitors, respectively). In mock-infected cells, little difference was observed in total MICA quantities following treatment with lysosomal inhibitors (Fig. 5B and D). Infection with BAC2 caused a reduction in the quantity of MICA, but MICA levels were restored by treatment with lysosomal inhibitors, some of which even induced MICA accumulation beyond the level seen with the mock-infected cells (Fig. 5B and D). In contrast, in cells infected with mutant BAC2 ΔUL148A, there was no accumulation of MICA upon treatment with lysosomal inhibitors (Fig. 5B and D). Therefore, while MICA levels in the vehicle-only treatment were higher in BAC2 ΔUL148A-infected cells than in BAC2-infected cells, this difference disappeared and was even reversed in lysosomal inhibitor-treated cells. Treatment with proteasomal inhibitors caused an accumulation of MICA in BAC2-infected cells and in BAC2 ΔUL148A-infected cells and failed to abrogate the difference between the two (Fig. 5C and E), indicating that this treatment inhibits a factor present in both viruses. These results suggest that MICA is downregulated in the presence of UL148A via lysosomal degradation.
DISCUSSION
HCMV harbors an astonishing repertoire of immune evasion mechanisms. Cells infected with HCMV demonstrate a reduction in MHC I expression, thus avoiding recognition by cytotoxic T lymphocytes. But this exposes infected cells to attack by NK cells, since they are inhibited by MHC class I proteins (27). Accordingly, mechanisms for evasion of NK cells are necessary for HCMV to successfully propagate. Indeed, in recent years more and more HCMV genes were discovered that target NK cell ligands in general and, specifically, ligands of the NKG2D activating receptor (28). Together with UL148A (described here), we now know of five HCMV mechanisms that operate to target MICA, including UL142, which also targets ULBP3, US18, US20, and US9 (15, 16, 18). MICB is also known to be downregulated by five HCMV factors: microRNA (miRNA) HCMV-miR-UL112, US12, US13, US20, and UL16 (29–31). Besides MICB, US12, US13, and US20 target ULBP2, and UL16 additionally targets ULBP1, ULBP2, and ULBP6. This variety of mechanisms, all specialized for downregulation of NKG2D ligands, demonstrates their importance for the recognition of HCMV-infected cells by the immune system.
One explanation as to why MICA is targeted by so many different HCMV genes might be its extreme allelic diversity. US9 selectively targets the MICA*008 allele (formerly considered resistant [18]); on the other hand, UL148A, US18, US20, and UL142 all selectively target full-length MICA alleles. It will therefore be interesting to test whether all full-length MICA alleles are similarly susceptible to UL148A, US18, US20, and UL142. In this regard, it is interesting to note the allele-specific preference for the targeting mechanism: US9 selectively targets MICA*008 to proteasomal degradation, whereas US18 and US20 (and now also UL148A) target full-length MICA alleles for lysosomal degradation (15, 18). It is possible that this divergence is due to the different biological properties of full-length MICA alleles, which differ from MICA*008 in their maturation pathway, cellular localization, membrane anchoring, and turnover rates (18).
Another explanation for the plethora of MICA-targeting HCMV mechanisms might be time-dependent control of MICA expression. In the case of UL148A, an effect on MICA expression was seen at an early time point (24 hpi), while US18 and US20 seemed to act during later stages of infection (72 hpi). Furthermore, it is possible that different HCMV MICA-targeting mechanisms have different, nonredundant, and unknown targets other than MICA.
The ULb′ region of the HCMV genome is lost upon passaging of clinical HCMV strains in cell culture, and despite studies of this region, only some of the genes therein have known functions. The genes with those functions include immune evasion genes as well as genes encoding chemokines and tumor necrosis factor (TNF) receptor homologs; hence, this region is considered of importance for virulence and immune evasion functions (20, 25, 32–34). With the discovery of UL148A, two of four now-known mechanisms for full-length MICA downregulation have been shown to be present within this region, further stressing its importance.
Since UL148A did not have an effect on MICA expression when overexpressed but downregulated MICA in the complementation assays, it is likely that it requires an additional viral factor. This complex mechanism, together with the fact that the presence of UL148A alone is not sufficient for downregulation of MICA, might explain why UL148A was not discovered as a MICA regulator earlier. Since we successfully performed the complementation assay in UL148A-overexpressing cells infected with VarS, we can already rule out the possibility that the entire ULb′ region encodes the viral partner(s). This unknown factor might be needed for activation of UL148A, localization of UL148A in the infected cell, or perhaps even recognition of MICA.
Our results also imply the existence of one or more unknown, UL148A-independent mechanisms targeting full-length MICA alleles. For one, MICA migrated at different sizes in HCMV-infected cells compared to mock-infected controls, independently of UL148A deletion, pointing to a viral mechanism modifying it. In addition, the ability of proteasome inhibitors to increase MICA levels in HCMV-infected cells suggests that despite the apparent preference for lysosomal degradation of full-length alleles, there might be a viral mechanism promoting their proteasomal degradation.
Apart from its function in NK cell activation, NKG2D is expressed on NKT cells, γδ T cells, and a subset of αβ T cells. Therefore, UL148A, as well as other MICA-targeting mechanisms, might have a broader significance in HCMV immune evasion of diverse cell types. The fact that HCMV devotes so many mechanisms to countering the NKG2D receptor, and, more specifically, to its ligand MICA, highlights the great importance of the stress-induced ligand system in the immune response to HCMV infection.
MATERIALS AND METHODS
Cells.
Human fibroblasts were obtained from primary cultures of foreskins from healthy donors. FLS1 and FLS3 fibroblasts were the kind gifts of F. Levi Schaffer (Hebrew University of Jerusalem). HFFs were used below passage 21. The following cell lines were used: RKO (CRL-2577), HCT116 (CCL-247), and A549 (CCL-185). All cell lines were obtained from the ATCC. NK cells were isolated from peripheral blood lymphocyte (PBL) samples and activated as previously described (35). All cell lines and fibroblasts were kept in Dulbecco's modified Eagle's medium (DMEM). Media were supplemented with 10% (vol/vol) fetal calf serum (FCS; Sigma-Aldrich) and with 1% (vol/vol) each of penicillin-streptomycin (pen-strep), sodium pyruvate, l-glutamine, and nonessential amino acids (Biological Industries).
Antibodies.
The following antibodies were used for flow cytometry. For primary antibodies, anti-MICA (clone 159227; R&D Systems), anti-MICB (clone 236511; R&D Systems), anti-ULBP1 (clone 170818; R&D Systems), anti-ULBP2/5/6 (clone 165903; R&D Systems), and anti-ULBP3 (clone 166514, R&D Systems) were used. For the secondary antibody, anti-mouse Alexa Fluor 647 (Jackson Laboratories) was used. For the conjugated antibody, anti-cytomegalovirus (anti-CMV) immediate early Alexa Fluor 488 (clone 8B1.2; Sigma-Aldrich) was used. The following antibodies were used for Western blotting. For primary antibodies, anti-HIS tag (AD1.1.10; R&D Systems), anti-MICA (clone EPR6568; Abcam), and anti-vinculin (clone EPR8185; Abcam) were used. For secondary antibodies, anti-mouse horseradish peroxidase (HRP) and anti-rabbit HRP, both purchased from Jackson laboratories, were used. Anti-NKG2D antibody (clone 149810; R&D Systems) was used for blocking assays.
Viruses and infection.
AD169VarS and AD169VarL were isolated as previously described (25).
BAC2 and BAC20 were cloned as previously described (25).
The recombinant HCMV deletion mutants were generated according to previously published procedures (36, 37). Briefly, a PCR fragment was generated (data not shown) using plasmid pSLFRTKn (38) as the template DNA. The PCR fragments containing a kanamycin resistance gene were inserted into the parental AD169-BAC2 strain (25) by homologous recombination in Escherichia coli. The inserted cassette replaces the target sequence which was defined by flanking sequences in the primers. Recombinant HCMVs were reconstituted from HCMV BAC DNA by Superfect (Qiagen) transfection into permissive MRC-5 cells.
The fraction of HCMV-infected cells was assessed by flow cytometry. Cells were resuspended at 24 hpi and then incubated on ice with the anti-CMV antibody (0.25 μg/well) for 30 min. Samples were used only if >75% infected and if the variance between samples was <15%.
Flow cytometry.
For flow cytometry, equivalent numbers of cells were plated and incubated overnight. Cells were resuspended and then incubated on ice with the primary antibody (0.2 μg/well) for 1 h, followed by a 30-min incubation on ice with the secondary antibody (0.75 μg/well). Histograms of cells transduced with a green fluorescent protein (GFP)-expressing lentivirus were gated on the GFP-positive (GFP+) population.
Lentivirus production.
Lentiviral vectors were produced in 293T cells using TransIT-LT1 transfection reagent (Mirus) for a transient three-plasmid transfection protocol as previously described (39). Cells were transduced in the presence of Polybrene (6 μg/ml). Transduction efficiency was evaluated by GFP levels, and only cell populations with >90% GFP+ cells were used. If necessary, cell sorting was performed to achieve the required efficiency.
Vectors and primers.
MICA*004 and MICA*008 were amplified from cDNA and inserted into lentiviral vector SIN18pRLL-hEFIap-E-GFP-WRPE, replacing the GFP with each of the sequences, as previously described (18). UL148A was amplified from cDNA of HCMV-infected cells (AD169VarL) and inserted into lentiviral vector pHAGE-DsRED(-)eGFP(+).
The following primers were used for the cloning of UL148A: UL148A forward (FW) (AAGCGGCCGCGCCGCCACCATGGGTTCCGAAAATCTCG), UL148A FW-Flag tag (AAGCGGCCGCGCCGCCACCATGGACTACAAAGACGATGACGACAAGGGTTCCGAAAATCTCGATC), UL148A reverse (REV) (AACTCGAGCTAGTAACACTCGTCCGACACTTC), UL148A REV-HIS tag (AACTCGAGCTAATGATGATGATGATGATGGTAACACTCGTCCGACACTTC), and UL148A REV-Flag tag (AACTCGAGCTACTTGTCGTCATCGTCTTTGTAGTCGTAACACTCGTCCGACACTTC).
NK cell killing assay.
For evaluation of NK cell cytotoxic activity against target cells, 35S release assays were performed as described previously (35), and the reaction mixtures were incubated for 5 h. Percentages of killing were calculated as follows: [counts per minute (cpm) (sample) − cpm (spontaneous release)]/[cpm (total release) − cpm (spontaneous release)] × 100.
Western blot analysis.
Equivalent numbers of cells were plated and incubated overnight. Cells were lysed in lysis buffer containing 0.6% sodium dodecyl sulfate (SDS), 10 mM Tris (pH 7.4), and the protease inhibitors aprotinin and phenylmethylsulfonyl fluoride (PMSF) (each at a 1:100 dilution). Lysates were then subjected to SDS polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The membrane was blocked in 5% skim milk–phosphate-buffered saline (PBS)–Tween 20 for 1 h and then incubated with a primary antibody overnight, washed 3 times in PBS-Tween 20, incubated with a secondary antibody for 0.5 h, and washed 3 times in PBS-Tween 20. Images were developed using an EZ-ECL kit (Biological Industries). Image Lab software was used for quantification.
Lysosome and proteasome inhibition.
For organelle inhibition, cells were incubated for 12 h with 100 μM leupeptin (Leu; Merck Millipore), 40 μM NH4Cl (Sigma-Aldrich), 0.5 μM concanamycin A (ConA; Sigma-Aldrich), 2.5 μM epoxomicin (Epx; A2S), or 5 μM MG132 (Calbiochem) or with a vehicle-only treatment. Cells treated only with vehicle were treated with the matching solvent (either DDW or DMSO) in the equivalent volume of the inhibitor applied.
Statistical methods.
For determinations of statistical significance, Student's t test was used. Error bars show standard errors of the means (SEM) from biological replicates.
ACKNOWLEDGMENTS
This work was supported by an Israel Cancer Research Fund professorship grant, by a DKFZ-MOST grant, by the Israel Science Foundation, and by the Ministry of Science and Technology.
The authors declare no conflict of interest.
REFERENCES
- 1.Davison AJ, Bhella D. 2007. Comparative genome and virion structure, p 177–203. In Human herpesviruses: biology, therapy, and immunoprophylaxis, Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 2.Griffiths PD. 2012. Burden of disease associated with human cytomegalovirus and prospects for elimination by universal immunisation. Lancet Infect Dis 12:790–798. doi: 10.1016/S1473-3099(12)70197-4. [DOI] [PubMed] [Google Scholar]
- 3.Sansoni P, Vescovini R, Fagnoni FF, Akbar A, Arens R, Chiu YL, Cičin-Šain L, Dechanet-Merville J, Derhovanessian E, Ferrando-Martinez S, Franceschi C, Frasca D, Fulöp T, Furman D, Gkrania-Klotsas E, Goodrum F, Grubeck-Loebenstein B, Hurme M, Kern F, Lilleri D, López-Botet M, Maier AB, Marandu T, Marchant A, Matheï C, Moss P, Muntasell A, Remmerswaal EBM, Riddell NE, Rothe K, Sauce D, Shin E-C, Simanek AM, Smithey MJ, Söderberg-Nauclér C, Solana R, Thomas PG, van Lier R, Pawelec G, Nikolich-Zugich J. 2014. New advances in CMV and immunosenescence. Exp Gerontol 55:54–62. doi: 10.1016/j.exger.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Lodoen MB, Lanier LL, Kaufmann SH, Walker BD. 2006. Natural killer cells as an initial defense against pathogens. Curr Opin Immunol 18:391–398. doi: 10.1016/j.coi.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitenshtein A, Charpak-Amikam Y, Yamin R, Bauman Y, Isaacson B, Stein N, Berhani O, Dassa L, Gamliel M, Gur C, Glasner A, Gomez C, Ben-Ami R, Osherov N, Cormack BP, Mandelboim O. 2016. NK cell recognition of Candida glabrata through binding of NKp46 and NCR1 to fungal ligands Epa1, Epa6, and Epa7 Cell Host Microbe 20:527–534. doi: 10.1016/j.chom.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orange JS. 2013. Natural killer cell deficiency. J Allergy Clin Immunol 132:515–525, quiz 526. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diefenbach A, Raulet DH. 2001. Strategies for target cell recognition by natural killer cells. Immunol Rev 181:170–184. doi: 10.1034/j.1600-065X.2001.1810114.x. [DOI] [PubMed] [Google Scholar]
- 8.Lanier LL. 1998. NK cell receptors. Annu Rev Immunol 16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 9.Shifrin N, Raulet DH, Ardolino M. 2014. NK cell self tolerance, responsiveness and missing self recognition. Semin Immunol 26:138–144. doi: 10.1016/j.smim.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geiger TL, Sun JC. 2016. Development and maturation of natural killer cells. Curr Opin Immunol 39:82–89. doi: 10.1016/j.coi.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanier LL. 2015. NKG2D receptor and its ligands in host defense. Cancer Immunol Res 3:575–582. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Bert N, Gasser S. 2014. Advances in NKG2D ligand recognition and responses by NK cells. Immunol Cell Biol 92:230–236. doi: 10.1038/icb.2013.111. [DOI] [PubMed] [Google Scholar]
- 13.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. 2013. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol 31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernández-Messina L, Reyburn HT, Valés-Gómez M. 2012. Human NKG2D-ligands: cell biology strategies to ensure immune recognition. Front Immunol 3:299. doi: 10.3389/fimmu.2012.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fielding CA, Aicheler R, Stanton RJ, Wang ECY, Han S, Seirafian S, Davies J, McSharry BP, Weekes MP, Antrobus PR, Prod'homme V, Blanchet FP, Sugrue D, Cuff S, Roberts D, Davison AJ, Lehner PJ, Wilkinson GWG, Tomasec P. 2014. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog 10:e1004058. doi: 10.1371/journal.ppat.1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chalupny NJ, Rein-Weston A, Dosch S, Cosman D. 2006. Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem Biophys Res Commun 346:175–181. doi: 10.1016/j.bbrc.2006.05.092. [DOI] [PubMed] [Google Scholar]
- 17.Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR. 2009. NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 83:12345–12354. doi: 10.1128/JVI.01175-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seidel E, Le VTK, Bar-On Y, Tsukerman P, Enk J, Yamin R, Stein N, Schmiedel D, Oiknine Djian E, Weisblum Y, Tirosh B, Stastny P, Wolf DG, Hengel H, Mandelboim O. 12 February 2015. Dynamic co-evolution of host and pathogen: HCMV downregulates the prevalent allele MICA*008 to escape elimination by NK cells. Cell Rep doi: 10.1016/j.celrep.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dargan DJ, Douglas E, Cunningham C, Jamieson F, Stanton RJ, Baluchova K, McSharry BP, Tomasec P, Emery VC, Percivalle E, Sarasini A, Gerna G, Wilkinson GWG, Davison AJ. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–1546. doi: 10.1099/vir.0.018994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, Mcgeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, Mcsharry BP, Wilkinson GWG, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J Gen Virol 85:1301–1312. doi: 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 22.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomasec P, Wang ECY, Davison AJ, Vojtesek B, Armstrong M, Griffin C, McSharry BP, Morris RJ, Llewellyn-Lacey S, Rickards C, Nomoto A, Sinzger C, Wilkinson GWG. 2005. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol 6:181–188. doi: 10.1038/ni1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanton RJ, Prod′homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, Bailer SM, Haas J, Antrobus R, Weekes MP, Lehner PJ, Vojtesek B, Miners KL, Man S, Wilkie GS, Davison AJ, Wang ECY, Tomasec P, Wilkinson GWG. 2014. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 16:201–214. doi: 10.1016/j.chom.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le VTK, Trilling M, Hengel H. 2011. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb′-encoded modulation of TNF-α signaling. J Virol 85:13260–13270. doi: 10.1128/JVI.06005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett NJ, Ashiru O, Morgan FJE, Pang Y, Okecha G, Eagle RA, Trowsdale J, Sissons JGP, Wills MR. 2010. Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J Immunol 185:1093–1102. doi: 10.4049/jimmunol.1000789. [DOI] [PubMed] [Google Scholar]
- 27.Halenius A, Gerke C, Hengel H. 2015. Classical and non -classical MHC I molecule manipulation by human cytomegalovirus: so many targets—but how many arrows in the quiver? Cell Mol Immunol 12:139–153. doi: 10.1038/cmi.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodier MR, Jonjić S, Riley EM, Juranić Lisnić V. 2018. CMV and natural killer cells: shaping the response to vaccination. Eur J Immunol 48:50–65. doi: 10.1002/eji.201646762. [DOI] [PubMed] [Google Scholar]
- 29.Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, Cosman D. 2003. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med 197:1427–1439. doi: 10.1084/jem.20022059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nachmani D, Lankry D, Wolf DG, Mandelboim O. 2010. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol 11:806–813. doi: 10.1038/ni.1916. [DOI] [PubMed] [Google Scholar]
- 31.Fielding CA, Weekes MP, Nobre LV, Ruckova E, Wilkie GS, Paulo JA, Chang C, Suárez NM, Davies JA, Antrobus R, Stanton RJ, Aicheler RJ, Nichols H, Vojtesek B, Trowsdale J, Davison AJ, Gygi SP, Tomasec P, Lehner PJ, Wilkinson GWG. 2017. Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. Elife 6:e22206. doi: 10.7554/eLife.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penfold ME, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ. 1999. Cytomegalovirus encodes a potent alpha chemokine. Proc Natl Acad Sci U S A 96:9839–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lüttichau HR. 2010. The cytomegalovirus UL146 gene product vCXCL1 targets both CXCR1 and CXCR2 as an agonist. J Biol Chem 285:9137–9146. doi: 10.1074/jbc.M109.002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montag C, Wagner JA, Gruska I, Vetter B, Wiebusch L, Hagemeier C. 2011. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor alpha (TNF-alpha) signaling by upregulating TNF-alpha receptor 1 cell surface expression. J Virol 85:11409–11421. doi: 10.1128/JVI.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandelboim O, Reyburn HT, Valés-Gómez M, Pazmany L, Colonna M, Borsellino G, Strominger JL. 1996. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J Exp Med 184:913–922. doi: 10.1084/jem.184.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med 196:805–816. doi: 10.1084/jem.20020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 38.Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, Hengel H. 2002. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J Virol 76:8596–8608. doi: 10.1128/JVI.76.17.8596-8608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, Goldman-Wohl D, Greenfield C, Yagel S, Hengel H, Altuvia Y, Margalit H, Mandelboim O. 2007. Host immune system gene targeting by a viral miRNA. Science 317:376–381. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]